

Abstract
Recent breakthroughs of cell phenotype reprogramming impose theoretical challenges on unravelling the complexity of large circuits maintaining cell phenotypes coupled at many different epigenetic and gene regulation levels, and quantitatively describing the phenotypic transition dynamics. A popular picture proposed by Waddington views cell differentiation as a ball sliding down a landscape with valleys corresponding to different cell types separated by ridges. Based on theories of dynamical systems, we establish a novel ‘epigenetic state network’ framework that captures the global architecture of cell phenotypes, which allows us to translate the metaphorical low-dimensional Waddington epigenetic landscape concept into a simple-yet-predictive rigorous mathematical framework of cell phenotypic transitions. Specifically, we simplify a high-dimensional epigenetic landscape into a collection of discrete states corresponding to stable cell phenotypes connected by optimal transition pathways among them. We then apply the approach to the phenotypic transition processes among fibroblasts (FBs), pluripotent stem cells (PSCs) and cardiomyocytes (CMs). The epigenetic state network for this case predicts three major transition pathways connecting FBs and CMs. One goes by way of PSCs. The other two pathways involve transdifferentiation either indirectly through cardiac progenitor cells or directly from FB to CM. The predicted pathways and multiple intermediate states are supported by existing microarray data and other experiments. Our approach provides a theoretical framework for studying cell phenotypic transitions. Future studies at single-cell levels can directly test the model predictions.
Keywords: gene regulatory network, nonlinear dynamics, non-equilibrium steady state
1. Introduction
How do mammalian cells that share the same genome exist in notably distinct phenotypes, exhibiting differences in morphology, gene expression patterns and epigenetic chromatin statuses? Furthermore, how do cells of different phenotypes differentiate reproducibly from a single fertilized egg? These fundamental questions are closely related to a deeply rooted paradigm in developmental biology that cell differentiation is irreversible. That is, once a cell differentiates into a terminal phenotype, the cell fate is determined. Waddington [1] suggested an epigenetic landscape picture for this irreversible process of establishing cell fates during development. It resembles a ball rolling down a slanted ‘landscape’ to the point of the lowest local elevation, where the axes and basins represent molecular concentrations and stable cell phenotypes, respectively, and decrease in the elevation corresponded to increased extent of differentiation. The Waddington epigenetic landscape is analogous to the energy landscape concept widely used in chemistry and physics studies, e.g. in protein physics each basin represents one stable protein conformation [2,3].
In contrast to this belief, initiated by a pioneering experiment done by Yamanaka's group [4], a growing body of research suggests the possibility of cell reprogramming that offers the potential of converting one type of cell into another [4–8]. Examples include converting fibroblasts (FBs) to cardiomyocytes (CMs) through induced pluripotent stem cells (iPSCs); ‘direct transdifferentiation’ (DTD) to CMs by overexpressing Gata4, Tbx5 and Mef2c [8]; and ‘indirect transdifferentiation’ (ITD) through a non-stem cell precursor by overexpressing stem cell markers Sox2, Oct4, c-Myc and Klf4, but preventing cells into iPSC state [6].
The existence of different cell types and the above-mentioned fascinating reprogramming experiments triggered theoretical studies on describing transitions between different cell phenotypes. In the literature, Waddington's landscape concept is again frequently referred to as an intuitive and transparent birds' eye view on understanding the reprogramming phenomena. The concept, though, only serves as a phenomenological metaphor lacking either mechanistic details or quantitatively predictive power [9]. Therefore, many efforts have been made towards quantifying Waddington's landscape concept. Using the Boolean network formalism, Kauffmann argued that each stable attractor of the regulatory network corresponds to a stable cell phenotype [10,11]. Recently, a number of theoretical studies have been made to define scalar quasi-potentials for a general nonlinear dynamic system with possible multiple attractors [12–18]. These studies start with the widely used differential equation-based approach towards modelling cellular processes. This differential equation-based approach integrates molecular biology details piece-by-piece, from which the system dynamics can be deduced quantitatively. Through constructing scalar landscapes, one tries to provide a global view of the system dynamics, which is necessary for studying phenotypic transition processes involving a large number of genes and lineages. However, these studies are subjected to the ‘curse of dimension’. That is, the computational cost increases quickly with the dimension of the system. A high-dimensional landscape is also difficult to visualize and analyse. Furthermore, quantitative information to constrain model parameters [11,15,19–21] is generally also lacking. There are proposals of using alternative approaches such as the statistical graphical models [22]. It may be fruitful to make connections of these different approaches.
To overcome the above difficulties related to dynamical system theories, using tools brought from protein and network sciences, in this paper we present a novel methodology offering quantitatively predictive power towards the cell reprogramming and cell phenotypic transitions in general, and apply it productively to an important experimental system, the reprogramming of FBs to iPSCs and CMs. With a minimal set of available biological input, the analysis provides a dynamic view of the reprogramming processes, leading to a number of testable predictions, many of which are supported by existing experimental results.
2. Results
2.1. Epigenetic state network model provides global and coarse-grained view of network dynamics
Our approach is inspired by studies of protein dynamics and glass transitions, where a well-defined energy landscape exists to quantify the interactions as a function of the atomic coordinates [2,3]. A basin of attraction is around a minimum on the landscape, which the system will move back to under a small perturbation (figure 1a). Between two neighbouring basins, there is a minimum energy path through a saddle point. Therefore, a landscape can be largely characterized by a collection of the basins and paths going through the saddle points. A system spends most of the time fluctuating within basins, and transitions between basins are rare events.
Figure 1.

State network as coarse-graining of the full dynamics. (a) On a real energy landscape, a minimal energy path through an unstable transition point (first-order saddle) connects two neighbouring basins of attraction. The landscape can be simplified to a network with basins as the nodes and connecting paths going through first-order saddles as the edges. (b) The vector field of a mutually inhibitive two-gene circuit (inset), where the nodes S1, S2 and S3 represent fixed-point attractors. The pie diagram of each attractor represents the corresponding expression pattern. The green diamond points correspond to the first-order saddles, surrounded by forward and backward optimal paths (grey (dark blue online) curves) connecting two neighbouring attractors. Notice that, for open systems, the optimal paths do not actually go through the saddles. (c) The ESN constructed from (a) by connecting neighbouring attractors. (d) Tuning the modelling parameters (see more details in the electronic supplementary material, tables S1 and S2) results in different regulatory dynamics (see nullcline plots), leading to distinct topologies of the associated ESNs. (Online version in colour.)
To generalize the above approach to describe the reprogramming processes, we adopt a set of coupled stochastic differential equations to describe the molecular interactions among the involved species (Material and methods),
| 2.1 |
where each component of x represents the expression level of each gene, the deterministic term F describes the gene interactions (λ specifies the maximum gene expression activity that is related to the chromatin state as discussed in more detail in Material and methods, and ζ reflects the intrinsic and environmental control parameters), and the stochasticity term satisfies  and
and 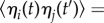
 , where the matrix D characterizes the strength of the stochastic noise, offering a quantitative description of gene regulation dynamics [11,23,24].
, where the matrix D characterizes the strength of the stochastic noise, offering a quantitative description of gene regulation dynamics [11,23,24].
In the absence of the noise term η, equation (2.1) reduces to a set of deterministic ordinary differential equations (ODEs), whose long-term behaviour is largely determined by its attractors [25]. For cell reprogramming, we are particularly interested in the fixed-point attractors that represent different stable cell phenotypes. The fact that the fixed-point attractors are stable under infinitesimal perturbation implies that the cell phenotypes are robust most of the time. Indeed, spontaneous phenotypic transitions are extremely rare. However, stochastic noise provides an unbounded perturbative force that drives the cell from one state to others [9]. A pair of most probable transition paths between two neighbouring attractors in general pass around (but not through [23]) a first-order saddle point (fixed point with only one component unstable) [26,27]. Therefore, the regulation dynamics described with differential equations can be approximated by a stochastic walk along a weighted network (epigenetic state network or ESN) whose nodes correspond to the fixed-point attractors, and edges (i.e. links between nodes) represent the most probable paths connecting two neighbouring attractors. Each edge carries a pair of transition rates kab and kba estimated by the Wentzell–Freidlin theory for sufficiently small noises [27,28] (electronic supplementary material, text section 1)
 |
2.2 |
and a similar expression for kba, where the integration is performed over the optimal path that minimizes the integral using the conjugate gradient algorithm. Therefore, equation (2.1) can be approximated by a master equation describing the network dynamics
| 2.3 |
where the ith element of z is the probability of finding the system in epigenetic state i, and K = {kab} is the transition matrix determined by equation (2.2). To apply the ESN approach to high-dimensional systems where the number of stationary points may grow exponentially with the dimensionality, we developed a conditional root-finding algorithm that allows us to efficiently locate fixed-point attractors and first-order saddles (Material and methods).
2.2. Epigenetic state network approach on a two-gene system
Figure 1b–d demonstrates the ESN approach applied to a two-gene (x1, x2) regulatory circuit, a simple prototype model sustaining multiple cell reprogramming paths [19,11]. Figure 1b shows a representative vector field in the phase space, showing that starting from an arbitrary (x1, x2) concentration the system relaxes to one of the three stable fixed points: one corresponding to a progenitor state during a differentiation process with both x1 and x2 high (S1), and another two differentiated states (with x1 high and x2 low (S2) or vice versa (S3)); together with three first-order saddle points and one unstable point. Two curves called separatrices passing through the saddle and unstable points separate the whole phase space into three regions centred around the three stable fixed points, corresponding to three basins in terms of landscape description. Further optimal path analysis reveals two paths connecting the two differentiated states S2 and S3: starting from S2, the first one first passes through the progenitor S1, corresponding to a dedifferentiation process, then differentiates into S3, and another is a direct path connecting the two phenotypes without going through the progenitor. For transitions between each pair of states, there is a pair of forward and backward optimal paths passing close to a first-order saddle point. The corresponding ESN in figure 1c captures all these basic topological features of this dynamical system in the phase space. Furthermore, figure 1d and electronic supplementary material, tables S1–S4 show that both the number of fixed-point attractors and network topology change with different kinetic parameters. Additional result can be found in the electronic supplementary material, text section 2. In this case, ESN provides an alternative representation of cell differentiation and reprogramming that captures the major dynamics of the underlying two-dimensional vector fields (figure 1c), consistent with traditional approaches such as bifurcation analysis [25]. However, the vector field of a high-dimensional system is not directly visible and the corresponding lower dimensional profile offers only partial information of the underlying landscape. The ESN approach coarse grains the essential characters of an arbitrary dimensional dynamic system into a low-dimensional network graph and thus can reveal the global features transparently. Especially, it is not straightforward to obtain information about the transition paths between two states from the bifurcation analysis.
2.3. Epigenetic state network approach on the fibroblast-induced pluripotent stem cell-cardiomyocyte system
To demonstrate the practical power of our approach, we next apply the ESN analysis to the FB-iPSC-CM system. This system has received much attention within the last few years [4–8], and experiments have shown FB to CM reprogramming through iPSCs, as well as DTD/ITD (figure 2a).
Figure 2.

Construction of the FB-iPSC-CM regulatory circuit. (a) Schematic of pluripotent reprogramming and TD for FB-iPSC-CM system, showing that FBs can be induced either to reprogram into pluripotent stem cells (PSCs) and later differentiate into CMs, or to transdifferentiate into CMs through a direct or indirect pathway. (b) The gene regulatory model circuit in this work, constructed based on the IPA (@Ingenuity) database. (Online version in colour.)
In a mammalian system, a fertilized egg first develops into an embryo. Embryonic stem cells (ESCs) that are pluripotent are derived from the inner cell mass of an early stage embryo called a blastocyst. Through the three primary germ layers, ectoderm, endoderm and mesoderm, ESCs can differentiate into each of the over 220 cell types found in the adult body. The iPSCs share some basic stemness features of ESCs, although some subtle differences have also been reported [29]. CMs are derivatives of the mesoderm germ line. The origins of FBs are more complex. In related experimental studies [30,31], the FBs are either mouse embryonic FBs (MEFs) or mouse tail-tip FBs, both of which are derivatives of the mesoderm germ line. Therefore in this work, we consider only intra-lineage cell reprogramming.
2.3.1. Ensemble averaged epigenetic state networks
In this work, we focus on the main regulatory molecules for each involved phenotype, constructing the regulatory circuit from the empirical observations (figure 2b; also see the electronic supplementary material, text section 3 for more details). The chromatin status determining the accessibility of associated regulators is the key factor that controls different cell phenotypes and is modified during the reprogramming process, possibly through some ‘plastic’ chromatin states [32]. For example, for the FB-iPSC reprogramming process, the FB regulators change from an open to a closed chromatin form, the PSC regulators from closed to open, while those of others (CM regulators here) remain closed. We are particularly interested in the following processes/chromatin states and the associated ESNs (see Material and methods and electronic supplementary material, figure S2): (i) FB-iPSC pluripotent reprogramming (PR-ESN), for which only the FB and pluripotent stem cell (PSC) regions are accessible and (ii) FB-CM transdifferentiation (TD-ESN), for which only the FB and CM regions are accessible. Note that as quantitative measurements of the reprogramming processes are largely limited nowadays the model we proposed here captures only the approximated biological information. Indeed, our goal here is to demonstrate the power of the proposed ESN methodology, rather than developing a complete regulatory circuit with a full quantitative description of the reprogramming process.
Here, we adopt a model ensemble approach [33]. That is, instead of working with a single set of parameters, we examine the model behaviours with an ensemble of parameter sets. The basic idea is that if a certain behaviour is observed over a broader range of parameters, one has higher confidence that it can be observed experimentally. Biologically, experimental studies at the single-cell level reveal non-genetic cellular heterogeneity, indicating an inherent variation of global gene expression patterns among the cells [34]. This heterogeneity also justifies that it is more relevant to perform the ESN analysis over an ensemble of parameters through Monte Carlo sampling, instead of one fixed set of kinetic constants, to reflect this cell-to-cell variation. To achieve ensemble space appropriate for all the processes considered we use the following boundary conditions. For PR-ESN, we set the chromatin region of CM regulators to remain closed and allow other chromatin parts to change during the process (i.e. we set λ = 0.1 for CM regulator maximum basal expression, and λ = 1 for others involving chromatin open/close changes). Moreover, both the FB and iPSC states have to occur within one single connected ESN cluster based on experimental observations; similarly, for TD-ESN we set λ = 0.1 for ESC regulators, and both the FB and CM states have to appear within a single connected ESN cluster (see more details in Material and methods and electronic supplementary material, text section 2).
2.3.2. Epigenetic state networks predict reprogramming intermediate states and pathways
Figure 3 shows the PR-ESN and TD-ESN averaged over 105 Monte Carlo realizations (electronic supplementary material, text section 3, figures S3 and S4), where the node and edge sizes are proportional to their occurrence probabilities. First, a large amount of cell states in PR-ESN (figure 3a) share similar expression patterns, reflecting different parameter sets used. We want to emphasize that for each parameter set, the number of possible states calculated from the model is small (see the electronic supplementary material, figure S4). TD-ESN also gives a number of states with varying gene expression levels. Second, there are a small number of states with high frequency of occurrence. While the states FB, SC and CM are what we require the model to have a priori, there are a few new states. For example, a pre-iPSC (PP) state appears as an intermediate during the reprogramming process, with all the regulators down. Another FC state appears as an intermediate state during TD with both FB and CM regulators on. Based on the basic assumption of the model ensemble idea, these are the predicted states that one probably observes experimentally.
Figure 3.

Epigenetic state networks of the FB-iPSC-CM system. (a) PR-ESN for FB → iPSC pluripotent reprogramming process, consisting of 295 states (nodes) and 1014 pathways (edges). (b) TD-ESN corresponding to the FB → CM TD process, consisting of 57 states and 133 edges. Both nodes' and edges' sizes are proportional to the occurrence probability of the associated states and pathways, respectively. The pie diagram of each node represents the expression pattern of the corresponding state, where different colours and pie diagram positions represent different regulator groups, and the brightness reflects the expression level. The ESNs are ensemble averaged, corresponding to overlaying ESNs of single cells, which show cell-to-cell variation. (Online version in colour.)
From an experimental perspective, these groups of cell states with similar expression patterns would appear as a cloud of data points forming a small number of clusters in a single-cell measurement, e.g. multiplex flow cytometry or single-cell quantitative PCR data. Currently, most existing measurements are at the bulk level, which have been averaged over a large number of cell patterns. In order to compare with these bulk measurements, we use the minimum spanning tree algorithm [35] to cluster the states in each ESN, and then calculate the relative expression level of each cluster by averaging the expressions over the states within the same cluster (figure 4a,b). The averaged expression patterns show that the regulator expressions of different cell phenotypes are largely exclusive to each other (e.g. FB and stem cell). This reflects the mutual inhibitions of regulators representing different cell phenotypes.
Figure 4.

Predicted and experimental relative expression levels of 15 key regulators, where Twist1, Snail1 and Snail2 correspond to the fibroblast regulators (FBRs) and Nkx2–5, Tbx5, Gata4 and Mef2c correspond to the cardiomyocyte regulators (CMRs), respectively. (a) Dominant states of pluripotent reprogramming predicted by coarse-graining PR-ESN. (b) Dominant states of direct TD predicted by coarse-graining TD-ESN. (c) The evolution of gene expression patterns during mouse embryonic fibroblast (MEF) reprogramming to iPSC (data from GSE19023), for 0 day post induction (dpi) (MEF state, top panel), 3 dpi (pre-iPSC (PP) state, middle panel) and the final mouse iPSC (MiPSC) state (bottom panel), respectively. (d) The gene expression patterns of human-induced pluripotent stem cell (HiPSC, top panel) differentiation to CM (bottom panel; data from GSE28191). (Online version in colour.)
The ESNs also predict a number of dominant, thus experimentally likely occurring, pathways. The PR-ESN consists of pathways connecting mostly the FB and the stem cell states (iPSC1 and iPSC2), dominated by a major path through a pre-iPSC (PP) state. The TD-ESN instead shows major parallel paths between the FB and CM states passing through the PP state, intermediate states with CM progenitor regulators on (cardiac progenitor, CP), or FC state (an intermediate state with both FB and CM regulators on). Counterintuitively, despite the FB and CM transcription factors interacting indirectly through progenitor regulators (electronic supplementary material, figure S2), TD-ESN shows a pathway connecting the FB and CM states through the FC state without passing through the upstream PP or CP states and is confirmed by experimental observations of direct TD [8]. This dynamic feature is only implicitly contained in the original regulatory circuit, but is revealed by the ESN explicitly.
2.3.3. Microarray data confirm predicted states and pathways
To facilitate comparison with experiments, we examine the published microarray data in [30,31,36] (see the electronic supplementary material, text section 3 for details). For pluripotent reprogramming, experimental observations show that the dynamical evolution from mouse embryonic FB (MEF, FB regulators turning on, all regulators turning off), to pre-iPSC (PP, silencing FB regulators without expressing PSC regulators) then mouse iPSC (MiPSC, PSC regulators turning on) states sequentially (figure 4c), confirming the prediction from the PR-ESN (figure 4a). Figure 4d reports the gene expression patterns measured in the differentiation experiments from human-induced pluripotent stem cell (HiPSC) into CM, showing clear expression patterns corresponding to the SC and CM states, respectively. For TD (figure 3), the TD-ESN predicts an intermediate state (FC) in which both the FB and CM regulators are turned on. The FB regulators are not monitored in existing experiments. Interestingly, we find reports on the mouse embryo cardio FB cells, which exist in vivo, have very similar expression pattern to the FC state [31]. This observation, while not confirming, suggests that existence of such intermediate state (FC) in the TD process is possible. Furthermore, recently Pereira et al. [37] actually reported intermediate states with mixed expression features during the progress of reprogramming mouse FBs into haematopoietic stem cells. Figure 5a summarizes a complete set of experimental agreements with the predicted cell states.
Figure 5.

List of experimental supports of predicted states (a) and pathways (b) of reprogramming and TD. (Online version in colour.)
The ESN predictions are supported by not only the experimentally observed cell states, but also more importantly, the major reprogramming pathways (figure 3). Figure 5b summarizes the predicted major pathways along with the supporting experimental results. For example, previous experiments found that the FB-to-iPSC reprogramming pathways are very likely to undergo the PP state, but it has also been observed that some cells bypass the PP state [38–40].
3. Discussion
3.1. Transitions among attractors describe cell reprogramming
Our ESN analysis provides a rigorous description of cell phenotypic transitions based on the dynamical systems theory. A cell regulatory network may have multiple attractors corresponding to different cell phenotypes. Here, we may quantitatively relate a cell phenotype to a certain gene expression pattern. Owing to stochasticity cells with the same phenotype show non-genetic cell-to-cell variations around a deterministic attractor. However, spontaneous fluctuations rarely result in transitions between different stable phenotypes. Similarly, a cell can often maintain its phenotype despite change of environment. This robust phenotypic stability is likely the consequence of natural selection, which is metaphorically represented by Waddington as basins separated by high barriers. However, for a cell with finite size, the ‘barrier height’ can only be finite, which opens the possibility of inducing phenotypic transitions with sufficiently large perturbations. Experimentally researchers have induced elevated intracellular protein levels of certain genes temporarily, through lentivirus transfection, mRNA or polypeptide injection [6,41]. These perturbations lift the system away from the attracting basins and allow it to relax to a different basin, resulting in changes of cell phenotypes.
Recently, Ferrell used bifurcation analysis to examine the Waddington landscape [42]. Our results also support the proposal of Ferrell that cell differentiation may take place through saddle node bifurcation, where undifferentiated and differentiated states may coexist, besides the pitchfork bifurcation illustrated in the original Waddington picture, where two differentiated states appear suddenly. Our approach shares some basic ideas and is applicable to systems of multiple degrees of freedom. A remarkable feature is that our approach goes beyond the conventional one and two parameter bifurcation analysis and provides a global view of the dynamic features of the system.
Our analysis not only reveals the possibility of cell reprogramming, but also implies how one can improve the processes. The optimal paths are the easiest paths for transitions from one phenotype to another one. Intuitively, the transition is more efficient if the external perturbation is applied along the optimal path than the perpendicular case. To be specific, consider the model system shown in figure 1a. To reprogram the phenotype from S2 to S3, one strategy is to suppress x1 and activate x2 simultaneously. However, a better strategy may be activating x2 first, driving the system to S1, then suppressing x1. Furthermore, a reverse procedure of suppressing x1 first then activating x2 may direct the system to take another reprogramming pathway bypassing S1. Therefore, in principle, one may control the reprogramming pathways by adopting different procedures. For a more complex reprogramming process such as the FB-iPSC-CM system, involvement of a number of intermediate states suggests time-dependent reprogramming strategies. Clearly, one requires more qualitative and quantitative information about the regulatory network and the dynamics to optimize the reprogramming processes.
As mentioned earlier, to induce cell reprogramming and TD, one typically over-expresses certain genes, or introduces the corresponding mRNAs or proteins synthesized extracellularly to the cells [6,41]. These externally induced perturbations are only temporal perturbations to cells and disappear eventually. On the other hand, the landscapes and the corresponding epigenetic states are defined by the intrinsic cellular steady states [12–15,43]. These perturbations therefore do not affect the landscape (and ESN here), but can be viewed as analogous to external force pushing the system out of the attractor. Knowledge of the landscape and ESN provide guidance on how to ‘exert’ the force to efficiently induce the transitions.
3.2. Proposed experimental tests
A gene regulatory circuit is reconstructed indirectly from various experimental results. On the other hand, the nodes of an ESN are direct experimental observables. Each node represents the expression (i.e. protein or mRNA) levels of genes under study at an attractive state. Therefore, we propose to measure the single-cell expression levels of selected genes during the course of reprogramming. We predict that these single-cell data form clusters corresponding to attractors. For example, we propose an experiment tracking both FB and CM regulators during the TD process to confirm our predicted FC state. Our analysis predicts that a reprogramming process may take place through a number of parallel pathways. This can also be tested experimentally by tracking the time courses at the single-cell level. We note some recent single-cell studies [40,44], which can serve as input for further theoretical studies.
3.3. Future development and application to general dynamical systems
In summary, tying with complex network analysis [45], the ESN approach provides a comprehensive mathematical framework of describing the cell reprogramming process from a global point of view, incorporating different cell types and collective changes of regulatory molecules coupled at both transcription and epigenetic levels.
In this work, our main focus is development of the methodology. Our application to the FB-iPSC-CM reprogramming system should be viewed as a first proof-of-principle test. We mainly use the requirement that the model should reproduce both the pluripotent reprogramming and TD phenomena to constrain the model parameters. Despite very limited inputs from experiments, we obtain a series of qualitative and semi-quantitative predictions confirmed by experimental observations, shedding a light on understanding these fundamentally important but complex processes through theoretical modelling. Once more data from experiments becomes available further detailed predictions from the ESN approach are promising. In particular, with the help of single-cell transcriptome and proteome time course measurement, the model aims to analyse the coupling among regulation mechanisms at different levels, providing the hope of optimizing transition dynamics in the cell phenotypic regulation. Epigenetic modification is a major part of a reprogramming process. In this work, we treat epigenetic modifications on the chromatins and gene expressions compositely. It will be more predictive to develop a model incorporating the molecular details of these processes. While for illustrative purposes in this work we only analyse a system with 10 degrees of freedom, the method can be readily applied to systems with higher dimensions. For a large network with multiple attractors, the rate limiting steps are transitions among the attractors. The ESN approach coarse grains the dynamics of a network to Markovian dynamics, thus permitting description of long time dynamics not feasible for direct simulations with stochastic differential equations.
The developed approach can be applied to study the dynamics of general stochastic nonlinear systems. It became a main theme in the past decade or so to study numerous complex systems in nature and social systems. A prominent finding is that most networks are not randomly organized, but repetitively show some scale-free or small-world topological features. These observations suggest strong correlation between network structures and functions. A clear new frontier for the field is adding the domain of dynamics for a full theory for complex systems [45]. Major obstacles, however, exist. For a large complex network, it is challenging to construct a mathematical description and analyse its dynamics. Without data of sufficient detail to determine the usually large number of model parameters, the prediction power of a model is questionable. The present coarse-grained state network approach provides an efficient way of studying long time dynamics of large-scale networks, similar to that in protein and glass studies. Future studies may expand the current approach to attractors beyond the fixed points.
4. Material and methods
4.1. Mathematical formalism
We use the following rate equation describing the gene expression dynamics of a species (see the electronic supplementary material, text section 1 for more discussions):
| 4.1 |
where  is the Wilson–Cowan function that has been widely used on modelling gene regulation dynamics [46–48] and
is the Wilson–Cowan function that has been widely used on modelling gene regulation dynamics [46–48] and  . The expression of G is a generic sigmoidal function with steepness (slope at wj = 0) that increases with σj. Each ωji is a real number in [−1, 1]; positive for the ‘activators’ and negative for the ‘inhibitors’ of node j. The sum, wj, is the net activation or inhibition on node j, and ωj0 determines whether node j is ‘on’ or ‘off’ when all input signals are 0. The sign pattern (−, 0, +) of the weight matrix ωji correlates with the network topology. The parameters γj's here, with values within [0,1], determine how quickly each variable approaches its goal value. Values of γj's do not affect the steady-state behaviour but the system dynamics. The parameters λj's reflect the maximum gene expression level determined by the chromatin status. λ is a constant with a maximum value equalling 1 indicating the chromatin region can be fully open and accessible to regulatory proteins and RNA polymerases. A reprogramming process involves changes at different levels including epigenetic and transcription levels. In general, one can treat the chromatin dynamics as stochastic transitions between discrete states, thus treat the overall reprogramming dynamics with a mixed continuum-discrete formalism, as in studies of other problems [49–52]. In this work, we treat the dynamics at epigenetic and transcription/translational levels compositely by the function G. Theoretically, it corresponds to the adiabatic representation as in quantum chemical dynamics [50].
. The expression of G is a generic sigmoidal function with steepness (slope at wj = 0) that increases with σj. Each ωji is a real number in [−1, 1]; positive for the ‘activators’ and negative for the ‘inhibitors’ of node j. The sum, wj, is the net activation or inhibition on node j, and ωj0 determines whether node j is ‘on’ or ‘off’ when all input signals are 0. The sign pattern (−, 0, +) of the weight matrix ωji correlates with the network topology. The parameters γj's here, with values within [0,1], determine how quickly each variable approaches its goal value. Values of γj's do not affect the steady-state behaviour but the system dynamics. The parameters λj's reflect the maximum gene expression level determined by the chromatin status. λ is a constant with a maximum value equalling 1 indicating the chromatin region can be fully open and accessible to regulatory proteins and RNA polymerases. A reprogramming process involves changes at different levels including epigenetic and transcription levels. In general, one can treat the chromatin dynamics as stochastic transitions between discrete states, thus treat the overall reprogramming dynamics with a mixed continuum-discrete formalism, as in studies of other problems [49–52]. In this work, we treat the dynamics at epigenetic and transcription/translational levels compositely by the function G. Theoretically, it corresponds to the adiabatic representation as in quantum chemical dynamics [50].
For the FB-iPSC-CM system analysed in this work, each variable xi represents the concentration (relative to its maximum) of one of the 10 chemical components: x1 = [NANOG], x2 = [OCT4], x3 = [SOX2], x4 = [KLF4], x5 = [brachyury], x6 = [MESP1], x7 = [Ripply2], x8 = [FB], x9 = [ISL1] and x10 = [CM].
4.2. Algorithm of searching fixed-point attractors and first-order saddles
The stationary points x0 of equation (4.1) satisfy F(x0) = 0. The traditional algorithm of searching the stationary points is based on the optimization of the auxiliary energy  The conventional Newton–Raphson (NR) method constructs a trajectory from an initial guess that converges towards a stationary point. At each step, the NR method adopts a moving step satisfying
The conventional Newton–Raphson (NR) method constructs a trajectory from an initial guess that converges towards a stationary point. At each step, the NR method adopts a moving step satisfying
| 4.2 |
obtained by optimizing the energy change
| 4.3 |
where J is the Jacobian matrix with its element defined as  . Yet, the NR method often converges to a stationary point other than a fixed-point attractor. The latter requires a negative definite Jacobian. To achieve this, instead of equation (4.2) we use a quasi-Newton updating step
. Yet, the NR method often converges to a stationary point other than a fixed-point attractor. The latter requires a negative definite Jacobian. To achieve this, instead of equation (4.2) we use a quasi-Newton updating step
| 4.4 |
where to-be-defined JQN is the quasi-Newton Jacobian that is negative definite everywhere. We first diagonalize  where
where  with the eigenvalues {hi} as its diagonal elements. We next construct
with the eigenvalues {hi} as its diagonal elements. We next construct  where
where
| 4.5 |
A similar quasi-Newton step known as eigenmode method [53] was previously developed in the chemistry community to seek energy minima in a potential energy landscape, in which J (the Hessian matrix) is symmetric and hence both Q and D are real-valued matrices. Our case is more general in the sense that the Jacobian J is not necessary to be symmetric and thus both Q and D may be complex valued. However, it can be proved that the constructed JQN is a real matrix and thus the quasi-Newton step (equation (4.4)) is well defined. If J is a symmetric matrix, equation (4.5) leads to the same formula in Tsai et al. [53]. In addition, when the searching trajectory falls into the basin (the region where the Jacobian is negative definite), equation (4.4) is consistent with the traditional NR step.
To search the first-order saddles, instead of applying equation (4.5) to all the eigen-directions at each step we pick one maximization direction i, and set
| 4.6 |
only for the diagonal component i, forcing the quasi-Newton steps to converge to a saddle point. We initially select the maximization direction i at random, and at each step calculate the overlap/correlation between the maximization direction at the previous step with all eigen-directions and choose the most overlapped eigen-direction to be maximized at this iteration.
4.3. Constructing the epigenetic state network
To construct the ESN, we enumerate first-order saddles using the above conditional root-finding algorithm. For each saddle, we seek two fixed-point attractors (basins) connected to it through the saddle's unstable direction. To do this, we apply a small random perturbation on the saddle and track the dynamic flow followed by the vector field. By connecting two neighbouring basins (nodes) through their common saddle (links), we obtain the ESN of the underling dynamic system. Finally, we apply the Wentzell–Freidlin theory (equation (2.2)) to assign appropriate transition rate (weight) to each link. Note that the network is bi-directional and the weight matrix will be asymmetric in general.
The 10 ODEs we use to describe the FB-iPSC-CM system have 62 unknown parameters that lack sufficient experimental input to determine their values. As a common problem on studying complex biological systems, our strategy here is, instead of using a single set of parameters, to analyse an ensemble of models [33] with different parameter sets that are related to the cell-to-cell non-genetic variation rooted in intrinsic and extrinsic stochasticity [9,34]. These model parameter sets, however, are required to satisfy some biological constraints (electronic supplementary material, text 2). The cell-to-cell variation justifies modelling the cellular systems with a distribution of parameter sets. Here in this work, we treat these parameters as frozen during the process, with the implicit assumption that variation of these parameters is slow. In physics, it corresponds to quenched disorder.
The overall Monte Carlo numerical procedure of obtaining the ensemble averaged ESN for the FB-iPSC-CM system is summarized in the electronic supplementary material, figure S3 and text section 3.
4.4. Coarse-graining epigenetic state networks
We measure for all three ESNs the transition rates for each pair of neighbouring states. The fact that the transition rate distribution is highly uneven indicates that the reprogramming dynamics is mostly dominated by those paths with large transition rates (or equivalently, small passage times; electronic supplementary material, figure S5). Therefore, we calculate the minimum spanning tree that captures the ‘backbone’ of the ESN by minimizing the average passage time (defined as the reciprocal of the transition rate).
Acknowledgements
We are grateful to Drs John Tyson, Fuchou Tang, Kathy Chen and Yue Teng for illuminating constructive discussions.
Funding statement
This work is supported by NSF DMS-0969417, EF-1038636 (to J.X.) and DGE-0966125. C.S. thanks NSF, ARL and ONR for support.
References
- 1.Waddington CH. 1957. The strategy of the genes: a discussion of some aspects of theoretical biology. London, UK: Allen & Unwin. [Google Scholar]
- 2.Wales D. 2004. Energy landscapes: applications to clusters, biomolecules and glasses. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 3.Frauenfelder H, Sligar SG, Wolynes PG. 1991. The energy landscapes and motions of proteins. Science 254, 1598–1603. ( 10.1126/science.1749933) [DOI] [PubMed] [Google Scholar]
- 4.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. ( 10.1016/j.cell.2006.07.024) [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. 2009. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 104, e30–e41. ( 10.1161/CIRCRESAHA.108.192237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Efe JA, Hilcove S, Kim J, Zhou H, Ouyang K, Wang G, Chen J, Ding S. 2011. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 13, 215–222. ( 10.1038/ncb2164) [DOI] [PubMed] [Google Scholar]
- 7.Xu H, Yi BA, Chien KR. 2011. Shortcuts to making cardiomyocytes. Nat. Cell Biol. 13, 191–193. ( 10.1038/ncb0311-191) [DOI] [PubMed] [Google Scholar]
- 8.Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. 2010. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. ( 10.1016/j.cell.2010.07.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balazsi G, van Oudenaarden A, Collins JJ. 2011. Cellular decision making and biological noise: from microbes to mammals. Cell 144, 910–925. ( 10.1016/j.cell.2011.01.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kauffman S. 1969. Homeostasis and differentiation in random genetic control networks. Nature 224, 177–178. ( 10.1038/224177a0) [DOI] [PubMed] [Google Scholar]
- 11.Huang S, Kauffman S. 2012. Complex gene regulatory networks—from structure to biological observables: cell fate determination. In Computational complexity: theory, techniques, and applications (ed. Meyers RA.), pp. 527–560. New York, NY: Springer; ( 10.1007/978-1-4614-1800-9_35) [DOI] [Google Scholar]
- 12.Wang J, Li C, Wang E. 2010. Potential and flux landscapes quantify the stability and robustness of budding yeast cell cycle network. Proc. Natl Acad. Sci. USA 107, 8195–8200. ( 10.1073/pnas.0910331107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ao P. 2004. Potential in stochastic differential equations: novel construction. J. Phys. A: Math. Gen. 37, L25 ( 10.1088/0305-4470/37/3/L01) [DOI] [Google Scholar]
- 14.Xing J. 2010. Mapping between dissipative and Hamiltonian systems. J. Phys. A Math. Theor. 43, 375003 ( 10.1088/1751-8113/43/37/375003) [DOI] [Google Scholar]
- 15.Zhou JX, Huang S. 2011. Understanding gene circuits at cell-fate branch points for rational cell reprogramming. Trends Genet. 27, 55–62. ( 10.1016/j.tig.2010.11.002) [DOI] [PubMed] [Google Scholar]
- 16.Li C, Wang J. 2013. Quantifying Waddington landscapes and paths of non-adiabatic cell fate decisions for differentiation, reprogramming and transdifferentiation. J. R. Soc. Interface 10, 20130787 ( 10.1098/rsif.2013.0787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Wang J. 2013. quantifying cell fate decisions for differentiation and re-programming of a human stem cell network: landscape and biological paths. PLoS Comput. Biol. 9, e1003165 ( 10.1371/journal.pcbi.1003165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K, Sasai M, Wang J. 2013. Eddy current and coupled landscapes for nonadiabatic and nonequilibrium complex system dynamics. Proc. Natl Acad. Sci. USA. 110, 14 930–14 935. ( 10.1073/pnas.1305604110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enver T, Pera M, Peterson C, Andrews PW. 2009. Stem cell states, fates, and the rules of attraction. Cell Stem Cell 4, 387–397. ( 10.1016/j.stem.2009.04.011) [DOI] [PubMed] [Google Scholar]
- 20.MacArthur BD, Ma'ayan A, Lemischka IR. 2009. Systems biology of stem cell fate and cellular reprogramming. Nat. Rev. Mol. Cell Biol. 10, 672–681. ( 10.1038/nrm2766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian XJ, Zhang H, Xing J. 2013. Coupled reversible and irreversible bistable switches underlying TGF β-induced epithelial to mesenchymal transition. Biophys. J. 105, 1079–1089. ( 10.1016/j.bpj.2013.07.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang R, Shoemaker R, Wang W. 2011. Systematic search for recipes to generate induced pluripotent stem cells. PLoS Comput. Biol. 7, e1002300 ( 10.1371/journal.pcbi.1002300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Zhang K, Xu L, Wang E. 2011. Quantifying the Waddington landscape and biological paths for development and differentiation. Proc. Natl Acad. Sci. USA 108, 8257–8262. ( 10.1073/pnas.1017017108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glass L, Kauffman SA. 1973. The logical analysis of continuous, non-linear biochemical control networks. J. Theor. Biol. 39, 103–129. ( 10.1016/0022-5193(73)90208-7) [DOI] [PubMed] [Google Scholar]
- 25.Strogatz SH. 2001. Nonlinear dynamics and chaos: with applications to physics, biology, chemistry, and engineering. Boulder, CO: Westview Press. [Google Scholar]
- 26.Maier R, Stein D. 1997. Limiting exit location distributions in the stochastic exit problem. SIAM J. Appl. Math. 57, 752–790. ( 10.1137/S0036139994271753) [DOI] [Google Scholar]
- 27.Freidlin M, Wentzell A. 1998. Random perturbations of dynamical systems. Berlin, Germany: Springer. [Google Scholar]
- 28.Aurell E, Sneppen K. 2002. Epigenetics as a first exit problem. Phys. Rev. Lett. 88, 048101 ( 10.1103/PhysRevLett.88.048101) [DOI] [PubMed] [Google Scholar]
- 29.Chin MH, et al. 2009. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell 5, 111–123. ( 10.1016/j.stem.2009.06.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heng JCD, et al. 2010. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 6, 167–174. ( 10.1016/j.stem.2009.12.009) [DOI] [PubMed] [Google Scholar]
- 31.Ieda M, Tsuchihashi T, Ivey K, Ross R, Hong T, Shaw RM, Srivastava D. 2009. Cardiac fibroblasts regulate myocardial proliferation through β1 integrin signaling. Dev. Cell 16, 233–244. ( 10.1016/j.devcel.2008.12.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orkin SH, Hochedlinger K. 2011. Chromatin connections to pluripotency and cellular reprogramming. Cell 145, 835–850. ( 10.1016/j.cell.2011.05.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown KS, Sethna JP. 2003. Statistical mechanical approaches to models with many poorly known parameters. Phys. Rev. E 68, 021904 ( 10.1103/PhysRevE.68.021904) [DOI] [PubMed] [Google Scholar]
- 34.Raj A, van Oudenaarden A. 2008. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. ( 10.1016/j.cell.2008.09.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macdonald P, Almaas E, Barabási AL. 2005. Minimum spanning trees of weighted scale-free networks. Europhys. Lett. 72, 308–314. ( 10.1209/epl/i2005-10232-x) [DOI] [Google Scholar]
- 36.Xu H, et al. 2011. Highly efficient derivation of ventricular cardiomyocytes from induced pluripotent stem cells with a distinct epigenetic signature. Cell Res. 22, 142–154. ( 10.1038/cr.2011.171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira CF, et al. 2013. Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell 13, 205–218. ( 10.1016/j.stem.2013.05.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silva J, Barrandon O, Nichols J, Kawaguchi J, Theunissen TW, Smith D. 2008. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol. 6, e253 ( 10.1371/journal.pbio.0060253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plath K, Lowry WE. 2011. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 12, 253–265. ( 10.1038/nrg2955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buganim Y, Faddah D, Cheng A, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. 2012. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222. ( 10.1016/j.cell.2012.08.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez F, Boue S, Izpisua Belmonte JC. 2011. Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat. Rev. Genet. 12, 231–242. ( 10.1038/nrg2937) [DOI] [PubMed] [Google Scholar]
- 42.Ferrell JE. 2012. Bistability, bifurcations, and Waddington's epigenetic landscape. Curr. Biol. 22, R458–R466. ( 10.1016/j.cub.2012.03.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graham R, Haken H. 1971. Generalized thermodynamic potential for Markov systems in detailed balance and far from thermal equilibrium. Z. Physik 243, 289–302. ( 10.1007/BF01394858) [DOI] [Google Scholar]
- 44.Polo JM, et al. 2012. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151, 1617–1632. ( 10.1016/j.cell.2012.11.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barabasi AL. 2009. Scale-free networks: a decade and beyond. Science 325, 412–413. ( 10.1126/science.1173299) [DOI] [PubMed] [Google Scholar]
- 46.Wilson HR, Cowan JD. 1972. Excitatory and inhibitory interactions in localized populations of model neurons. Biophys. J. 12, 1–24. ( 10.1016/S0006-3495(72)86068-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tyson JJ, Baumann WT, Chen C, Verdugo A, Tavassoly I, Wang Y, Weiner LM, Clarke R. 2011. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 11, 523–532. ( 10.1038/nrc3081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu Y, Glaros T, Zhu M, Wang P, Wu Z, Tyson JJ, Li L, Xing J. 2012. Network topologies and dynamics leading to endotoxin tolerance and priming in innate immune cells. PLoS Comput. Biol. 8, e1002526 ( 10.1371/journal.pcbi.1002526) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agmon N, Hopfield JJ. 1983. Transient kinetics of chemical reactions with bounded diffusion perpendicular to the reaction coordinate: intramolecular processes with slow conformational changes. J. Chem. Phys. 78, 6947–6959. ( 10.1063/1.444643) [DOI] [Google Scholar]
- 50.Marcus RA. 1999. Electron transfer past and future. In Advances in chemical physics: electron transfer—from isolated molecules to biomolecules, part 1, vol. 106 (eds Prigogine J, Rice SA.), pp. 1–6. Hoboken, NJ: John Wiley; ( 10.1002/9780470141656.ch1) [DOI] [Google Scholar]
- 51.Walczak AM, Onuchic JN, Wolynes PG. 2005. Absolute rate theories of epigenetic stability. Proc. Natl Acad. Sci. USA 102, 18 926–18 931. ( 10.1073/pnas.0509547102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xing J. 2007. Nonequilibrium dynamic mechanism for allosteric effect. Phys. Rev. Lett. 99, 168103 ( 10.1103/PhysRevLett.99.168103) [DOI] [PubMed] [Google Scholar]
- 53.Tsai CJ, Jordan KD. 1993. Use of an eigenmode method to locate the stationary points on the potential energy surfaces of selected argon and water clusters. J. Phys. Chem. 97, 11 227–11 237. ( 10.1021/j100145a019) [DOI] [Google Scholar]
- 54.Stadtfeld M, Hochedlinger K. 2010. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 24, 2239–2263. ( 10.1101/gad.1963910) [DOI] [PMC free article] [PubMed] [Google Scholar]