

Key Points
Generated a reference transcriptome for ex vivo, cultured, and stimulated mast cells, contrasted against a broad collection of primary cells.
Identified BMPs as function-modulating factors for mast cells.
Abstract
Mast cells (MCs) mature exclusively in peripheral tissues, hampering research into their developmental and functional programs. Here, we employed deep cap analysis of gene expression on skin-derived MCs to generate the most comprehensive view of the human MC transcriptome ever reported. An advantage is that MCs were embedded in the FANTOM5 project, giving the opportunity to contrast their molecular signature against a multitude of human samples. We demonstrate that MCs possess a unique and surprising transcriptional landscape, combining hematopoietic genes with those exclusively active in MCs and genes not previously reported as expressed by MCs (several of them markers of unrelated tissues). We also found functional bone morphogenetic protein receptors transducing activatory signals in MCs. Conversely, several immune-related genes frequently studied in MCs were not expressed or were weakly expressed. Comparing MCs ex vivo with cultured counterparts revealed profound changes in the MC transcriptome in in vitro surroundings. We also determined the promoter usage of MC-expressed genes and identified associated motifs active in the lineage. Befitting their uniqueness, MCs had no close relative in the hematopoietic network (also only distantly related with basophils). This rich data set reveals that our knowledge of human MCs is still limited, but with this resource, novel functional programs of MCs may soon be discovered.
Introduction
Although mast cells (MCs) are commonly associated with the elicitation of immunoglobulin E–mediated allergic inflammation,1,2 the spectrum of possible MC functions has greatly expanded over the last few years to now cover processes in which MCs assume potentially detrimental (autoimmune and inflammatory diseases, obesity, or tumors) or potentially beneficial roles (anti-inflammatory/tolerogenic effects, other types/stages of cancer, or antimicrobial defense).3-10 With the exception of type I allergy (for which a large body of evidence also exists for humans), the roles that MCs play in human health and disease are undefined, because MC research to date strongly relies on studies in the mouse, even though MCs differ considerably between species.11
Research into the full range of functional programs of a cell requires information on its specific gene-activity profile. Previous transcriptome profiling efforts concentrated either on MCs alone (eg, stimulated vs baseline12-14 and MCs cultured from different sources15,16 or at different times of development17) or on comparisons between MCs and other leukocytes (either experimentally18 or in silico19). Although these studies provided valuable insights into the transcriptional landscape of MCs, they did not allow direct comparisons with nonimmune cells and were limited by the MC model(s) employed.
In a natural setting, MCs develop exclusively in peripheral tissues like skin, gut, and lung. For research purposes, however, MCs are typically first expanded in culture, though it is currently unknown how well cultured MCs are representative of their in vivo counterparts.
Here, we used cap analysis of gene expression (CAGE) technology,20 mapping transcription start sites (TSSs) and their relative expression levels, to obtain a comprehensive view of the human MC transcriptome. The major improvements are as follows: (1) we use skin MCs immediately ex vivo (and compare them directly with cultured skin-derived MCs); (2) transcriptome profiling is based on a quantitative technique combining cap trapping with high-throughput next-generation sequencing; (3) MCs are embedded in the Functional Annotation of Mammalian Genome 5 (FANTOM5) project, which provides transcriptome data on a large collection of 975 samples,21 so that gene activity in MCs can be put immediately into perspective; and (4) we provide additional information on preferential promoter usage and associated motif activity. Our study reveals that a multitude of nonannotated genes are particularly active in the lineage, several of which are only detectable in MCs ex vivo. Collectively, these findings underline that our current knowledge of human MCs is fairly limited. This paper can serve as a catalyst to spur further research into the functional programs of MCs and the specific roles these cells assume in health and disease in humans. This work is part of the FANTOM5 project. Data downloads, genomic tools, and copublished manuscripts are summarized online at http://fantom.gsc.riken.jp/5/.
Methods
Skin MC purification and culture
MC purification was performed as described previously.22,23 The skin was obtained from cosmetic breast-reduction surgeries with informed consent of the patients. All experiments were conducted according to the Declaration of Helsinki Principles and approved by the ethics committee of the Charité Universitätsmedizin Berlin.
Briefly, skin was cut into strips and treated with dispase (Becton Dickinson, Heidelberg, Germany) at 4°C overnight. After removal of the epidermis, the dermis was chopped into small pieces and digested with collagenase (Worthington, Lakewood, NJ), hyaluronidase (Sigma, Deisenhofen, Germany), and DNase I (Roche, Basel, Switzerland) for 1 hour at 37°C. After 3 steps of filtration, the remaining tissue was subjected to a second digestion step. MC purification from the dispersates was achieved by selection with anti–human c-Kit microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and an automated magnetic-activated cell sorting separation device. Viability (trypan-blue exclusion) and purity (acidic toluidine-blue staining) exceeded 99%. A total of 0.8 to 1.1 × 107 cells (from 1 donor) were immediately lysed in TRIzol and further processed for HeliScopeCAGE. MCs from 3 donors were used for the ex vivo analysis (donors 2, 3, and 4). To obtain the expanded samples, MCs were cultured for 4 to 5 weeks (donors 1, 5, and 8), as described previously.24 Stimulation was achieved by FcεRI crosslinking with AER-37 at 2 µg/mL (eBioscience, San Diego, CA). Only samples showing >60% of activation (degranulation; CD107a upregulation) were included as stimulated samples in the deep-CAGE analysis.
CAGE data, clustering, and gene assignment
The MC CAGE data and the entire FANTOM5 data set have been submitted to the DNA Data Bank of Japan database under accession numbers DRA000991 (samples from donors 1-4) and DRA001026 (samples from donors 5 and 8). Additional supplementary processed data files are available online at http://fantom.gsc.riken.jp/5/suppl/Motakis_et_al_2013. The details of the CAGE library generation and clustering are explained in the FANTOM5 main manuscript.21 Briefly, ∼4 million CAGE tags for each library were aligned to the genome (Hg19), neighboring tags were grouped into clusters, and decomposition-based peak identification (DPI) was applied to decompose clusters into independently regulated subregions (DPI peaks). The DPI peaks were annotated based on known transcript 5′ ends within 500 bases and summarized into regions (supplemental Methods, available on the Blood Web site).
The methods for RNA extraction, bioinformatics analysis, MC treatments, flow cytometry, histamine measurement, reverse-transcription quantitative polymerase chain reaction, and enzyme-linked immunosorbent assay are described in supplemental Methods.
Results
CAGE expression profiles of known MC marker genes
From the initial 184 827 FANTOM5 regions,21 15 643 annotated genes and 17 872 unannotated transcribed regions (33 515 in total) were expressed by MCs (supplemental Methods). Using this data set, we first focused on well-defined signature genes of the MC lineage, which were highly expressed by MCs and otherwise detected in a limited spectrum of cell types (Table 1). The most specific lineage markers were the MC-specific proteases. The profile was largely consistent with the expectation, although it has never been demonstrated for so many samples in direct comparison. There was no expression of lineage markers of other skin cells in the MC samples (THY1 for fibroblasts, KDR/CDH5 for endothelial cells, KRT10/IVL for keratinocytes, TYR for melanocytes, and CD1A/CD207 for Langerhans cells), demonstrating an exceptional degree of MC purity.
Table 1.
Expression pattern of genes encoding well-defined MC markers
| Gene | MC ex vivo* | MC cultured† | Cultured + stimulated‡ | (Next) Primary sample§ | Mean all|| | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D2 | D3 | D4 | D1 | D8 | D5 | D1 | D8 | D5 | |||
| KIT | 3 272 | 3 633 | 3268 | 4 426 | 2906 | 3 125 | 2094 | 1 261 | 2195 | 548 (melanocyte) | 16 |
| IL1RL1 | 656 | 1 156 | 418 | 487 | 340 | 423 | 1631 | 488 | 1073 | 423 (lung) | 8 |
| FCER1A | 811 | 657 | 1087 | 2 699 | 1774 | 2 405 | 1211 | 1 670 | 2983 | 3272 (immature LC) | 19 |
| MS4A2 | 1 163 | 1 717 | 1284 | 2 173 | 1109 | 2 827 | 1580 | 533 | 1759 | 60 (basophil) | 1 |
| ENPP3 | 312 | 454 | 273 | 890 | 700 | 920 | 3209 | 1 116 | 1084 | 157 (basophil) | 3 |
| HDC | 994 | 1 091 | 864 | 1 516 | 772 | 925 | 496 | 400 | 707 | 255 (basophil) | 5 |
| TPSAB1-TPSB2 | 2 536 | 1 332 | 2650 | 671 | 250 | 991 | 0 | 57 | 492 | 76 (stimulated MΦ) | 0.1 |
| TPSD1 | 552 | 783 | 558 | 317 | 972 | 1 585 | 40 | 518 | 925 | 5 (gallbladder) | 0.2 |
| CMA1 | 1 841 | 1 919 | 1240 | 631 | 119 | 1 210 | 502 | 203 | 624 | 8 (left atrium) | 0.2 |
| CPA3 | 5 057 | 6 001 | 2824 | 7 089 | 7295 | 7 883 | 7305 | 5 884 | 4386 | 239 (CD34+ diff. ery.) | 4 |
| CTSG | 10 520 | 12 133 | 6902 | 14 981 | 7274 | 16 961 | 7000 | 10 752 | 9271 | 1278 (CD34+ BM) | 37 |
| HPGDS | 1 245 | 1 220 | 1220 | 4 097 | 3088 | 3 545 | 3489 | 2 587 | 3182 | 333 (immature LC) | 5 |
| LTC4S | 199 | 270 | 226 | 1 830 | 3261 | 1 684 | 81 | 1 478 | 329 | 194 (immature LC) | 7 |
BM, bone marrow; CD34+ diff. ery. = CD34+ cells differentiated along the erythroid pathway; D, donor; LC, Langerhans cell; MΦ, macrophage.
Donor 2/donor 3/donor 4 (directly upon isolation from skin).
Donor 1/donor 8/donor 5 (MCs expanded in culture for 4-5 weeks).
Donor 1 stimulated for 2.5 hours/donor 8 stimulated for 5 hours/donor 5 stimulated for 16 hours.
Nontransformed cell/tissue sample with the (second) highest expression of the gene.
Mean expression of all FANTOM5 non-MC samples. The values are given in (rounded) tags per million tags.
Reproducibility among samples was statistically assessed by principal component and correlation analysis, further highlighting the excellent quality of the data set, supported by an estimated biological coefficient of variation of 0.3846 for the MC samples.25
Identification of novel genes exclusively or preferentially active in MCs
The simultaneous availability of genome-wide transcriptome data for a large collection of samples allowed the search for additional genes with MC-restricted activity. The genes depicted in Table 2 had fold change = mean (MCs) / mean (all non-MC FANTOM5 samples) > 100, representing novel bona fide MC markers. The (putative) functions of the genes were searched in public databases (http://www.proteinatlas.org; http://www.genecards.org) if no specific references are given. Many genes identified herein have no annotated function so far.
Table 2.
Genes with an MC-restricted expression pattern newly identified by FANTOM5
| Gene | MC ex vivo* | MC cultured† | Cultured + stimulated‡ | (Next) Primary sample§ | Mean all|| | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D2 | D3 | D4 | D1 | D8 | D5 | D1 | D8 | D5 | |||
| MRGPRX2 | 1059 | 938 | 949 | 4 | 32 | 220 | 2 | 21 | 146 | 4 (fetal skin) | 0.1 |
| RGS13 | 942 | 1090 | 902 | 385 | 747 | 604 | 620 | 472 | 1636 | 89 (appendix) | 2 |
| C1orf150 | 1231 | 822 | 1059 | 1680 | 482 | 1256 | 6440 | 1949 | 2779 | 313 (basophil) | 5 |
| SIGLEC6 | 673 | 518 | 1190 | 1887 | 1634 | 1614 | 360 | 1166 | 1422 | 265 (placenta) | 4 |
| ERVFRD-1 | 65 | 89 | 75 | 734 | 176 | 315 | 1745 | 463 | 916 | 355 (retinal pigment epithelial cell), 132 (placenta) | 2 |
| SVOPL | 31 | 52 | 40 | 173 | 105 | 217 | 104 | 93 | 113 | 18 (CD34+ HPC) | 0.6 |
| C20orf118 | 46 | 80 | 69 | 100 | 27 | 172 | 19 | 22 | 107 | 46 (small intestine) | 0.4 |
| VWA5A | 1400 | 1180 | 1308 | 1646 | 1469 | 1908 | 453 | 749 | 2079 | 257 (amniotic membrane) | 14 |
D, donor; HPC, hematopoietic progenitor cell.
Donor 2/donor 3/donor 4 (directly upon isolation from skin).
Donor 1/donor 8/donor 5 (MCs expanded in culture for 4-5 weeks).
Donor 1 stimulated for 2.5 hours/donor 8 stimulated for 5 hours/donor 5 stimulated for 16 hours.
Nontransformed cell/tissue sample with (second) highest expression of the gene.
Mean expression of all non-MC FANTOM5 samples. The values are given in (rounded) tags per million tags; if <1, 1 decimal place is given.
c1orf150 was exclusively active in MCs and basophils, whereas expression of C20orf118 was confined to MCs alone. The function of both genes is unknown.
ERVFRD-1 (HERV-FRD) originates from an ancient infection by retroviruses and can induce cell-cell fusion, a phenomenon found in placenta.26 Its preferential expression in MCs raises the possibility of fusogenic capacity, as described for selected cell types.27
MRGPRX2 was exclusively active in MCs ex vivo and exhibited the highest fold change among candidates. It mediates MC degranulation by nonimmunologic secretagogues28 and binds cathelicidin.29 Thus, MRGPRX2 may play important roles in skin MC activation.
Though not anticipated to be MC specific, RGS13 has well-understood functions in MCs, operating as a negative regulator of MC signaling.30
SIGLEC6 is generally viewed as placenta specific31 but was found here to be substantially more active in MCs than in placenta.
SVOPL has no known function but is related to the synaptic vesicle protein SVOP.
VWA5A is functionally undefined but ranks in the top position among genes influencing longevity.32
MCs are excellently suited to delineate the biological significance of these poorly defined genes in future studies.
In addition, we identified a substantial host of genes with highest activity in MCs and enrichment by at least 10-fold compared with the mean of all 893 FANTOM5 samples (supplemental Table 2). Although several genes are associated with MC biology (such as granule biosynthesis/function33,34), the function of others is currently ignored. This indicates that our view of MCs is largely incomplete.
FANTOM5 profiling data are excellently suited to stimulate research into new functional programs of MCs: BMPR1A as a modulator of MC function
Of the genes preferentially expressed by MCs, BMPR1 appeared attractive for further analysis because of its well-documented role in hematopoietic stem cells (HSCs),35 with which MCs share common features (see below).
We initially verified that skin cells produced BMPR ligands (BMP2/4), so that MCs were exposed to these mediators in their natural surroundings. Because BMPR activation stimulates lineage-specific transcription factors in HSCs,36 we tested the impact of BMPR stimulation on the expression of markers and transcription factors (TFs) of the lineage.37 FcεRIα and GATA-1 transcripts were selectively increased by BMP signaling (Figure 1A), probably through direct activation of lineage-specific TFs by Smads.37
Figure 1.

BMPR1 regulates MC effector functions and increases MC survival. (A) MCs were stimulated with BMP4 at 20 ng/mL for the times indicated and the expression of selected MC marker genes quantified by reverse-transcription quantitative polymerase chain reaction and normalized to β-actin. Data are shown in relation to cells kept in the absence of bone morphogenetic protein (BMP) for the same times. (B) Cultured MCs (last addition of SCF 5-7 days earlier) were washed and replated in fresh media with or without BMP4 (20 ng/mL) for 24 hours, after which time histamine release was elicited by FcεRI crosslinking with different concentrations of AER-37. IgER, immunoglobulin E receptor. (C) Cultured MCs were stimulated by AER-37 (0.1 μg/mL) for 2 hours, washed, and replated in fresh media with or without BMP4 (20 ng/mL) ± stem cell factor (SCF) (100 ng/mL) for 48 hours. After this time, cells were subjected to a second round of stimulation by AER-37. MCs are largely refractory to a second stimulation for prolonged times, but BMP accelerates this recovery. Although SCF alone also accelerates recovery, the effects from BMP and SCF are additive. (D) MCs after isolation were cultured in the presence or absence of the indicated factors for 10 days, and recovery of viable cells was assessed. The numbers indicate growth factor concentrations in ng/mL. All data in this figure are the mean ± standard error of the mean from 5-7 independent assays. *P < .05; **P < .01; ***P < .001.
In addition, BMP4 signaling increased the sensitivity to FcεRI aggregation (Figure 1B). After stimulation, MCs become refractory to a second stimulus, but BMP4 assisted in MC recovery from refractoriness, increasing responsiveness to subsequent FcεRI crosslinking after the first stimulus (Figure 1C).
Ligation of BMPR also prolonged MC survival, and this effect required low concentrations of BMP2 (Figure 1D; SCF is given as positive control). In summary, rational use of the FANTOM5 data sets allowed us to uncover BMPR1A as a new functional receptor of human MCs that crosstalks with the FcεRI pathway.
Genes with unexpectedly weak activity in MCs
Next, we focused on genes commonly investigated in MCs. There is controversy as to whether MCs serve as antigen-presenting cells.38 Here, MCs were found to express negligible levels of the different MHCII members (Table 3). Additionally, ex vivo MCs expressed virtually no chemokine receptors. The inconsistency with previous reports39 may be a result of the MC subset employed, because in vitro–expanded MCs showed increased expression of several receptors (Table 3).
Table 3.
Transcripts of important immune receptors underrepresented in MCs
| MC ex vivo* | MC cultured† | Cultured + stimulated‡ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | D2 | D3 | D4 | D1 | D8 | D5 | D1 | D8 | D5 | First primary sample§ | Mean all|| |
| Receptors involved in antigen presentation | |||||||||||
| HLA-DRA | 45 | 30 | 25 | 0.1 | 0 | 0 | 0 | 0 | 0.1 | 26 120 (migratory LC) | 798 |
| HLA-DPA1 | 28 | 20 | 16 | 8.8 | 22 | 18 | 10 | 27 | 16 | 13 278 (migratory LC) | 408 |
| HLA-DPB1 | 16 | 13 | 17 | 2 | 4.6 | 7.5 | 0.9 | 13 | 4.2 | 13 319 (migratory LC) | 239 |
| HLA-DQA1 | 0.5 | 1 | 6.2 | 0 | 0.4 | 0 | 0 | 0 | 0 | 17 246 (migratory LC) | 143 |
| HLA-DQA2 | 26 | 1.4 | 4 | 0 | 0 | 0 | 0 | 0 | 0.1 | 3 963 (migratory LC) | 38 |
| Chemokine receptors | |||||||||||
| CCR1 | 0.2 | 0 | 0 | 0 | 0 | 0 | 0 | 0.7 | 0 | 705 (stim. monocyte) | 18 |
| CCR2 | 0 | 0.4 | 0.2 | 0 | 1.2 | 0.1 | 0.3 | 0.7 | 0 | 178 (monocyte) | 5.3 |
| CCR3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 190 (whole blood) | 2.3 |
| CCR4 | 2.6 | 0.5 | 1.4 | 21 | 6.6 | 80 | 1.3 | 3.5 | 27 | 1 384 (memory Treg) | 19 |
| CCR5 | 0 | 0 | 0.2 | 0.4 | 2.5 | 0.7 | 0 | 0.7 | 0.8 | 199 (stim. monocyte) | 7.8 |
| CCRL2 | 0 | 0 | 0.2 | 0.4 | 0.2 | 1.2 | 3.4 | 0.7 | 1 | 470 (stim. monocyte) | 8 |
| CXCR3 | 0.7 | 0.4 | 0.5 | 4.2 | 0.2 | 5.6 | 4.1 | 7.7 | 40 | 359 (plasmacytoid DC) | 5.9 |
| IL8RA/CXCR1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 216 (whole blood) | 13 |
| Pattern recognition receptors | |||||||||||
| TLR1 | 0.9 | 1.2 | 1.2 | 0.3 | 1.9 | 0.4 | 0 | 0.7 | 2.4 | 434 (neutrophil) | 12 |
| TLR2 | 4.7 | 3.9 | 2.3 | 0 | 1 | 0 | 0 | 0 | 0.4 | 896 (eosinophil) | 29 |
| TLR3 | 1.4 | 0.7 | 0.5 | 3.9 | 2.5 | 3.4 | 0.3 | 0.7 | 3.6 | 114 (placenta) | 6.5 |
| TLR4 | 3.5 | 11 | 5.4 | 75 | 22 | 44 | 28 | 16 | 28 | 1 085 (stim. monocyte) | 55 |
| TLR7 | 0.9 | 0.4 | 0.8 | 0.1 | 0.2 | 0 | 0 | 0 | 1.5 | 343 (plasmacytoid DC) | 7.7 |
| TLR9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 162 (plasmacytoid DC) | 1.9 |
DC, dendritic cell; LC, Langerhans cell; stim., stimulated; Treg, regulatory T cell.
Donor 2/donor 3/donor 4 (directly upon isolation from skin).
Donor 1/donor 8/donor 5 (MCs expanded in culture for 4-5 weeks).
Donor 1 stimulated for 2.5 hours/donor 8 stimulated for 5 hours/donor 5 stimulated for 16 hours.
Nontransformed cell/tissue sample with (second) highest expression of the gene.
Mean expression of all non-MC FANTOM5 samples. The values are given in (rounded) tags per million tags; if <10, 1 decimal place is given.
Finally, the expression pattern of TLRs, by which MCs supposedly contribute to immune surveillance, was re-examined.8,9 Surprisingly, skin MCs expressed minute amounts of TLR genes if compared directly with other immune cells (Table 3). Although TLRs may activate MCs in selected microenvironments, the data conclusively explain our previous inability to detect TLR2 and TLR4 in MCs ex vivo or to activate them by specific ligands (M.B. and S.G., unpublished data).
MCs change their transcriptome in in vitro surroundings
We found significant changes in the transcriptome of cultured vis-à-vis ex vivo MCs. Out of 33 515 expressed regions, 5826 differentially expressed targets were identified (1802 RefSeq annotated; Figure 2A). A total of 85.5% of all differentially expressed (and 70.1% of the annotated) targets were downregulated, implying that various transcripts, which accumulate in MCs in vivo, are not maintained outside of the tissue. Functional classification by Gene Ontology revealed that cultured MCs were enriched in selective enzymes (oxidoreductases and transferases), indicating metabolic shifts occurring simultaneously with cell-cycle progression24 (Figure 2B). Accordingly, genes involved in energy metabolism and biosynthetic pathways (respiratory chain, cholesterol/nucleotide biosynthesis, pentose phosphate shunt; supplemental Table 3) were overrepresented, and so were cell-cycle regulators. In contrast, TF genes strongly dominated in the ex vivo samples (Figure 2A-B). Because TFs determine the expression of gene networks, it is reasonable that their expression coordinates the activity of multiple downstream genes, explaining the greater diversity in the gene repertoire active in MCs in the tissue.
Figure 2.
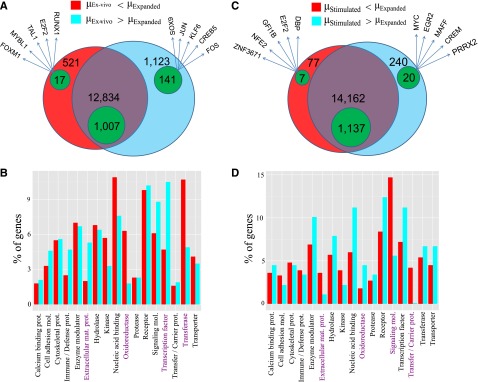
MCs change their transcriptome in in vitro surroundings and upon FcεRI crosslinking. Comparison of the different MC samples by Venn diagrams (A,C) and protein class Gene Ontology analysis (B,D). (A) Venn diagram with the number of RefSeq genes expressed at lower level in ex vivo MCs (red/green-within-red areas) and at higher level in ex vivo MCs (cyan/green-within-cyan areas) as well as those nondifferentially regulated (purple/green-within-purple areas). The genes are specified in supplemental Table 3. The green areas denote the number of TF genes in each group. Five indicative TFs from the differentially regulated lists are also given. (B) Significant protein class terms (α = 5%) of the differentially regulated groups. Pronounced differences are highlighted in purple. (C) Venn diagram with the number of genes expressed at lower/higher level in stimulated as opposed to expanded MCs in analogy to panel A. The genes are specified in supplemental Table 4. (D) Significant protein class terms in stimulated vs expanded MCs in analogy with panel B.
Strikingly, several genes of other blood lineages were induced in cultured MCs (multilineage markers along with the platelet gene ITGA2B and the erythroid regulator GFI1B40), suggesting that MCs may undergo partial de- or transdifferentiation in vitro. This was further supported by the de novo appearance of genes from other tissues, in particular UTS2 (expressed by cultured MCs and parts of the nervous system) and MAOA (highest in cultured MCs and adipocytes).
Changes in the MC transcriptome following activation
The most commonly studied (and clinically relevant) activation pathway for MCs is FcεRI aggregation. Here, we found 260 upregulated and 84 downregulated targets (supplemental Table 1; Figure 2C). Signaling components and carrier proteins specifically decreased, whereas oxidoreductases increased upon stimulation (Figure 2D). These changes likely contribute to desensitization (ie, unresponsiveness to a second stimulus) and survival, because a striking number of genes were associated with apoptosis and cell-cycle progression (with proapoptotic genes being coordinately downregulated and antiapoptotic genes upregulated). Therefore, stimulated cells seem to protect themselves from apoptosis but also halt the cell cycle to fully recover from the incisive event of stimulation (supplemental Table 4).
On the other hand, soluble mediators were the most potently induced genes following FcεRI aggregation (eg, CCL1 and IL-5; supplemental Table 4). Receptors responsible for the communication with T cells were likewise upregulated (supplemental Figure 2A shows OX40L and TNFRSF9/ILA at the protein level). Apart from several exceptions, there was good overall agreement with previous profiling efforts, implying that this pathway is fairly consistent among MC subsets.12-16
IL-31 was appreciably expressed in only one of the stimulated MC samples, suggesting particularly tight control. Testing MCs from 10 individuals, we found highly variable IL-31 protein levels among donors (supplemental Figure 2B). This result adds to the importance of MCs in the pruritus network,41 whereas interindividual variability may contribute to diseases characterized by chronic itch.
In addition to the essentially expected pattern of soluble mediators and coreceptors, unexpected genes were likewise induced by stimulation. The most striking examples were organ-specific genes, such as XIRP1 (heart42) and AQP2 (kidney43).
In summary, activation of MCs induces not only expected transcripts but also selected markers of unrelated tissues.
Hierarchical clustering and heatmap analysis
We employed hierarchical clustering to identify patterns of genes coexpressed with significant TFs (see Babina et al36 and references therein) and with the newly discovered marker MRGPRX2.
Gata1/Gata2 cluster
GATA1/GATA2 belong to the large cluster shown in Figure 3A. A variety of genes with MC-selective expression that tended to be upregulated in culture were found in this group (color distribution in Figure 3A; red predominates in the MC samples on the left). In addition to GATA1/GATA2, this cluster contains additional TF genes (eg, HOXB2, LYL1, and SOX13), and numerous genes of this cluster are validated targets of these TFs (eg, GATA2/KIT44 and GATA1/HOXB245). Further TF-gene pairings may be uncovered based on this analysis.
Figure 3.

Clusters of MC-relevant genes. The hierarchical clustering of 49 FANTOM5 hematopoietic lineage samples using all data and following extraction of subclusters for (A) GATA1/GATA2 (tree shown in panel A is that same as in panels B and C), (B) MITF, and (C) MRGPRX2. The algorithm is described in the supplemental Methods. More detailed, higher-resolution versions are provided as supplemental Figures 3-5.
MITF cluster
In contrast to GATA1/GATA2, the MITF cluster contained virtually no genes with MC-restricted expression (Figure 3B). Genes found here were active not only in monocytes/MCs but also in melanocytes (IDH1, NQO1, and TSPAN4), although melanocytes were not included in the cluster analysis, providing independent validation of the clustering procedure. The osteoclast/melanoblast marker GPNMB, a well-defined MITF-target,46 was captured by our clustering analysis.
MRGPRX2 cluster
The MRGPRX2 cluster turned out to be particularly informative, because it encompassed newly identified genes exclusively or particularly active in MCs as well as several TFs (TAL1, PBX1, MEIS2, and ERG; Figure 3C).
Indeed, PBX1 and MEIS2 are known to cooperate, regulating a variety of genes, and they can also form complexes with KLF447 (a TF with particularly high activity in MCs, as discovered here). PBX1 is involved in development and Mullerian-duct formation,48 and AMHR2 expression (receptor for anti–Mullerian hormone and part of the cluster) was unexpectedly highest in MCs. These TFs may therefore well contribute to the MC-selective gene signature and warrant further exploration. In keeping with the much higher expression of MRGPRX2 in ex vivo MCs, the majority of the genes in this cluster were preferentially active in ex vivo MCs (Figure 3C; red predominates among the MC samples on the right).
More detailed, higher-resolution versions of these 3 clusters are provided in supplemental Figures 3-5.
MC-specific promoters and motif activity
The above analyses concentrated on gene-expression patterns in MCs vs non-MCs to identify gene-activity programs that distinguish MCs from other cells. Contrary to microarrays, however, CAGE not only quantifies gene expression but also precisely maps TSSs, thereby identifying the specific promoter(s) used (included in supplemental Tables 3 and 4). In addition to genes overexpressed by MCs, we found selective promoters particularly active in MCs also in more broadly expressed genes. Supplemental Table 5 gives promoters hyperactive in the lineage (expression 50-fold higher in MCs vs mean of non-MCs) and contrasts them against the overall expression of the respective genes; at the top of these lists are examples (LHX3, STX3, and C11orf49 for ex vivo MCs) for which MC selectivity of a particular promoter surpassed MC selectivity at the gene level by more than 1000-fold.
We also found that promoter preference can change in dependence of the microenvironment. EXOC6B is an interesting example, because it forms part of the exocytosis machinery (substantially more active in cultured vis-à-vis ex vivo MCs24). EXOC6B was highly expressed by MCs, with no change between subsets at the gene level but striking difference at the promoter level (supplemental Figure 6), perhaps giving rise to transcripts with altered translational activity or stability. Similar promoter swaps were detected in several other genes upon culture (supplemental Table 6), whereas no such genes could be identified upon stimulation.
The mapping of TSSs in MC transcripts allowed identification of motifs preferentially enriched in active MC promoters. By using the previously published motif activity response analysis approach,49 which fits the CAGE expression profiles to computationally predicted regulatory sites for TFs, we found important regulators that explain the CAGE data variation and predict the regulatory role and importance of each regulator (supplemental Methods). Supplemental Table 7 ranks these motifs according to their activity.49 Interestingly, the motif most active in ex vivo MCs turned out to be PATZ1, which so far has been associated with spermatogenesis and testis development.50 In fact, PATZ1 expression was higher in MCs than in most other blood cells, and MCs unexpectedly expressed several testicular genes. Another interesting motif was FOXJ1, with fundamental roles in developmental processes,51,52 whereas nothing is known about its role in mastopoiesis. The FOXJ1 target gene calpastatin51 correlated with FOXJ1 expression in MCs. The preferential expression and activity of several FOX family TFs (FOXI2 and FOXJ1) suggests that they may have fundamental yet hitherto-overlooked roles in the MC lineage.
Although multiple regulatory levels exist between the transcription of a TF gene and activity of the respective protein in the nucleus, various TFs with particularly active motifs were also highly expressed by MCs (eg, KLF4, CREB1, and ELF1), indicating that at least one part of their activity stems from transcriptional regulation.
MCs in the hematopoietic network
There is consensus that human MCs are derived from HSCs, but their relationship with other bone-marrow derived cells is barely defined, and MCs were not included in efforts examining the global architecture of hematopoiesis.53 Thus, the FANTOM5 data set provides the unique opportunity to position MCs relative to other lineages. To this end, principal component analysis was applied to 49 samples for the purpose of deriving components representing large fractions of data variability. PC1-3 cumulatively explains >80% of the variance (supplemental Methods). PC1, the dominant component, segregated the samples mainly by source (blood, tissue [ie, bone marrow/skin], or culture), whereas PC2 segregated the samples by cell type (lymphoid, myeloid long-lived/plastic, or myeloid short-lived/end stage) (Figure 4A left). Accordingly, ex vivo MCs clustered with Langerhans cells and HSCs (cells from skin or immediately after exit from bone marrow), whereas cultured MCs clustered with other cultured cells in PC1. In PC2, MCs were found to belong to the myeloid/plastic subset. Of particular interest, however, was the separation of MCs from all other samples in PC3, potentially explaining the uniqueness of MCs (Figure 4A right). No other cell showed a separation of this kind. In keeping with principal component analysis, MCs had likewise no close neighbor by Pearson correlation (Figure 4B). The best concordance for ex vivo MCs was again found with HSCs (perhaps as a result of similar TF patterns54) and Langerhans cells. MCs showed a more-than-expected relation with all lymphocytes (Figure 4A-B), which fits previous observations.55 Therefore, MCs may be tentatively positioned between myeloid and lymphoid cells (Figure 4A), which would favor an early separation of the MC lineage.
Figure 4.
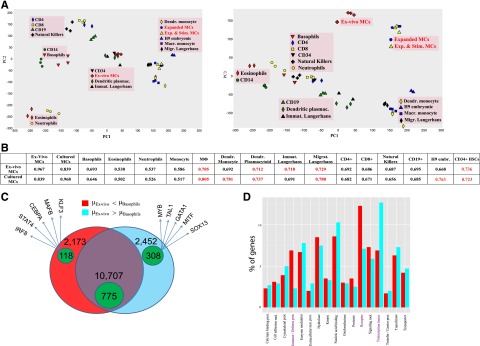
MCs in the hematopoietic network. (A) Principal component analysis of 49 blood samples of the FANTOM5 data set (H9 embryonic stem cells are included for further comparison) as described in supplemental Methods. The first 3 principal components (PCs) explain 82.3% of the total variance (see supplemental Methods). (B) Means of all pairwise Pearson correlation coefficients for each sample pair as described in supplemental Methods. The mean coefficients of MCs vs other blood samples that are >0.7 are depicted in red. (C) Venn diagram with the number of genes expressed at higher level (red/green-within-red areas), and at lower level in basophils vs ex vivo MCs (cyan/green-within-cyan areas) and nondifferentially expressed RefSeq genes (purple/green-within-purple areas). The genes are specified in supplemental Table 8. The green areas denote the number of TF genes in each group. Five indicative TFs are given for each cell. (D) Significant protein class terms (α = 5%) of the differentially expressed groups.
The similarity between MCs and basophils seemed fairly limited. Because developmental relationships between the cells in humans have remained obscure, we analyzed this relationship in greater detail. Of a total of 15 332 annotated (non-TF) genes, 4625 were differentially expressed, with slightly more genes being overrepresented in MCs (Figure 4C). The almost equal distribution at the level of non-TF genes was skewed in favor of MCs when TF genes were regarded (72.3% higher in MCs, 27.7% higher in basophils), further highlighting that greater TF diversity is a hallmark of MCs.
A closer inspection of the differential genes (supplemental Table 8) revealed that MCs overexpressed not only typical lineage markers but also genes shared between MCs and other cell/tissue subsets, including the brain (eg, CALB2, DIP2C, EPB41L1, KIAA1549, L1CAM, and NTM). Basophils, on the other hand, expressed a gene set typically associated with immune/inflammatory responses (eg, S100 family, TREM, and HLA-DRA). The most differential genes on comparison with MCs were expressed by other myelocytes, not just by basophils, putting basophils closer to other blood cells (Figure 4A). In fact, barely any truly basophil-specific genes could be identified, in stark contrast with MCs (also underscored by the divergence in the number of specific promoters: 39 in basophils vs 542 in ex vivo MCs; supplemental Table 5).
Taken together, basophils represent typical myeloid cells dedicated to host defense and immune modulation (Figure 4D). Conversely, MCs express more genes exclusively active in the lineage or shared by cells/tissues not primarily dedicated to immune function. As a result, various genes are specifically overexpressed by MCs, but only few are specific for basophils. Overall, this speaks against a common dual precursor of MCs/basophils in humans, in contrast to what was found in the mouse.56 In fact, mouse MCs have many characteristics of (human) basophils, including dependency on IL-3 for their development.11
Discussion
FANTOM5 has generated the most comprehensive transcriptome data collection using sophisticated technical and bioinformatics tools. The first-time-ever inclusion of human MCs in a global expression atlas has unequivocally demonstrated the individual character of MCs and suggested their participation in as-yet-undiscovered physiological processes. The unique character of MCs is owed to several “private genes” expressed strongly by MCs but only weakly (or not at all) by other constituents of the human body. Both well-characterized MC markers (Table 1) and genes newly discovered as MC specific in this study (Table 2) fall into this category. Moreover, MCs express a plethora of other genes at the highest levels (supplemental Table 2) and are enriched in entire networks (eg, G-protein–coupled receptors, associated signaling intermediates/negative regulators and G-protein–regulated TFs as well as genes related to the TGF network).
In addition, MCs not only express a multitude of genes not associated with the lineage before but also use MC-specific promoters to express non–MC-specific genes. This finer level of resolution is only achievable with deep sequencing.
Although MCs are derived from HSCs, their distinctiveness is underlined by the absence of several transcripts, which normally accumulate in (innate) immune cells, by the absence of a near neighbor in the hematopoietic network and by the profound difference vis-à-vis basophils, despite their overlap regarding few genes not shared by other cells. Although basophils seem typical myelocytes dedicated to host defense and immune regulation, several overexpressed transcripts of MCs are actually signature genes of (unrelated) organs, especially brain (which may be explained by the similarity in their exocytosis machineries and the presence of neurotransmitter/neurotransmitter receptors in MCs) and reproductive tissues (epididymis, testis, ovary, and placenta [eg, ADAM12, HERV-FRD, SIGLEC6, SPAG8, SPATA16, and WNK3]). Considering the extremely limited expression of some of these genes, their activity in MCs is remarkable, and research into their implication in the lineage may identify unanticipated functions of both the cells and the genes.
The similarity with reproductive tissues, together with the relationship of MCs with HSCs and even embryonic stem cells (Figure 4A-B), implies that MCs may be equipped with a certain degree of “stemness” and that their plasticity goes beyond what is currently assumed.38
In fact, we show directly that MCs experience profound changes when exposed to an altered microenvironment (Figure 2A-B). Because human MC research relies almost exclusively on cultured MCs, gene-expression patterns and functional properties of the lineage may have been missed so far.
Understanding the molecular pathways effective in MCs is a prerequisite to modulate deregulated MC activities. The unique gene signature of MCs offers the possibility to specifically target these cells in therapeutic settings without serious side effects. Therefore, our study also offers an excellent basis for the identification of MC-specific targets for pharmacologic intervention.
In summary, FANTOM5 has created an outstanding resource for the MC community. The presented data sets will undoubtedly spur further research to decipher the programs that orchestrate lineage commitment, differentiation, maintenance, and functional spectra of this unique cell subset.
Supplementary Material
Acknowledgments
The authors thank all members of the FANTOM5 consortium for contributing to generation of samples and analysis of the data set, GeNAS for data production, and Ms. Annett von Grüner and Mr. Jayson Harshbarger for excellent technical assistance.
FANTOM5 was made possible by a research grant for RIKEN Omics Science Center from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Y.H.) and an Innovative Cell Biology by Innovative Technology (Cell Innovation Program) grant from the MEXT, Japan (Y.H.). This work was supported by a research grant from MEXT to RIKEN Preventive Medicine and Diagnosis Innovation Program and a grant from MEXT to RIKEN Center for Life Science Technologies, the European Centre for Allergy Research Foundation (ECARF) and the Global Allergy and Asthma European Network (GA2LEN), supported by the Sixth EU Framework programme for research contract n° FOOD-CT-2004-50637.
Footnotes
There is an Inside Blood Commentary on this article in this issue.
This article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.R.R.F. and M.B. designed the study; S.G. isolated the MCs and performed most experiments; M.B. performed several experiments; Y.I. isolated RNA; E.M. performed all bioinformatics analysis of MCs; M.I. was responsible for data production; T.L. was responsible for tag mapping; M.d.H. helped with the motif activity analysis; H.K. managed the data handling; P.C., Y.H., and A.R.R.F. were responsible for FANTOM5 management and concept; M.B. analyzed and interpreted the data and wrote the manuscript; and S.G., E.M., T.Z., and A.R.R.F. helped with interpretation and the writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Magda Babina, Department of Dermatology and Allergy, Charité Campus Mitte, Universitätsmedizin Berlin, Chariteplatz 1, D-10117 Berlin, Germany; e-mail: magda.babina@charite.de (for mast cell biology); and Alistair Forrest, Genome Information Analysis Team, Division of Genomic Technologies, RIKEN Center for Life Science Technologies, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, 230-0045 Japan; e-mail alistair.forrest@gmail.com (for FANTOM5 and CAGE profiling).
References
- 1.Brown JM, Wilson TM, Metcalfe DD. The mast cell and allergic diseases: role in pathogenesis and implications for therapy. Clin Exp Allergy. 2008;38(1):4–18. doi: 10.1111/j.1365-2222.2007.02886.x. [DOI] [PubMed] [Google Scholar]
- 2.Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18(5):693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Navi D, Saegusa J, Liu FT. Mast cells and immunological skin diseases. Clin Rev Allergy Immunol. 2007;33(1-2):144–155. doi: 10.1007/s12016-007-0029-4. [DOI] [PubMed] [Google Scholar]
- 4.Sayed BA, Christy A, Quirion MR, Brown MA. The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol. 2008;26:705–739. doi: 10.1146/annurev.immunol.26.021607.090320. [DOI] [PubMed] [Google Scholar]
- 5.Kneilling M, Röcken M. Mast cells: novel clinical perspectives from recent insights. Exp Dermatol. 2009;18(5):488–496. doi: 10.1111/j.1600-0625.2009.00860.x. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15(8):940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta. 2009;1796(1):19–26. doi: 10.1016/j.bbcan.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol. 2010;40(7):1843–1851. doi: 10.1002/eji.201040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann AM, Abraham SN. New roles for mast cells in pathogen defense and allergic disease. Discov Med. 2010;9(45):79–83. [PubMed] [Google Scholar]
- 10.Khazaie K, Blatner NR, Khan MW, et al. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011;30(1):45–60. doi: 10.1007/s10555-011-9286-z. [DOI] [PubMed] [Google Scholar]
- 11.Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7(2):93–104. doi: 10.1038/nri2018. [DOI] [PubMed] [Google Scholar]
- 12.Nakajima T, Inagaki N, Tanaka H, et al. Marked increase in CC chemokine gene expression in both human and mouse mast cell transcriptomes following Fcepsilon receptor I cross-linking: an interspecies comparison. Blood. 2002;100(12):3861–3868. doi: 10.1182/blood-2002-02-0602. [DOI] [PubMed] [Google Scholar]
- 13.Sayama K, Diehn M, Matsuda K, et al. Transcriptional response of human mast cells stimulated via the Fc(epsilon)RI and identification of mast cells as a source of IL-11. BMC Immunol. 2002;3:5. doi: 10.1186/1471-2172-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayapal M, Tay HK, Reghunathan R, et al. Genome-wide gene expression profiling of human mast cells stimulated by IgE or FcepsilonRI-aggregation reveals a complex network of genes involved in inflammatory responses. BMC Genomics. 2006;7:210. doi: 10.1186/1471-2164-7-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iida M, Matsumoto K, Tomita H, et al. Selective down-regulation of high-affinity IgE receptor (FcepsilonRI) alpha-chain messenger RNA among transcriptome in cord blood-derived versus adult peripheral blood-derived cultured human mast cells. Blood. 2001;97(4):1016–1022. doi: 10.1182/blood.v97.4.1016. [DOI] [PubMed] [Google Scholar]
- 16.Kashiwakura J, Yokoi H, Saito H, Okayama Y. T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J Immunol. 2004;173(8):5247–5257. doi: 10.4049/jimmunol.173.8.5247. [DOI] [PubMed] [Google Scholar]
- 17.Dahl C, Saito H, Kruhøffer M, Schiøtz PO. Identification of tryptase- and chymase-related gene clusters in human mast cells using microarrays. Allergy. 2006;61(3):276–280. doi: 10.1111/j.1398-9995.2005.00950.x. [DOI] [PubMed] [Google Scholar]
- 18.Liu SM, Xavier R, Good KL, et al. Immune cell transcriptome datasets reveal novel leukocyte subset-specific genes and genes associated with allergic processes. J Allergy Clin Immunol. 2006;118(2):496–503. doi: 10.1016/j.jaci.2006.04.040. [DOI] [PubMed] [Google Scholar]
- 19.Saito H, Matsumoto K, Okumura S, et al. Gene expression profiling of human mast cell subtypes: an in silico study. Allergol Int. 2006;55(2):173–179. doi: 10.2332/allergolint.55.173. [DOI] [PubMed] [Google Scholar]
- 20.Kanamori-Katayama M, Itoh M, Kawaji H, et al. Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res. 2011;21(7):1150–1159. doi: 10.1101/gr.115469.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forrest ARR, Kawaji H, Rehli M, et al. A promoter level mammalian expression atlas. Nature doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed]
- 22.Babina M, Guhl S, Stärke A, Kirchhof L, Zuberbier T, Henz BM. Comparative cytokine profile of human skin mast cells from two compartments—strong resemblance with monocytes at baseline but induction of IL-5 by IL-4 priming. J Leukoc Biol. 2004;75(2):244–252. doi: 10.1189/jlb.0403157. [DOI] [PubMed] [Google Scholar]
- 23.Guhl S, Franke R, Schielke A, et al. Infection of in vivo differentiated human mast cells with hantaviruses. J Gen Virol. 2010;91(Pt 5):1256–1261. doi: 10.1099/vir.0.019505-0. [DOI] [PubMed] [Google Scholar]
- 24.Guhl S, Artuc M, Neou A, Babina M, Zuberbier T. Long-term cultured human skin mast cells are suitable for pharmacological studies of anti-allergic drugs due to high responsiveness to FcεRI cross-linking. Biosci Biotechnol Biochem. 2011;75(2):382–384. doi: 10.1271/bbb.100745. [DOI] [PubMed] [Google Scholar]
- 25.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9(2):321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 26.Esnault C, Priet S, Ribet D, et al. A placenta-specific receptor for the fusogenic, endogenous retrovirus-derived, human syncytin-2. Proc Natl Acad Sci USA. 2008;105(45):17532–17537. doi: 10.1073/pnas.0807413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terada N, Hamazaki T, Oka M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416(6880):542–545. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- 28.Tatemoto K, Nozaki Y, Tsuda R, et al. Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem Biophys Res Commun. 2006;349(4):1322–1328. doi: 10.1016/j.bbrc.2006.08.177. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian H, Gupta K, Guo Q, Price R, Ali H. Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: resistance to receptor phosphorylation, desensitization, and internalization. J Biol Chem. 2011;286(52):44739–44749. doi: 10.1074/jbc.M111.277152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bansal G, DiVietro JA, Kuehn HS, et al. RGS13 controls g protein-coupled receptor-evoked responses of human mast cells. J Immunol. 2008;181(11):7882–7890. doi: 10.4049/jimmunol.181.11.7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkman-Van der Linden EC, Hurtado-Ziola N, Hayakawa T, et al. Human-specific expression of Siglec-6 in the placenta. Glycobiology. 2007;17(9):922–931. doi: 10.1093/glycob/cwm065. [DOI] [PubMed] [Google Scholar]
- 32.Walter S, Atzmon G, Demerath EW, et al. A genome-wide association study of aging. Neurobiol Aging. 2011;32(11):2109.e15-e28. [DOI] [PMC free article] [PubMed]
- 33.Forsberg E, Pejler G, Ringvall M, et al. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature. 1999;400(6746):773–776. doi: 10.1038/23488. [DOI] [PubMed] [Google Scholar]
- 34.Merickel A, Edwards RH. Transport of histamine by vesicular monoamine transporter-2. Neuropharmacology. 1995;34(11):1543–1547. doi: 10.1016/0028-3908(95)00148-y. [DOI] [PubMed] [Google Scholar]
- 35.Robin C, Durand C. The roles of BMP and IL-3 signaling pathways in the control of hematopoietic stem cells in the mouse embryo. Int J Dev Biol. 2010;54(6-7):1189–1200. doi: 10.1387/ijdb.093040cr. [DOI] [PubMed] [Google Scholar]
- 36.Babina M, Schülke Y, Kirchhof L, et al. The transcription factor profile of human mast cells in comparison with monocytes and granulocytes. Cell Mol Life Sci. 2005;62(2):214–226. doi: 10.1007/s00018-004-4480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trompouki E, Bowman TV, Lawton LN, et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147(3):577–589. doi: 10.1016/j.cell.2011.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12(11):1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halova I, Draberova L, Draber P. Mast cell chemotaxis - chemoattractants and signaling pathways. Front Immunol. 2012;3:119. doi: 10.3389/fimmu.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Randrianarison-Huetz V, Laurent B, Bardet V, Blobe GC, Huetz F, Duménil D. Gfi-1B controls human erythroid and megakaryocytic differentiation by regulating TGF-beta signaling at the bipotent erythro-megakaryocytic progenitor stage. Blood. 2010;115(14):2784–2795. doi: 10.1182/blood-2009-09-241752. [DOI] [PubMed] [Google Scholar]
- 41.Yosipovitch G, Papoiu AD. What causes itch in atopic dermatitis? Curr Allergy Asthma Rep. 2008;8(4):306–311. doi: 10.1007/s11882-008-0049-z. [DOI] [PubMed] [Google Scholar]
- 42.Wang Q, Lin JL, Wu KH, et al. Xin proteins and intercalated disc maturation, signaling and diseases. Front Biosci (Landmark Ed) 2012;17:2566–2593. doi: 10.2741/4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology. 2005;146(12):5063–5070. doi: 10.1210/en.2005-0868. [DOI] [PubMed] [Google Scholar]
- 44.Maeda K, Nishiyama C, Ogawa H, Okumura K. GATA2 and Sp1 positively regulate the c-kit promoter in mast cells. J Immunol. 2010;185(7):4252–4260. doi: 10.4049/jimmunol.1001228. [DOI] [PubMed] [Google Scholar]
- 45.Vieille-Grosjean I, Huber P. Transcription factor GATA-1 regulates human HOXB2 gene expression in erythroid cells. J Biol Chem. 1995;270(9):4544–4550. doi: 10.1074/jbc.270.9.4544. [DOI] [PubMed] [Google Scholar]
- 46.Ripoll VM, Meadows NA, Raggatt LJ, et al. Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene. 2008;413(1-2):32–41. doi: 10.1016/j.gene.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 47.Bjerke GA, Hyman-Walsh C, Wotton D. Cooperative transcriptional activation by Klf4, Meis2, and Pbx1. Mol Cell Biol. 2011;31(18):3723–3733. doi: 10.1128/MCB.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnabel CA, Selleri L, Cleary ML. Pbx1 is essential for adrenal development and urogenital differentiation. Genesis. 2003;37(3):123–130. doi: 10.1002/gene.10235. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki H, Forrest AR, van Nimwegen E, et al. FANTOM Consortium. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet. 2009;41(5):553–562. doi: 10.1038/ng.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fedele M, Franco R, Salvatore G, et al. PATZ1 gene has a critical role in the spermatogenesis and testicular tumours. J Pathol. 2008;215(1):39–47. doi: 10.1002/path.2323. [DOI] [PubMed] [Google Scholar]
- 51.Gomperts BN, Gong-Cooper X, Hackett BP. Foxj1 regulates basal body anchoring to the cytoskeleton of ciliated pulmonary epithelial cells. J Cell Sci. 2004;117(Pt 8):1329–1337. doi: 10.1242/jcs.00978. [DOI] [PubMed] [Google Scholar]
- 52.Jacquet BV, Salinas-Mondragon R, Liang H, et al. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development. 2009;136(23):4021–4031. doi: 10.1242/dev.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novershtern N, Subramanian A, Lawton LN, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson NK, Foster SD, Wang X, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7(4):532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 55.Taghon T, Yui MA, Rothenberg EV. Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat Immunol. 2007;8(8):845–855. doi: 10.1038/ni1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. 2012;37(1):25–33. doi: 10.1016/j.immuni.2012.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.