

Abstract
Background
The RAS/ERK and PI3K/AKT pathways induce oncogenic gene expression programs and are commonly activated together in cancer cells. Often, RAS/ERK signaling is activated by mutation of the RAS or RAF oncogenes, and PI3K/AKT is activated by loss of the tumor suppressor PTEN. In prostate cancer, PTEN deletions are common, but, unlike other carcinomas, RAS and RAF mutations are rare. We have previously shown that over-expression of “oncogenic” ETS transcription factors, which occurs in about one-half of prostate tumors due to chromosome rearrangement, can bypass the need for RAS/ERK signaling in the activation of a cell migration gene expression program. In this study we test the role of RAS/ERK and PI3K/AKT signaling in the function of oncogenic ETS proteins.
Results
We find that oncogenic ETS expression negatively correlates with RAS and RAF mutations in prostate tumors. Furthermore, the oncogenic ETS transcription factors only increased cell migration in the absence of RAS/ERK activation. In contrast to RAS/ERK, it has been reported that oncogenic ETS expression positively correlates with PI3K/AKT activation. We identified a mechanistic explanation for this finding by showing that oncogenic ETS proteins required AKT signaling to activate a cell migration gene expression program through ETS/AP-1 binding sequences. Levels of pAKT correlated with the ability of oncogenic ETS proteins to increase cell migration, but this process did not require mTORC1.
Conclusions
Our findings indicate that oncogenic ETS rearrangements cause a cell migration gene expression program to switch from RAS/ERK control to PI3K/AKT control and provide a possible explanation for the high frequency of PTEN, but not RAS/RAF mutations in prostate cancer.
Keywords: Prostate cancer, ETS, RAS/ERK, PI3K/AKT, Cell migration
Background
The RAS/RAF/MEK/ERK (RAS/ERK) and PI3K/AKT signaling pathways regulate gene expression programs that promote cell growth, proliferation, motility, and survival [1,2]. Mutations that cause constitutive RAS/ERK or PI3K/AKT signaling are among the most common alterations in human cancer and both pathways are often activated in the same tumor [3,4]. PI3K/AKT activation is common in prostate cancer, often due to loss of a suppressor of the pathway, PTEN [5]. However, unlike other carcinomas, prostate cancers rarely have activating mutations in RAS or RAF [6], and thus, the mechanisms that allow transcriptional activation of RAS/ERK target genes in this malignancy are not fully understood.
RAS/ERK signaling can be initiated by tyrosine kinase receptors that activate RAS, followed by the RAF/MEK/ERK kinase cascade, resulting in phosphorylated ERK (pERK). pERK, in turn, phosphorylates transcription factors, including some members of the ETS family, leading to increased transcriptional activation of target genes [7]. PI3K phosphorylates phosphoinositides leading to activation of downstream proteins such as the kinase AKT [8]. PTEN, a phosphatase, can reverse this process and acts as a tumor suppressor. Activated AKT has multiple functions, one being the activation of the mTOR containing signaling complex mTORC1, which alters translational control of gene expression. AKT also activates the mTORC2 complex, which provides positive feedback by phosphorylating and activating AKT. The RAS/ERK and PI3K/AKT pathways are highly interconnected. For example, RAS can activate PI3K, and AKT can phosphorylate and inhibit RAF [9,10].
A rearrangement of chromosome 21 that results in fusion of the TMPRSS2 and ERG genes occurs in approximately 50% of prostate tumors [11]. TMPRSS2:ERG joins the 5′ regulatory regions and 5′ UTR of TMPRSS2, which is highly expressed in prostate, to the open reading frame of ERG, resulting in expression of either a full-length, or N-terminally truncated version of ERG, an ETS family transcription factor that is not normally expressed in prostate cells. Similar fusions that over-express the ETS genes ETV1, ETV4, and ETV5 occur in another 10% of prostate tumors [11-13]. Expression of these oncogenic ETS family members in prostate cells drives cellular invasion and migration [14,15] and promotes the transition from neoplasia to carcinoma [16]. We previously reported that over-expression of ERG or ETV1 can activate a gene expression program that drives cell migration [15]. Genes in this program are regulated by a RAS-responsive enhancer sequence consisting of neighboring ETS and AP-1 transcription factor binding sites. In normal prostate cells, these genes can be activated by RAS/ERK signaling, likely via ERK phosphorylation of an ETS protein bound to the ETS/AP-1 sequence. There are 12–15 ETS transcription factors expressed in normal prostate that are candidates for this role [17]. Our previous data support a model that when ERG, ETV1, ETV4, or ETV5 are over-expressed in prostate cells, they can replace the ETS family member(s) normally bound to ETS/AP-1 sites and activate the RAS-inducible cell migration gene expression program in the absence of RAS/ERK signaling [15]. Thus over expression of one of these four “oncogenic” ETS genes can mimic RAS/ERK pathway activation.
The two most common genomic aberrations in prostate cancer are PTEN deletion and the TMPRSS2/ERG rearrangement [11,18,19]. Whereas a RAS mutation in other carcinomas might activate both ERK and PI3K signaling, we propose that prostate tumors have an alternative way to activate these pathways: PTEN deletion (PI3K/AKT activation) coupled with oncogenic ETS-overexpression (activation of RAS/ERK target genes). Supporting this hypothesis, PTEN deletion is more common in prostate tumors with TMPRSS2-ERG rearrangements, than in those without [16,20], and in mouse models, ERG over-expression results in adenocarcinoma only when accompanied by a second mutation that activates the PI3K/AKT pathway [16,20,21].
Here we test the relationship between oncogenic ETS expression and both the RAS/ERK and PI3K/AKT pathways. We provide the first comprehensive analysis of oncogenic ETS protein expression in prostate cancer cell lines. We then show that the status of both the RAS/ERK and PI3K/AKT pathways can change the ability of over-expressed ETS proteins to promote prostate cell migration. Significantly, we find that oncogenic ETS expression makes cell migration less dependent on RAS/ERK signaling, but increases the importance of PI3K/AKT signaling. We provide evidence that this switch in the signaling pathway requirement is due to AKT-dependent, but mTORC1-independent, regulation of oncogenic ETS function through ETS/AP-1 binding sequences. Therefore, switching the ETS protein at ETS/AP-1 sequences changes the ability of signaling pathways to regulate a critical oncogenic gene expression program.
Results
Oncogenic ETS gene rearrangement occurs in tumors lacking RAS/ERK mutations
If oncogenic ETS gene rearrangements replace RAS/ERK activation, we predict that RAS/ERK mutations will occur only in ETS rearrangement negative tumors. To test this hypothesis, we examined the results of three recently published studies [6,22,23] that both sequence exons and identify chromosome rearrangements in prostate tumors (Table 1). Together these studies examine 266 prostate tumors. One-half (133) have ERG or ETV1 chromosome rearrangements. We searched for either gene fusions, or point mutations in canonical RAS/ERK pathway genes (RAS, RAF, MEK, and ERK encoding genes). Eight tumors had such aberrations, and all eight were negative for oncogenic ETS rearrangements. This indicates that, while genomic alterations in RAS/ERK pathway components are rare in prostate cancer, there is a statistically significant (P = 0.007; Fisher’s exact test) mutual exclusivity of these alterations and ETS rearrangements. It has been previously reported that PI3K/AKT activation via PTEN deletion positively correlates with ETS gene rearrangements [16,20]. A search for PTEN loss in these 266 tumors (Table 1) confirms these findings and indicates that PTEN loss is more than twice as likely in tumors with ETS gene rearrangements than in those without (P = 0.0008; Fisher’s exact test). In conclusion, ERG and ETV1 gene rearrangements positively correlate with PTEN loss and negatively correlate with RAS/ERK mutations in tumors.
Table 1.
Correlation of RAS/ERK pathway mutations, PTEN loss, and oncogenic ETS expression in prostate tumors
| ETS - | ETS + | Total | |
|---|---|---|---|
| PTEN loss |
19 |
43 |
62 |
| RAS/ERK mutation |
8 |
0 |
8 |
| APC mutation |
6 |
5 |
11 |
| Total | 133 | 133 | 266 |
A summary of the 266 tumors analyzed by Taylor et al.[22], Grasso et al. [6], and Baca et al. [23]. ETS status indicates presence (+) or absence (-) of an ERG rearrangement in Taylor et al. and either an ERG or ETV1 rearrangement in Grasso et al. and Baca et al. RAS/ERK mutation includes point mutations from all three studies or verified gene rearrangements resulting in transcript fusions in Grasso et al. or Baca et al. for HRAS, KRAS, NRAS, RAF1, ARAF, BRAF, MAP2K1, MAP2K2, MAPK1, or MAPK3. APC mutations are included as a control to show that low frequency mutations are not always enriched in one category.
Prostate cancer cell lines as models of oncogenic ETS function
To test the effect of RAS/ERK signaling and PI3K/AKT signaling on oncogenic ETS function in prostate cell lines, we must first determine which cell lines have these characteristics. Although some prostate cancer cell lines, such as VCaP (ERG) and LNCaP (ETV1) are reported to have oncogenic ETS gene rearrangements [11,14], the full extent of oncogenic ETS protein expression, including fusion-independent expression, in commonly used prostate cancer cell lines has not been determined. To identify the expression level of the four oncogenic ETS proteins, we first tested available antibodies using purified recombinant proteins (Figure 1A). We identified antibodies to ERG, ETV1, ETV4, and ETV5 that could detect each protein at femtomolar levels. Because ETV1, ETV4, and ETV5 are homologous proteins, the sensitivity and specificity of these antibodies were compared. ETV1 and ETV4 antibodies were specific, but the ETV5 antibody recognized ETV4 and ETV5 equally. We then examined oncogenic ETS protein levels, along with phosphorylated ERK (pERK: RAS/ERK pathway) and phosphorylated AKT (pAKT: PI3K/AKT pathway) levels in six prostate cancer cell lines (Figure 1B and Additional file 1: Figure S1A). DU145 cells, which have a KRAS gene rearrangement [24], did not have high levels of any oncogenic ETS protein, or pAKT, but did have pERK, consistent with the small fraction of prostate cancers with RAS/ERK pathway mutations (Table 1). Of the remaining five prostate cancer cell lines, four had high expression of a single oncogenic protein. These included ERG in VCaP, consistent with a TMRPSS2/ERG rearrangement [11], ETV1 in MDA-PCa-2B, consistent with an ETV1 gene rearrangement [14], and ETV4 in PC3, consistent with high ETV4 mRNA [25]. ETV4 protein was also present at high levels in CWR22Rv1. Of the four lines with high oncogenic ETS protein expression, all had high levels of pAKT, but only one (CWR22Rv1) had high levels of pERK, consistent with the analysis of prostate tumors in Table 1. Surprisingly, despite an ETV1 gene rearrangement [14], and high ETV1 mRNA levels [25], ETV1 protein was not observed in LNCaP cells. However, this is consistent with results from Vitari et al. who showed low ETV1 protein levels in LNCaP cells due to proteasomal targeting by the COP1 E3 ubiquitin ligase [26].
Figure 1.

Prostate cell lines vary in oncogenic ETS expression and RAS/ERK pathway activation. (A) The sensitivity and specificity of antibodies detecting oncogenic ETS proteins were tested by immunoblot of the indicated amount of purified full-length proteins. (B) Immunoblots show levels of four oncogenic ETS proteins, pAKT (PI3K/AKT activation), pERK (RAS/ERK activation), total ERK, and tubulin control in six prostate cancer cell lines (left) and three cell lines derived from normal prostate (right). (C) Immunoblots show levels of ETV4 and pERK in the indicated cell lines with or without U0126 (10 μM, 10 hr). The same cell extracts are loaded on one gel above the dashed line, and a second gel below. ETV4 is only visible in DU145 cells after a very long exposure, hence it is not observed in (A).
Long exposures could identify pERK, pAKT, and some ETS proteins at low levels in immunoblots from most cell lines. To more quantitatively establish the “high-level” threshold shown in Figure 1B, ETS proteins in cell extracts were compared with purified standards (Additional file 1: Figure S1B). All “high-level” expression for ETS proteins exceeded 50,000 proteins per cell, and was highest at 330,000 proteins per cell for ERG in VCaP. Low-level ETS expression was 10,000 proteins per cell (ETV4 in DU145) or less (Additional file 1: Figure S1B and data not shown).
It is possible that oncogenic ETS expression and signaling pathway activation could influence each other. To test this, RWPE-1 (RWPE) cells derived from normal prostate [27] or variations of this line that express either Ki-RAS (RWPE-KRAS, also known as RWPE-2) or ERG (RWPE-ERG) were compared. ERG levels in RWPE-ERG cells were similar to VCaP cells (Additional file 1: Figure S1C). None of the oncogenic ETS were expressed at high levels in RWPE or RWPE-KRAS cells, and only ERG was expressed in RWPE-ERG cells (Figure 1B). As expected, KRAS increased both pERK and pAKT levels (Figure 1B). Interestingly, over-expression of ERG also resulted in activation of AKT and a small increase in pERK (Figure 1B). In other cell types, the RAS/ERK pathway activates ETV1, ETV4, and ETV5 expression [28]. Therefore, high ETV4 expression in CWR22Rv1 cells could be the result of ERK activation. To test this, CWR22Rv1 and DU145 cells were treated with the MEK inhibitor U0126 for 24 hours. In both cell lines, U0126 decreased pERK levels, but did not alter levels of ETV4 (Figure 1C). Therefore, RAS/ERK activation does not drive oncogenic ETS expression in prostate cancer cell lines, however in at least one context (ERG in RWPE) an oncogenic ETS could induce the phosphorylation of both AKT and, to a lesser degree, ERK.
Oncogenic ETS proteins and KRAS drive prostate cell migration, but not synergistically
We next tested the role of signaling pathways in the ability of oncogenic ETS proteins to drive cell migration. Because cancer derived cell lines have many mutations and copy number alterations that affect cellular phenotypes, we used the RWPE-ERG and RWPE-KRAS cell lines to compare the ability of oncogenic ETS and RAS signaling to promote cell migration in the same cellular background. RWPE-ERG and RWPE-KRAS cells migrated 5- and 10-fold more than RWPE cells (Figure 2A and Additional file 2: Figure S2), indicating that both ERG and KRAS induce cell migration. Similar to our previous findings [15], overexpression of oncogenic ETS proteins ETV1, ETV5, and ERG, but not other ETS proteins (FLI1 and SPDEF), promoted RWPE cell migration (Figure 2B and Additional file 2: Figure S2). In contrast, when the same ETS proteins were over-expressed in RWPE-KRAS cells, none of the oncogenic ETS proteins induced additional cell migration (Figure 2C and Additional file 2: Figure S2), suggesting that these ETS proteins and KRAS were functioning to activate the same pathway. These findings are consistent with our model that oncogenic ETS proteins can mimic RAS activation in cell lines lacking RAS activity, and are distinct from ETS proteins expressed in normal prostate.
Figure 2.
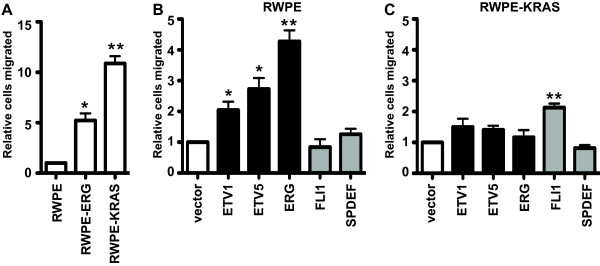
ETS expression and RAS activation induce migration of prostate cells via the same pathway. (A) A transwell assay measured relative number of migrating RWPE cells expressing ERG or activated KRAS relative to normal RWPE cells (first lane). (B, C) Transwell assays measured migration of (B) RWPE cells, or (C) RWPE-KRAS cells expressing oncogenic (Black bars) or non-oncogenic (Grey bars) ETS proteins. Number of cells migrated is reported relative to the same cell line transduced with an empty vector (white bar). Mean and SEM of three biological replicates (each mean of two technical replicates) are shown for (A) and five biological replicates for (B) and (C). P-values compare indicated value to the hypothetical mean (1) and are calculated by t test: * < 0.05, ** < 0.005, unmarked > 0.05.
A role for the PI3K/AKT pathway in oncogenic ETS function
To identify signaling pathways required for the oncogenic function of ETS factors, a microarray analysis of ETV4 knockdown in PC3 prostate cancer cells [25] was compared to the Connectivity Map database [29] that contains microarray data of PC3 cells treated with 1309 small molecules, including many signaling pathway inhibitors. Similarities between the gene expression profile of a signaling pathway inhibitor and ETV4 knockdown would predict a role for that pathway in oncogenic ETS function. The top two, and three of the top five small molecules that induced gene expression changes most similar to ETV4 knockdown were inhibitors of either PI3K or mTOR, a downstream effector of PI3K (Table 2). These data suggest that in PC3 cells, PI3K and ETV4 activate a similar gene expression program.
Table 2.
Drugs that alter PC3 gene expression most similarly to ETV4 depletion
| Rank | Drug | Target | P value |
|---|---|---|---|
| 1 |
Sirolimus (Rapamycin) |
mTOR |
<0.00001 |
| 2 |
LY-294002 |
PI3K |
<0.00001 |
| 3 |
Trichostatin A |
HDAC |
0.00002 |
| 4 |
Alexidine |
Tyrosine Phosphatase |
0.00004 |
| 5 | Wortmannin | PI3K | 0.00006 |
Gene expression changes from small molecule treatments of PC3 cells in the Connectivity Map database [29] were compared to gene expression changes previously reported for ETV4 depletion in PC3 cells [25]. Small molecules that elucidated changes most similar to ETV4 depletion are rank ordered by P value.
To test if the PI3K pathway is required for an oncogenic ETS protein to promote the cell migration phenotype, RWPE-ERG and RWPE-KRAS cells were treated with the PI3K inhibitor, LY294002. LY294002 reduced AKT phosphorylation in both lines, consistent with PI3K inhibition (Figure 3A). Strikingly, PI3K inhibition completely abrogated cell migration induced by ERG, but not cell migration induced by KRAS (Figure 3B and Additional file 2: Figure S2). In fact RWPE-KRAS cells actually migrated more when PI3K was inhibited. This increased migration may be due to relief of RAF inhibition by AKT [9], as RWPE-KRAS cells had higher pMEK levels after treatment by LY294002 (Figure 3A). To confirm the role of PI3K, a second PI3K inhibitor, ZSTK474, was also tested (Figures 3A and 3B). Like LY294002, ZSTK474 significantly reduced migration of RWPE-ERG cells, but not RWPE-KRAS cells. Cell migration induced by other oncogenic ETS factors, ETV1, and ETV5, was also abrogated by PI3K inhibition (Figure 3C and Additional file 2: Figure S2). A second cell migration assay, the scratch assay, confirmed that PI3K inhibition reduced migration caused by ERG expression, but not migration caused by KRAS (Figure 3D and Additional file 3: Figure S3). An AKT inhibitor had a similar effect (Figure 3D and Additional file 3: Figure S3), indicating that PI3K is functioning via AKT activation. These results indicate that overexpression of an oncogenic ETS gene can switch the control of prostate cell migration from the RAS/ERK pathway to the PI3K/AKT pathway.
Figure 3.

An active PI3K/AKT pathway is required for oncogenic ETS, but not KRAS, to induce prostate cell migration. (A) An immunoblot shows the levels of pAKT, pMEK (activator of ERK), or tubulin (control) after LY294002 (20 μM; 24 h) or ZSTK474 (2 μM; 24 h) treatment in RWPE-ERG or RWPE-KRAS cells. (B) A transwell assay measured cell migration of RWPE prostate cells with or without ERG and KRAS overexpression and in the presence or absence of the PI3K inhibitors LY294002 (20 μM) or ZSTK474 (2 μM). The number of migrated cells is shown as the mean and SEM of six biological replicates (except for ZSTK474 treated cells which have three replicates) relative to RWPE-empty vector. (C) A transwell assay, as in (A), tested the role of PI3K inhibition on ETV1 and ETV5 expressing RWPE cells and shows the mean and SEM of three biological replicates. (D) Results of the scratch assay performed in the presence or absence of LY294002 (20 μM) and AKT inhibitor VIII (10 μM) in RWPE-ERG (Grey bar) and RWPE-KRAS (white bar) cells. The percentage of scratch filled is shown as the mean and SEM of three biological replicates (each mean of three technical replicates) relative to no treatment. P-values are calculated by t test: * < 0.05, ** < 0.005, *** < 0.0005, unmarked > 0.05.
We next tested if the PI3K pathway was regulating the ability of ERG to activate the transcription of RAS- and ERG-responsive target genes near enhancers that are co-occupied by ETS and AP-1 proteins. The expression levels of two such genes, ARHGAP29, and SMAD3, were measured by quantitative reverse transcription PCR (qRT-PCR) (Figure 4A and B). Both ARHGAP29 and SMAD3 have roles in cell migration and/or cell morphology [30,31], are direct targets of oncogenic ETS proteins and AP-1 by ChIP-seq [15], and are activated by KRAS and oncogenic ETS expression (Figure 4A and B). Similar to the cell migration phenotype, the activation of both genes was significantly attenuated by PI3K inhibition in RWPE-ERG cells, but not in RWPE-KRAS cells (Figure 4A and B). Therefore cell migration changes are consistent with changes in the expression of these two oncogenic ETS target genes.
Figure 4.

The PI3K pathway can alter the expression of cell migration genes via ETS/AP-1 sites in oncogenic ETS overexpressing cells. mRNA expression of (A)ARHGAP29 or (B)SMAD3, in the presence and absence of PI3K inhibitor (LY294002, 20 μM), in RWPE-ERG and RWPE-KRAS cells was measured by qRT-PCR and compared to RWPE cells. Mean and SEM of seven biological replicates are shown. (C) Firefly luciferase activity from a vector with the indicated sequences (3 copies of neighboring ETS and AP-1 binding sequences or versions of the same with point mutations) is shown relative to Renilla luciferase from a control vector transfected in RWPE cells. The ERK pathway is inhibited by UO126 where indicated. Mean and SEM of six biological replicates (each mean of two technical replicates) are shown. Luciferase reporter activity measured as in (C) is shown in (D) RWPE-ERG, or (E) RWPE-KRAS cells as activity in LY294002 treated cells (20 μM) relative to untreated. P-values are calculated by t test: n.s > 0.10, * < 0.05.
These results indicate that the PI3K/AKT pathway functions through ERG to regulate expression of cell migration genes. We next used a reporter assay to test if these gene expression changes were mediated by the ETS/AP-1 binding sequences we found in the enhancers of oncogenic ETS target genes. Three copies of an ETS/AP-1 consensus sequence were cloned upstream of a minimal promoter driving firefly luciferase. Luciferase expression from this vector was higher when the ERK pathway was active, indicating that this pathway regulates the reporter construct (Figure 4C). Point mutations in either the ETS or AP-1 binding sequences completely eliminated luciferase expression indicating that both binding sites are required for activity (Figure 4C). The PI3K inhibitor, LY294002, caused a significant decrease in the activity of this reporter in RWPE-ERG cells (Figure 4D), but significantly increased activity in RWPE-KRAS cells (Figure 4E), consistent with the cell migration findings. Therefore, the PI3K pathway can alter the expression of cell migration genes via ETS/AP-1 sites.
The role of AKT in oncogenic ETS function is not via mTORC1
PI3K/AKT signaling has a number of cellular functions including the activation of the mTOR-containing complexes mTORC1 and mTORC2 [8]. mTORC1 includes the Raptor protein and regulates gene expression via translational control. mTORC2 includes the Rictor protein and provides positive feedback by phosphorylating and activating AKT. To test the role of mTOR-containing complexes in oncogenic ETS function, shRNAs were used to knockdown mTOR, Raptor, and Rictor, in RWPE-ERG cells (Figure 5A). Loss of Raptor resulted in an increase in cell migration, indicating that mTORC1 is not required for the ability of PI3K/AKT to promote cell migration (Figure 5B and Additional file 2: Figure S2). Loss of mTOR had little effect on RWPE-ERG migration, while loss of Rictor decreased migration (Figure 5B and Additional file 2: Figure S2). Because the major role of the Rictor-containing mTORC2 complex is thought to be the phosphorylation of AKT, we hypothesized that these results were due to changes in AKT phosphorylation. Consistent with previous findings [32-34], Raptor knockdown increased AKT phosphorylation, and Rictor knockdown decreased AKT phosphorylation (Figure 5C). Therefore, the effect of mTOR containing complexes on RWPE-ERG cell migration can be explained indirectly by changes to pAKT levels, rather than by a direct role.
Figure 5.

PI3K/AKT signaling in oncogenic ETS function is not through mTORC1. (A) shRNA knock down of mTOR, Raptor (mTORC1 complex) and Rictor (mTORC2 complex) in RWPE-ERG cells was confirmed by qRT-PCR analysis. Mean and SEM of two biological replicates (each mean of two technical replicates) are shown. (B) A transwell assay measured cell migration of RWPE-ERG cells stably expressing the indicated shRNA relative to a negative control (shRNA targeting luciferase, which is not expressed in this cell line). Results are the mean and SEM of four independent experiments, each the mean of two technical replicates. (C) Immunoblot showing the expression level of pAKT and tubulin in RWPE-ERG cells expressing the indicated shRNA. P-values are calculated by t test: *** < 0.0005.
Discussion
PTEN deletion and the TMPRSS2:ERG rearrangement are the two most common genomic aberrations in prostate tumors. These alterations result in activation of the PI3K/AKT pathway and expression of the transcription factor ERG in prostate cells. Expression of ERG alone in prostate epithelia does not induce adenocarcinoma, but ERG is oncogenic when expressed in combination with PI3K/AKT activation [16,20,21], indicating an important synergy between these pathways. Our results identify a mechanistic connection between the expression of oncogenic ETS, such as ERG, and activation of the PI3K/AKT pathway. We show that AKT activation is required for oncogenic ETS proteins to increase transcription of genes critical for cellular migration - a pathway that promotes progression of a neoplasia to an adenocarcinoma. Interestingly, in cells lacking oncogenic ETS expression, these genes are activated by the RAS/ERK pathway through enhancer ETS/AP-1 binding motifs, and are likely activated by mutations in this pathway in other cancers. We show that oncogenic ETS protein expression replaces RAS/ERK regulation of these genes with PI3K/AKT regulation. Our results are consistent with a recent finding that in mice the over-expression of ERG in prostate epithelia only results in significant changes in gene expression when PTEN is deleted [35]. Together these findings provide an explanation for why the PI3K/AKT pathway is activated more often than the RAS/ERK pathway in prostate cancers, but not in other carcinomas that lack ETS gene fusions.
We provide the first comprehensive analysis of oncogenic ETS, pERK and pAKT protein levels in prostate cancer cell lines (Figure 1B). These results indicate that commonly used prostate cancer cell lines recapitulate patterns of oncogenic ETS expression observed in tumors, such as a positive correlation between oncogenic ETS expression and PI3K/AKT pathway activation, and negative correlation between oncogenic ETS expression and RAS/ERK pathway mutations. CWR22Rv1 provided one exception to these correlations, as it expressed ETV4, pERK, and pAKT. This may reflect a unique role for ETV4, since a recent report indicates that expression of ETV4, but not other oncogenic ETS genes correlates with both PI3K and RAS signaling in prostate tumors [36]. Prostate tumors rarely have multiple ETS gene rearrangements [37], leading to the hypothesis that oncogenic ETS genes have overlapping functions and therefore there is no advantage to the tumor to express more than one. Figure 1 indicates that oncogenic ETS proteins, even when expressed in a fusion-independent manner, show the same pattern, supporting this redundancy model. This analysis also revealed that ERG expression strongly increased pAKT levels, which may provide a positive-feedback loop by increasing ERG function (Figure 1B). This contrasts with findings in mice, where ERG did not increase pAKT [16]. It may be that the effect of ERG on this pathway, and thus the necessity of PTEN deletion for increased pathway activation, varies by cellular background. In summary, the cell line profiling presented here provides a basis for using these lines to model the complex crosstalk of oncogenic ETS expression and signaling in various prostate tumors.
The requirement of AKT for transcriptional activation by an ETS factor is novel. This could be due to AKT directly phosphorylating ETS or AP-1 at ETS/AP-1 sequences. AKT is known to modify transcription factors, such as those from the FOXO family [38]. It is also possible that AKT is working through downstream signaling factors. We have ruled out mTORC1, but AKT can modify many other signaling proteins. These AKT-regulated proteins include a number of factors specific to neurons, such as the GABA-A receptor [39], Huntingtin [40], and Ataxin1 [41]. Interestingly, one of the normal functions of the “oncogenic” ETS proteins ETV1 and ETV4 is to cause certain neurons to outgrow and invade the spinal cord during development [42,43]. Furthermore, PI3K/AKT signaling [44], and ETV1 and ETV4 expression [45] can both promote survival of neurons in the absence of neuronal growth factors. Therefore, processes that are oncogenic in prostate epithelia could reflect normal synergy between AKT and these ETS factors in neurons.
The ability to switch the signaling pathway that controls prostate cell migration by altering expression of oncogenic ETS transcription factors provides an interesting example of a mechanism for modulating a gene expression program. Cells can change transcription factor activity via expression levels, or localization. This can gradually alter the fraction of time that a transcription factor occupies a binding site compared to a competing transcription factor. If these competing factors respond to distinct signaling pathways, the effect of this process will depend on the status of each pathway. This allows both transcription factors and signaling pathways to have distinct functions in different cellular backgrounds. In the case of prostate cancer, this work indicates that oncogenic ETS status may be an important factor when deciding to target RAS/ERK or PI3K/AKT signaling during treatment.
Conclusions
Here we demonstrate that the aberrant expression of an oncogenic ETS transcription factor in prostate cells can switch the regulation of a cell migration gene expression program from RAS/ERK to PI3K/AKT control. This provides a mechanistic rationale for the correlation between PI3K signaling and ERG expression in prostate tumors and identifies a novel mode of ETS regulation that might be exploited by future therapeutics.
Methods
Cell culture and viral transduction
All cell lines were authenticated by the University of Arizona Genetics Core using PowerPlex 16HS Assay (Promega) with > 80% match to eight core STR loci [46], with the exception of LNCaP, which was obtained from ATCC immediately prior to use. Cell lines were cultured according to ATCC recommendations as follows; RWPE (RWPE-1) and RWPE-KRAS (RWPE-2): Keratinocyte SFM (Invitrogen), LNCaP and CWR22Rv1: RPMI 1640 (Mediatech-Cellgro) with 10% fetal bovine serum (FBS) [Sigma], PC3: F12K medium (Mediatech-Cellgro) with 10% FBS. 293 EBNA, HEK-293 T, DU145 and VCaP: Dulbecco’s modification Eagle (DMEM) [Sigma] with 10% FBS, MDA-PCa-2b: BRFF-HPC1 (Athena Enzyme Systems) with 20% FBS. All media were supplemented with 1% Penicillin/Streptomycin (Mediatech-Cellgro).
ETS proteins with N-terminal 3xFlag tags were stably expressed in RWPE via retrovirus as described previously [15]. Plasmids for lentiviral shRNA knockdowns were obtained from AddGene, mTOR (#1855), Raptor (#1857) and Rictor (#1853), are from Sarbassov et al.[33]. Lentivirus was produced by co-transfection of pLKO.1 constructs in HEK293T cells with pMDLg/pRRE, pRSV-Rev and pMD2.G envelope plasmids from Dull et al.[47] and AddGene.
Transwell migration and In vitro scratch assays
Transwell migration assays were done as described previously [15]. In brief, 5×104 cells were introduced to the transwell (8 μM pore size; BD Bioscience) and incubated for 63 h, except for RWPE-KRAS cells summarized in Figure 2C, which were incubated for 54 h. Migrated cells are reported as the mean of four representative fields per membrane, and the mean of two technical replicates (membranes) per biological replicate.
For in vitro scratch assays, cells were plated in 35 mm plates and grown to full confluence, and the cultures were scratched by pipette tip. Migration into the open area was documented at 40 h post-scratching by microscopy. Free area was measured using TScratch software (cse-lab.ethz.ch/software) [48].
Measuring protein and RNA
RNA levels were measured by quantitative reverse transcription-PCR as described previously [15], using primers in Additional file 4: Table S1.
Whole-cell extracts of equivalent cell number were separated by SDS-PAGE and blotted to nitrocellulose. Antibodies for immunoblotting were: ERK (sc-94) and pERK (sc-7383) from Santa Cruz Biotechnology, ETV5 (ab102010) and ETV1 (ab81086) from Abcam, pAKT (S473) and pMEK (9121) from Cell Signaling, Tubulin (T-9026) from Sigma, ETV4 (ARP32263_P050) from Aviva Systems Biology, and ERG (9FY) from Biocare Medical.
Purification of His-tagged ETS proteins for antibody validation was as described previously [49]. DNA binding activity was verified by EMSA. Concentration was calculated by comparison to BSA standards on Coomassie stained 10% SDS-PAGE gels.
Luciferase assays
Luciferase assays used a Dual Luciferase Reporter Assay System (Promega) according to manufacturer instructions with some modifications. Wild type and mutant ETS/AP-1 sequences (Additional file 4: Table S1) were cloned upstream of the firefly luciferase-pGL4.25 (Promega) plasmid cut with HindIII and NheI. The Renilla luciferase gene was sub-cloned from pRL-null to pGL4.25 plasmid by replacing firefly sequence. Cells were plated at ~50% confluency in a 6 well plate (2.5×105 cells/well) 24 hrs before transfection. Cells were transfected with 1 μg of firefly and renilla plasmid using TransIT Prostate Transfection Kit (Mirus). After 24 hours, media was removed, cells were resuspended in 250 μL 1×PLB, and disrupted by three freeze/thaw cycles. Luciferase activity was measured in 20 μL of cell lysate using Appliskan Multimode Microplate reader (Thermo Scientific). Firefly values were normalized to renilla values.
Abbreviations
RAS/ERK: RAS/RAF/MEK/ERK pathway; pERK: Phosphorylated ERK; pAKT: phosphorylated AKT.
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
NS did the experiments reported in Figures 1, 3, and 5. JAB carried out expression profiling and reporter assays in Figure 4. MWF did the migration assays in Figure 2. TJJ interpreted data and assisted in writing. PCH interpreted data and wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Quantitative assessment of ETS protein levels.
Representative images cell migration assays.
Representative images of scratch assays.
Oligonucleotide primer sequences.
Contributor Information
Nagarathinam Selvaraj, Email: nagaselv@indiana.edu.
Justin A Budka, Email: jubudka@umail.iu.edu.
Mary W Ferris, Email: mwferris@indiana.edu.
Travis J Jerde, Email: tjjerde@iupui.edu.
Peter C Hollenhorst, Email: pchollen@indiana.edu.
Acknowledgements
We thank Indiana University colleague Heather Hundley for critical reading of the manuscript. We thank Shyh-Han Tan and Albert Dobi from the Center for Prostate Disease Research for samples and advice regarding the ERG antibody. This work was supported by (Research Scholar Award; RSG-13-215-01-DMC) from the American Cancer Society to P.C.H and by NIH-NIDDK DK092366-01A1 to TJJ. This work was also supported by funds from the Walther Cancer Foundation and the Showalter Trust.
References
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Uzgare AR, Isaacs JT. Enhanced redundancy in Akt and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004;64:6190–6199. doi: 10.1158/0008-5472.CAN-04-0968. [DOI] [PubMed] [Google Scholar]
- Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci U S A. 2006;103:14477–14482. doi: 10.1073/pnas.0606836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC, McIntosh LP, Graves BJ. Genomic and biochemical insights into the specificity of ETS transcription factors. Annual Rev Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, Mehra R, Chinnaiyan AM. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25:2147–2157. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, Cordon-Cardo C, Gerald W, Pandolfi PP. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC, Jones DA, Graves BJ. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004;32:5693–5702. doi: 10.1093/nar/gkh906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray IC, Stewart LM, Phillips SM, Hamilton JA, Gray NE, Watson GJ, Spurr NK, Snary D. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br J Cancer. 1998;78:1296–1300. doi: 10.1038/bjc.1998.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95:5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, Gerald WL, Sawyers CL. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465–12470. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, Theurillat JP, Soong TD, Nickerson E, Auclair D, Tewari A, Beltran H, Onofrio RC, Boysen G, Guiducci C, Barbieri CE, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Ramos AH, Winckler W. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XS, Shankar S, Dhanasekaran SM, Ateeq B, Sasaki AT, Jing X, Robinson D, Cao Q, Prensner JR, Yocum AK, Wang R, Fries DF, Han B, Asangani IA, Cao X, Li Y, Omenn GS, Pflueger D, Gopalan A, Reuter VE, Kahoud ER, Cantley LC, Rubin MA, Palanisamy N, Varambally S, Chinnaiyan AM. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discovery. 2011;1:35–43. doi: 10.1158/2159-8274.CD-10-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC, Paul L, Ferris MW, Graves BJ. The ETS gene ETV4 is required for anchorage-independent growth and a cell proliferation gene expression program in PC3 prostate cells. Genes Cancer. 2011;1:1044–1052. doi: 10.1177/1947601910395578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Leong KG, Newton K, Yee C, O'Rourke K, Liu J, Phu L, Vij R, Ferrando R, Couto SS, Mohan S, Pandita A, Hongo JA, Arnott D, Wertz IE, Gao WQ, French DM, Dixit VM. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature. 2011;474:403–406. doi: 10.1038/nature10005. [DOI] [PubMed] [Google Scholar]
- Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis. 1997;18:1215–1223. doi: 10.1093/carcin/18.6.1215. [DOI] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myagmar BE, Umikawa M, Asato T, Taira K, Oshiro M, Hino A, Takei K, Uezato H, Kariya K. PARG1, a protein-tyrosine phosphatase-associated RhoGAP, as a putative Rap2 effector. Biochem Biophys Res Comm. 2005;329:1046–1052. doi: 10.1016/j.bbrc.2005.02.069. [DOI] [PubMed] [Google Scholar]
- Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, Scher HI, Zheng D, Sawyers CL. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;8:1023–1029. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytes A, Mitrofanova A, Kinkade CW, Lefebvre C, Lei M, Phelan V, Lekaye HC, Koutcher JA, Cardiff RD, Califano A, Shen MM, Abate-Shen C. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc Natl Acad Sci U S A. 2013;110:E3506–E3515. doi: 10.1073/pnas.1303558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson MA, Lafargue CJ, Macdonald TY, Pflueger D, Kitabayashi N, Santa-Cruz AM, Garsha KE, Sathyanarayana UG, Riley JP, Yun CS, Nagy D, Kosmeder JW, Pestano GA, Tewari AK, Demichelis F, Rubin MA. Testing mutual exclusivity of ETS rearranged prostate cancer. Lab Invest. 2010;91:404–412. doi: 10.1038/labinvest.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/S0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/S0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/S1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/S0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. doi: 10.1016/S0092-8674(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Livet J, Sigrist M, Stroebel S, de Paola V, Price SR, Henderson CE, Jessell TM, Arber S. ETS gene Pea3 controls the central position and terminal arborization of specific motor neuron pools. Neuron. 2002;35:877–892. doi: 10.1016/S0896-6273(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes-Davis A, Reid YA, Kline MC, Storts DR, Strauss E, Dirks WG, Drexler HG, MacLeod RA, Sykes G, Kohara A, Nakamura Y, Elmore E, Nims RW, Alston-Roberts C, Barallon R, Los GV, Nardone RM, Price PJ, Steuer A, Thomson J, Masters JR, Kerrigan L. Match criteria for human cell line authentication: where do we draw the line? Int J Cancer. 2013;132:2510–2519. doi: 10.1002/ijc.27931. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geback T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- Hollenhorst PC, Chandler KJ, Poulsen RL, Johnson WE, Speck NA, Graves BJ. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet. 2009;5:e1000778. doi: 10.1371/journal.pgen.1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantitative assessment of ETS protein levels.
Representative images cell migration assays.
Representative images of scratch assays.
Oligonucleotide primer sequences.