

Introduction
Necrotizing enterocolitis (NEC) is an emergency affecting the bowel in neonates, most often in Very Low Birth Weight (VLBW) premature newborns. The incidence of NEC in VLBW infants approaches and sometimes exceeds 10% in neonatal intensive care units around the world [1]. This incidence has not significantly changed in the past two decades. However, with improved survival of extremely premature newborns over the past few decades, the prevalence of NEC and its complications have increased [2]. NEC can be often managed medically, but 20–40% of infants need surgical management [3]. Infants that require surgical intervention are at higher risk of mortality and longer-term morbidity including complications such as stoma prolapse or stricture, short bowel syndrome, and impaired long-term growth and neurodevelopmental outcomes [4, 5].
Despite a high volume of research in clinical as well as laboratory settings, we still possess only a limited understanding of the pathophysiological mechanisms of this devastating illness. Multiple etiologic factors including immaturity of the preterm newborn intestinal tract, formula feeding, infections, and ischemia have been associated with NEC. A combination of these risk factors, perhaps based on genetic predisposition, possibly lead to the mucosal and epithelial injury that is the initiating event of NEC. Intestinal epithelial integrity depends on a fine balance between proliferation and differentiation of epithelium from intestinal stem cells (ISC) that line the intestinal crypts and cellular loss by apoptosis near the villi tips. Signaling mediated by a variety of pathways plays a key role in creating and maintaining this balance and also in creating a mucosal barrier that serves to reinforce intestinal epithelial integrity. The aim of this article is to provide an in-depth review of intestinal epithelial barrier development and structure, and how they are affected by inflammatory signaling and regulatory pathways, leading to NEC.
Development of the intestinal barrier
Early embryonic development
Containment of absorption from the environment in a closed tube was a key evolutionary step in the development of multicellular organisms. The formation of a specialized digestive organ allowed the development of other cells into additional specialized organ systems. This is unlike primitive organisms, which cannot regulate their absorptive surface. Similar to most epithelial surfaces in vertebrates, the epithelium of the small intestine is derived from the endoderm. Exposure to the TGF-β related growth factor, Nodal and Sox-2 allows endodermal cells to express Hhex thereby becoming committed to the anterior endoderm. Conversely, cdx-2 expression in the posterior endoderm leads to upregulation of transcriptional factors that are required for intestinal development, resulting in the development of small and large intestines [6, 7]. After successive transformations through pseudostratified squamous epithelium and stratified epithelium to the more mature columnar epithelium, cells begin to undergo differentiation accompanied by villus and secondary lumen formation. Combined with mesenchymal condensation and proliferation this process leads to the development of the crypt-villus units, as the basic building blocks of the small intestinal epithelium. In the large intestine, villi are absent and mature colonocytes form the colonic surface epithelium instead. Significant mesenchymal-endodermal interactions occur throughout the process of intestinal development. Endodermal derived PDGF and Hedgehog signals (mediated by the FoxL1 transcription factor) induce mesenchymal growth into nascent villi, and in turn, mesenchyme-derived signaling pathways such as Wnt/β-catenin-Tcf3 and BMP initiate development and differentiation of the epithelial cells [8, 9]. Scattered islands of proliferating cells become more organized in inter-villus spaces, and provide niches populated by precursor stem cells to form the intestinal crypts of Lieberkuhn. Wnt and BMP signaling pathways also play important roles in crypt development – inhibition of these pathways leads to disordered and abnormal crypt formation.
Cellular differentiation
In the small intestine, crypt base columnar (CBC) stem cells produce progenitor cells that develop into epithelial cells of various types. Initial formation of villi is associated with the first visible signs of epithelial differentiation into absorptive enterocytes, and secretory goblet and enterochromaffin cell types. Crypt development leads to the emergence of another distinctive intestinal secretory epithelial cell type called the Paneth cells, which play a central role in various functions of the GI tract. Numerous signaling pathways and transcription factors have been identified to be involved in the intricate mechanisms that underlie intestinal epithelial cell differentiation.
Following a cycle of self-renewing asymmetrical division and one or more subsequent transit amplifications steps in the crypt stem cell niche, newly formed intestinal epithelial cells differentiate into at least seven different cell types. Of these seven, five are relatively well characterized and two remain to be relatively obscure. The five relatively well characterized members are enterocytes, goblet cells, Paneth cells, enteroendocrine cells and M cells, whereas the more obscure members are the cup cells and tuft cells. As shown in figure 1, CBC cells produce progenitor cells that are variably induced by atonal homologue-1 (atoh1) or hairy enhances of split-1 (hes1) to differentiate into the various secretory cell types or into absorptive enterocytes respectively [10, 11]. Both Atoh-1 and Hes-1 repress and regulate each other. Atoh1 allows intestinal expression of the Neurogenin3 (neurog3) transcription factor that induces differentiation of enteroendocrine cells. Growth factor independent factor 1 (gf1) inhibits this pathway to permit differentiation into goblet/Paneth cells, a pathway that is modulated by the SAM pointed domain containing ets transcription factor (spdef) [12]. Thus relative functional activity of the neurog3/gf1 factors decides allocation of cell fates between these alternate pathways of differentiation. Other transcription factors and proteins have also been implicated - SRY-box containing gene 9 (Sox9) and CDX-2 (a homeobox transcription factor) in the formation and proliferation of the Paneth cell population, Forkhead box protein A1 and A2 (FOXA1, FOXA2) in goblet and enteroendocrine cell differentiation, and Kruppel-like factor 4 (Klf4) in goblet cell development [13–15]. Hes1 also induces differentiation of absorptive enterocytes. Absorptive enterocytes are poorly developed in E47-like factor 3 (Elf3) knock-out (KO) mice indicating Elf3’s importance in enterocyte development [16, 17]. Notch signaling pathways play a very important role in intestinal epithelial cell differentiation, and both Atoh1 and Hes1 activity are regulated by Notch ligands such as Dll1 and Dll4. Loss of Notch activity leads to conversion of intestinal epithelial cells into goblet cells [18]. TLR4 signaling via Notch is increased in intestinal tissue samples from patients with NEC, and numbers of goblet cells were reduced [19]. Additionally, TLR4 signaling-dependent Notch signaling suppresses goblet cell differentiation, and this mechanism was required for induction of experimental NEC in mice [20] indicating that these mechanisms have significant roles in the pathogenesis of NEC.
Figure 1.
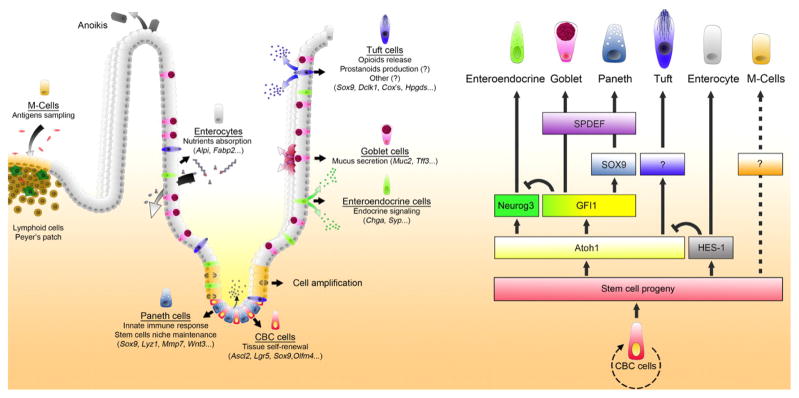
Schematic of intestinal epithelial differentiation. Adapted from Gerbe at al 2011. Following the generation of new cells, intestinal epithelial cells must undergo differentiation, which occurs simultaneously with their migration out of the stem cell niche. Transcription factors that drive this differentiation as well as functions of the terminally differentiated, specialized epithelial cells are shown.
Ontogeny
The basic structure of the epithelium is laid down by the end of the first trimester. Goblet cells, enteroendocrine cells and absorptive cells with microvilli are seen starting from 8 weeks. The crypt-villus axis is established around this time, and villi begin to develop in a proximal-distal fashion around 10–12 weeks of gestation. Mature crypts appear at 12 – 19 weeks. Intercellular tight junctions (although not fully functional yet) become detectable by 10–12 weeks but mature enterocytes develop only by 22 weeks. Goblet cells start producing mucin by 12 weeks [21]. Paneth cells are seen after week 12 and start producing defensins by week 13. The intestinal immune system with functional Peyer’s patches becomes established by week 20, but becomes fully mature only after 29 weeks of gestation [22]. The premature newborn intestinal barrier therefore contains most of the structural elements that make up the mature anatomic intestinal barrier, but many of these components are still functionally immature. The immature barrier is permeable to macromolecules that allows for exchange of bioactive molecules between the amniotic fluid and fetus in utero [23]. This excess permeability needs to be abolished quickly after birth to prevent entry of noxious and injurious stimuli [24]. This happens in term infants in the first postnatal week and is augmented by breast milk and probiotics [25, 26]. Preterm infants replicate this phenomenon, but in a less efficient pattern, i.e. infants at 26–29 weeks GA were shown to exhibit normal levels of permeability soon after birth similar to term infants, but this changes around 3–4 weeks of life when their gut permeability increases temporarily to fetal levels for unknown reasons. Its intriguing that this happens around the clinically expected peak period for incidence of NEC [27]; exploring the significance and mechanisms behind these permeability changes may be key to preventing NEC.
Structure of the intestinal barrier
Humans are exposed to and interact with diverse antigens and numerous microorganisms every day of their existence. As the largest mucosal surface and immune organ system in the human body, the gastrointestinal tract plays a very important role in interfacing humans with their environment. Maintenance of intestinal mucosal integrity prevents entrance or absorption of harmful microbes and toxins; however, the epithelial barrier must also allow sufficient and selective permeability, as well as provide active transport, in order to absorb and retain nutrients. Intestinal epithelial structure and the contribution of its cellular, junctional and non-cellular elements (active transport processes, passive permeability and synthesis and secretion of bioactive components) to the creation of this balanced and complex luminal barrier are reviewed below along with potential disturbances in these elements that can lead to NEC.
Cellular Elements
The intestinal epithelial lining is made up of a single layer of multiple cell types, all derived from the ISCs. These mucosal cells serve multiple functions: nutrient absorption, preventing entry of pathogenic organisms and unprocessed antigens, innate inflammatory signaling, secretion of molecules that contribute to the mucosal barrier, antigen presentation to underlying immune cells, and production of endocrine signaling molecules. Portions of the epithelial layer that are immediately adjacent to the mucosa associated lymphoid tissue (MALT), such as Peyer’s patches, have their own crypt cells that differentiate into M-cells and columnar epithelial cells which cover the surface of this specialized lymphoid tissue.
Absorptive enterocytes make up 80% of the intestinal epithelial cells. While their major function is absorption of nutrients, they are also responsible for maintenance of the intestinal barrier. Similar to all other epithelial cell types, a principal characteristic of enterocytes is cellular polarity. Their membranes are divided into apical and basal/lateral or basolateral domains, separated from each other by the tight junction. This polarity is generated and maintained by specialized membrane trafficking routes and compartments (reviewed by Roignot et al) [28]. The apical plasma membrane has a specialized lipid composition, and includes transporters, receptors and enzymes that are unique to this domain and enable it to perform its specialized functions. Additionally, the apical plasma membrane forms microvilli (“brush border”) that lie underneath the mucous layer and increase the overall absorptive surface multifold. They also organize the overlying epithelial lining fluid and mucin layer. The inter-microvillar space is loaded with antimicrobial peptides and adsorbed proteolytic enzymes. Both of these, together with the mucins, limit microbial adherence and invasion. The basal/lateral surfaces also contain a specific complement of receptors, transporters and enzymes that are required for the specialized functions of this domain. Particularly abundant and relevant component in this domain is the polymeric immunoglobulin receptor (PIGR) that perform secretory IgA transcytosis across the cell to be secreted into the intestinal lumen (reviewed by Mostov) [29]. Absorptive enterocytes can also express neonatal Fc receptors (FcRn), which can mediate bidirectional transport of immunoglobulins between the luminal compartment and the submucosa (reviewed by Baker et al) [30]. This system is much better characterized in rodents, but there is evidence for the expression of FcRn in the human fetal and neonatal intestinal epithelium [31].
Several aspects of NEC pathogenesis have been described that relate to almost all of the known functions of absorptive enterocytes listed above. The apical membrane of enterocytes is loaded with lactase as well as glucose and galactose transporters necessary for digestion of lactose and efficient absorption of the resulting glucose and galactose. NEC almost never occurs in neonates before the beginning of enteral feeding and it has been postulated that stagnation of undigested and/or unabsorbed nutrients contribute to bacterial overgrowth that may be pathogenic. A relative lactase deficiency, or a decreased expression of glucose and galactose transporters have been considered as underlying causes [32]. The highest level of expression for these molecules is in the jejunum, yet their deficiency predictably would result in a cumulative effect toward the distal end of the small intestine. This spatial separation of cause and effect, together with the limited availability of intestinal - typically, ileal - specimens from human NEC, poses a severe limitation on our ability to address this hypothesis. There is however, at least limited evidence for benefit from formula with pre-digested lactose in the prevention of NEC - along with increased weight gain [33, 34]. Additionally, there is evidence in animal models that sugar malabsorption can result in experimental NEC [35]. The function of FcRn as a very important determinant of neonatal health has been demonstrated clearly in rodents, but human-specific importance in the GI system is yet to be demonstrated.
Microfold (M) cells are located in follicle-associated epithelium (FAE) that is part of the mucosal immune system. They possess unique microfolds that allow microbes to gain entry and their basolateral surfaces have pocket-like structures that harbor B and T lymphocytes, macrophages and/or dendritic cells. This structure enables M-cells to absorb antigen molecules by phagocytosis and then present them to the antigen-presenting immune cells. Thus, they act as functional openings in the intestinal mucosal barrier [14]. Increase in M-cell number has been noted during intestinal inflammation. This might serve to increase antigen presentation to immune cells leading to protective immune responses [36]. Despite their potentially important role in regulating immune tolerance and antimicrobial immune responses, there is virtually no data on the developmental changes of M cells in the fetal and neonatal small intestine or, specifically, in necrotizing enterocolitis.
Goblet cells are the major secretory epithelial cells in both the small and large intestines. They are the source of numerous factors that are essential for maintenance of the barrier function such as mucin, trefoil peptides, resistin-like molecules-β and Fcγ binding protein [37]. Additionally, a transcellular route of antigen presentation has been described for goblet cells. In an asphyxia/cold stress model of NEC in rats, goblet cell number, function and mucus layer thickness was shown to be decreased in areas of gut affected by NEC compared to normal areas. In addition to restoring other functions such as paracellular permeability and tight junction integrity, administration of the trophic factor EGF was also shown to increase mucus layer thickness and aid the proliferation of goblet cells. These changes correlated with a reversal of NEC-like changes in the same model [38]. Ischemia-induced mucus barrier loss and bacterial penetration are quickly reduced by goblet cell proliferation in both human and mice models, at least in the colon [39]. More recent studies using mice with intestinal epithelial-targeted disruption of the TLR4 gene in a rodent model of NEC indicate that activation of notch signaling by TLR4 results in goblet cell depletion. Mechanistically, this was mediated by the TRIF-dependent arm of TLR4 signaling and it was required for the development of experimental NEC [20].
Paneth cells are unusual among mature epithelial cells in that they are located in the crypt bases rather than in the villi, are seen only in the small epithelium, and have a lifespan of 30 days or more compared to the 2–5 day lifespan of the rest [40]. One of their important functions is to serve as a critical protective niche for stem cells in the crypt bases. They are also responsible for producing many of the anti-microbial peptides such as defensins, cathelicidins, Ang4, and regenerating islet-derived protein 3 gamma (RegIIIγ) in response to invasion by pathogens as is evidenced by immunostaining for these molecules and the massive protein secretory apparatus including rough endoplasmic reticulum and Golgi apparatus that are seen in these cells [41, 42]. Other products include Lysozyme C and phospholipase A2 which break down peptidoglycan and phosphoglycerides found in bacterial cell walls. These secretory products reduce prolonged antigen exposure and the accompanying inflammation. They also secrete trophic factors such as EGF, TGF-α, Wnt3 and Dll4 that promote maintenance of the intestinal stem cell population [43].
Lately, there has been significant attention to Paneth cell activation and/or depletion as a potential significant contributor to NEC pathogenesis. There are multiple lines of evidence for changes in Paneth cell numbers between tissue samples collected from patients undergoing bowel resection for NEC and various “controls”. A serious difficulty in interpreting these findings is the varying degree of necrosis in the surgically removed bowel sections, the varying lengths of time between the acute necrotic process and the actual surgical intervention and the variety of bowel specimens that were considered as controls for the sake of comparison. Despite the difficulty of interpretation, this remains to be a worthwhile are of future investigation… that have substantiated the mechanistic role of Paneth cell “injury” in rodent models of NEC. From the point of the present review, it is notable that Paneth cells, in addition to their innate defense role, have been suggested to serve as “reserve stem cells” as they retain an ability to be re-programmed “ to stem cell-like phenotypes [44]. Thereby, they may play a role in the ability of the intestinal epithelium to recover from injury.
Tuft cells and enteroendocrine cells
In rodents tuft cells in the intestine develop postnatally and are increasing in number at weaning. However, in the human intestine they appear at 20–22 weeks of gestation [45]. They are the predominant intestinal epithelial cells that possess cyclooxygenase activity and may serve as a source of prostaglandins, which are important regulators of inflammatory processes [46]. However, relatively little is known about tuft cells and their role in disease processes. Although there is no specific information available regarding any changes of tuft cell numbers or pertaining to a potential role in NEC it is notable that cyclooxygenase inhibitor drugs increase the risk of intestinal injury. Given that tuft cells are the major source of cyclooxygenase activity, investigating their potential role in NEC would be warranted. Enteroendocrine cells are the source of several hormones such as gastrin, histamine, serotonin, cholecystokinin (CCK), somatostatin and glucagon-like peptides (GLP1 and 2). These hormones have significant signaling functions both locally in the GI tract and systemically. Locally, they are important for GI motility, luminal secretions, and regulation of immune and inflammatory processes [47]; several of these hormones also regulate epithelial turnover.
Intercellular junctions
In addition to the intestinal epithelial cells themselves, the spaces and junctions between them are equally important in forming the epithelial barrier, and regulating its permeability properties. The intercellular junctions in the intestinal epithelium form the lateral junctions between cells and are made up of at least 3 distinct elements: adherens junctions, tight junctions and desmosomes [48]. The basic architecture and building block of the epithelial junction complex is illustrated in figure 2 [49]. Adherens junctions and tight junctions together form the apical junctional complex (AJC). Desmosomes are localized somewhat more basally and usually are referred to as lateral junctions. Adherens junctions (AJs) consist mostly of intracellular catenin-F-actin complexes linked to transmembrane cadherins that form complexes with similar E-cadherins of neighboring cells, thus creating links between adjoining cells. Another element in this link consists of cytoskeletal F-actin binding proteins called afadins interacting with immunoglobulin-like transmembrane proteins called Nectins that in turn interact with nectins from neighboring cells, serving to strengthen the linkage created by E-cadherins [48]. In addition to providing a mechanism for cells to adhere to each other, the junctional proteins are linked to critical signaling systems on the cytoplasmic side, which regulate fundamental cell survival pathways [50]. Tight junctions (TJs) can be visualized as areas of actual contact between adjoining cells. These are formed by multiprotein complexes made up of occludins, claudins, junctional-adhesions molecule-A and tricellulin. These proteins interact both with proteins from the same cell (cis) and with proteins from neighboring cells (trans) in both homotypical and heterotypical fashion to form circumferential ring like structures that interface with cytoskeletal scaffolds including F-actin [51, 52]. TJs protect against deleterious invasion of the epithelium by antigens, microbes and toxins and maintenance of cytoskeletal integrity. Additionally, they are important for maintaining epithelial polarity by preventing movement of membrane lipids and proteins between the apical and basolateral domains. Similar to adherens junctions, the intracellular domain of the tight junction complex is linked to signal transduction pathways. These, in turn, play significant roles in regulating proliferation, survival, migration and differentiation of epithelial cells [53]. Furthermore, the specific makeup of tight junctions, and the regulation of their molecular components have major roles in regulating the selectivity and magnitude of paracellular permeability. Desmosomes are the third type of intercellular junctions whose role and importance in barrier maintenance is less understood than the apical junctional complex.
Figure 2.

Figure 2 the epithelial junctional complex. Adapted from Laukoetter et al [49].
The structural and functional integrity of tight junctions is not static in nature and can be influenced by numerous factors. Osmotic load, along with the availability of glucose and sodium in the gut lumen, has been shown to vastly increase TJ permeability in rat jejunum [54–56]. Injury by oxidants is known to disrupt the actin cytoskeleton leading to “leaky” junctions [57]. Proteomic studies in induced NEC lesions in preterm piglets have shown that proteins involved in cytoskeletal and cell integrity and motility are found in increased quantities in their proteome [58]. Actin and tubulin have been shown to be part of the differential proteomic responses to enteral feedings detected in plasma when preterm piglets are raised in germ-free vs. conventional environments [59]. The gram-negative endotoxin LPS causes cytoskeletal rearrangements in human small intestinal mesenchymal cells and actin disruptors severely affect the release of IL-6 from small intestinal human fibroblasts. Actins are known mediators of epithelial healing responses, especially after injurious stimuli from gram negative stimuli [60], and after treatment with antibiotics [61]. Nitric oxide (NO), which has roles at multiple levels in the pathogenesis of NEC, is known to regulate intestinal permeability by changing the expression and localization of tight junction proteins ZO-1 and 2, occludin and claudin [62]. Probiotic mixtures prevent or reduce ileitis through a TNF-α dependent mechanism in mice with increased intestinal permeability by increasing occludin and tight junction proteins and decreasing the expression of claudin [25]. Finally, although Dextran sulfate sodium (DSS) colitis models are associated with tight junction damage related increases in intestinal permeability, many of these models also show evidence of widespread epithelial cell damage, indicating that tight junction loss or dysfunction alone may not be enough to cause disease. However, it is also plausible that chronic increases in intestinal permeability due to tight junction loss may underlie an altered “alert state” of the mucosa-associated lymphoid tissue, which results in a disproportionate inflammatory response.
Non-cellular Elements
Mucin
All mucosal surfaces are covered by a gelatinous secretion called mucus. Mucus is made up of water (95%) with lipids, phospholipids, cholesterol and proteins [63]. It is viscous and elastic and this is primarily due to mucins, large glycoproteins that are produced in the intestines by goblet cells [64]. Carbohydrates including N-acetylgalactosamine and N-acetylfucosamine in moderately branching structures make up 80% of mucin structure, attached to a protein core that is rich in serine, threonine and proline repeats [65]. Many types of mucins have been identified, but MUC2 is the major secreted mucin found in the small intestine [66, 67]. The depth of the mucus barrier varies in different parts of the intestinal tract, with two layers in the stomach and colon and one layer in the small intestine [68]. The viscosity of the mucus layer also varies according to concentration of secreted mucin, and this can be modulated by a variety of factors, including ambient pH and ionic strength. Mucin-associated oligosaccharides adhere tightly to the apical surfaces of epithelial cells, isolating potential pathogens and thus creating a barrier that prevents many pathogenic microbes from reaching the epithelial surface [69]. Lysozyme, defensins, and immunoglobulins, enzymes and other proteins that reside in the mucus layer contribute to the defensive properties of the mucus layer [70]. Gastrointestinal mucins are degraded via complex pathways. Although proteases slowly dissolve the mucus gel to highly glycosylated mucin fragments, continuous mucin production and secretion from goblet cells ensures maintenance of the gel layer.
Until now 21 different mucin genes (MUC) have been identified. Based on their location, structure and function those mucins have been subdivided into secretory and membrane bound forms. Although membrane bound mucins can be located on the surface of the epithelium, their function and role in the gut is yet to be defined. Secretory mucins are primarily responsible for mucus gel formation. Out of the 21 MUC genes, MUC2 is the major mucin reported to be expressed throughout the intestinal epithelium by 12 weeks of gestation. It is highly expressed in the goblet cells and found in the columnar epithelium of the intestine and colon. MUC2 was the first human mucin gene to be cloned and completely sequenced. MUC2 gene KO mice have thinner mucus layers and develop spontaneous colitis. MUC2 rich mucus layer alteration has been shown to be necessary for the induction of DSS- induced colitis in animal models. This highlights the important roles played by the mucus layer in mitigating microbial invasion and inflammation in the GI tract [71, 72]. In young suckling rats, sialyltransferase activity predominates in the production of the immature mucus layer. In contrast, fucosyltransferase is the major enzyme responsible for producing glycoconjugates that are characteristic of the mature mucus layer. Thus, a difference in mucus glycosylation pattern may lead to differences in bacterial colonization patterns [73] [74]. Newborn intestinal mucus contains more protein and less carbohydrate compared to adults and this may affect properties ranging from viscosity to antigenicity, effects that may be even more pronounced in preterm newborns [75]. Overall, developmental differences in the quantity and quality of mucin produced have important contributions towards mucosal injury in premature newborns.
Trefoil proteins
Goblet cells in the intestinal mucosa produce a 59-amino acid peptide called intestinal trefoil factor (ITF). ITF is secreted onto the surface of the epithelium in both the small and the large intestine [76]. Trefoil stands for Trifolium, or three-leaved plant, due to the resemblance of ITF crystal structure to such pants. The peptide chains of ITF isoforms contain a conserved trefoil motif of 40–45 amino acid residues. Six cysteine residues in this motif hold the intervening peptide sequences in a three looped structure with the aid of disulfide bonds. Because of its three looped structure, ITF is resistant to acidic pH and proteolytic digestion in the gastrointestinal tract.
In humans, three trefoil peptides have been discovered i.e. pS2 (TFF1), human intestinal trefoil peptide- hITF (TFF2) and human spasmolytic polypeptide- hSP (TFF3). Genes of those three peptides have been localized to a cluster on chromosome 21. Under normal physiological conditions, all three peptides are predominantly expressed in the epithelium of the gastrointestinal tract. hITF, in particular, is expressed in the duodenum and colon. Trefoil factors play an important role in the process of restitution, a process by which uninjured cells surrounding an area of mucosal damage migrate across the injured surface to rapidly restore epithelial integrity. Animal models of intestinal mucosal injury have shown an increase of up to 1000fold of trefoil protein. During repair of a damaged intestinal epithelial layer, goblet cells secrete ITF proteins. Trefoil proteins have shown cooperative interactions with mucin glycoprotein in reducing detergent and toxin induced mucosal hyper-permeability in intestinal cell monolayer models.[77] The importance of trefoil factors is illustrated by experimental models of (DSS) induced colitis, in which ITF-deficient mice had significantly increased DSS colitis compared to wild-type mice. Restitution was significantly decreased in these mice and improved with administration of ITF, after which epithelial cells form continuous sheets with very few gaps. ITFs possibly exert this protective effect by acting on the E-cadherin catenin complex [78] and by inhibiting apoptosis [79–81].
There is at least some evidence that TFFs may play a beneficial role in preventing NEC. TFFs are abundant in human milk, which is protective against NEC, at the highest concentration in the colostrum, then rapidly declining after birth [82]. Experimentally, oral TFF administration has been shown to ameliorate NEC-like histological changes and inflammation in a commonly used rodent model of NEC [83].
Antimicrobial Factors
Paneth cells, as well as mucosa-associated immune cells release a number of antimicrobial factors; reviewed by Ho et al [84]. Chief among them are small cationic peptides called α and β defensins, which are the most commonly found endogenous antimicrobials in the gastrointestinal tract. Two human α isoforms (HD5 and HD6) and the mouse α isoform cryptidin are exclusively expressed by Paneth cells. Human and mouse β defensins are expressed throughout the GI epithelium. Of these, HBD1 (human) and mDefB10 (mouse) are expressed constitutively, while HBD2, 3 and 4 (human) and MBD3 (mouse) are strongly induced under inflammatory conditions [85]. Cathelicidins, another class of Paneth-cell derived antimicrobial peptides, are also present in the amniotic fluid and milk. Their expression is stimulated by bacterial products such as butyrate and endotoxin [86].
Two antiproteases that are secreted by the GI epithelium have been endowed by antimicrobial properties as well. These are elafin and secretory leukocyte proteinase inhibitor (SLPI). Both are upregulated under inflammatory conditions and have been thought to stimulate mucosal healing [87, 88]. Other antimicrobial peptides that are expressed in lesser or higher quantities by epithelial cells in the GI tract include: bactericidal/permeability increasing protein (BPI), hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein (HIP/PAP), and lysozyme [89]. Breast milk derived Lactoferrin also has a significant role in gut immunity.
Enteric defensin expression is apparent at 24 weeks and gradually increases towards term in the developing intestine [90, 91]. Salzman et al [90] showed that infants with NEC exhibit increased level of defensin (HD5 and HD6) mRNA expression and protein levels as compared to controls comprised of ileal atresia or meconium ileus. Paneth cell numbers were also increased in the more preterm infants with NEC compared to controls without NEC. In another study, patients with NEC were gestational age-matched with controls and repeat samples were collected from the same patients at the time of ostomy takedown [92]. In this cohort, there was no difference in Paneth cell numbers and antimicrobial gene expression between controls and NEC, but there was a significant increase in both Paneth cell numbers and in antimicrobial peptides after recovery from NEC. Yet another study of fecal levels of HBD1 and HBD2 showed an increase in moderate NEC, but no increase in patients with severe NEC [93]. In this study there was an increased level of HBD1 and HBD2 expression after recovery from NEC. Given the divergent study populations, methods and antimicrobial genes tested it is not possible to form a coherent conclusion regarding the role of antimicrobial peptides in the pathogenesis of NEC. However, the data are overall consistent with a protective role for these innate defense molecules in NEC. It is plausible that a relative deficiency of antimicrobial peptides and Paneth cells at low gestational ages may play a role in NEC pathogenesis.
Another major component of the defensive barrier is secretory IgA which is produced by plasma cells that populate the lamina propria of the GI tract. IgA protects against a host of inflammatory agents including viruses and bacterial endotoxin through the immune exclusion mechanism [94]. Secretory IgA (sIgA) contains the extracellular domain of PIGR, also called secretory component or μ chain, which is cleaved from the rest of the receptor when the receptor-IgA complex is exocytosed at the apical membrane. This μ chains endows sIgA with anti-inflammatory functions and characteristics that are additive to that of serum IgA without the secretory component; reviewed by Johansen and Kaetzel [95].
sIgA has been recognized as a critically important component of mucosal defense and some of the protective effects of human colostrum and milk against NEC have been attributed to its sIgA content. A relative deficiency of sIgA in the premature neonate has been implicated as a potential risk factor for NEC. A study conducted in the 1980-s concluded that oral supplementation of immunoglobulins may prevent NEC [96]. This was the only reasonably well-designed study so far that used an immunoglobulin preparation that was composed of mainly IgA. An overall conclusion reached through meta-analysis indicates that oral supplementation of immunoglobulins provides no benefit in the prevention of NEC, or any other major neonatal outcomes of prematurity. However, it is notable that trials so far have used mostly IgG; even the study that showed benefit used IgA without the critical secretory component that makes sIgA functionally distinct from unprocessed IgA. It is yet to be determined if specific supplementation of sIgA would provide any benefit.
Miscellaneous factors
Periodic and coordinated movements orchestrated by the enteric nervous system and carried out by the intestinal muscle layers are responsible for efficient passage of gastrointestinal content. Efficient passage of GI content is important for nutrient absorption. Additionally, proper motility prevents the stagnation of nutrients that may lead to bacterial overgrowth. Migratory motor complexes (MMCs) are the elementary waves of activity that trigger peristalsis. These are incompletely developed until 32 weeks of gestation and very uncoordinated in infants less than 27 weeks of gestation [97]. MMCs are mediated by cyclical surges in the hormone motilin and pancreatic polypeptide. Neither of these hormones is fully established until after birth and may contribute to the absence of these motor complexes in more immature infants [98]. Ischemic damage affecting the enteric nervous system and surrounding glial cells has been noted in autopsy specimens from infants that died due to NEC [99]. Specific ablation of glial cells causes massive changes similar to NEC and Crohn’s disease in animal models [100]. Heparin-binding EGF (HB-EGF) has been shown to be required for the normal development of the enteric nervous plexus in rodents [101]. Given that HB-EGF also protects rodents from experimental NEC [102], these findings establish the possibility of a causal relationship between development of the enteric nervous plexus and NEC.
Intestinal epithelial self-renewal and repair
The intestinal stem cells (ISCs) niche: While their exact location is still an area of debate, crypts in both the large and small intestine contain stem cells that express the leucine-rich G protein-coupled receptor 5 (Lgr5) [103]. Other ISC markers include Bmi1+, Ascl2, Olfactomedin-4 (Olfm4) and Musashi-1 (Msi-1). ISC cells that express these markers often also exhibit Lgr5+ positivity, so it is unclear whether such cells represent distinct sub-groups with unique functions [104, 105]. Intestinal stem cells are surrounded by Paneth cells, which provide a protective and permissive niche for these cells to proliferate and differentiate. This niche is also made up of other elements – enteric neurons, intraepithelial lymphocytes, and the basement membrane. Similar to the in utero development of the GI tract, interaction between mesenchymal cells such as myofibroblasts and stem cells is crucial for the maintenance of the stem cell niche throughout life [106].
Stem cell self-renewal
The intestinal epithelium is among the most actively self-renewing tissues in mammals. Stem cells occupying intestinal crypts are monoclonal and are equally capable of populating the crypts they occupy, implying that there is no “single dominant stem cell” [107]. CBC stem cells sustain the turnover of epithelial cells by undergoing cell division either symmetrically (2 daughter stem cells or 2 daughter non-stem progenitor cells) or asymmetrically (1 stem cell that remains in the crypt and a transit-amplifying (TA) cell. TA cells go through multiple cell division cycles to produce more progenitor cells. These migrate upwards along the crypt base and villus surface while undergoing further differentiation into the mature cell types of the epithelium. The restricted space within crypt bases plays an important role in deciding the relative fates of daughter stem cells. Progeny that lose contact with Paneth cells migrate upwards to continue the process of differentiation while cells that maintain contact continue to reside in the crypt base to maintain stem cell reserves [108]. Proliferation of stem cells is still not well understood, but it is clear that factors such as Wnt, BMP, Hedgehog and Notch all play important roles in regulating this process. Injurious stimuli such as radiation damage to stem cell population can cause TA cells near the crypt base to lose their differentiation and revert back to Lgr5+ stem cells. This safeguard allows for replenishment of stem cell numbers, to a limited extent [44]. A recent study has shown that intraperitoneal administration of stem cells from amniotic fluid in a rat model of NEC causes these cells to home to areas of mucosal injury in the intestine. This experimental “stem cell therapy” reduced the incidence of experimental NEC and improved the survival of rat pups. AFS cells homed to intestinal villi and induced increased stromal cell COX-2 expression [109]. Similarly, bone marrow-derived mesenchymal stem cells administered either intraperitoneally or intravenously homed to sites of NEC like injury and improved outcomes in a rat model of NEC [102, 110].
Epithelial proliferation
The basal rate of proliferation in the intestinal epithelium is driven by the coordinate action of extracellular mediators such as Wnt, homotypic cell adhesion by the E-cadherin β-catenin system and signaling via integrin-extracellular matrix interactions. These basal mechanisms are further augmented or attenuated by hormones, growth factors and inflammatory mediators. Homotypic cell adhesion via E-cadherin stabilizes β-catenin in adherens junctions. In the absence of Wnt signaling the remaining β-catenin in the cytoplasm is phosphorylated, targeted to the β-catenin degradation complex and degraded, preventing its translocation to the nucleus. In the absence of nuclear-translocated β-catenin, expression levels of key signaling molecules of cell survival and cell cycle progression are diminished, leading to cell cycle arrest. Activation Frizzled or LRP receptors by the extracellular ligand Wnt prevents proteosomal degradation of β-catenin, which activates transcription of target genes leading to stem cell proliferation [111]. While Wnt is produced by Paneth cells, this has been shown to a dispensable factor for epithelial proliferation in-vivo, indicating that other sources of Wnt exist in the adult mammalian intestinal tract [112]. In vitro studies of Lgr5+ ISCs have shown that provision of growth factors including EGF, R-Spondin1, Jagged and Noggin are necessary for the production of crypt-villus organoid structures as well as for the production of epithelial cells of different lineages for prolonged periods of time. Other factors are produced by the mesenchymal cells in vivo. Mesenchymal cells also produce Wnt-2b, an alternative and replacement to Paneth cell-derived Wnt [113]. Proliferation of cells within the confined spaces of the crypts is thought to be the primary driving force behind the migration of these cells along the crypt-villus axis and into the villi tips [114].
Toll-like receptor 4 (TLR4), the receptor for Gram-negative bacterium-derived LPS has a significant role in the pathogenesis of NEC in animal models. The biology of TLR4 and its implications for NEC have been thoroughly reviewed elsewhere in the same issue (Lu and Hackam). However, it is notable that there is a significant discord between the roles of TLR4 signaling in NEC and in adult models of mucosal injury. Overall, there is a substantial body of evidence indicating that TLR4-dependent inflammatory signals are required for efficient repair in adult murine models of colitis. In these adult rodent models of disease, loss of TLR4 signaling results in deficient repair and overall worse disease outcomes. This is in contrast to findings in neonatal rodent models of necrotizing enterocolitis [19, 20, 115, 116]. It is also notable that in rodent models of NEC TLR4-dependent mechanisms inhibit stem cell proliferation [116, 117], which is in contrast to TLR4-dependent increased proliferation in adult models [118, 119]. In colitis-associated increased mitotic activity, the inflammatory regulation of proliferation was dependent on TLR4 signaling and COX-2 [118, 120]. These findings relate back to the potential but mostly unexplored role of tuft cells in the inflammatory regulation and homeostasis of the epithelial barrier [46]. In the adult models decreased stem cell proliferation and deficient repair after injury results in overall worsened disease outcomes. TLR4 has been shown to activate β-catenin in intestinal neoplasia models as well [119]. The simplest explanation for the differences between neonatal and adult models is a developmental change that shifts the TLR4-dependent inflammatory signaling from pro-apoptotic to pro-proliferative in nature. Alternatively, the different observations may reflect on differences between colon and small intestine, since the dominant manifestation of NEC and colitis are affecting different parts of the GI tract. Yet another explanation is a difference between the relative contribution of innate and adaptive immunity and their respective mediators in neonatal and adult models. Nevertheless, the details of underlying mechanisms are yet to be characterized and likely hold important clues regarding disease pathogenesis.
The apparently beneficial roles of inflammatory signals in tissue repair are not unique to colitis. Inflammatory signals have been shown to play roles in recovery after tissue injury in organs other than the gut by regulating stem cell proliferation locally and by recruiting stem cells to sites of injury. For instance, inflammation-induced stem cell proliferation and repair have been described in the CNS [121–123].
Epithelial migration
The time course is 2–4 days from stem cell proliferation in the crypt and apoptosis at the villus tip. During this time, epithelial cells have to migrate the distance of 600 – 800 μm, approximately the width of 60–100 cells. This migratory process requires the interaction of the cells with the extracellular matrix via integrins, signaling via a multitude of kinases and an active involvement of the cytoskeleton. There is a great deal of overlap between signaling mechanisms that regulate cellular proliferation, migration and apoptosis. As a general rule of thumb, the same mechanisms that stimulate cellular proliferation also stimulate migration, whereas signals that induce epithelial apoptosis also inhibit migration. Inflammatory signals contribute to this balance with an intriguing mix of stimulatory and inhibitory effects on epithelial migration. Innate, TLR4-dependent signaling inhibits enterocyte migration both in tissue culture models and in rodent models of disease [19, 124]. On the other hand, Leukotriene D4 [125], interleukin 22 [126], and tumor necrosis factor alpha (TNF-α) can either increase or inhibit migration in a dose dependent fashion [127]. Therefore, inflammation cannot simply be viewed as a universally undesirable condition as it has both deleterious and beneficial aspects on the epithelial barrier. It is more likely that the quantity, timing, location and specific aspects of the inflammatory response, and whether these parameters are commensurate with the physiologic requirements will determine desirable or undesirable effects. TLR4-dependent innate inflammatory signaling has been shown to be unequivocally damaging in rodent models of NEC, both in terms of inhibiting epithelial proliferation, migration and goblet cell development [19, 20, 115–117, 124, 128, 129]. The mechanisms and details of these TLR4 mediated deleterious effects are detailed by Lu and Hackam elsewhere.
Apoptosis
Mature epithelial cells that reach tips of the villi persist there for only a short time before they are shed into the lumen. Epithelial cells at villus tips undergo anoikis, a special form of apoptosis seen in anchorage-dependent cells that lose their attachments to their underlying matrix. In this normal process, loss of cell-matrix attachments, is thought to be the cause of cell death, rather than death causing the detachment [130]. Caspase-3 activation is involved in the mechanism of anoikis, and blockade of caspase-3 activity leads to decreased apoptotic cell corpses at the villi [131]. However, mice deficient in caspase/FADD domains exhibited no visible alterations in their epithelial structure. Thus, this mechanism may not be indispensable to normal epithelial homeostasis in the gut [132, 133]. On the other hand, increased apoptosis appears to play an important role in the pathogenesis of NEC. There is evidence for an increased rate of enterocyte apoptosis in human and experimental NEC; in animal models, specific apoptosis inhibition protects from experimental NEC [134–137]. In this pathological scenario, apoptotic nuclei appear to localize within the epithelial layer away from the villus tip, which may be distinct from the normal process of anoikis that occurs at the villus tips during cell detachment.
Healing and repair
While multiple pathogenetic mechanisms cause injury to the intestinal epithelium on a constant basis, the capacity of the barrier for self-repair and restitution is remarkable. Epithelial cells adjacent to a wound exhibit first-response “stopgap” changes in their structure and polarity. They become flattened leading to elongated cells and extend lamellopodia, thus changing their morphology from columnar to fibroblastic. Apical actin filaments linked to the zonulae adherens contract and link to cell to one another. This process creates a “purse string” like contraction that narrows and closes the gap in the monolayer [138, 139]. Further healing depends on multiple processes. All four components, proliferation, migration, differentiation and apoptosis need to function properly and in cohesion to restore epithelial integrity [140]. Proliferation of intestinal epithelial cells has been well-studied in the Drosophila gut model which closely resembles the mammalian gastrointestinal system [141]. Drosophila ISCs in intestinal crypts exhibit marked proliferative responses in response to bacteria and other injurious stimuli. This response is mediated by JAK-STAT and JNK signaling and requires EGFR ligands that are produced by overlying villi enterocytes that undergo apoptosis in response to the injury [142–144]. The process is turned off by BMP-2 signaling which, when overactive, can interfere with ISC proliferation and prevent intestinal epithelial repair. This indicates that optimal proliferative responses to injury depend upon striking the right balance between these opposing signaling pathways [145].
A host of growth factors that normally are present in human milk have been employed in animal models and in tissue culture models to test their efficacy for prevention of experimental NEC or bowel injury in hypoxia-reoxygenation. These include EGF [134], IGF [146], HB-EGF [102, 110], TGF β [147], and EPO [148]. All of these, when tested, exhibited a combined effect on epithelial survival, migration and apoptosis, resulting in decreased injury, improved repair in vivo and in vitro. It is yet to be determined whether any of these agents are safe and efficacious in humans, but they certainly provide novel therapeutic avenues to explore.
Summary
As detailed above, the principal components of the epithelial barrier are multiple epithelial cell types, junctional complexes between the epithelial cells and epithelial secretory products. There are additional constituents contributed by the cells of the mucosa-associated immune system, such as secretory immunoglobulins. When this barrier is intact it protects against intestinal injury. It is generally accepted that a collapse of this barrier plays a significant role in the pathogenesis of NEC. The mature gastrointestinal tract is an environment where the only thing that can be considered constant is change - Cells that line the mucosal layer undergo continuous replacement and ratios of different cell types in this epithelial layer are subject to rapid alterations. The chemical and physical composition of non-cellular constituents of the barrier is also highly adaptable. Given the very nature of gastrointestinal function, repair and maintenance after injury is quite frequent even under conditions that may not be considered specifically pathogenic. In this context, it is important to note that NEC is a disease of the developing gastrointestinal tract. The immaturity of this developing barrier and that of the regulatory mechanisms responsible for its maintenance as well as dis-regulated inflammation are all very important contributors to the pathogenesis of NEC. Yet, most would agree that we are only beginning to understand the details. The mechanisms of epithelial renewal, adaptation, injury and repair in fully developed organisms are all subjects to current intense research efforts and much progress has been made in the recent decade. Much less is known about late fetal development and perinatal adaptation to extra-uterine life by this highly dynamic entity. Future researches targeting late fetal and perinatal developmental aspects of epithelial dynamics are likely to lead to seminal insight into the pathogenesis of NEC. Another aspect of the epithelial barrier that holds important clues about pathogenesis and potential therapy is the innate antimicrobial defense. Further studies regarding Paneth cells, the regulation of their numbers and developmental regulation of antimicrobial genes are all promising areas. It is yet to be determined whether oral sIgA administration would be more efficacious in comparison to IgA, as it would be predicted based on the inherently different immunological properties of the serum and secretory forms of this immunoglobulin. The role played by M-cells and tuft cells in intestinal barrier function, as well as further clarification of the nuances in the equilibrium between pro and anti-inflammatory mediators in barrier regulation will shed even more light on this topic. What also needs to be done is to exploit this knowledge to devise practical strategies to help protect infants against this deleterious illness. It is hoped that this article will have served well as a summary of our current understanding of the intestinal barrier as it relates to NEC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holman RC, Stoll BJ, Clarke MJ, Glass RI. The epidemiology of necrotizing enterocolitis infant mortality in the united states. American journal of public health. 1997;87:2026–2031. doi: 10.2105/ajph.87.12.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henry MC, Moss RL. Necrotizing enterocolitis. Annual review of medicine. 2009;60:111–124. doi: 10.1146/annurev.med.60.050207.092824. [DOI] [PubMed] [Google Scholar]
- 3.Guthrie SO, Gordon PV, Thomas V, Thorp JA, Peabody J, Clark RH. Necrotizing enterocolitis among neonates in the united states. J Perinatol. 2003;23:278–285. doi: 10.1038/sj.jp.7210892. [DOI] [PubMed] [Google Scholar]
- 4.Horwitz JR, Lally KP, Cheu HW, Vazquez WD, Grosfeld JL, Ziegler MM. Complications after surgical intervention for necrotizing enterocolitis: A multicenter review. J Pediatr Surg. 1995;30:994–999. doi: 10.1016/0022-3468(95)90328-3. [DOI] [PubMed] [Google Scholar]
- 5.Schulzke SM, Deshpande GC, Patole SK. Neurodevelopmental outcomes of very low-birth-weight infants with necrotizing enterocolitis: A systematic review of observational studies. Arch Pediatr Adolesc Med. 2007;161:583–590. doi: 10.1001/archpedi.161.6.583. [DOI] [PubMed] [Google Scholar]
- 6.Lewis SL, Tam PP. Definitive endoderm of the mouse embryo: Formation, cell fates, and morphogenetic function. Developmental dynamics : an official publication of the American Association of Anatomists. 2006;235:2315–2329. doi: 10.1002/dvdy.20846. [DOI] [PubMed] [Google Scholar]
- 7.Suh E, Traber PG. An intestine-specific homeobox gene regulates proliferation and differentiation. Molecular and cellular biology. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller MF, Cohen ED, Baggs JE, Lu MM, Hogenesch JB, Morrisey EE. Wnt ligands signal in a cooperative manner to promote foregut organogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15348–15353. doi: 10.1073/pnas.1201583109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batts LE, Polk DB, Dubois RN, Kulessa H. Bmp signaling is required for intestinal growth and morphogenesis. Developmental dynamics : an official publication of the American Association of Anatomists. 2006;235:1563–1570. doi: 10.1002/dvdy.20741. [DOI] [PubMed] [Google Scholar]
- 10.VanDussen KL, Samuelson LC. Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Developmental biology. 2010;346:215–223. doi: 10.1016/j.ydbio.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulvaney J, Dabdoub A. Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: Function, regulation, and context dependency. J Assoc Res Otolaryngol. 2012;13:281–293. doi: 10.1007/s10162-012-0317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noah TK, Kazanjian A, Whitsett J, Shroyer NF. Sam pointed domain ets factor (spdef) regulates terminal differentiation and maturation of intestinal goblet cells. Experimental cell research. 2010;316:452–465. doi: 10.1016/j.yexcr.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, Blache P, Jay P. Sox9 regulates cell proliferation and is required for paneth cell differentiation in the intestinal epithelium. The Journal of cell biology. 2007;178:635–648. doi: 10.1083/jcb.200704152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye DZ, Kaestner KH. Foxa1 and foxa2 control the differentiation of goblet and enteroendocrine l- and d-cells in mice. Gastroenterology. 2009;137:2052–2062. doi: 10.1053/j.gastro.2009.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghaleb AM, McConnell BB, Kaestner KH, Yang VW. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the kruppel-like factor 4 gene. Developmental biology. 2011;349:310–320. doi: 10.1016/j.ydbio.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by hes-1. Nature genetics. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 17.Ueo T, Imayoshi I, Kobayashi T, Ohtsuka T, Seno H, Nakase H, Chiba T, Kageyama R. The role of hes genes in intestinal development, homeostasis and tumor formation. Development. 2012;139:1071–1082. doi: 10.1242/dev.069070. [DOI] [PubMed] [Google Scholar]
- 18.VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, Tran IT, Maillard I, Siebel C, Kolterud A, Grosse AS, Gumucio DL, Ernst SA, Tsai YH, Dempsey PJ, Samuelson LC. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, Dubowski TD, Sodhi CP, Hackam DJ. A critical role for tlr4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. Journal of immunology (Baltimore, Md. 2007;179:4808–4820. doi: 10.4049/jimmunol.179.7.4808. [DOI] [PubMed] [Google Scholar]
- 20.Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, Prindle T, Jr, Russo AM, Afrazi A, Good M, Brower-Sinning R, Firek B, Morowitz MJ, Ozolek JA, Gittes GK, Billiar TR, Hackam DJ. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143:708–718. e701–705. doi: 10.1053/j.gastro.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lebenthal A, Lebenthal E. The ontogeny of the small intestinal epithelium. JPEN Journal of parenteral and enteral nutrition. 1999;23:S3–6. doi: 10.1177/014860719902300502. [DOI] [PubMed] [Google Scholar]
- 22.Rumbo M, Schiffrin EJ. Ontogeny of intestinal epithelium immune functions: Developmental and environmental regulation. Cellular and molecular life sciences : CMLS. 2005;62:1288–1296. doi: 10.1007/s00018-005-5033-3. [DOI] [PubMed] [Google Scholar]
- 23.Wallingford JC, Milunsky A, Underwood BA. Vitamin a and retinol-binding protein in amniotic fluid. The American journal of clinical nutrition. 1983;38:377–381. doi: 10.1093/ajcn/38.3.377. [DOI] [PubMed] [Google Scholar]
- 24.Walker WA. Gastrointestinal host defence: Importance of gut closure in control of macromolecular transport. Ciba Foundation symposium. 1979:201–219. doi: 10.1002/9780470720530.ch12. [DOI] [PubMed] [Google Scholar]
- 25.Corridoni D, Pastorelli L, Mattioli B, Locovei S, Ishikawa D, Arseneau KO, Chieppa M, Cominelli F, Pizarro TT. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a tnf-dependent mechanism. PLoS One. 2012;7:e42067. doi: 10.1371/journal.pone.0042067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colome G, Sierra C, Blasco J, Garcia MV, Valverde E, Sanchez E. Intestinal permeability in different feedings in infancy. Acta paediatrica (Oslo, Norway : 1992) 2007;96:69–72. doi: 10.1111/j.1651-2227.2007.00030.x. [DOI] [PubMed] [Google Scholar]
- 27.Beach RC, Menzies IS, Clayden GS, Scopes JW. Gastrointestinal permeability changes in the preterm neonate. Arch Dis Child. 1982;57:141–145. doi: 10.1136/adc.57.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roignot J, Peng X, Mostov K. Polarity in mammalian epithelial morphogenesis. Cold Spring Harb Perspect Biol. 2013:5. doi: 10.1101/cshperspect.a013789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostov KE. Transepithelial transport of immunoglobulins. Annual review of immunology. 1994;12:63–84. doi: 10.1146/annurev.iy.12.040194.000431. [DOI] [PubMed] [Google Scholar]
- 30.Baker K, Qiao SW, Kuo T, Kobayashi K, Yoshida M, Lencer WI, Blumberg RS. Immune and non-immune functions of the (not so) neonatal fc receptor, fcrn. Semin Immunopathol. 2009;31:223–236. doi: 10.1007/s00281-009-0160-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, Lencer WI, Blumberg RS. Human neonatal fc receptor mediates transport of igg into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004;20:769–783. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 32.Buddington RK, Bering SB, Thymann T, Sangild PT. Aldohexose malabsorption in preterm pigs is directly related to the severity of necrotizing enterocolitis. Pediatric research. 2008;63:382–387. doi: 10.1203/PDR.0b013e318165bfed. [DOI] [PubMed] [Google Scholar]
- 33.Erasmus HD, Ludwig-Auser HM, Paterson PG, Sun D, Sankaran K. Enhanced weight gain in preterm infants receiving lactase-treated feeds: A randomized, double-blind, controlled trial. The Journal of pediatrics. 2002;141:532–537. doi: 10.1067/mpd.2002.127499. [DOI] [PubMed] [Google Scholar]
- 34.Tan-Dy CR, Ohlsson A. Lactase treated feeds to promote growth and feeding tolerance in preterm infants. Cochrane Database Syst Rev. 2013;3:CD004591. doi: 10.1002/14651858.CD004591.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thymann T, Moller HK, Stoll B, Stoy AC, Buddington RK, Bering SB, Jensen BB, Olutoye OO, Siggers RH, Molbak L, Sangild PT, Burrin DG. Carbohydrate maldigestion induces necrotizing enterocolitis in preterm pigs. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1115–1125. doi: 10.1152/ajpgi.00261.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith MW, Peacock MA. “M” cell distribution in follicle-associated epithelium of mouse peyer’s patch. The American journal of anatomy. 1980;159:167–175. doi: 10.1002/aja.1001590205. [DOI] [PubMed] [Google Scholar]
- 37.Kodaka T, Kuroiwa M, Higashi S. Structural and distribution patterns of surface ‘prismless’ enamel in human permanent teeth. Caries research. 1991;25:7–20. doi: 10.1159/000261336. [DOI] [PubMed] [Google Scholar]
- 38.Clark JA, Doelle SM, Halpern MD, Saunders TA, Holubec H, Dvorak K, Boitano SA, Dvorak B. Intestinal barrier failure during experimental necrotizing enterocolitis: Protective effect of egf treatment. Am J Physiol Gastrointest Liver Physiol. 2006;291:G938–949. doi: 10.1152/ajpgi.00090.2006. [DOI] [PubMed] [Google Scholar]
- 39.Grootjans J, Hundscheid IH, Lenaerts K, Boonen B, Renes IB, Verheyen FK, Dejong CH, von Meyenfeldt MF, Beets GL, Buurman WA. Ischaemia-induced mucus barrier loss and bacterial penetration are rapidly counteracted by increased goblet cell secretory activity in human and rat colon. Gut. 2013;62:250–258. doi: 10.1136/gutjnl-2011-301956. [DOI] [PubMed] [Google Scholar]
- 40.Bjerknes M, Cheng H. The stem-cell zone of the small intestinal epithelium. I. Evidence from paneth cells in the adult mouse. The American journal of anatomy. 1981;160:51–63. doi: 10.1002/aja.1001600105. [DOI] [PubMed] [Google Scholar]
- 41.Ouellette AJ. Paneth cells and innate mucosal immunity. Curr Opin Gastroenterol. 2010;26:547–553. doi: 10.1097/MOG.0b013e32833dccde. [DOI] [PubMed] [Google Scholar]
- 42.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted paneth cell. Cellular and molecular life sciences : CMLS. 2002;59:156–170. doi: 10.1007/s00018-002-8412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ. Intestinal label-retaining cells are secretory precursors expressing lgr5. Nature. 2013;495:65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 45.Sato A. Tuft cells. Anat Sci Int. 2007;82:187–199. doi: 10.1111/j.1447-073X.2007.00188.x. [DOI] [PubMed] [Google Scholar]
- 46.Gerbe F, van Es JH, Makrini L, Brulin B, Mellitzer G, Robine S, Romagnolo B, Shroyer NF, Bourgaux JF, Pignodel C, Clevers H, Jay P. Distinct atoh1 and neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. The Journal of cell biology. 2011;192:767–780. doi: 10.1083/jcb.201010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grube D. The endocrine cells of the digestive system: Amines, peptides, and modes of action. Anatomy and embryology. 1986;175:151–162. doi: 10.1007/BF00389591. [DOI] [PubMed] [Google Scholar]
- 48.Staehelin LA. Three types of gap junctions interconnecting intestinal epithelial cells visualized by freeze-etching. Proceedings of the National Academy of Sciences of the United States of America. 1972;69:1318–1321. doi: 10.1073/pnas.69.5.1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–407. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: Orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 51.Gumbiner B. Structure, biochemistry, and assembly of epithelial tight junctions. The American journal of physiology. 1987;253:C749–758. doi: 10.1152/ajpcell.1987.253.6.C749. [DOI] [PubMed] [Google Scholar]
- 52.Niessen CM. Tight junctions/adherens junctions: Basic structure and function. The Journal of investigative dermatology. 2007;127:2525–2532. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 53.Anderson JM, Van Itallie CM, Fanning AS. Setting up a selective barrier at the apical junction complex. Current opinion in cell biology. 2004;16:140–145. doi: 10.1016/j.ceb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 54.Laker MF, Menzies IS. Increase in human intestinal permeability following ingestion of hypertonic solutions. J Physiol. 1977;265:881–894. doi: 10.1113/jphysiol.1977.sp011750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madara JL. Increases in guinea pig small intestinal transepithelial resistance induced by osmotic loads are accompanied by rapid alterations in absorptive-cell tight-junction structure. The Journal of cell biology. 1983;97:125–136. doi: 10.1083/jcb.97.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez M, Barber A, Ponz F. Effect of osmolarity on the epithelial paracellular permeability in rat jejunum. Revista espanola de fisiologia. 1996;52:103–112. [PubMed] [Google Scholar]
- 57.Banan A, Zhang Y, Losurdo J, Keshavarzian A. Carbonylation and disassembly of the f-actin cytoskeleton in oxidant induced barrier dysfunction and its prevention by epidermal growth factor and transforming growth factor alpha in a human colonic cell line. Gut. 2000;46:830–837. doi: 10.1136/gut.46.6.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hang P, Sangild PT, Sit WH, Ngai HH, Xu R, Siggers JL, Wan JM. Temporal proteomic analysis of intestine developing necrotizing enterocolitis following enteral formula feeding to preterm pigs. J Proteome Res. 2009;8:72–81. doi: 10.1021/pr800638w. [DOI] [PubMed] [Google Scholar]
- 59.Jiang P, Sangild PT, Siggers RH, Sit WH, Lee CL, Wan JM. Bacterial colonization affects the intestinal proteome of preterm pigs susceptible to necrotizing enterocolitis. Neonatology. 2011;99:280–288. doi: 10.1159/000317807. [DOI] [PubMed] [Google Scholar]
- 60.Chakravortty D, Nanda Kumar KS. Bacterial lipopolysaccharide induces cytoskeletal rearrangement in small intestinal lamina propria fibroblasts: Actin assembly is essential for lipopolysaccharide signaling. Biochimica et biophysica acta. 2000;1500:125–136. doi: 10.1016/s0925-4439(99)00098-8. [DOI] [PubMed] [Google Scholar]
- 61.Jiang P, Jensen ML, Cilieborg MS, Thymann T, Wan JM-F, Sit W-H, Tipoe GL, Sangild PT. Antibiotics increase gut metabolism and antioxidant proteins and decrease acute phase response and necrotizing enterocolitis in preterm neonates. PLoS ONE. 2012;7:e44929. doi: 10.1371/journal.pone.0044929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han X, Fink MP, Delude RL. Proinflammatory cytokines cause no*-dependent and -independent changes in expression and localization of tight junction proteins in intestinal epithelial cells. Shock (Augusta, Ga) 2003;19:229–237. doi: 10.1097/00024382-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 63.Clamp JR, Ene D. The gastric mucosal barrier. Methods and findings in experimental and clinical pharmacology. 1989;11 (Suppl 1):19–25. [PubMed] [Google Scholar]
- 64.Specian RD, Oliver MG. Functional biology of intestinal goblet cells. The American journal of physiology. 1991;260:C183–193. doi: 10.1152/ajpcell.1991.260.2.C183. [DOI] [PubMed] [Google Scholar]
- 65.Bell SL, Xu G, Khatri IA, Wang R, Rahman S, Forstner JF. N-linked oligosaccharides play a role in disulphide-dependent dimerization of intestinal mucin muc2. The Biochemical journal. 2003;373:893–900. doi: 10.1042/BJ20030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hansson GC. Role of mucus layers in gut infection and inflammation. Current opinion in microbiology. 2012;15:57–62. doi: 10.1016/j.mib.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the muc2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proceedings of the National Academy of Sciences of the United States of America. 2011;108 (Suppl 1):4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Atuma C, Strugala V, Allen A, Holm L. The adherent gastrointestinal mucus gel layer: Thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol. 2001;280:G922–929. doi: 10.1152/ajpgi.2001.280.5.G922. [DOI] [PubMed] [Google Scholar]
- 69.Ambort D, van der Post S, Johansson ME, Mackenzie J, Thomsson E, Krengel U, Hansson GC. Function of the cysd domain of the gel-forming muc2 mucin. The Biochemical journal. 2011;436:61–70. doi: 10.1042/BJ20102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGuckin MA, Linden SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9:265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 71.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. Muc2-deficient mice spontaneously develop colitis, indicating that muc2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 72.Dharmani P, Leung P, Chadee K. Tumor necrosis factor-α and muc2 mucin play major roles in disease onset and progression in dextran sodium sulphate-induced colitis. PLoS ONE. 2011;6:e25058. doi: 10.1371/journal.pone.0025058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chu SH, Walker WA. Developmental changes in the activities of sialyl- and fucosyltransferases in rat small intestine. Biochimica et biophysica acta. 1986;883:496–500. doi: 10.1016/0304-4165(86)90289-8. [DOI] [PubMed] [Google Scholar]
- 74.Dai D, Nanthkumar NN, Newburg DS, Walker WA. Role of oligosaccharides and glycoconjugates in intestinal host defense. Journal of pediatric gastroenterology and nutrition. 2000;30 (Suppl 2):S23–33. [PubMed] [Google Scholar]
- 75.Snyder JD, Walker WA. Structure and function of intestinal mucin: Developmental aspects. International archives of allergy and applied immunology. 1987;82:351–356. doi: 10.1159/000234225. [DOI] [PubMed] [Google Scholar]
- 76.Suemori S, Lynch-Devaney K, Podolsky DK. Identification and characterization of rat intestinal trefoil factor: Tissue- and cell-specific member of the trefoil protein family. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:11017–11021. doi: 10.1073/pnas.88.24.11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kindon H, Pothoulakis C, Thim L, Lynch-Devaney K, Podolsky DK. Trefoil peptide protection of intestinal epithelial barrier function: Cooperative interaction with mucin glycoprotein. Gastroenterology. 1995;109:516–523. doi: 10.1016/0016-5085(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 78.Durer U, Hartig R, Bang S, Thim L, Hoffmann W. Tff3 and egf induce different migration patterns of intestinal epithelial cells in vitro and trigger increased internalization of e-cadherin. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2007;20:329–346. doi: 10.1159/000107519. [DOI] [PubMed] [Google Scholar]
- 79.Bossenmeyer-Pourie C, Kannan R, Ribieras S, Wendling C, Stoll I, Thim L, Tomasetto C, Rio MC. The trefoil factor 1 participates in gastrointestinal cell differentiation by delaying g1-s phase transition and reducing apoptosis. The Journal of cell biology. 2002;157:761–770. doi: 10.1083/jcb200108056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kayademir T, Silva Edos S, Pusch C, Beck S, Machado JC, Gott P. A novel 25 bp tandem repeat within the human trefoil peptide gene tff2 in 21q22. 3: Polymorphism and mammalian evolution. European journal of human genetics : EJHG. 1998;6:121–128. doi: 10.1038/sj.ejhg.5200166. [DOI] [PubMed] [Google Scholar]
- 81.Playford RJ, Marchbank T, Chinery R, Evison R, Pignatelli M, Boulton RA, Thim L, Hanby AM. Human spasmolytic polypeptide is a cytoprotective agent that stimulates cell migration. Gastroenterology. 1995;108:108–116. doi: 10.1016/0016-5085(95)90014-4. [DOI] [PubMed] [Google Scholar]
- 82.Vestergaard EM, Nexo E, Wendt A, Guthmann F. Trefoil factors in human milk. Early human development. 2008;84:631–635. doi: 10.1016/j.earlhumdev.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 83.Zhang BH, Yu HG, Sheng ZX, Luo HS, Yu JP. The therapeutic effect of recombinant human trefoil factor 3 on hypoxia-induced necrotizing enterocolitis in immature rat. Regulatory peptides. 2003;116:53–60. doi: 10.1016/s0167-0115(03)00177-0. [DOI] [PubMed] [Google Scholar]
- 84.Ho S, Pothoulakis C, Koon HW. Antimicrobial peptides and colitis. Current pharmaceutical design. 2013;19:40–47. doi: 10.2174/13816128130108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ouellette AJ. Paneth cell alpha-defensins in enteric innate immunity. Cell Mol Life Sci. 2011;68:2215–2229. doi: 10.1007/s00018-011-0714-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Campbell Y, Fantacone ML, Gombart AF. Regulation of antimicrobial peptide gene expression by nutrients and by-products of microbial metabolism. Eur J Nutr. 2012;51:899–907. doi: 10.1007/s00394-012-0415-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Motta JP, Magne L, Descamps D, Rolland C, Squarzoni-Dale C, Rousset P, Martin L, Cenac N, Balloy V, Huerre M, Frohlich LF, Jenne D, Wartelle J, Belaaouaj A, Mas E, Vinel JP, Alric L, Chignard M, Vergnolle N, Sallenave JM. Modifying the protease, antiprotease pattern by elafin overexpression protects mice from colitis. Gastroenterology. 2011;140:1272–1282. doi: 10.1053/j.gastro.2010.12.050. [DOI] [PubMed] [Google Scholar]
- 88.Si-Tahar M, Merlin D, Sitaraman S, Madara JL. Constitutive and regulated secretion of secretory leukocyte proteinase inhibitor by human intestinal epithelial cells. Gastroenterology. 2000;118:1061–1071. doi: 10.1016/s0016-5085(00)70359-3. [DOI] [PubMed] [Google Scholar]
- 89.Ho S, Pothoulakis C, Koon HW. Antimicrobial peptides and colitis. Curr Pharm Des. 2013;19:40–47. doi: 10.2174/13816128130108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salzman NH, Polin RA, Harris MC, Ruchelli E, Hebra A, Zirin-Butler S, Jawad A, Porter EM, Bevins CL. Enteric defensin expression in necrotizing enterocolitis. Pediatr Res. 1998;44:20–26. doi: 10.1203/00006450-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 91.Richter M, Topf HG, Groschl M, Frohlich T, Tzschoppe A, Wenzl TG, Kohler H. Influence of gestational age, cesarean section, and type of feeding on fecal human beta-defensin 2 and tumor necrosis factor-alpha. Journal of pediatric gastroenterology and nutrition. 2010;51:103–105. doi: 10.1097/MPG.0b013e3181cd26f9. [DOI] [PubMed] [Google Scholar]
- 92.Puiman PJ, Burger-Van Paassen N, Schaart MW, De Bruijn AC, De Krijger RR, Tibboel D, Van Goudoever JB, Renes IB. Paneth cell hyperplasia and metaplasia in necrotizing enterocolitis. Pediatric research. 2011;69:217–223. doi: 10.1203/PDR.0b013e3182092a9a. [DOI] [PubMed] [Google Scholar]
- 93.Jenke AC, Zilbauer M, Postberg J, Wirth S. Human beta-defensin 2 expression in elbw infants with severe necrotizing enterocolitis. Pediatric research. 2012;72:513–520. doi: 10.1038/pr.2012.110. [DOI] [PubMed] [Google Scholar]
- 94.Macpherson AJ, Geuking MB, McCoy KD. Homeland security: Iga immunity at the frontiers of the body. Trends Immunol. 2012;33:160–167. doi: 10.1016/j.it.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 95.Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and iga transport: New advances in environmental factors that stimulate pigr expression and its role in mucosal immunity. Mucosal Immunol. 2011;4:598–602. doi: 10.1038/mi.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eibl MM, Wolf HM, Furnkranz H, Rosenkranz A. Prevention of necrotizing enterocolitis in low-birth-weight infants by iga-igg feeding. The New England journal of medicine. 1988;319:1–7. doi: 10.1056/NEJM198807073190101. [DOI] [PubMed] [Google Scholar]
- 97.Berseth CL. Gestational evolution of small intestine motility in preterm and term infants. J Pediatr. 1989;115:646–651. doi: 10.1016/s0022-3476(89)80302-6. [DOI] [PubMed] [Google Scholar]
- 98.Jadcherla SR, Klee G, Berseth CL. Regulation of migrating motor complexes by motilin and pancreatic polypeptide in human infants. Pediatric research. 1997;42:365–369. doi: 10.1203/00006450-199709000-00018. [DOI] [PubMed] [Google Scholar]
- 99.Wedel T, Krammer HJ, Kuhnel W, Sigge W. Alterations of the enteric nervous system in neonatal necrotizing enterocolitis revealed by whole-mount immunohistochemistry. Pediatric pathology & laboratory medicine : journal of the Society for Pediatric Pathology, affiliated with the International Paediatric Pathology Association. 1998;18:57–70. [PubMed] [Google Scholar]
- 100.Bush TG. Enteric glial cells. An upstream target for induction of necrotizing enterocolitis and crohn’s disease? Bioessays. 2002;24:130–140. doi: 10.1002/bies.10039. [DOI] [PubMed] [Google Scholar]
- 101.Zhou Y, James I, Besner GE. Heparin-binding epidermal growth factor-like growth factor promotes murine enteric nervous system development and enteric neural crest cell migration. Journal of Pediatric Surgery. 2012;47:1865–1873. doi: 10.1016/j.jpedsurg.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen CL, Yu X, James IO, Zhang HY, Yang J, Radulescu A, Zhou Y, Besner GE. Heparin-binding egf-like growth factor protects intestinal stem cells from injury in a rat model of necrotizing enterocolitis. Laboratory investigation; a journal of technical methods and pathology. 2012;92:331–344. doi: 10.1038/labinvest.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 104.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shaker A, Rubin DC. Intestinal stem cells and epithelial-mesenchymal interactions in the crypt and stem cell niche. Transl Res. 2010;156:180–187. doi: 10.1016/j.trsl.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Winton DJ, Blount MA, Ponder BA. A clonal marker induced by mutation in mouse intestinal epithelium. Nature. 1988;333:463–466. doi: 10.1038/333463a0. [DOI] [PubMed] [Google Scholar]
- 108.Doupe DP, Jones PH. Cycling progenitors maintain epithelia while diverse cell types contribute to repair. Bioessays. 2013;35:443–451. doi: 10.1002/bies.201200166. [DOI] [PubMed] [Google Scholar]
- 109.Zani A, Cananzi M, Fascetti-Leon F, Lauriti G, Smith VV, Bollini S, Ghionzoli M, D’Arrigo A, Pozzobon M, Piccoli M, Hicks A, Wells J, Siow B, Sebire NJ, Bishop C, Leon A, Atala A, Lythgoe MF, Pierro A, Eaton S, De Coppi P. Amniotic fluid stem cells improve survival and enhance repair of damaged intestine in necrotising enterocolitis via a cox-2 dependent mechanism. Gut. 2013 doi: 10.1136/gutjnl-2012-303735. [DOI] [PubMed] [Google Scholar]
- 110.Yang J, Watkins D, Chen CL, Bhushan B, Zhou Y, Besner GE. Heparin-binding epidermal growth factor-like growth factor and mesenchymal stem cells act synergistically to prevent experimental necrotizing enterocolitis. Journal of the American College of Surgeons. 2012;215:534–545. doi: 10.1016/j.jamcollsurg.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical wnt signals are essential for homeostasis of the intestinal epithelium. Genes & development. 2003;17:1709–1713. doi: 10.1101/gad.267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roth S, Franken P, Sacchetti A, Kremer A, Anderson K, Sansom O, Fodde R. Paneth cells in intestinal homeostasis and tissue injury. PLoS One. 2012;7:e38965. doi: 10.1371/journal.pone.0038965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Farin HF, van Es JH. Clevers Hb: Redundant sources of wnt regulate intestinal stem cells and promote formation of paneth cells. Gastroenterology. 2012 doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 114.Heath JP. Epithelial cell migration in the intestine. Cell biology international. 1996;20:139–146. doi: 10.1006/cbir.1996.0018. [DOI] [PubMed] [Google Scholar]
- 115.Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS. The roles of bacteria and tlr4 in rat and murine models of necrotizing enterocolitis. J Immunol. 2006;177:3273–3282. doi: 10.4049/jimmunol.177.5.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sodhi CP, Shi XH, Richardson WM, Grant ZS, Shapiro RA, Prindle T, Jr, Branca M, Russo A, Gribar SC, Ma C, Hackam DJ. Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology. 2010;138:185–196. doi: 10.1053/j.gastro.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, Good M, Afrazi A, Marino R, Slagle D, Ma C, Branca MF, Prindle T, Jr, Grant Z, Ozolek J, Hackam DJ. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. The Journal of biological chemistry. 2012;287:37296–37308. doi: 10.1074/jbc.M112.375881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT. Cox-2 is regulated by toll-like receptor-4 (tlr4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–877. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Santaolalla R, Sussman DA, Ruiz JR, Davies JM, Pastorini C, España CL, Sotolongo J, Burlingame O, Bejarano PA, Philip S, Ahmed MM, Ko J, Dirisina R, Barrett TA, Shang L, Lira SA, Fukata M, Abreu MT. Tlr4 activates the β-catenin pathway to cause intestinal neoplasia. PLoS ONE. 2013:8. doi: 10.1371/journal.pone.0063298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng L, Riehl TE, Stenson WF. Regulation of colonic epithelial repair in mice by toll-like receptors and hyaluronic acid. Gastroenterology. 2009;137:2041–2051. doi: 10.1053/j.gastro.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Calza L, Giardino L, Pozza M, Bettelli C, Micera A, Aloe L. Proliferation and phenotype regulation in the subventricular zone during experimental allergic encephalomyelitis: In vivo evidence of a role for nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3209–3214. doi: 10.1073/pnas.95.6.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park DH, Eve DJ, Musso J, 3rd, Klasko SK, Cruz E, Borlongan CV, Sanberg PR. Inflammation and stem cell migration to the injured brain in higher organisms. Stem Cells Dev. 2009;18:693–702. doi: 10.1089/scd.2009.0008. [DOI] [PubMed] [Google Scholar]
- 123.Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Baron-Van Evercooren A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ. Endotoxin inhibits intestinal epithelial restitution through activation of rho-gtpase and increased focal adhesions. The Journal of biological chemistry. 2004;279:24592–24600. doi: 10.1074/jbc.M313620200. [DOI] [PubMed] [Google Scholar]
- 125.Paruchuri S, Broom O, Dib K, Sjolander A. The pro-inflammatory mediator leukotriene d4 induces phosphatidylinositol 3-kinase and rac-dependent migration of intestinal epithelial cells. The Journal of biological chemistry. 2005;280:13538–13544. doi: 10.1074/jbc.M409811200. [DOI] [PubMed] [Google Scholar]
- 126.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, Diepolder H, Marquardt A, Jagla W, Popp A, Leclair S, Herrmann K, Seiderer J, Ochsenkuhn T, Goke B, Auernhammer CJ, Dambacher J. Il-22 is increased in active crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;290:G827–838. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 127.Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, Whitehead R, Polk DB. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Physiol Cell Physiol. 2003;284:C953–961. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 128.Dai S, Sodhi C, Cetin S, Richardson W, Branca M, Neal MD, Prindle T, Ma C, Shapiro RA, Li B, Wang JHC, Hackam DJ. Extracellular high mobility group box-1 (hmgb1) inhibits enterocyte migration via activation of toll-like receptor-4 and increased cell-matrix adhesiveness. J Biol Chem. 2010;285:4995–5002. doi: 10.1074/jbc.M109.067454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Qureshi FG, Leaphart C, Cetin S, Li J, Grishin A, Watkins S, Ford HR, Hackam DJ. Increased expression and function of integrins in enterocytes by endotoxin impairs epithelial restitution. Gastroenterology. 2005;128:1012–1022. doi: 10.1053/j.gastro.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 130.Gunther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut. 2013;62:1062–1071. doi: 10.1136/gutjnl-2011-301364. [DOI] [PubMed] [Google Scholar]
- 131.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes & development. 2010;24:2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. Fadd prevents rip3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 133.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates tnf-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Clark JA, Lane RH, Maclennan NK, Holubec H, Dvorakova K, Halpern MD, Williams CS, Payne CM, Dvorak B. Epidermal growth factor reduces intestinal apoptosis in an experimental model of necrotizing enterocolitis. American journal of physiology Gastrointestinal and liver physiology. 2005;288:G755–762. doi: 10.1152/ajpgi.00172.2004. [DOI] [PubMed] [Google Scholar]
- 135.Ford H, Watkins S, Reblock K, Rowe M. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. 1997;32:275–282. doi: 10.1016/s0022-3468(97)90194-9. [DOI] [PubMed] [Google Scholar]
- 136.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatric research. 2004;55:622–629. doi: 10.1203/01.PDR.0000113463.70435.74. [DOI] [PubMed] [Google Scholar]
- 137.Oste M, Van Haver E, Thymann T, Sangild P, Weyns A, Van Ginneken CJ. Formula induces intestinal apoptosis in preterm pigs within a few hours of feeding. JPEN Journal of parenteral and enteral nutrition. 2010;34:271–279. doi: 10.1177/0148607109337540. [DOI] [PubMed] [Google Scholar]
- 138.Fath KR, Mamajiwalla SN, Burgess DR. The cytoskeleton in development of epithelial cell polarity. Journal of Cell Science. 1993;106:65–73. doi: 10.1242/jcs.1993.supplement_17.10. [DOI] [PubMed] [Google Scholar]
- 139.Waller DA, Thomas NW, Self TJ. Epithelial restitution in the large intestine of the rat following insult with bile salts. Virchows Archiv A, Pathological anatomy and histopathology. 1988;414:77–81. doi: 10.1007/BF00749741. [DOI] [PubMed] [Google Scholar]
- 140.Iizuka M, Konno S. Wound healing of intestinal epithelial cells. World Journal of Gastroenterology. 2011;17:2161–2171. doi: 10.3748/wjg.v17.i17.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lucchetta EM, Ohlstein B. The drosophila midgut: A model for stem cell driven tissue regeneration. Wiley Interdisciplinary Reviews: Developmental Biology. 2012;1:781–788. doi: 10.1002/wdev.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beebe K, Lee WC, Micchelli CA. Jak/stat signaling coordinates stem cell proliferation and multilineage differentiation in the drosophila intestinal stem cell lineage. Dev Biol. 2010;338:28–37. doi: 10.1016/j.ydbio.2009.10.045. [DOI] [PubMed] [Google Scholar]
- 143.Amcheslavsky A, Jiang J, Ip YT. Tissue damage-induced intestinal stem cell division in drosophila. Cell Stem Cell. 2009;4:49–61. doi: 10.1016/j.stem.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Buchon N, Broderick NA, Chakrabarti S, Lemaitre B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in drosophila. Genes & development. 2009;23:2333–2344. doi: 10.1101/gad.1827009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guo Z, Driver I, Ohlstein B. Injury-induced bmp signaling negatively regulates drosophila midgut homeostasis. J Cell Biol. 2013;201:945–961. doi: 10.1083/jcb.201302049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ozen S, Akisu M, Baka M, Yalaz M, Sozmen EY, Berdeli A, Kultursay N. Insulin-like growth factor attenuates apoptosis and mucosal damage in hypoxia/reoxygenation-induced intestinal injury. Biol Neonate. 2005;87:91–96. doi: 10.1159/000081897. [DOI] [PubMed] [Google Scholar]
- 147.Shiou SR, Yu Y, Guo Y, Westerhoff M, Lu L, Petrof EO, Sun J, Claud EC. Oral administration of tgf-beta1 protects the immature gut from injury via smad-dependent suppression of epithelial nf-kappab signaling and pro-inflammatory cytokine production. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.M113.503946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shiou SR, Yu Y, Chen S, Ciancio MJ, Petrof EO, Sun J, Claud EC. Erythropoietin protects intestinal epithelial barrier function and lowers the incidence of experimental neonatal necrotizing enterocolitis. The Journal of biological chemistry. 2011;286:12123–12132. doi: 10.1074/jbc.M110.154625. [DOI] [PMC free article] [PubMed] [Google Scholar]