

Abstract
Human immunodeficiency virus type 1 (HIV-1) infection remains a worldwide epidemic, and innovative therapies to combat the virus are needed. Developing a host-oriented antiviral strategy capable of targeting the biomolecules that are directly or indirectly required for viral replication may provide advantages over traditional virus-centric approaches. We used quantitative proteomics by SWATH-MS in conjunction with bioinformatic analyses to identify host proteins, with an emphasis on nucleic acid binding and regulatory proteins, which could serve as candidates in the development of host-oriented antiretroviral strategies. Using SWATH-MS, we identified and quantified the expression of 3608 proteins in uninfected and HIV-1-infected monocyte-derived macrophages. Of these 3608 proteins, 420 were significantly altered upon HIV-1 infection. Bioinformatic analyses revealed functional enrichment for RNA binding and processing as well as transcription regulation. Our findings highlight a novel subset of proteins and processes that are involved in the host response to HIV-1 infection. In addition, we provide an original and transparent methodology for the analysis of label-free quantitative proteomics data generated by SWATH-MS that can be readily adapted to other biological systems.
Keywords: DHX15, HIV, HDAC, macrophage, MDM, NCOR, spliceosome, SWATH, YBOX1, z-test
Introduction
As obligate intracellular parasites, viruses are dependent on hijacking host cell machinery and processes for viral propagation and dissemination, all while avoiding detection and clearance by host immune cells. Human immunodeficiency virus type 1 (HIV-1), the causative agent of acquired immune deficiency syndrome (AIDS), encodes only nine genes and infection results in the rapid takeover of host cell machinery, while not only evading, but destroying, the host immune system. HIV-1 infects CD4+ cells, including helper T-cells and mononuclear phagocytes (monocytes, macrophages, and dendritic cells).1−4 Although HIV-1 is associated with widespread destruction of T-cells, monocytes and macrophages continue to persist after infection and these cells have an integral role in viral production, dissemination, and latency.5−8 Thus, it stands to reason that characterizing the molecular host response of macrophages to HIV-1 infection may provide novel avenues for therapeutic intervention against the virus. Global analyses of the transcriptome9−11 and the proteome12−14 of HIV-1-infected macrophages have revealed substantial alterations in biomolecule expression, suggesting a role for altered expression and/or function of nucleic acid binding and regulatory proteins, which include transcription and translation regulatory proteins.
The underlying aim of our investigation was to identify altered host factors involved in HIV-1 infection of the macrophage that could serve as novel candidates in the development of host-oriented antiretroviral strategies, similar to how the discovery of the homozygous CCR5Δ32 allele as being protective against HIV-1 infection led to the development of Maraviroc15 a CCR5 receptor antagonist that prevents HIV-1 entry. We hypothesized that chronic infection of macrophages with HIV-1 results in the reprogramming of nucleic acid binding and regulatory protein expression. We tested this hypothesis using a SWATH-MS16 (Sequential Windowed data independent Acquisition of the Total High-resolution Mass Spectra) quantitative proteomics approach in conjunction with bioinformatic analyses to reveal unique insights for how the virus alters biological processes. Alterations were observed in groups of proteins affecting key mechanisms, such as RNA processing and transcription regulation. The proteins identified and quantified in this report represent an original set of host factors that warrant continued investigation in the development of host-oriented antiviral strategies.
Materials and Methods
Safety Considerations
All cell culture and viral infection procedures were performed in a biosafety level 2+ (BSL-2+) laboratory operated under BSL-3 precautions and procedures. When necessary, virus was disinfected using 1:10 bleach in water solution. Virus was inactivated prior to removal of samples from the BSL-2+ laboratory using 4% sodium dodecyl sulfate (SDS).
Cell Culture and HIV-1 Infection
Primary monocytes were obtained by leukophoresis from donors that were HIV-1, HIV-2 and hepatitis seronegative and monocytes were purified (∼97% pure) by countercurrent centrifugal elutriation.17 All cells were collected from individuals that had provided written informed consent on research protocols approved by the UNMC institutional review board. Primary monocytes were plated using a density of 1 million cells/mL in monocyte differentiation media, which consisted of macrophage serum-free media (Life Technologies; Grand Island, NY) supplemented with 10 ng/mL colony stimulating factor-1 (CSF-1, PreproTech; Rocky Hill, NJ), 1× Nutridoma-SP (Roche Biotechnology; Basel, Switzerland), 1× antibiotic-antimycotic (Life Technologies) and 0.01 M HEPES buffer (Life Technologies). On day 4 postplating, one-half of the media was exchanged with fresh, prewarmed monocyte differentiation media.
On day 7 postplating, media was reduced to one-half and cells were inoculated with HIV-1ADA using a multiplicity of infection (MOI) of 1 viral particle per cell. Uninfected cells were mock infected using PBS in macrophage maintenance media (same formulation as monocyte differentiation media, but without the addition of CSF-1). After 4 h of primary infection, the media was removed, cells were washed using warmed phosphate-buffered saline (PBS), and fresh macrophage maintenance media was applied. Media was exchanged every 48 h.
Sample Preparation: The Monocyte-Derived Macrophage (MDM) Reference Spectral Library
Samples used to generate the MDM reference spectral library were subjected to subcellular enrichment using the Qproteome Nuclear Protein kit for soluble nuclear proteins or the Qproteome Cell Compartment kit for total nuclear proteins (Qiagen; Valencia, CA). For each nuclear enrichment procedure, samples were pooled equivalently using 25 μg of protein from each donor (n = 4) for each treatment condition. Each pooled sample was processed using filter-assisted sample preparation (FASP).18 Peptides were fractionated by isoelectric point by OFFGEL electrophoresis using pH 3–10 OFFGEL strips (Agilent, Santa Clara, CA).12 Fractionated peptides were cleaned and prepared for mass spectrometry using C18 spin-columns following manufacturer protocols (Thermo Fisher; Rockford, IL).
Sample Preparation: Data-Independent Acquisition (DIA) Mass Spectrometry of Experimental Samples
Samples used for DIA mass spectrometry were collected exactly 120 h (5 days) postinfection. Cells were washed with ice-cold PBS, scraped, pelleted, and stored at −80 °C until processed. All samples were processed in unison to minimize technical variability. Each cell pellet was thawed on ice and resuspended in cell lysis buffer containing 4% (w/v) SDS, 0.1 M dithiothreitol (DTT), 0.1 M Tris-HCl, and 100 units/mL Benzonase Nuclease (Merck KGaA; Darmstadt, Germany), pH 7.6. Lysates were vortexed at room temperature using maximum speed for 5 min and were then boiled at 95 °C for 5 min. Protein quantification was performed using the Pierce 660 nm Protein Assay supplemented with 50 mM ionic detergent compatible reagent (both from Thermo Fisher) following manufacturer’s protocols. On the basis of protein quantifications, each experimental sample was aliquoted into 25 μg samples for processing using the FASP method.18 Peptides were cleaned using Oasis mixed cation exchange cartridge following manufacturer’s protocols (Waters; Milford, MA). Peptides were quantified by absorbance at 205 nm19 on the NanoDrop2000 (Thermo Scientific; Wilmington, DE), and 2 μg of protein was taken for a final cleaning step using C18 Zip-Tips (Millipore; Billerica, MA).
Mass Spectrometry
All samples were analyzed by reverse-phase high-pressure liquid chromatography electrospray ionization tandem mass spectrometry (RP-HPLC-ESI-MS/MS) using a commercial 5600 Triple-TOF (AB Sciex; Concord, Canada) mass spectrometer operating in high-sensitivity mode. The mass spectrometer was coupled with an Eksigent NanoLC-Ultra 1D plus (Eksigent; Dublin, CA) and nanoFlex cHiPLC system (Eksigent). RP-HPLC was performed via a trap and elute configuration using Nano cHiPLC columns (Eksigent); the trap column (200 μm × 0.5 mm) and the analytical column (75 μm × 15 cm) were both manufacturer (Eksigent)-packed with 3 μm ChromSP C-18 media (300 Å). The nanospray needle voltage was set to 2400 V in HPLC-MS mode. Reverse-phase LC solvents included solvent A (0.1% (v/v) formic acid in HPLC water) and solvent B (95% (v/v) acetonitrile with 0.1% (v/v) formic acid).
All samples were loaded using a stepwise flow rate of 10 μL/min for 8.5 min and 2 μL/min for 1 min using 100% solvent A. Samples were eluted from the analytical column at a flow rate of 0.3 μL/min using a linear gradient of 5% solvent B to 35% solvent B over duration of 180 min. The column was regenerated by washing with 90% solvent B for 15 min and re-equilibrated with 5% solvent B for 15 min. Autocalibration of spectra occurred after acquisition of every 4 samples using dynamic LC–MS and MS/MS acquisitions of 25 fmol β-galactosidase.
Samples used to generate the SWATH-MS spectral library were subjected to traditional, data-dependent acquisition (DDA). For these experiments, the mass spectrometer was operated such that a 250-ms survey scan (TOF-MS) was performed and the top 50 ions were selected for subsequent MS/MS experiments using an accumulation time of 50 ms per MS/MS experiment for a total cycle time of 2.75 s. The selection criteria for parent ions included an intensity of greater than 100 counts/s, charge state from +2 to +5, mass tolerance of 50 mDa and were not present on a dynamic exclusion list. Once an ion had been fragmented by MS/MS, its mass and isotopes were excluded from further MS/MS fragmentation for 15 s. Ions were fragmented in the collision cell using rolling collision energy.
Experimental samples were subjected to cyclic DIA of mass spectra using 25-Da swaths in a similar manner to previously established methods.16,20 For these experiments, the mass spectrometer was operated such that a 50-ms survey scan (TOF-MS) was performed and subsequent MS/MS experiments were performed on all precursors. These MS/MS experiments were performed in a cyclic manner using an accumulation time of 96 ms per 25-Da swath (34 swaths total) for a total cycle time of 3.314 s. Ions were fragmented for each MS/MS experiment in the collision cell using rolling collision energy.
Generating the MDM Reference Spectral Library
All DDA mass spectrometry files were searched in unison using ProteinPilot software v. 4.2 (AB Sciex) with the Paragon algorithm. Samples were input as unlabeled samples with the following parameters: methyl methanethiosulfonate (MMTS) cysteine alkylation, digestion by trypsin and no special factors. The search was conducted using a through identification effort of a UniProt Swiss-Prot database (November 2012 release) containing human and HIV-1 proteins, as well as common laboratory contaminants. False discovery rate analysis was also performed. The output of this search is a .group file and was used as the MDM reference spectral library. The .group file contains the following information that is required for targeted data extraction: protein name and UniProt accession, stripped peptide sequence, modified peptide sequence, Q1 and Q3 ion detection, retention time, relative intensity, precursor charge, fragment type, score, confidence, and decoy result.
Targeted Data Extraction
Spectral alignment and targeted data extraction of DIA samples was performed using PeakView v.1.2 (AB Sciex) using the MDM reference spectral library generated above. All DIA files were loaded and exported in .txt format in unison using an extraction window of 20 min and the following parameters: 8 peptides, 5 transitions, peptide confidence of >99%, exclude shared peptides, and XIC width set at 50 ppm. This export results in the generation of three distinct files containing the quantitative output for (1) the area under the intensity curve for individual ions, (2) the summed intensity of individual ions for a given peptide, and (3) the summed intensity of peptides for a given protein. The protein data was used for all data analysis and is provided in Supplementary data set 2. Laboratory contaminants and reversed sequences were removed from the data set prior to statistical analysis.
Statistical Analysis Using z-Transformation
Each SWATH-MS experiment (defined here as one condition for one donor) was transformed independently of other experiments, as described in Cheadle et al.21 The raw intensity for each protein was transformed by taking the natural log (ln) of the intensity followed by assignment of z-score (eq 1), where x is the experimental value, μ is the mean of all experimental values and σ is the standard deviation of all experimental values. Next, the Δz was calculated for each protein in a pairwise manner for each donor (Δz = zHIV – zControl) and the average Δz across all donors was calculated. The z-test was then conducted for each protein using the paired sample z-test (eq 2), where Δzavg is the average Δz across all donors, D is the hypothesized mean (null hypothesis) of pairwise differences, σd is the standard deviation of the pairwise differences per protein, and √n is the square root of the sample size (number of biological donors). The p-value for the computed z-test statistic was assigned using the standard normal distribution. All statistics were performed in IBM SPSS Statistics v. 21 (IBM; Armonk, NY).
| 1 |
| 2 |
Bioinformatic Analyses
Functional analysis, pathway overrepresentation, and centrality analysis were performed using an array of complementary, open-access bioinformatic tools.
The DAVID bioinformatic resource 6.7 (http://david.abcc.ncifcrf.gov/)22,23 was used for functional analysis of proteins and analysis was performed using keywords from the protein information resource (SP_PIR_KEYWORDS), UniProt sequence feature (UP_SEQ_FEATURE), and gene ontology (GO) terms (GOTERM_BP_FAT, GOTERM_BP_ALL, GOTERM_MF_FAT and GOTERM_MF_ALL). Functional analysis of protein using PANTHER v. 8.0 (www.pantherdb.org)24−26 was performed using the protein class tool, which is based on PANTHER index terms and is complementary, but not identical, to GO terms and PIR keywords.
Protein–protein interactions among all identified transcription regulators were investigated using STRING 9.05 (http://string-db.com)27,28 using only experimental evidence with a confidence of greater than 0.4 (medium confidence). Orphan proteins (unconnected proteins) and satellite networks (networks detached from the largest network) were removed and the network information was exported for visualization and analysis in Cytoscape v. 3.0.2 (www.cytoscape.org).29 Centrality analysis of the protein–protein interaction network was performed in Cytoscape using the CentiScaPe 2.0 plug-in.30 Centrality analysis was conducted by computing node eccentricity, radiality and closeness within the network. Computed values were then assigned rank and the summed rank was used for the total centrality measure.
Pathway overrepresentation analysis was performed using the Reactome v. 46 curated pathway database analyze data tool (www.reactome.org).31−33 Complementary pathway analysis in DAVID was performed using biological biochemical image database (BBID), biocarta (BIOCARTA), and Kyoto Encyclopedia of Genes and Genomes (KEGG_PATHWAY). The KEGG pathway34,35 (v. 68.0) for the spliceosome was colored using the KEGG mapper color pathway tool (www.genome.jp/kegg/). The Quick GO browser (accessed December 4, 2013)36 was used to download all protein annotations for the spliceosome (GO, 0005681; “spliceosomal complex”) specific to the human (tax, 9606) and restricted to only those proteins with direct evidence (evidence, Inferred from Direct Assay (IDA)).
Generation of the Transcription Regulator Reference Database
Previously reported peer-reviewed data sets containing transcription factor genes,37 transcription factor gene loci,38 and known and putative transcription regulators25 were merged and converted to UniProt (Swiss-Prot) accession numbers using the UniProt ID mapping tool (www.uniprot.org/mapping/). The UniProt accession output for each mapping exercise was merged, duplicate values were removed, reviewed entries (Swiss-Prot) were retained, and protein information was retrieved using the UniProt retrieve tool (www.uniprot.org/retrieve/). Selection of transcription regulators identified by SWATH-MS was achieved by alignment of UniProt accession numbers from the SWATH-MS data to the transcription regulator reference database.
p24 Immunocytochemistry
Immunocytochemistry was performed for experimental (DIA mass spectrometry samples) samples immediately following harvest using the EnVision+ system-HRP (DAB) immunohistochemistry kit (Dako; Carpinteria, CA) following manufacturer’s protocols. Briefly, cells were fixed using 4% (w/v) paraformaldehyde in PBS for 15 min at room temperature and then permeabilized using 1% (v/v) Triton X-100 in PBS for 30 min at 37 °C followed by 10 min at room temperature. Samples were blocked using 1× Tris-buffered saline supplemented with 0.05% (v/v) Tween-20 (TBST) for 60 min at room temperature. Next, a peroxidase block was performed for 15 min. Primary antibody to HIV p24 (clone 38/7.47, Abcam; Cambridge, MA) was applied at a concentration of 0.5 μg/mL in TBST for 1 h at room temperature. Peroxidase-labeled and polymerized secondary antibody was then applied for 15 min. Staining for p24 was visualized by applying DAB+ substrate-chromogen solution for 5 min. Nuclei were visualized by applying Mayer’s Hematoxylin-Lillie’s modification (Dako) for 60 s followed by 37 mM ammonia for 5 min. Images were captured using the Nikon Eclipse TS100 inverted microscope using a 20× objective (200× total magnification). Brightness, contrast and levels were uniformly corrected across the entire image using Adobe Photoshop CS4 and were performed to aid in calculations of percent infected cells, which was calculated as the percent of HIV-1 p24 positive cells ± the standard deviation across biological replicates.
Results And Discussion
Investigations of the host response of macrophages to HIV-1 infection (and other viruses) have primarily utilized macrophage-like immortalized cell lines, such as U93739 and THP-1,40 rather than primary cells. Although these cell lines have the benefit of rapid expansion and lower variability compared to primary MDM, substantial proteomic differences have been documented in cell lines as compared to their primary cell counterparts.41 To avoid the limitations brought about by the use of cell lines, we used primary monocytes differentiated to macrophages in our examination of the altered cellular changes induced by HIV-1 infection. To avoid bias from individual donors, we performed seven experiments with independent donors to achieve generalizable results. As assessed by immunocytochemistry detection of the HIV-1 p24 capsid protein, we achieved a robust infection of MDM (99.0% ± 0.58%; percent infected cells ± standard deviation across biological replicates) at 5 days postinfection and thereby decreased the potential for proteomic-masking effects brought about by a mixed culture of infected and uninfected cells. In addition, we observed the formation of multinucleated giant cells, which serves as a hallmark for productive HIV-1 infection of macrophages in vitro and in vivo and marks a stage of infection in which cell death is not prominent.17
We characterized the proteomic alterations in the host response of HIV-1-infected MDM using a biphasic SWATH-MS approach (Supplementary Scheme 1). An experimentally derived, nonquantitative MDM reference spectral library was generated using traditional DDA mass spectrometry (Supplementary data set 1). Nuclear protein-enriched samples obtained by subcellular fractionation (Figure 1A) were used to generate a library that contained 3030 distinct protein groups (3743 proteins before grouping) identified with greater than 99% confidence (Figure 1B) and passed global false discovery rate (FDR) from fit analysis using a critical FDR of 1% (Figure 1C). Bioinformatic analysis of the 3030 protein groups using PANTHER revealed enrichment for nucleic acid binding proteins and for transcription factors (Figure 1D). Taken together, these findings indicated that the experimentally generated MDM reference library contained only high confidence proteins and verified our expectations of robust coverage of nucleic acid binding proteins.
Figure 1.

Generation of an MDM reference spectral library. (A) The experimental approach for generation of an MDM reference spectral library used two distinct set of samples: total nuclear proteins and soluble nuclear proteins. (B) ProteinPilot analyzed nearly 1 million spectra to produce an MDM reference spectral library containing 3030 protein groups (3743 proteins total) with greater than 99% confidence and all these proteins passed global FDR from fit analysis (C). The 3030 protein groups were analyzed by PANTHER (D) for protein class and revealed enrichment for nucleic acid binding proteins and for transcription factors.
Following the development of an MDM reference spectral library, the identification and quantification of proteins from experimental samples was performed using cyclic DIA mass spectrometry (Supplementary Scheme 1). The experimental samples used for DIA were MDM cultured in vitro and either infected with HIV-1ADA or mock-infected with PBS. The MDM were obtained from seven independent biological donors to maximize the power of our proteomic testing while balancing experimental feasibility.42 To reduce experimental variability, we used minimally processed whole-cell lysates, in which proteins were digested by trypsin and peptides separated one-dimensionally for 180 min on a reverse-phase LC gradient. DIA data for each experimental sample was submitted in unison to the PeakView software for targeted data extraction, which resulted in the quantitative export of 3608 unique proteins (proteins without shared peptides, Supplementary data set 2).
The raw intensity data (peak intensity, area under the peak, etc.) generated by mass spectrometry is inherently skewed and, as such, requires normalization prior to parametric statistical testing.42 To achieve this normal distribution, proteomic data for each SWATH-MS experiment was natural log (ln) transformed and then normalized by z-score, as described in Cheadle et al.21 This transformation minimized distortions introduced from sample preparation and data acquisition (Figure 2A).21 A paired sample z-test, which is conceptually equivalent to the paired sample t test, was used to identify differences in protein expression between conditions while accounting for variation between biological replicates on a protein-by-protein basis. This analysis identified 420 proteins as significantly altered (p < 0.05) in MDM during HIV-1 infection (Figure 2B, Supplementary data set 2).
Figure 2.

Mass-spectrometry based proteomics by SWATH-MS. (A) Data transformation. Average values (top, raw intensity; middle, natural log transformed; bottom, Z-score) for each protein were plotted as uninfected vs infected and the coefficient of determination (R2) was calculated. Each transformation improved the goodness of fit as assessed by R2 value. (B) Statistical analysis. A heat map of Δz (zHIV-1 – zcontrol) values for the 420 significantly altered proteins revealed extensive biological variability for individual proteins. Numbers above each lane represent each donor, and n-bar is the average of all biological donors (average Δz). Orange represents an up-regulation (+1) of a protein for an individual donor, whereas blue represents a down-regulation (−1). Likewise, red represents an up-regulation (+1) of a protein based on average Δz and green represents a down-regulation (−1). Proteins are sorted by descending values of average Δz. (C) Comparison-based validation. Protein expression values generated using SWATH-MS (lane 1, average Δz values) were matched to protein expression values provided by Kraft-Terry et al.12 (lane 2); the HIV-1, Human Interaction database (lane 3); Barrero et al.13 (lane 4); and Pathak et al.14 (lane 5). For lanes 1–5, protein expression values are red for increased expression (+1) and green for decreased expression (−1) in HIV-1-infected MDM. In addition, comparison was conducted for identification of proteins by SWATH-MS against HIV-1 dependency factors as established by Konig et al.43 (lane 6), Brass et al.44 (lane 7) and Zhou et al.45 (lane 8). Identifications of proteins from each study are shown in blue.
Proteomics-based quantitative expression data obtained from previously published studies of HIV-1-infected macrophages (MDM or macrophage-like cell lines)12−14 was used to evaluate our SWATH-MS data using a side-by-side comparison. Expression-specific findings reported by the HIV-1, Human Protein Interaction Database46−48 were also included in the comparison and provided data obtained from a multitude of cellular models (designated as “Mix” in Figure 2C, lane 3). Although 125 of the 420 differentially expressed proteins identified by SWATH-MS are reported to interact with HIV-1 proteins as reported by the HIV-1, Human Protein Interaction Database (indicated by asterisk (*) in Supplementary data set 2), only 13 proteins from the HIV-1, Human Protein Interaction Database display global differential expression during infection and matched the SWATH-MS results. In combination, using the proteomic data sets as well as the HIV-1, Human Protein Interaction Database, we observed conservation for the direction of expression changes among 45 differentially expressed proteins (Figure 2C, lanes 1–5). In addition, our SWATH-MS proteomic data set contained 29 significantly altered HIV-1 dependency factors as identified in RNA interference studies43−45 (Figure 2C, lanes 6–8). Nineteen proteins in our data set displayed conflicted expression with previous studies (Supplementary Figure 1). Notably, a higher degree of conserved directionality changes was observed with the data sets from primary macrophages as compared to cell lines. All in all, using this comparative approach, we were able to corroborate previously reported proteomic findings, identify expression changes in known HIV-1 dependency factors, and highlight alterations in expression of human proteins known to interact with HIV-1 proteins (Supplementary data set 2). We postulate that the remaining 245 proteins with no documented association with HIV-1 infection, but differentially expressed as determined by SWATH-MS, represent novel discoveries and continued investigation is merited.
The probable biological functions of the proteins identified in the SWATH-MS data was explored using a multifaceted bioinformatic approach focused on known molecular functions, pathways, and protein–protein interactions. Functional analysis using DAVID and PANTHER (Supplementary data sets 2 and 3, respectively) revealed enrichment for proteins involved in RNA binding and processing (Figure 3A). Pathway analysis using DAVID and Reactome (Supplementary data sets 4 and 5, respectively) complemented these findings by identifying a possible role for mRNA processing via the spliceosomal pathway. Visualization of the canonical spliceosomal pathway revealed a dysregulation of proteins for many of the known components of the spliceosome in HIV-1-infected MDM (Figure 3B). Alterations to specific spliceosomal functions and components were investigated (Supplementary data set 7) and among the proteins displaying increased expression (Table 1), YBOX1 and DHX15 are predicted HIV-1 dependency factors43 and unique to the U11/U12 spliceosome.49 On the basis of these results, we postulate that HIV-1 infection of the macrophage results in dysregulation of the spliceosome, which may subsequently influence viral pathogenesis by affecting alternative splicing of host proteins that are required in the host response to viral infection or in normal physiological functions of the macrophage (the role of the spliceosome in human disease is reviewed in Faustino and Cooper50).
Figure 3.

Bioinformatic analyses expose alterations in nucleic acid binding and regulatory proteins. (A) PANTHER analysis revealed enrichment for nucleic acid binding proteins (top). Expansion of this family of proteins identified RNA binding proteins (center) involved in mRNA processing (bottom) as substantially enriched. In addition, transcription factors (top) and other DNA binding proteins (center) were also enriched. (B) KEGG pathway analysis revealed dysregulation of spliceosomal components. Robust coverage of proteins was observed for proteins involved in spliceosomal processing of mRNA (yellow for identifications) and several spliceosomal components were differentially regulated in HIV-1 infected MDM (green = decreased expression in HIV-1-infected cells, pink = increased expression). The corresponding protein ID for each gene name provided by KEGG is available in Supplementary data set 2.
Table 1. Spliceosomal Proteins Displaying Increased Expression in HIV-1-Infected MDM.
| UniProt ID | symbol | HIV-1 dependency factor | GO namea | reference | SWATH-MS Δz | Z-test p-value |
|---|---|---|---|---|---|---|
| P67809 | YBOX1 | * | U12-type spliceosomal complex | Will et al.49 | 0.60 | 2.05 × 10–2 |
| P22626 | ROA2 | spliceosomal complex | Neubauer et al.51 | 0.51 | 2.12 × 10–3 | |
| P22626 | ROA2 | catalytic step 2 spliceosome | Jurica et al.52 | 0.51 | 2.12 × 10–3 | |
| Q8IYB3 | SRRM1 | catalytic step 2 spliceosome | Jurica et al.52 | 0.29 | 9.36 × 10–3 | |
| O43143 | DHX15 | * | U12-type spliceosomal complex | Will et al.49 | 0.15 | 4.74 × 10–2 |
Gene Ontology (GO) name as inferred by direct assay (IDA).
Functional enrichment analysis also revealed that nucleic acid binding proteins and transcription regulators were enriched among differentially expressed proteins (Figure 3) and centrality analysis of protein–protein interactions was used to understand the potential consequences of altered transcription regulator protein expression. Since many transcription regulators work in complexes, centrality analysis was performed on all transcription regulator proteins identified by SWATH-MS regardless of differential expression. Transcription regulator proteins were selected from our SWATH-MS data set using an in-house generated transcription regulator reference database containing 2725 known and putative human transcription regulator proteins obtained from three peer-reviewed data sets (Supplementary data set 8).25,37,38 This targeted enrichment revealed 510 transcription regulator proteins identified in our SWATH-MS analysis, including 67 proteins that were differentially expressed (Supplementary data set 2). A network of experimentally verified protein–protein interactions was generated using STRING and uploaded to Cytoscape (Figure 4A) and used for centrality analysis (Supplementary data set 9). Among the 67 differentially expressed transcription regulators, 42 were included in the network and the remainders were excluded because of a lack of experimental evidence for protein–protein interactions within the data set. Centrality analysis of the entire network highlighted a particular importance for NCOR1, NCOR2 and HDAC2 (upper 10th percentile for summed rank, Figure 4B), which suggests that perturbation of these proteins may have substantial downstream consequences to the entire system.
Figure 4.

Centrality analysis reveals critical nodes among interacting transcription regulators. (A) Cytoscape visualization of a STRING-generated network composed of experimentally verified protein–protein interactions among the transcription regulators identified in HIV-1 infected and uninfected MDM. Proteins in blue were identified, but not significantly altered. Proteins in red displayed increased expression in HIV-1-infected MDM, whereas proteins in green displayed decreased expression. The most central differentially expressed proteins (HDAC2, NCOR1, NCOR2) have darkened color and the node shapes are circular. (B) Centrality analysis using the node parameters of closeness, eccentricity, and radiality. Proteins were assigned a rank for each centrality measure, and the summed rank was used to sort proteins from higher centrality (left) to lower centrality (right). Differentially expressed proteins in the upper 10th percentile for summed rank are indicated with an asterisk (*). Red protein names indicate increased expression in HIV-1-infected MDM, whereas green protein names indicate decreased expression. The complete centrality analysis is provided in Supplementary data set 8.
In macrophages, it has been documented that NCOR1 and NCOR2 proteins regulate the expression of proinflammatory genes via NF-κB signaling by forming complexes that include the HDAC family of proteins.53−55 Our observation for the altered expression of NCOR1, NCOR2 and HDAC2 proteins in HIV-1-infected macrophages is consistent with the dysregulated production of pro-inflammatory molecules observed for these cells.56,57 In addition, NCOR2 is a predicted HIV-1 dependency factor45 and single nucleotide polymorphisms of the NCOR2 gene have been correlated with a heightened risk for HIV-1 infection among exposed individuals.58 Likewise, HDAC2 differential expression and regulation has been correlated with resistance to HIV-1 infection among exposed individuals.59 Unlike NCOR2 and HDAC2, which have been implicated in the infectivity of HIV-1, NCOR1 is reported to be a positive mediator of HIV-1 latency.60 Taken together, our findings support the importance of these transcription regulators, and further mechanistic investigations for the function of these proteins in HIV-1 infection of the macrophage are warranted.
Conclusion
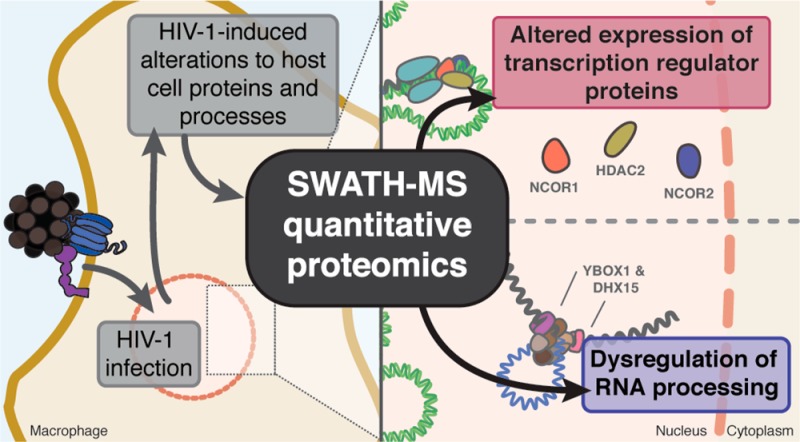
The development of additional host-oriented antiretroviral strategies is of utmost importance because of the continual emergence of antiretroviral resistant strains of HIV-1. Despite extensive screening for HIV-1-dependency factors,43−45 little headway has been made in the development of novel host-oriented antiviral strategies, which we believe may be in part from a lack of translatability from immortalized cell lines to the primary immune cells naturally affected by the virus. As such, in this investigation, we performed proteomic screening using SWATH-MS on primary immune cells infected in vitro with HIV-1 and we provide evidence for novel biological processes and proteins perturbed in HIV-1-infected macrophages (Figure 5). In addition to these findings, we provide an original and transparent methodology for the analysis of SWATH-MS quantitative proteomics data that can readily be adapted to most any cellular system.
Figure 5.

A proposed model for nucleic acid binding and regulatory protein dysregulation in HIV-1 infection of MDM. Upon HIV-1 infection and replication (A), cell-signaling cascades are activated resulting in differential activation of transcription regulator proteins and subsequent alterations in biomolecule expression impacting the transcriptome and the proteome (B). We have provided evidence for the reprogramming of host protein expression in response to HIV-1 infection at the level of nucleic acid binding and regulatory proteins involved in transcription (C) and RNA processing (D). Proteins predicted to be HIV-1 dependency factors are indicated with an asterisk (*).
Acknowledgments
The authors would like to thank Brenda Morsey for her assistance in cell culture, Melinda Wojtkiewicz of the Mass Spectrometry and Proteomics Core Facility (MSPCF) at UNMC for her assistance with mass spectrometry, and Dr. Michael Belshan (Creighton University; Omaha, NE) for his critical review of this manuscript. Financial support was provided by National Institutes of Health grants R01 DA030962, P30 MH062261 and P20 RR016469. In addition, N. A. Haverland is supported by a University of Nebraska Medical Center Graduate Studies Fellowship.
Glossary
Abbreviations
- BSL3
biosafety level 3
- CSF-1
colony stimulating factor 1 (aka macrophage colony stimulating factor, MSCF)
- DDA
data-dependent acquisition
- DIA
data-independent acquisition
- DTT
dithiothreitol
- FASP
filter assisted sample preparation
- FDR
false discovery rate
- GO
gene ontology
- HIV-1
Human Immunodeficiency Virus Type 1
- MDM
monocyte-derived macrophage
- MMTS
methyl methanethiosulfonate
- MOI
multiplicity of infection
- PBS
phosphate buffered saline
- RP-HPLC-ESI-MS/MS
reverse-phase high-pressure liquid chromatography electrospray ionization tandem mass spectrometry
- SDS
sodium dodecyl sulfate
- SWATH-MS
SWATH mass spectrometry
- TBST
Tris-buffered saline with Tween-20
Supporting Information Available
One figure, one scheme, and nine data sets are provided in zip file. Supplementary Figure 1 documents conflicted proteins in the comparison-based validation among proteomic data sets. Supplementary Scheme 1 provides a visual explanation of our SWATH-MS approach. Supplementary data set 1 contains all information for peptides and proteins used in compiling the MDM spectral library. Supplementary data set 2, tab #1 provides all SWATH-MS data at the protein level, including data transformations and statistical testing. The data set was truncated to significantly altered proteins in tab #2 and also contains information for protein qualities, such as transcription regulator; HIV-1, human protein interaction; comparison-based validation; and HIV-1 dependency factors. Supplementary data set 3 provides the complete DAVID analysis, including search criteria, DAVID functional enrichment clustering output, and the proteins involved in RNA processing. Supplementary data set 4 provides the complete PANTHER analysis for protein class. Supplementary data set 5 includes Reactome analysis for canonical pathways. Supplementary data set 6 contains results from DAVID pathway analysis. Supplementary data set 7 contains all experimentally verified proteins that are components of the spliceosome. Supplementary data set 8 is the transcription regulator reference database compiled from Ravasi et al.,37 Vaquerizas et al.,38 and Mi et al.25 Supplementary data set 9 is the centrality analysis using all transcription regulators identified in HIV-1-infected and uninfected MDM. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lifson J. D.; Reyes G. R.; McGrath M. S.; Stein B. S.; Engleman E. G. AIDS retrovirus induced cytopathology: giant cell formation and involvement of CD4 antigen. Science 1986, 23247541123–7. [DOI] [PubMed] [Google Scholar]
- Ho D. D.; Rota T. R.; Hirsch M. S. Infection of monocyte/macrophages by human T lymphotropic virus type III. J. Clin. Invest. 1986, 7751712–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgleish A. G.; Beverley P. C.; Clapham P. R.; Crawford D. H.; Greaves M. F.; Weiss R. A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 3125996763–7. [DOI] [PubMed] [Google Scholar]
- Alexaki A.; Liu Y.; Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 65388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig S.; Gendelman H. E.; Orenstein J. M.; Dal Canto M. C.; Pezeshkpour G. H.; Yungbluth M.; Janotta F.; Aksamit A.; Martin M. A.; Fauci A. S. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 1986, 23347681089–93. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Cross G. D.; Callaway C. S.; McDougal J. S. In vitro infection of human monocytes with human T lymphotropic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV). J. Immunol. 1986, 1371323–9. [PubMed] [Google Scholar]
- Gartner S.; Markovits P.; Markovitz D. M.; Kaplan M. H.; Gallo R. C.; Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 1986, 2334760215–9. [DOI] [PubMed] [Google Scholar]
- Coleman C. M.; Wu L. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 2009, 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J.; Chain B. M.; Miller R. F.; Webb B. L.; Barclay W.; Towers G. J.; Katz D. R.; Noursadeghi M. HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS 2009, 23172255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh R.; Florence E.; Vlieghe E.; Boonefaes T.; Grooten J.; Houthuys E.; Tran H. T.; Gali Y.; De Baetselier P.; Vanham G.; Raes G. Transcriptome analysis of monocyte-HIV interactions. Retrovirology 2010, 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. N.; Kohler J. J.; Coberley C. R.; Sleasman J. W.; Goodenow M. M. HIV-1 activates macrophages independent of Toll-like receptors. PLoS One 2008, 312e3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft-Terry S. D.; Engebretsen I. L.; Bastola D. K.; Fox H. S.; Ciborowski P.; Gendelman H. E. Pulsed stable isotope labeling of amino acids in cell culture uncovers the dynamic interactions between HIV-1 and the monocyte-derived macrophage. J. Proteome Res. 2011, 1062852–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero C. A.; Datta P. K.; Sen S.; Deshmane S.; Amini S.; Khalili K.; Merali S. HIV-1 Vpr modulates macrophage metabolic pathways: a SILAC-based quantitative analysis. PLoS One 2013, 87e68376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak S.; De Souza G. A.; Salte T.; Wiker H. G.; Asjo B. HIV induces both a down-regulation of IRAK-4 that impairs TLR signalling and an up-regulation of the antibiotic peptide dermcidin in monocytic cells. Scand. J. Immunol. 2009, 703264–76. [DOI] [PubMed] [Google Scholar]
- MacArthur R. D.; Novak R. M. Reviews of anti-infective agents: maraviroc: the first of a new class of antiretroviral agents. Clin. Infect. Dis. 2008, 472236–41. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Rost H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, 116O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman H. E.; Orenstein J. M.; Martin M. A.; Ferrua C.; Mitra R.; Phipps T.; Wahl L. A.; Lane H. C.; Fauci A. S.; Burke D. S.; et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 1988, 16741428–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 65359–62. [DOI] [PubMed] [Google Scholar]
- Scopes R. K. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 1974, 591277–82. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Huttenhain R.; Surinova S.; Gillet L. C.; Mouritsen J.; Brunner R.; Navarro P.; Aebersold R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 2013, 1381247–56. [DOI] [PubMed] [Google Scholar]
- Cheadle C.; Vawter M. P.; Freed W. J.; Becker K. G. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003, 5273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4144–57. [DOI] [PubMed] [Google Scholar]
- Huang da W.; Sherman B. T.; Lempicki R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 3711–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P. D.; Kejariwal A.; Guo N.; Mi H.; Campbell M. J.; Muruganujan A.; Lazareva-Ulitsky B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006, 34Web Server issueW645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H.; Muruganujan A.; Thomas P. D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41D1D377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H.; Muruganujan A.; Casagrande J. T.; Thomas P. D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 881551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B.; Lehmann G.; Bork P.; Huynen M. A. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28183442–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A.; Szklarczyk D.; Frankild S.; Kuhn M.; Simonovic M.; Roth A.; Lin J.; Minguez P.; Bork P.; von Mering C.; Jensen L. J. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41Database issueD808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline M. S.; Smoot M.; Cerami E.; Kuchinsky A.; Landys N.; Workman C.; Christmas R.; Avila-Campilo I.; Creech M.; Gross B.; Hanspers K.; Isserlin R.; Kelley R.; Killcoyne S.; Lotia S.; Maere S.; Morris J.; Ono K.; Pavlovic V.; Pico A. R.; Vailaya A.; Wang P. L.; Adler A.; Conklin B. R.; Hood L.; Kuiper M.; Sander C.; Schmulevich I.; Schwikowski B.; Warner G. J.; Ideker T.; Bader G. D. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007, 2102366–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scardoni G.; Petterlini M.; Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25212857–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milacic M.; Haw R.; Rothfels K.; Wu G.; Croft D.; Hermjakob H.; #039; Eustachio P.; Stein L. Annotating Cancer Variants and Anti-Cancer Therapeutics in Reactome. Cancers 2012, 441180–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Eustachio P. Pathway databases: making chemical and biological sense of the genomic data flood. Chem. Biol. 2013, 205629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D. Building models using Reactome pathways as templates. Methods Mol. Biol. 2013, 1021, 273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.The KEGG database. Novartis Found. Symp. 2002, 247, 91–101; discussion 101–3, 119–28, 244–52. [PubMed] [Google Scholar]
- Kanehisa M.; Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28127–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D.; Dimmer E.; Huntley R.; Barrell D.; O’Donovan C.; Apweiler R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics 2009, 25223045–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T.; Suzuki H.; Cannistraci C. V.; Katayama S.; Bajic V. B.; Tan K.; Akalin A.; Schmeier S.; Kanamori-Katayama M.; Bertin N.; Carninci P.; Daub C. O.; Forrest A. R.; Gough J.; Grimmond S.; Han J. H.; Hashimoto T.; Hide W.; Hofmann O.; Kamburov A.; Kaur M.; Kawaji H.; Kubosaki A.; Lassmann T.; van Nimwegen E.; MacPherson C. R.; Ogawa C.; Radovanovic A.; Schwartz A.; Teasdale R. D.; Tegner J.; Lenhard B.; Teichmann S. A.; Arakawa T.; Ninomiya N.; Murakami K.; Tagami M.; Fukuda S.; Imamura K.; Kai C.; Ishihara R.; Kitazume Y.; Kawai J.; Hume D. A.; Ideker T.; Hayashizaki Y. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 2010, 1405744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas J. M.; Kummerfeld S. K.; Teichmann S. A.; Luscombe N. M. A census of human transcription factors: function, expression and evolution. Nature Rev. Genet. 2009, 104252–63. [DOI] [PubMed] [Google Scholar]
- Sundstrom C.; Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 1976, 175565–77. [DOI] [PubMed] [Google Scholar]
- Tsuchiya S.; Kobayashi Y.; Goto Y.; Okumura H.; Nakae S.; Konno T.; Tada K. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982, 4241530–6. [PubMed] [Google Scholar]
- Pan C.; Kumar C.; Bohl S.; Klingmueller U.; Mann M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell. Proteomics 2009, 83443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp N. A.; Lilley K. S. Design and analysis issues in quantitative proteomics studies. Proteomics 2007, 7Suppl. 142–50. [DOI] [PubMed] [Google Scholar]
- Konig R.; Zhou Y.; Elleder D.; Diamond T. L.; Bonamy G. M.; Irelan J. T.; Chiang C. Y.; Tu B. P.; De Jesus P. D.; Lilley C. E.; Seidel S.; Opaluch A. M.; Caldwell J. S.; Weitzman M. D.; Kuhen K. L.; Bandyopadhyay S.; Ideker T.; Orth A. P.; Miraglia L. J.; Bushman F. D.; Young J. A.; Chanda S. K. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass A. L.; Dykxhoorn D. M.; Benita Y.; Yan N.; Engelman A.; Xavier R. J.; Lieberman J.; Elledge S. J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 3195865921–6. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Xu M.; Huang Q.; Gates A. T.; Zhang X. D.; Castle J. C.; Stec E.; Ferrer M.; Strulovici B.; Hazuda D. J.; Espeseth A. S. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 45495–504. [DOI] [PubMed] [Google Scholar]
- Fu W.; Sanders-Beer B. E.; Katz K. S.; Maglott D. R.; Pruitt K. D.; Ptak R. G. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009, 37Database issueD417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinney J. W.; Dickerson J. E.; Fu W.; Sanders-Beer B. E.; Ptak R. G.; Robertson D. L. HIV-host interactions: a map of viral perturbation of the host system. AIDS 2009, 235549–54. [DOI] [PubMed] [Google Scholar]
- Ptak R. G.; Fu W.; Sanders-Beer B. E.; Dickerson J. E.; Pinney J. W.; Robertson D. L.; Rozanov M. N.; Katz K. S.; Maglott D. R.; Pruitt K. D.; Dieffenbach C. W. Cataloguing the HIV type 1 human protein interaction network. AIDS Res. Hum. Retroviruses 2008, 24121497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C. L.; Schneider C.; Hossbach M.; Urlaub H.; Rauhut R.; Elbashir S.; Tuschl T.; Luhrmann R. The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. RNA 2004, 106929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino N. A.; Cooper T. A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 174419–37. [DOI] [PubMed] [Google Scholar]
- Neubauer G.; King A.; Rappsilber J.; Calvio C.; Watson M.; Ajuh P.; Sleeman J.; Lamond A.; Mann M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet. 1998, 20146–50. [DOI] [PubMed] [Google Scholar]
- Jurica M. S.; Licklider L. J.; Gygi S. R.; Grigorieff N.; Moore M. J. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 2002, 84426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C. K.; Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 105365–76. [DOI] [PubMed] [Google Scholar]
- Mottis A.; Mouchiroud L.; Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes & development 2013, 278819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G.; Glass C. K. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends in endocrinology and metabolism: TEM 2006, 178321–7. [DOI] [PubMed] [Google Scholar]
- Gendelman H. E.; Friedman R. M.; Joe S.; Baca L. M.; Turpin J. A.; Dveksler G.; Meltzer M. S.; Dieffenbach C. A selective defect of interferon alpha production in human immunodeficiency virus-infected monocytes. J. Exp. Med. 1990, 17251433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtmayerova H.; Nottet H. S.; Nuovo G.; Raabe T.; Flanagan C. R.; Dubrovsky L.; Gendelman H. E.; Cerami A.; Bukrinsky M.; Sherry B. Human immunodeficiency virus type 1 infection alters chemokine beta peptide expression in human monocytes: implications for recruitment of leukocytes into brain and lymph nodes. Proc. Natl. Acad. Sci. U.S.A. 1996, 932700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinn L. W.; Tang M.; Kessing B. D.; Lautenberger J. A.; Troyer J. L.; Malasky M. J.; McIntosh C.; Kirk G. D.; Wolinsky S. M.; Buchbinder S. P.; Gomperts E. D.; Goedert J. J.; O’Brien S. J. Genetic associations of variants in genes encoding HIV-dependency factors required for HIV-1 infection. J. Infect. Dis. 2010, 202121836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R. C.; Sivro A.; Kimani J.; Jaoko W.; Plummer F. A.; Ball T. B. Epigenetic control of IRF1 responses in HIV-exposed seronegative versus HIV-susceptible individuals. Blood 2011, 11792649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan M.; Schiralli Lester G. M.; Lee C.; Missra A.; Wasserman G. A.; Steffen M.; Gilmour D. S.; Henderson A. J. Negative elongation factor (NELF) coordinates RNA polymerase II pausing, premature termination, and chromatin remodeling to regulate HIV transcription. J. Biol. Chem. 2013, 2883625995–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.