

Abstract
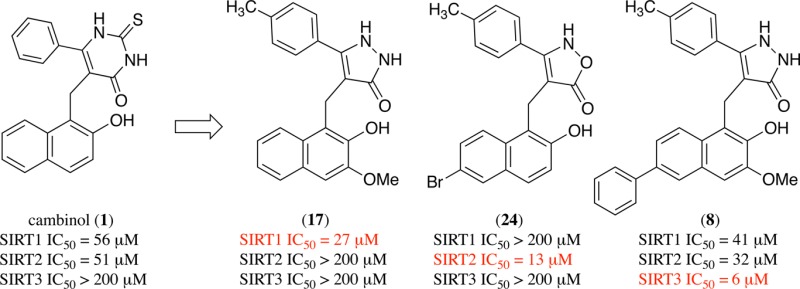
Sirtuins are a family of NAD+-dependent protein deacetylases that play critical roles in epigenetic regulation, stress responses, and cellular aging in eukaryotic cells. In an effort to identify small molecule inhibitors of sirtuins for potential use as chemotherapeutics as well as tools to modulate sirtuin activity, we previously identified a nonselective sirtuin inhibitor called cambinol (IC50 ≈ 50 μM for SIRT1 and SIRT2) with in vitro and in vivo antilymphoma activity. In the current study, we used saturation transfer difference (STD) NMR experiments with recombinant SIRT1 and 20 to map parts of the inhibitor that interacted with the protein. Our ongoing efforts to optimize cambinol analogues for potency and selectivity have resulted in the identification of isoform selective analogues: 17 with >7.8-fold selectivity for SIRT1, 24 with >15.4-fold selectivity for SIRT2, and 8 with 6.8- and 5.3-fold selectivity for SIRT3 versus SIRT1 and SIRT2, respectively. In vitro cytotoxicity studies with these compounds as well as EX527, a potent and selective SIRT1 inhibitor, suggest that antilymphoma activity of this compound class may be predominantly due to SIRT2 inhibition.
Introduction
Identification of new therapeutic compound classes and validation of new therapeutic targets remain major hurdles in drug discovery. In the past decade, human sirtuins (homologues of yeast Silent Information Regulator Two or Sir-2) have emerged as targets for cancer chemotherapy as well as for neurodegenerative and aging-related disorders such as Huntington’s disease, Alzheimer’s disease, and diabetes.2 Although strong evidence exists for sirtuins having a central role in these debilitating diseases, their validation as targets for therapeutic intervention using small molecule modulators has been controversial.3−5 The most publicized efforts at modulation of sirtuin activity have been with the plant polyphenol resveratrol.6 This purported sirtuin activator was shown to have highly beneficial effects in animal models of metabolic disorders (e.g., diabetes) and lifespan extension using experimental models that have since been largely shown to be flawed.7−9 EX-527, a potent and selective SIRT1 inhibitor (SIRT1: human sirtuin isoform 1), was found to be devoid of chemotherapeutic effect; however, cambinol, tenovin-1, tenovin-6, and salermide, nonselective SIRT1/SIRT2 inhibitors, were found to have significant antitumor activity.1,10−12 Combined use of a nonselective sirtuin inhibitor niacinamide (nicotinamide) and a pan-type I/II HDAC (i.e., zinc-dependent histone deacetylases) inhibitor vorinostat yielded encouraging results in a recent diffuse large B-cell lymphoma phase I clinical trial further validating sirtuins as antilymphoma drug targets.13 Additionally, SRT1720, a potent direct SIRT1 activator that was originally developed for its potential in lifespan extension or antiaging activity, was later found to be beneficial in a rat diabetes model employing a mechanism which may involve indirect activation of SIRT1.14 The recent functional characterization of other sirtuin isoforms such as SIRT3, SIRT5, SIRT6, and SIRT7 has further complicated the field as it is becoming increasingly clear that in addition to SIRT1 and 2, these isoforms may also play major roles in aging (SIRT3, SIRT6) as well as in cell-proliferation disorders (SIRT7).15 Additional controversies regarding artifacts of popular in vitro assays to identify novel small molecule modulators of sirtuin activity have also hampered the validation of these enzymes for pharmacological intervention.16
Previously, in an attempt to identify isoform selective sirtuin inhibitors, we carried out a phenotypic screen using an NCI chemical library that resulted in discovery of cambinol (5-[(2-hydroxy-1-naphthyl)methyl]-6-phenyl-2-thioxo-2,3-dihydro-4(1H)-pyrimidinone) 1.1 Titration experiments using SIRT2 and an acetyl-peptide substrate showed that cambinol was competitive with the peptide substrate but noncompetitive with NAD+ suggesting that its binding site partially overlaps with that of the substrate. Cambinol inhibits SIRT1 and SIRT2 with mid-micromolar IC50 and induces hyperacetylation of p53, a known SIRT1 target, and α-tubulin, a SIRT2 target, in cell-based deacetylation assays.17,18 In addition, cambinol was found to be an effective chemosensitizer for the DNA damaging agent etoposide both in p53-proficient and p53-deficient cancer cell lines. As a single agent, cambinol was found to be selectively toxic to BCL6 expressing B-cell lymphoma cells suggesting that it could be used as a lead compound for antilymphoma drug development.1 Cambinol however suffers from major drawbacks including (1) moderate potency, as it did not induce tumor regression, but a decrease in tumor growth rate, and (2) poor solubility. Since the discovery of cambinol, we have focused on optimization of cambinol’s potency and validation of specific sirtuin isoforms (e.g., SIRT1 and SIRT2) as targets for B-cell lymphoma chemotherapy. The cambinol pharmacophore has been well studied (Figure 1). The β-naphthol nucleus is absolutely essential for activity, and the aryl ring is also important as replacing it with five-membered or a heterocyclic ring results in significant loss of potency.1,19−22 The pyrimidinedione (thiouracil) heteroaryl ring requires a balance of hydrogen bond donor and acceptor groups to maintain potency.
Figure 1.

Cambinol analogues. (a) cambinol; (b) schematic; (c) pyrazolone analogues (e.g., 8 and 17); and (d) isoxazolone analogues (e.g., 24).
To further develop the structure activity relationship (SAR), we investigated substituting the pyrimidinedione ring (heteroaryl group, Figure 1b) with five-membered heterocycles (pyrazolone 1c and isoxazolone 1d) as well as altering the functional groups on the naphthyl and aryl rings. To guide us with the specific positioning of the functional group to observe increased potency, we conducted saturation transfer difference (STD) NMR experiments using 20 (Figure 2). The data obtained from STD NMR experiments identified portions of the ligand that interact with the receptor and suggest sites available for additional elaboration. Cell-based toxicity assays show a significant correlation between SIRT2 inhibition and cytotoxicity in the Namalwa Burkitt’s lymphoma cell line.
Figure 2.

1H NMR spectrum and STD spectrum of (20) and SIRT1. The chemical structure of (20) using naphthalene numbering for clarity. Relative saturation transfer (C-7 proton =100%).
Results
Syntheses on new cambinol analogues closely parallel that of the parent compound, cambinol (Scheme 1). Target compounds are prepared by a three-step sequence from 2-hydroxy-1-naphthaldehyde starting materials. For cambinol, base-catalyzed Knoevenagel condensation with ethyl-benzoylacetate with 2-hydroxy-1-naphthaldehyde affords the α,β-unsaturated β-ketoester (Scheme 1). Sodium borohydride reduction of the α,β-unsaturated olefin followed by base-promoted 2-thiouracil formation with thiourea yields cambinol. For naphthyl and quinolyl derivatives of cambinol, hydroxy-naphthaldehyde and hydroxy-quinaldehyde starting materials were prepared by titanium-catalyzed Reiche formylation reaction (1-carbaldehyde-2-hydroxynaphthalene derivatives) from the corresponding phenols, and the Duff reaction of hexamethylenetetramine (HMTA) with 6-hydroxy-2-quinolones in the presence of trifluoroacetic acid (TFA) afforded the corresponding 5-carbaldehyde-6-hydroxy-2-quinolones, respectively (Schemes 1 and 2). The pyrazolone analogues were prepared by reacting the appropriate β-ketoester with hydrazine, while syntheses of the isoxazolone analogues used corresponding hydroxylamine compounds as nucleophiles (Scheme 1).
Scheme 1. Synthesis of Naphthyl Pyrimidinedione, Pyrazolone, and 5-Isoxazolone Sirtuin Inhibitors (1–18, 24–26).

Reagents and conditions: (a) Cl2CHOMe, TiCl4, CH2Cl2, 0–20 °C, 24 h; (b) aryloyl ethyl acetoacetate, piperidine, EtOH, reflux, 2 h; (c) NaBH4, pyridine, 20 °C, 2 h; (d) thiourea, NaOEt, EtOH, reflux, 18 h; (e) hydrazine, DMF, 20 °C, 1.5 h; (f) hydroxylamine, DMF, 60 °C, 18 h;
Scheme 2. Synthesis of Quinolyl Pyrazolone Sirtuin Inhibitors (19–23).

Reagents and conditions: (a) HMTA, TFA, 100 °C, 2 h; (b) POX3, DMF, temp, time X = Cl, Br; (c) aryloyl ethyl acetoacetate, piperidine, EtOH, reflux, 2 h; (d) NaBH4, pyridine, 20 °C, 24 h; (e) hydrazine, DMF, 20 °C, 1.5 h.
SIRT1, SIRT2, and SIRT3 enzyme inhibition studies, carried out using the SIRT-Glo assay (Promega Corp., Madison, WI) at a single inhibitor concentration (50 μM), revealed a range of activities against each target (Tables 1, 2, and 3). Cambinol, the parent of this class of agents, was found to be an nonselective SIRT1, SIRT2 inhibitor with 54% SIRT1 inhibition, 46% SIRT2 and 16% SIRT3 inhibition at a single 50 μM concentration (Table 1), and dose response titration against these enzymes with cambinol gave IC50 values very similar to published values (SIRT1 IC50 = 56 μM, SIRT2 IC50 = 51 μM) and >200 μM SIRT3 IC50.1,19
Table 1. Naphthyl Pyrimidinedione (1) and Pyrazolone (2–18) Sirtuin Inhibitorsa.
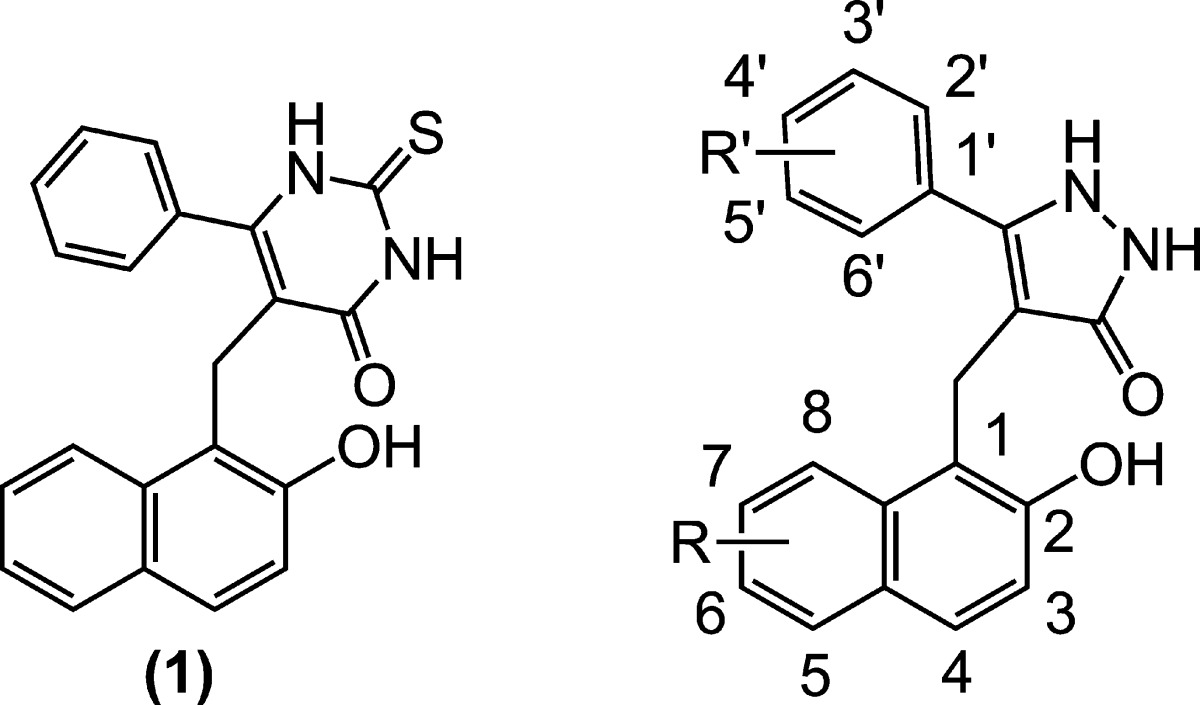
| compound | R | R′ | SIRT1 (% inh) | SIRT2 (% inh) | SIRT3 (% inh) |
|---|---|---|---|---|---|
| cambinol (1) | cambinol | cambinol | 54 | 46 | 16 |
| 2 | H | H | 48 | 18 | 9 |
| 3 | H | 3-Br | 8 | 25 | n.d. |
| 4 | H | 4-Br | 36 | 33 | 22 |
| 5 | H | 4-CH3 | 58 | 27 | 21 |
| 6 | H | 4-F | 24 | 29 | 9 |
| 7 | H | 4-CF3 | 48 | 29 | 14 |
| 8 | 6-phenyl | 4-CH3 | 66 | 73 | 82 |
| 9 | 6-CH3O | 4-CH3 | 64 | 53 | 35 |
| 10 | 6-CN | 4-CH3 | 25 | 21 | n.d. |
| 11 | 6-CH3 | 4-CH3 | 39 | 19 | 12 |
| 12 | 6-C2H5 | 4-CH3 | 45 | 23 | 7 |
| 13 | 6-Br | H | 59 | 47 | 8 |
| 14 | 6-Br | 4-CH3 | 62 | 9 | 21 |
| 15 | 7-Br | 4-CH3 | 42 | 38 | 13 |
| 16 | 3-Br | 4-CH3 | 49 | 20 | 14 |
| 17 | 3-CH3O | 4-CH3 | 78 | 19 | 14 |
| 18 | 6-Br | 4-CH3O | 40 | 30 | 18 |
Percentage sirtuin inhibition at 50 μM compound; n.d. = not determined.
Table 2. Quinolyl Pyrazolone Sirtuin Inhibitorsa.
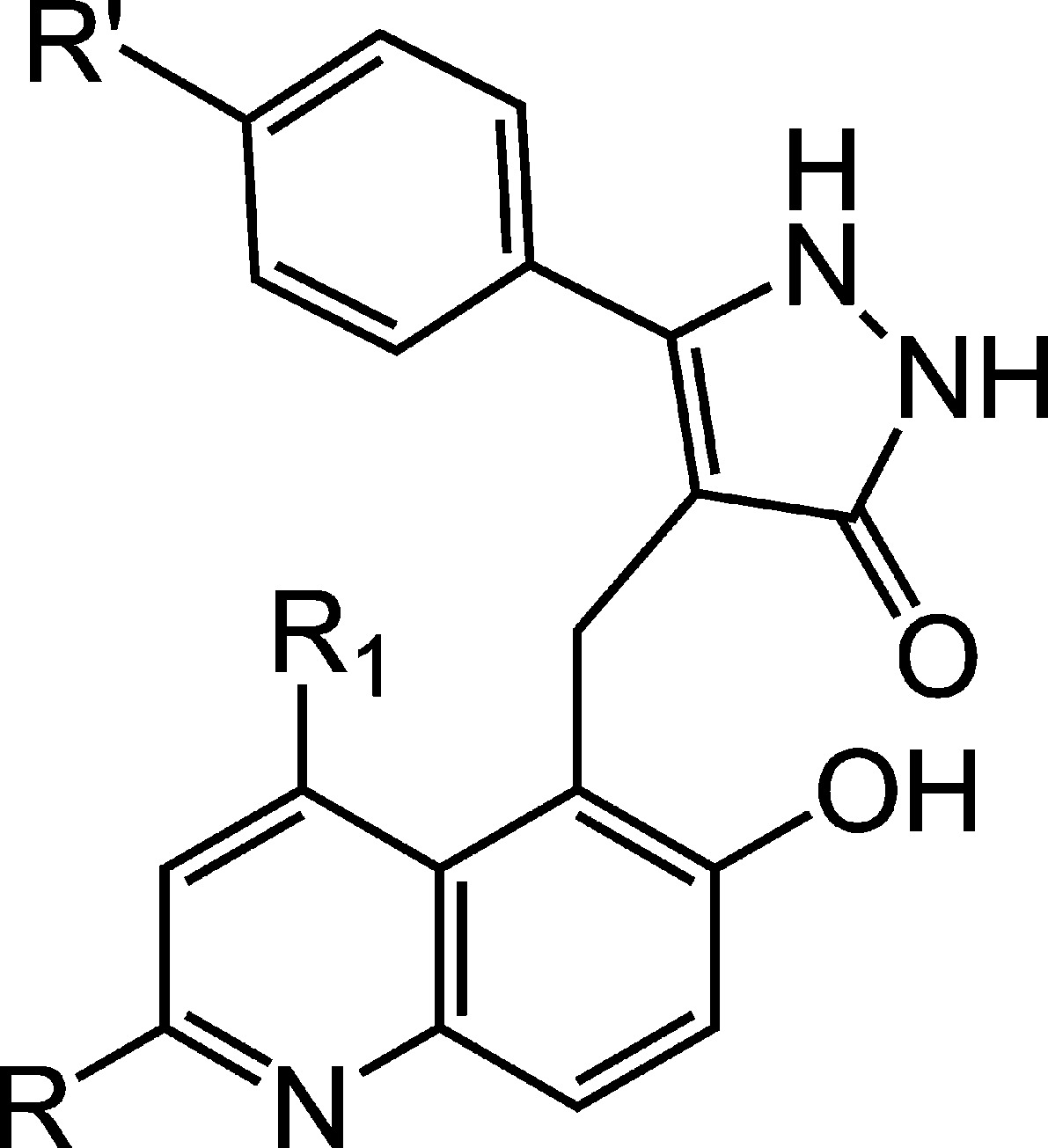
| compound no. | R | R1 | R′ | SIRT1 (% inh) | SIRT2 (% inh) | SIRT3 (% inh) |
|---|---|---|---|---|---|---|
| 19 | H | H | CH3 | 39 | 23 | 16 |
| 20 | Cl | H | CH3 | 45 | 17 | 20 |
| 21 | Br | H | CH3 | 51 | 38 | 42 |
| 22 | Cl | CH3 | H | 27 | 24 | 32 |
| 23 | Cl | H | isopropyl | 85 | 37 | 32 |
Percentage sirtuin inhibition at 50 μM compound.
Table 3. Naphthyl 5-Isoxazolone Sirtuin Inhibitorsa.
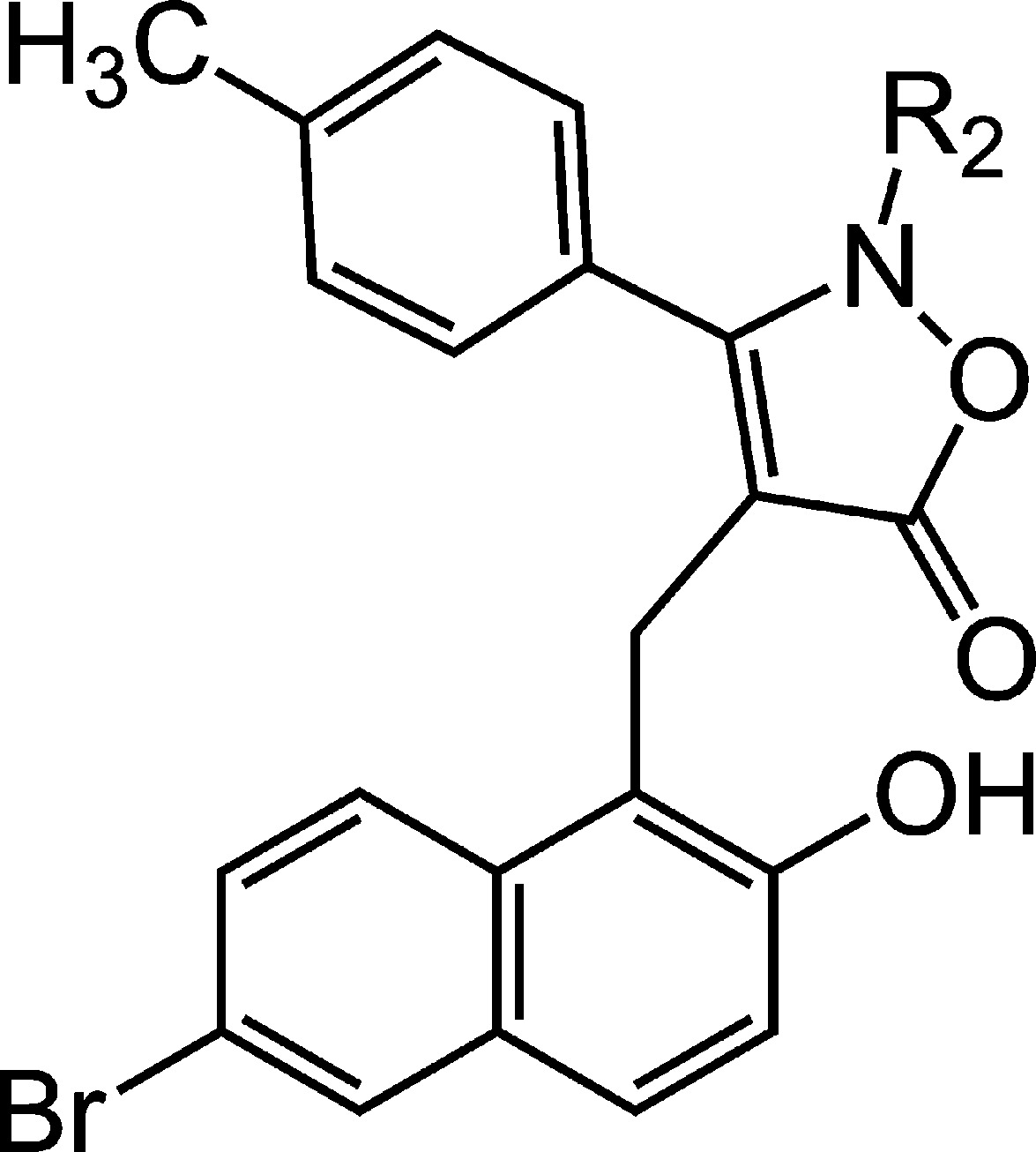
| compound no. | R2 | SIRT1 (% inh) | SIRT2 (% inh) | SIRT3 (% inh) |
|---|---|---|---|---|
| 24 | H | 5 | 87 | 18 |
| 25 | CH3 | 11 | 48 | 17 |
| 26 | benzyl | 2 | 45 | 24 |
Percentage sirtuin inhibition at 50 μM compound.
We carried out an STD NMR experiment using recombinant SIRT1 and compound 20 (Figure 2).23 The relative saturation transfer enhancement at positions we were able to evaluate is shown in Figure 2. The results show that C-7 proton (naphthalene numbering is used for clarity since most of the compounds discussed in this section are naphthalene derivatives) gives the strongest STD signal (100% relative intensity) and forms a close contact with the protein. The aryl C-4′ methyl protons (66% signal intensity) and C-8 proton (74% signal intensity) form contacts with the protein, but these may be suboptimal. Consistent with this, compounds 1 (cambinol), 2, 6, 13, and 22 that contain a small substituent at C-4′ (i.e., hydrogen or fluorine) are relatively poor and nonselective sirtuin inhibitors. The most selective compounds 17 and 24 have methyl groups at C-4′ suggesting that SIRT1 and SIRT2 residues that interact with this group are well conserved. Likewise, compounds that contain larger than hydrogen (i.e., methyl or isopropyl) substituents at the C-4′ position of the aryl moiety tend to favor SIRT1 inhibition with the bulkiest substituent (i.e., isopropyl, compound 23) having the highest selectivity (>3-fold, Table 4). The STD values for the -methylene protons and the NHs of the pyrazolone moiety could not be determined because of the overlap with the water signal and the fast exchange with solvent deuterons, respectively. STD values for the C-3 hydrogen and for protons in the phenyl ring (C-2′, C-3′, C-5′, C-6′) could not be accurately determined due to overlapping signals. The most significant determinant of the selectivity between SIRT1 and SIRT2 in the present compound series is the H-bond donor or acceptor at the 2 position of the heteroaryl unit (i.e., N–H or oxygen) in 17 and 24. The reversal of hydrogen bond directionality results in a reversal of selectivity with the hydrogen-bond donor compounds 17 displaying up to a 7.8-fold SIRT1 selectivity and the hydrogen bond acceptor 24 a 15.4-fold SIRT2 selectivity with approximately similar overall inhibitory activity against the preferred isoform: 26 μM IC50 for 17 against SIRT1 and 13 μM IC50 for 24 against SIRT2. Substitution of a hydrophobic aromatic group at C-6 of the naphthalene group gave the most potent SIRT3 inhibitor (compound 8, SIRT3 IC50 6 μM). This compound however was relatively nonselective with SIRT1 and SIRT2 IC50 of 41 and 32 μM, respectively. The potency of C-6 phenyl substituted naphthalene across the SIRT isoforms suggests a conserved hydrophobic pocket present in all three proteins.
Table 4. Isoform-Selective SIRT Inhibitorsa.
| compound no. | SIRT1 IC50 (μM)a | SIRT2 IC50 (μM)a | SIRT3 IC50 (μM)a |
|---|---|---|---|
| 1 | 56 | 51 | >200 |
| 8 | 41 | 32 | 6 |
| 14 | 37 | >200 | >200 |
| 17 | 26 | >200 | >200 |
| 23 | 21 | 68 | 122 |
| 24 | >200 | 13 | >200 |
Concentration giving 50% inhibition of sirtuin activity.
Our previous studies with cambinol demonstrated that SIRT1 inhibition could be quantified in cells by determining the level of p53 acetylation following induction of DNA damage by the cytotoxic topoisomerase II poison etoposide.1 We treated NCI-H460 cells with 1 μM etoposide in the presence or absence of cambinol 1 or 14, a SIRT1-selective inhibitor (Tables 1 and 4). As expected, the more potent SIRT1 inhibitor 14 shows a significantly higher level of p53 acetylation at a given concentration (Figure 3). Likewise, SIRT2 inhibition results in hyper-acetylation of α-tubulin. The SIRT2-selective inhibitor 24 induced a rapid and robust dose-dependent increase in α-tubulin acetylation in NCI-H460 cells (Figure 4).
Figure 3.
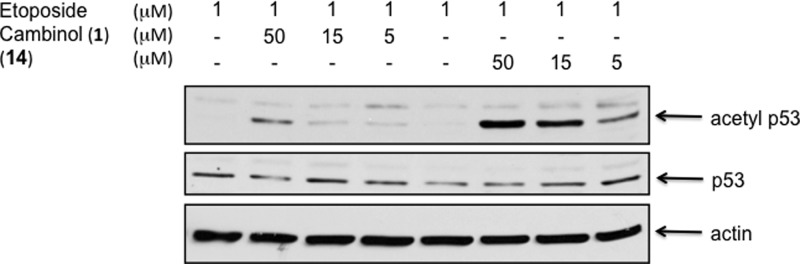
Inhibition of SIRT1-mediated deacetylation of p53 in the presence of DNA damage. Determination of p53 acetylation in NCI-H460 cells by Western blot following 24 h drug treatment.
Figure 4.
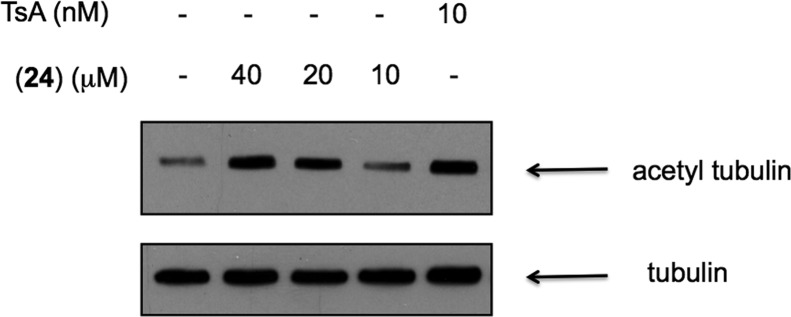
Inhibition of SIRT2-mediated deacetylation of α-tubulin. Determination of α-tubulin acetylation in NCI-H460 cells by Western blot following 4 h treatment with trichostatin A (TsA) and (24).
Part of the motivation behind these studies was to determine whether SIRT1, SIRT2 or SIRT3 inhibition, or possibly a subset of these enzymes, was responsible for cambinol’s antilymphoma activity. Cell-based sirtuin activity assays were carried out by determining the acetylation status of well established SIRT1 (p53) and SIRT2 (α-tubulin) in the presence of isoform selective inhibitors. Cytotoxicity assays were carried out with the human Burkitt’s lymphoma cell line Namalwa grown under standard conditions.1 Cells were plated in 96-well plates, treated with the test compound or DMSO control for 72 h, and viable cells were quantified by luminescence using CellTiterGlo (Promega Corp., Madison, WI). Cell viability assays showed that both SIRT1- and SIRT2-selective series exhibit antiproliferative, cytotoxic activity against the Namalwa Burkitt’s lymphoma cell line, while the SIRT3-selective compound was less toxic. In the Namalwa cell line, cell death occurs by induction of apoptosis as evidenced by dose-dependent appearance of annexin V-positive cells as indication of early stage apoptosis (Figure 5). In addition, while there is a strong correlation between SIRT2 inhibition and Namalwa cytotoxicity (r = 0.56, p = 0.0014) (Figure 6), neither SIRT1 (r = −0.11) nor SIRT3 (r = 0.21) (data not shown) inhibition correlates with Namalwa cytotoxicity. Three compounds, the SIRT1-selective 17, SIRT2-selective 24 and SIRT3-selective 8, were tested against an expanded panel of Burkitt’s lymphoma (Dakiki, Daudi, Mutu, Oku, Ramos and Namalwa), diffuse large B-cell lymphoma (SU-DHL4 and OCI-Ly8-LAM53), nontransformed Epstein–Barr virus (EBV) immortalized B-cell lines (B1 and B2), and epithelial cancer cell lines (HCT116-colon, MCF7-breast, NCI-H460-nonsmall cell lung cancer and OVCAR3-ovarian) (Table 5). The SIRT2-selective inhibitor 24 exhibited potent cytotoxicity in both lymphoma and epithelial cancer cell lines with IC50 ranging from 3 to 7 μM relative to the nontransformed B-cell lines (IC50 22–28 μM).
Figure 5.
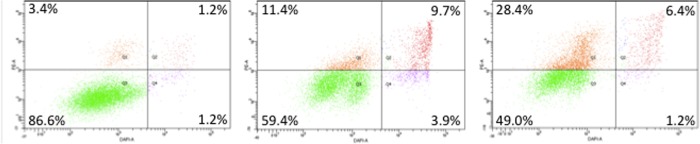
Induction of apoptosis in Namalwa cells treated with 24. FACS analysis of Namalwa cells treated with DMSO (left), 10 μM (24) (center) and 25 μM (24) (right) for 16 h. Cells were stained with annexin V-PE (y-axis) and DAPI (x-axis). Cells entering an early phase of apoptosis are present in the upper left panel.
Figure 6.
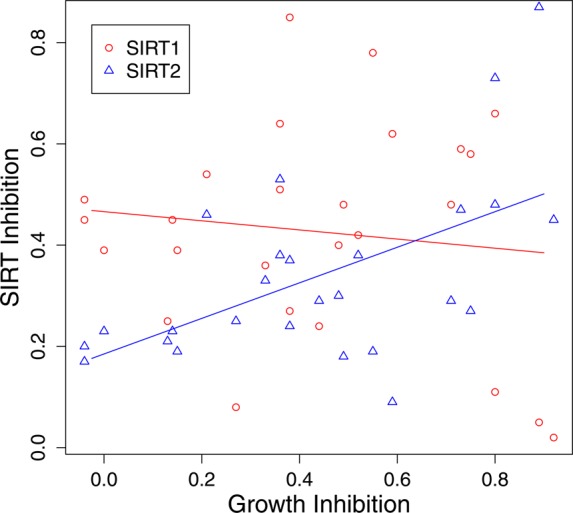
Correlation between sirtuin inhibition and Namalwa Burkitt’s lymphoma cell line growth inhibition. SIRT1 (red circles) and SIRT2 (blue triangles) inhibition (y-axis) versus Namalwa growth inhibition (x-axis).
Table 5. Cell Line Growth Inhibitiona.
| compound no. | Dakiki | Daudi | Mutu | Oku | Ramos | Namalwa | SU-DHL4 | LAM53 |
|---|---|---|---|---|---|---|---|---|
| 8 | 68 | n.d. | 33 | 51 | 28 | 40 | 27 | 34 |
| 17 | 29 | 21 | 15 | 11 | 15 | 15 | 77 | 32 |
| 24 | 7 | 5 | 3 | 5 | 7 | 4 | 5 | 7 |
| compound no. | B1 | B2 | HCT116 | MCF7 | NCIH460 | OVCAR3 |
|---|---|---|---|---|---|---|
| 8 | 36 | 47 | 58 | 74 | 55 | 68 |
| 17 | 41 | 58 | >80 | 32 | 61 | 13 |
| 24 | 22 | 28 | 8 | 5 | 3 | 10 |
Concentration giving 50% growth inhibition following 72 h drug treatment. Dakiki, Daudi, Mutu, Oku, Ramos, Namalwa are Burkitt’s lymphoma cell lines. SU-DHL4 and LAM53(OCI-Ly8-LAM53) are diffuse large B-cell lymphoma lines. B1 and B2 are EBV-immortalized nontransformed B-cell lines. HCT116 (colon), MCF7 (breast), NCI-H460 (lung), and OVCAR3 (ovarian) are epithelial cancer cell lines.
Discussion and Conclusions
Cambinol, (5-[(2-hydroxy-1-naphthyl)methyl]-6-phenyl-2-thioxo-2,3-dihydro-4(1H)-pyrimidinone) 1, is a nonselective sirtuin inhibitor with equivalent inhibitory activity against SIRT1 and SIRT2. It also is a reasonably potent antitumor agent in vitro and in vivo with particular activity against Burkitt’s lymphoma cell lines.1 In an effort to delineate the contribution of SIRT1 and SIRT2 inhibition in this antitumor activity, we sought to develop cambinol analogues with improved potency and selectivity. Studies by Medda et al. and Rotili et al. have partly addressed the structure activity relationships of six-membered pyrimidinedione-containing (i.e., cambinol-like) compounds.19,24 In an effort to investigate an alternative chemical space, we prepared a series of five-membered pyrazolone- and 5-isoxaolone-containing compounds. The five-membered ring pyrazolone compounds (i.e., 2–23) exhibit a range of specificities but generally favor SIRT1 inhibition. The most selective of these compounds, 14 and 17, exhibit a greater than 5-fold and 7-fold, respectively, preference for SIRT1 over SIRT2. The 5-isoxazolone compounds (i.e., 24–26) favor SIRT2 inhibition with a greater than 15-fold preference shown by 24.
In order to gain a molecular understanding of the observed selectivity, we sought to identify portions of the ligand involved in the interaction with SIRT1 protein. The STD NMR experiment with recombinant SIRT1 and 20, a relatively nonselective but highly soluble ligand, has identified positions of the ligand as potential sites for improving selectivity and potency (Figure 2). The efficiency of saturation transfer was lowest at C-8 of the quinolone ring system (naphthalene numbering) and at the 4-methyl group of the aryl substituent suggesting that these portions of the ligand interact with the protein surface but in a suboptimal manner. Consistent with this finding, compounds with large substituents at the C-4 position yielded more potent inhibitors (e.g., 20 versus 23, Table 2) in agreement with the structural model proposed by Medda et al.19 Saturation transfer was most efficient at C-4 and C-7 of the quinoline ring suggesting close ligand/protein interactions; however, the C-8 proton showed a weaker interaction. Examination of the structural variation at these positions suggests a reasonable explanation of the observed selectivities. The binding site for the C-4 phenyl position appears to tolerate medium-sized substituents equally well in SIRT1 and SIRT2, with bulkier groups such as isopropyl showing an improved preference for SIRT1. The strongest determinant of selectivity in the current compound series is the hydrogen bond donor (i.e., N–H at the pyrazolone 2 position in 17) or hydrogen bond acceptor (i.e., O at the dihydro 1,2-oxazol-5-one 1 position in 24). Remarkably, the reversal of directionality of the hydrogen bond donor/acceptor pair leads to a 7.8-fold SIRT1 selectivity in the case of the former 24 and a 15.4-fold SIRT2 selectivity for the latter, with approximately equipotent overall inhibitory activity against the preferred isoform: 26 μM IC50 for 17 against SIRT1 and 13 μM IC50 for 24 against SIRT2. Future studies will attempt to optimize interactions at C-4′ and C-8. Placement of a bulky hydrophobic phenyl group at the naphthalene C-6 position yielded 8, a potent although relatively nonselective SIRT3 inhibitor. Elaboration of the C-6 substituent provides a means to increase overall potency. The SAR trends observed in the current compound series provide a strong starting point for development of potent isoform-selective as well as pan-sirtuin inhibitors.
While both SIRT1 and SIRT2-selective compounds exhibit some degree of cytotoxicity, a strong correlation between SIRT2 inhibition and Namalwa cytotoxicity (r = 0.56, p = 0.0014) suggests that SIRT2 inhibition may be primarily responsible for the observed antilymphoma activity. Lack of correlation between SIRT1 inhibition and cytotoxicity observed in our study is consistent with the findings that EX-527, a potent and selective SIRT1 inhibitor, does not exhibit activity against B-cell lymphoma cell lines, as reported previously by others for epithelial tumor cells.25 It is possible, however, that inhibition of SIRT1 may play a role in other tumor contexts, as suggested by the findings by Rotili et al.24,26 In addition to Burkitt’s lymphoma cell lines, we tested 8, 17, and 24 against normal immortalized lymphoblastic lines (B1 and B2), diffuse large B-cell lymphoma lines as well as a panel of solid tumor cell lines (Table 5). Interestingly, colon (HCT116) breast (MCF7) and nonsmall cell lung carcinoma (NCI-H460) also showed significant sensitivity to SIRT2 inhibition. The data presented in this manuscript make a strong case for further development of sirtuin inhibitors as anticancer agents.
Experimental Section
Protein Expression
Human SIRT1 cDNA was cloned into the pET-4a vector and transformed into BL21-DE3 Escherichia coli cells. Expression of SIRT1 protein fusion with hexa-histidine tag (C-terminal) was induced with IPTG (5 mM). Recombinant protein was purified from bacterial lysates using the Ni-NTA column (Clontech Laboratories Inc. Mountain View, CA) according to manufacturer’s protocols.
Enzyme Inhibition Assays
SIRT1 was expressed and purified as described above. SIRT2 and SIRT3 were purchased from Cayman Chemical (Ann Arbor, MI). Enzyme inhibition assays were performed in 96-well plates using the SIRT-Glo Assay (Promega Corp., Madison WI) according to the manufacturer’s instructions. Compounds were dissolved in 100% DMSO and were tested over a five 3-fold dilution concentration range (final DMSO concentration 0.25%). IC50 values were determined in triplicate and reported values are averages of two independent experiments.
Cell Viability Assays
Cell lines were obtained from ATCC (Manasas, VA) and were grown under standard conditions. For viability assays, cells were dispensed into 96-well plates and treated with test compounds in 100% DMSO (final DMSO concentration 0.25%). Cells were incubated with test compounds (or DMSO controls) for 72 h, and viability was determined using the CellTiterGlo Assay (Promega Corp., Madison WI) according to manufacturer’s instructions. Assays were carried out in triplicate reported values are averages of two independent experiments.
Western Blot
NCI-H460 human lung carcinoma cells (ATCC) were grown under standard conditions. Drug treatment and Western blots were carried out as previously described.1 Antibodies for acetylated α-tubulin (clone 6-11B-1, Sigma, St. Louis, MO), α-tubulin (clone DM1A, Calbiochem, San Diego, CA), actin (Roche, Indianapolis, IN), acetylated p53 (Cell Signaling, Danvers, MA), p53 (Cell Signaling, Danvers, MA) and acetylated lysine (polyclonal antibody; Cell Signaling, Danvers, MA) were used at dilutions recommended by the manufacturer.
Flow Cytometry
Namalwa cells were treated with 10 and 25 μM (24) or solvent (DMSO) for 16 h, washed in phosphate buffered saline, and suspended at approximately 200 000 cells in 200 μL of binding buffer containing 5 μL of annexin V-PE (BD Biosciences, San Jose, CA) for measuring phosphatidylserine, and 4′,6-diamidino-2-phenylindole (DAPI) at a concentration of 2.5 μg/mL, for nucleic acids. After 15 min incubation at room temperature, cell fluorescence was analyzed on a FACScanR (Becton Dickinson, Franklin Lakes, NJ).
General Chemistry
Unless specified otherwise, reagents were obtained from Sigma-Aldrich and used without additional purification. 6-Hydroxy-2(1H)-quinolinone and 4-methylquinoline-2,6-diol were obtained from Bioblocks Inc. 3-(4-isopropyl-phenyl)-3-oxo-propionic acid ethyl ester was obtained from Vitas M Laboratories. Thin layer chromatography was carried out using Merck 60 F254 silica gel plates using appropriate solvent mixtures. Solvents were ACS reagent grade and anhydrous solvents (Aldrich and Acros) were used as received. Medium pressure chromatography was carried out using Biotage Isolera with Silicycle HP cartridges. LCMS was performed using an Agilent 1100 HPLC system equipped with a Waters XTerra MS C18 5 um, 4.6 × 50 mm column, an Agilent photodiode array detector and an in-line Agilent 6130 single quadrupole mass spectrometer. The LC method involved gradient elution from 0 to 95% acetonitrile in water (0.1% formic acid) over 6 min. Final purity of compounds was determined using ACE 3, C8–300, 150 × 3.0 mm column with the above-mentioned gradient over 15 min. Agilent ChemStation software was used to develop methods and calculate percentage purity. All compounds reported are at least 95% pure.
NMR Experiments
All NMR experiments were performed at 25 °C on a Varian Unity Plus 300 spectrometer or Varian Unity-Inova 500 MHz spectrometer equipped with a 5 mm triple-resonance 1H(13C/15N), z-axis pulsed-field gradient probe head. For characterization purposes, samples consisted of a ∼5 mM solution of each compound in chloroform-d (99.8% D, Cambridge Isotopes), dimethyl sulfoxide-d6 (99.9% D, Cambridge Isotopes), benzene-d6 (99.5% D, Cambridge Isotopes) or acetone-d6 (99.9% D, Cambridge Isotopes), and the spectra were referenced to residual solvent peaks at 7.27, 2.50, 7.16, and 2.05 ppm, respectively. 1H-1D spectra were acquired at a resolution of 16k complex points in the time domain with 32 accumulations each (sw = 6000 Hz, d1 = 3 s).
Saturation Transfer Difference (STD) NMR
All STD experiments were performed in D2O solution to eliminate the influence of exchangeable protein protons. The purified SIRT1 stock solution (12 μM) was dialyzed against deuterated phosphate buffered saline (PBS). The exchange buffer was prepared by lyophilizing a PBS solution at pH 7.4 and redissolving it in D2O (99.9% D, Cambridge Isotopes). NMR samples contained 1.5 μM SIRT1 and 200 μM compound 20 in PBS in D2O pH 7.4 (uncorrected for D2O) with 1% dimethyl sulfoxide-d6 as a cosolvent. STD NMR experiments were performed at 25 °C as previously described.27 Briefly, two free induction decay (FID) data sets were collected in an interleaved manner to minimize temporal fluctuations with the protein irradiation frequency set on-resonance (−0.5 ppm) and off-resonance (40 ppm), respectively (sw =6000 Hz, 16 steady state scans, 2048 transients, 4k complex points, d1 = 3 s). Protein saturation was obtained using a train of individual 50 ms long, frequency selective Gaussian radio frequency (rf) pulses separated by an interpulse delay of 1 ms. The number of selective pulses was set to 50, leading to a total saturation time (τsat) of 2.5 s. Gradient Tailored Excitation (WATERGATE) scheme was employed to suppress the residual water signal.28 Suppression of the background signals arising from the protein was not required. The FID acquired with off-resonance irradiation generated the reference spectrum (Ioff) whereas the difference FID (off-resonance–on-resonance) yielded the STD spectrum (ISTD = Ioff – Ion). Spectra were processed with an exponential apodization function (1 Hz line-broadening) and zero-filling to 8k complex points before Fourier transformation and baseline correction with a third order Bernstein polynomial fit. The STD measurements were done in duplicate, and all data were processed and analyzed using MNova 8.1 processing software (Mestrelab Research, Santiago de Compostela, Spain).
Chemistry
General Procedure I for Preparation of Substituted 2-Hydroxy-1-Naphthalene Carbaldehydes
The procedure was adapted from the protocol for formylation of electron rich phenols reported by Garcia et al.29 A solution of TiCl4 (18 mmol) and dichloromethyl ether (9 mmol) in anhydrous dichloromethane (10 mL) was stirred at 0 °C for 15 min. A solution of the corresponding substituted naphthol (9 mmol) in either CH2Cl2 or 1,2-dichloroethane (30 mL) was added dropwise, and the reaction was warmed to room temperature. The reaction was allowed to stir overnight after which it was quenched by adding 1 N HCl (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL), and the organic layers were then combined, dried with Na2SO4, and reduced to dryness to afford a residue which was further purified using medium pressure chromatography to yield the pure aldehyde.
a. 6-Bromo-2-hydroxynaphthalene-1-carbaldehyde (51)
A reddish brown residue initially obtained was further purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 1.5 g (6 mmol, 67%) as a white powder. 1H NMR (500 MHz, chloroform-d) δ 13.12 (s, 1H), 10.79 (s, 1H), 8.24 (d, J = 8.8 Hz, 1H), 7.97 (d, J = 2.4 Hz, 1H), 7.91 (d, J = 9.3 Hz, 1H), 7.70 (dd, J = 8.8, 2.4 Hz, 1H), 7.19 (d, J = 9.3 Hz, 1H). LRMS: m/z = 249.1 (M – H)−.
b. 2-Hydroxy-6-phenylnaphthalene-1-carbaldehyde (52)
6-Bromo-2-hydroxynaphthalene-1-carbaldehyde (1.39 mmol), phenyl boronic acid (2.09 mmol), bis(dibenzylideneacetone)palladium(0) (0.0139 mmol), and tricyclohexylphosphine (0.033 mmol) were suspended in dioxane (7 mL). To this mixture was added potassium phosphate tribasic (1.27 M), and the solution was heated at 100 °C for 6 h. The reaction was cooled, loaded onto a small silica column, and eluted with EtOAc (20 mL), and the filtrate was collected. The extracts were washed with water, dried using Na2SO4, and reduced to dryness to afford a dark brown residue. The residue was then purified by medium pressure chromatography using gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 140 mg (0.56 mmol, 40%) as a white powder. 1H NMR (500 MHz, chloroform-d) δ 13.16 (s, 1H), 10.85 (s, 1H), 8.42 (d, J = 8.8 Hz, 1H), 8.05 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 2.4 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 7.3 Hz, 2H), 7.51 (t, J = 7.8 Hz, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.19 (d, J = 8.8 Hz, 1H), LRMS [ES]+: m/z = 249.1 (M + H)+.
c. 2-Hydroxy-6-methoxynaphthalene-1-carbaldehyde (53)
A black residue was obtained which further purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 500 mg (2.47 mmol, 43%). 1H NMR (500 MHz, chloroform-d) δ 12.90 (s, 1H), 10.79 (s, 1H), 8.27 (d, J = 9.3 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.30 (dd, J = 9, 2.7 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 7.13 (d, J = 2.7 Hz, 1H), 3.93 (s, 3H). LRMS [ES]+: m/z = 203.1 (M + H)+.
d. 5-Formyl-6-hydroxynaphthalene-2-carbonitrile (54)
The reaction was carried out in 1,2-dichloroethane. The residue obtained after extraction was purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 72 mg (0.365 mmol, 42%). 1H NMR (500 MHz, chloroform-d) δ 13.36 (s, 1H), 10.82 (s, 1H), 8.45 (d, J = 8.8 Hz, 1H), 8.19 (d, J = 2 Hz, 1H), 8.05 (d, J = 9.3 Hz, 1H), 7.80 (dd, J = 8.8, 2 Hz, 1H), 7.30 (d, J = 9.3 Hz, 1H). LRMS [ES]+: m/z = 198.1 (M + H)+.
e. 2-Hydroxy-6-methylnaphthalene-1-carbaldehyde (55)
6-Methyl-2-hydroxy-1-naphthalene was synthesized from 6-methoxy-2-naphthaldehyde as reported previously.30 Formylation was carried using the above-mentioned general procedure. The reaction residue was purified using a gradient EtOAc/hexane solvent system (10–25% EtOAc) yielding 386 mg (2.07 mmol, 81%) as a pale yellow solid. 1H NMR (500 MHz, chloroform-d) δ 13.03 (s, 1H), 10.81 (s, 1H), 8.26 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 9.3 Hz, 1H), 7.59 (d, J= 2.0, 1H), 7.46 (dd, J = 8.8, 2 Hz, 1H), 7.12 (d, J = 9.3 Hz, 1H), 2.51 (s, 3H). LRMS [ES]+: m/z = 187.1 (M + H)+.
f. 6-Ethyl-2-hydroxynaphthalene-1-carbaldehyde (56)
6-Ethyl-2-hydroxy-1-naphthalene was synthesized from 6-methoxy-2-aceto-naphthone as reported previously.30 Formylation was carried using the above-mentioned general procedure. The residue obtained was purified using a gradient EtOAc/hexane solvent system (10–25% EtOAc) yielding 322 mg (1.61 mmol, 67%). 1H NMR (500 MHz, chloroform-d) δ 13.07 (s, 1H), 10.85 (s, 1H), 8.28 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 9.3 Hz, 1H), 7.60 (d, J= 2.0, 1H), 7.51 (dd, J = 7.8, 2 Hz, 1H), 7.12 (d, J = 9.3 Hz, 1H), 2.82 (q, J = 7.8 Hz, 2H), 1.34 (t, J = 7.8 Hz, 3H). LRMS [ES]+: m/z = 201.1 (M + H)+.
g. 7-Bromo-2-hydroxynaphthalene-1-carbaldehyde (57)
A reddish brown residue was initially obtained which further purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 860 mg (3.42 mmol, 76%) as a white powder. 1H NMR (500 MHz, chloroform-d) δ 13.19 (s, 1H), 10.74 (s, 1H), 8.50 (s, 1H), 7.95 (d, J = 9.3 Hz, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.54 (dd, J = 8.8, 1.7 Hz, 1H), 7.17 (d, J = 9.3 Hz, 1H). LRMS: m/z = 249.1 (M – H)−.
h. 3-Bromo-2-hydroxynaphthalene-1-carbaldehyde (58)
A reddish brown residue was initially obtained was further purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 850 mg (3.38 mmol, 76%) as a white powder. 1H NMR (500 MHz, chloroform-d) δ 13.82 (s, 1H), 10.76 (s, 1H), 8.35 (d, J = 8.8 Hz, 1H), 8.31 (s, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.66 (ddd, J = 8.8, 7.0, 1.2 Hz, 1H), 7.48 (ddd, J = 8.8, 7.0, 1.2 Hz, 1H). LRMS [ES]+: m/z = 251.0 (M + H)+.
i. 2-Hydroxy-3-methoxynaphthalene-1-carbaldehyde (59)
A reddish brown residue was initially obtained which further purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 75 mg (0.37 mmol, 36%) as a white powder. 1H NMR (500 MHz, chloroform-d) δ 13.46 (s, 1H), 10.74 (s, 1H), 8.23 (d, J = 8.3 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.48 (ddd, J = 8.3, 7.6, 1.2 Hz, 1H), 7.41 (ddd, J = 8.3, 7.6, 1.2 Hz, 1H), 7.30 (s, 1H), 4.01 (s, 3H). LRMS [ES]+: m/z = 203.1 (M + H)+.
General Procedure (II) for Preparation of 2-Benzoyl-3H-benzo[f]chromen-3-ones
The compounds were synthesized using the established synthetic route for cambinol and its analogues reported earlier.19 To a solution of substituted hydroxyl naphthaldehydes (5 mmol) in ethanol (5 mL) were added the corresponding ethyl benzoyl acetates (5 mmol). Piperidine (5 drops) was added, and the reaction was heated under reflux for 2 h. The reaction was allowed to cool, and the yellowish precipitate obtained was collected by filtration and washed with ethanol several times to get the condensation product. Synthesis of 2-benzoyl-3H-benzo[f]chromen-3-one (29) has been previously reported.19
a. 2-(3-Bromobenzoyl)-3H-benzo[f]chromen-3-one (30)
Yield 700 mg (3.7 mmol, 74%). 1H NMR (500 MHz, chloroform-d) δ 9.00 (d, J = 0.6 Hz, 1H), 8.34–8.28 (m, 1H), 8.18–8.12 (m, 1H), 8.08–8.03 (m, 1H), 8.01–7.95 (m, 1H), 7.86–7.73 (m, 3H), 7.65 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 7.58–7.52 (m, 1H), 7.43–7.35 (m, 1H). LRMS [ES]+: m/z = 379.09 (M + H)+.
b. 2-(4-Bromobenzoyl)-3H-benzo[f]chromen-3-one (31)
Yield 500 mg (1.32 mmol, 72%). 1H NMR (500 MHz, chloroform-d) δ 8.98 (d, J = 0.7 Hz, 1H), 8.33–8.27 (m, 1H), 8.17–8.11 (m, 1H), 8.00–7.94 (m, 1H), 7.82–7.72 (m, 3H), 7.68–7.59 (m, 3H), 7.57–7.51 (m, 1H). LRMS [ES]+: m/z = 379.09 (M + H)+.
c. 2-(4-Methylbenzoyl)-3H-benzo[f]chromen-3-one (32)
Yield 506 mg (1.59 mmol, 66%). 1H NMR (500 MHz, chloroform-d) δ 8.89 (s, 1H), 8.26 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 9.0 Hz, 1H), 7.98–7.90 (m, 1H), 7.87–7.80 (m, 2H), 7.72 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.61 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.52 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 7.9 Hz, 1H), 7.18 (s, 1H), 2.44 (s, 3H). LRMS [ES]+: m/z = 315.10 (M + H)+.
d. 2-(4-Fluorobenzoyl)-3H-benzo[f]chromen-3-one (33)
The precipitate was recrystallized overnight from ethyl acetate and dried under vacuum yielding 490 mg (1.57 mmol, 1H NMR (500 MHz, chloroform-d) δ 8.95 (s, 1H), 8.29 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 9.0 Hz, 1H), 8.00–7.92 (m, 3H), 7.74 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.63 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.53 (d, J = 9.0 Hz, 1H), 7.22–7.13 (m, 2H). LRMS [ES]+: m/z = 319.1 (M + H)+.
e. 2-(4-Trifluoromethylbenzoyl)-3H-benzo[f]chromen-3-one (34)
The precipitate was recrystallized from ethyl acetate and dried under a vacuum to yield 450 mg (1.21 mmol, 53%). 1H NMR (500 MHz, chloroform-d) δ 9.06 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.99 (ddt, J = 13.6, 8.1, 0.8 Hz, 3H), 7.81–7.73 (m, 3H), 7.65 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.54 (dd, J = 9.0, 0.7 Hz, 1H). LRMS [ES]+: m/z = 369.1 (M + H)+.
f. 2-(4-Methylbenzoyl)-8-phenyl-3H-benzo[f]chromen-3-one (35)
Yield 204 mg (0.51 mmol, 68%). 1H NMR (500 MHz, chloroform-d) δ 8.91 (s, 1H), 8.34 (d, J = 8.7 Hz, 1H), 8.19–8.12 (m, 2H), 7.99 (dd, J = 8.7, 1.9 Hz, 1H), 7.88–7.82 (m, 2H), 7.76–7.70 (m, 2H), 7.59–7.48 (m, 3H), 7.47–7.39 (m, 1H), 7.31 (d, J = 7.9 Hz, 2H), 2.45 (s, 3H). LRMS [ES]+: m/z = 391.1 (M + H)+.
g. 8-Bromo-2-benzoyl-3H-benzo[f]chromen-3-one (36)
Yield 200 mg (0.52 mmol, 74%) 1H NMR (500 MHz, chloroform-d) δ 10.771 (s, 1H), 8.23 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 2.0 Hz, 1H), 7.89 (d, J = 9.1 Hz, 1H), 7.70–7.71 (m, J = 9.0 Hz, 3H), 7.68 (d, J = 2.0 Hz, 1H), 7.31–7.22 (m, 2H), 7.17 (d, J = 9.1 Hz, 1H). LRMS [ES]+: m/z = 379.09 (M + H)+.
h. 8-Methoxy-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (37)
Yield 95 mg (0.27 mmol, 70%). 1H NMR (500 MHz, chloroform-d) δ 8.82 (s, 1H), 8.15 (d, J = 9.2 Hz, 1H), 7.99 (d, J = 9.0 Hz, 1H), 7.86–7.80 (m, 2H), 7.50 (dd, J = 9.0, 0.7 Hz, 1H), 7.37 (dd, J = 9.2, 2.6 Hz, 1H), 7.33–7.23 (m, 3H), 3.96 (s, 3H), 2.44 (s, 3H). LRMS [ES]+: m/z = 345.1 (M + H)+.
i. 2-(4-Methylbenzoyl)-3-oxo-3H-benzo[f]chromene-8-carbonitrile (38)
Yield 113 mg of light pink solid (0.33 mmol, 73%). 1H NMR (500 MHz, benzene-d6) δ 8.01 (s, 1H), 7.91–7.82 (m, 2H), 7.24 (d, J = 1.7 Hz, 1H), 7.03 (dd, J = 8.7, 1.7 Hz, 1H), 6.94 (d, J = 8.7 Hz, 1H), 6.92–6.88 (m, 3H), 6.79–6.75 (m, 1H), 1.96 (s, 3H). LRMS [ES]+: m/z = 340.1 (M + H)+.
j. 8-Ethyl-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (39)
Yield 260 mg (0.76 mmol, 70%). 1H NMR (500 MHz, chloroform-d) δ 8.87 (s, 1H), 8.18 (d, J = 8.6 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.88–7.80 (m, 2H), 7.73 (d, J = 2.0 Hz, 1H), 7.58 (dd, J = 8.6, 1.8 Hz, 1H), 7.49 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 2.86 (q, J = 7.6 Hz, 2H), 2.44 (s, 3H), 1.35 (t, J = 7.6 Hz, 3H). LRMS [ES]+: m/z = 343.1 (M + H)+.
k. 8-Methyl-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (40)
Yield 250 mg (0.76 mmol, 71%). 1H NMR (500 MHz, chloroform-d) δ 8.87 (s, 1H), 8.15 (d, J = 8.6 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.87–7.80 (m, 2H), 7.72 (d, J = 1.8 Hz, 1H), 7.55 (dd, J = 8.6, 1.7 Hz, 1H), 7.49 (dd, J = 9.0, 0.7 Hz, 1H), 7.33–7.26 (m, 2H), 2.58–2.54 (m, 3H), 2.44 (s, 3H). LRMS [ES]+: m/z = 329.1 (M + H)+.
l. 8-Bromo-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (41)
Yield 650 mg (1.65 mmol, 83%). 1H NMR (500 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.57 (d, J = 9.0 Hz, 1H), 8.40 (d, J = 2.1 Hz, 1H), 8.28 (d, J = 9.1 Hz, 1H), 7.91–7.81 (m, 3H), 7.71 (d, J = 9.1 Hz, 1H), 7.38–7.32 (m, 2H), 2.40 (s, 3H). LRMS [ES]+: m/z = 393.0 (M + H)+.
m. 9-Bromo-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (42)
Yield 180 mg (0.45 mmol, 72%). 1H NMR (500 MHz, acetone-d6) δ 9.11 (s, 1H), 8.84 (dd, J = 1.8, 0.8 Hz, 1H), 8.34–8.28 (m, 1H), 8.06 (d, J = 8.7 Hz, 1H), 7.97–7.90 (m, 2H), 7.78 (dd, J = 8.7, 1.9 Hz, 1H), 7.63 (dd, J = 9.0, 0.7 Hz, 1H), 7.37 (ddt, J = 8.0, 1.5, 0.7 Hz, 2H), 2.44 (s, 3H). LRMS [ES]+: m/z = 393.1 (M + H)+.
n. 5-Bromo-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (43)
Yield 80 mg (0.20 mmol, 34%). 1H NMR (500 MHz, chloroform-d) δ 8.85 (s, 1H), 8.38 (s, 1H), 8.27–8.21 (m, 1H), 7.91–7.81 (m, 3H), 7.73 (ddd, J = 8.4, 6.9, 1.3 Hz, 1H), 7.63 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 7.34–7.27 (m, 2H), 2.45 (s, 3H). LRMS [ES]+: m/z = 393.1 (M + H)+.
o. 5-Methoxy-2-(4-methylbenzoyl)-3H-benzo[f]chromen-3-one (44)
Yield 200 mg (0.58 mmol, 65%). 1H NMR (500 MHz, chloroform-d) δ 8.86 (s, 1H), 8.20–8.14 (m, 1H), 7.83 (dq, J = 8.3, 2.0 Hz, 3H), 7.61–7.53 (m, 2H), 7.44 (s, 1H), 7.29 (d, J = 7.9 Hz, 2H), 4.10 (s, 3H), 2.44 (s, 3H). LRMS [ES]+: m/z = 345.1 (M + H)+.
p. 8-Bromo-2-(4-methoxybenzoyl)-3H-benzo[f]chromen-3-one (45)
Yield 95 mg (0.23 mmol, 63%). 1H NMR (500 MHz, chloroform-d) δ 8.79 (s, 1H), 8.16–8.09 (m, 2H), 8.01 (d, J = 9.1 Hz, 1H), 7.96–7.89 (m, 2H), 7.79 (dd, J = 8.9, 2.1 Hz, 1H), 7.59–7.53 (m, 1H), 7.01–6.94 (m, 2H), 3.90 (s, 3H). LRMS [ES]+: m/z = 409.0 (M + H)+.
General Procedure (III) for Preparation of Pyrazolone-Based Analogues
The corresponding 2-benzoyl ketocoumarin (0.5 mmol) was dissolved in dry pyridine (3 mL). To this solution was added NaBH4 (0.625 mmol), and the reaction was stirred at room temp for 2 h. The mixture was then poured in cold 2 M hydrochloric acid (10 mL), which resulted in a white precipitate. The precipitate was washed several times with water, dried under a vacuum to yield the corresponding 2-benzoyl-1,2-dihydrocoumarin which was taken to the next step without purification. To a stirring solution of 2-benzoyl-1,2 dihydrocoumarin (0.1 mmol) in DMF (0.2 mL) was added hydrazine (0.125 mmol), and the mixture was stirred for 1.5 h at room temperature. Water (5 mL) was then added, and the aqueous layer was extracted with EtOAc (15 mL). The organic layer was separated, dried with Na2SO4, and reduced to dryness to afford a yellowish brown crude product, which was further purified using medium pressure chromatography.
a. 4-[(2-Hydroxynaphthalen-1-yl)methyl]-5-phenyl-2,3-dihydro-1H-pyrazol-3-one (2)
The solid product was recrystallized from a mixture of ethyl acetate and n-heptane. Yield 100 mg (0.316 mmol, 64%). 1H NMR (300 MHz, acetone-d6) δ 7.64 (dd, J = 8.0, 1.4 Hz 1H), 7.53 (d, 8.8 Hz, 1H), 7.51–7.46 (m, 3H), 7.40–7.32, (m, 3H), 7.15–7.01 (m, 3H), 4.11 (s, 2H). LRMS [ES]+: m/z = 317.1 (M + H)+.
b. 5-(3-Bromophenyl)-4-[(2-hydroxynaphthalen-1-yl)methyl]-2,3-dihydro-1H-pyrazol-3-one (3)
The crude product was purified by medium pressure chromatography using a gradient MeOH:EtOAc solvent system (1–10% MeOH). Yield 25 mg (0.063 mmol, 48%). 1H NMR (500 MHz, acetone-d6) δ 7.83–7.56 (m, 4H), 7.56–7.44 (m, 2H), 7.39 (t, J = 7.9 Hz, 1H), 7.20–7.04 (m, 3H), 4.27 (s, 2H). LRMS [ES]+: m/z = 395.01 (M + H)+.
c. 5-(4-Bromophenyl)-4-[(2-hydroxynaphthalen-1-yl)methyl]-2,3-dihydro-1H-pyrazol-3-one (4)
The crude product was purified by medium pressure chromatography using a gradient MeOH–EtOAc solvent system (1–10% MeOH). Yield 15 mg (0.037 mmol, 49%). 1H NMR (500 MHz, DMSO-d6) δ 11.67 (s, 1H), 9.88 (s, 1H), 7.68–7.60 (m, 2H), 7.51 (dd, J = 20.3, 8.5 Hz, 3H), 7.35 (d, J = 8.5 Hz, 2H), 7.14 (dt, J = 6.8, 4.0 Hz, 2H), 7.06 (d, J = 8.8 Hz, 1H), 4.11 (s, 2H). LRMS [ES]+: m/z = 395.01 (M + H)+.
d. 4-[(2-Hydroxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (5)
The crude product was purified by medium pressure chromatography using a gradient MeOH:EtOAc solvent system (1–10% MeOH). Yield 22 mg (0.066 mmol, 42%). 1H NMR (500 MHz, DMSO-d6) δ 11.54 (s, 1H), 10.13 (s, 2H), 7.68–7.62 (m, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.48 (t, J = 8.1 Hz, 1H), 7.38 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 7.7 Hz, 2H), 7.16–7.01 (m, 3H), 4.10 (s, 2H), 2.34 (s, 3H). LRMS [ES]+: m/z = 331.20 (M + H)+.
e. 5-(4-Fluorophenyl)-4-[(2-hydroxynaphthalen-1-yl)methyl]-2,3-dihydro-1H-pyrazol-3-one (6)
Yield 60 mg (0.18 mmol, 58%). 1H NMR (500 MHz, DMSO-d6) δ 10.00 (s, 1H), 9.89 (s, 1H), 7.68–7.61 (m, 2H), 7.59–7.46 (m, 2H), 7.41 (dd, J = 8.4, 5.6 Hz, 2H), 7.26–7.20 (m, 2H), 7.14 (tt, J = 6.3, 2.5 Hz, 2H), 7.05 (d, J = 8.8 Hz, 1H), 4.09 (s, 2H). LRMS [ES]+: m/z = 335.10 (M + H)+.
f. 4-[(2-Hydroxynaphthalen-1-yl)methyl]-5-[4-(trifluoromethyl)phenyl]-2,3-dihydro-1H-pyrazol-3-one (7)
Yield 60 mg (0.15 mmol, 57%). 1H NMR (500 MHz, DMSO-d6) δ 11.82 (s, 1H), 9.78 (s, 1H), 7.67–7.56 (m, 6H), 7.51 (d, J = 8.8 Hz, 1H), 7.14 (td, J = 6.9, 4.6 Hz, 2H), 7.04 (d, J = 8.8 Hz, 1H), 4.16 (s, 2H). LRMS [ES]+: m/z = 385.10 (M + H)+.
g. 4-[(2-Hydroxy-6-phenylnaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (8)
The crude product was purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 20 mg (0.05 mmol, 50%). 1H NMR (500 MHz, DMSO-d6) δ 8.28 (d, J = 1.9 Hz, 1H), 8.10–7.97 (m, 4H), 7.93–7.77 (m, 3H), 7.50 (t, J = 7.6 Hz, 2H), 7.44–7.33 (m, 4H), 4.16 (s, 2H), 2.39 (s, 3H). LRMS [ES]+: m/z = 407.1 (M + H)+.
h. 4-[(2-Hydroxy-6-methoxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (9)
The crude product was purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 10 mg (0.027, 39%). 1H NMR (500 MHz, DMSO-d6) δ 9.87 (s, 1H), 9.62 (s, 1H), 7.43- 7.40 (m, 4H), 7.20 (d, J = 7.5 Hz, 1H), 7.11–7.02 (m, 3H), 6.73–6.67 (m, 1H), 4.06 (s, 2H), 2.34 (s, 3H). LRMS [ES]+: m/z = 361.2 (M + H)+.
i. 6-Hydroxy-5-([5-(4-methylphenyl)-3-oxo-2,3-dihydro-1H-pyrazol-4-yl]methyl)naphthalene-2-carbonitrile (10)
The crude product was purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (1–10% EtOAc) yielding 10 mg (0.028, 40%). 1H NMR (500 MHz, DMSO-d6) δ 9.49 (s, 1H), 8.28 (d, J = 1.8 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.34 (m, 4H), 7.18 (m, 3H), 4.11 (s, 2H), 2.31 (s, 3H). LRMS [ES]+: m/z = 356.2 (M + H)+.
j. 4-[(2-Hydroxy-6-methylnaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (11)
The crude product was purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (10–25% EtOAc) yielding 42 mg (0.12 mmol, 80%). 1H NMR (300 MHz, DMSO-d6) δ 7.52–7.35 (m, 3H), 7.16 (m, 3H), 6.86 (d, J = 8.9 Hz, 1H), 4.07 (s, 2H), 2.62–2.51 (m, 3H), 2.32 (s, 3H). LRMS [ES]+: m/z = 345.2 (M + H)+.
k. 4-[(6-Ethyl-2-hydroxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (12)
The crude product was purified by medium pressure chromatography using a gradient EtOAc/hexane solvent system (10- 25% EtOAc). Yielding 37 mg (0.10 mmol, 71%). 1H NMR (300 MHz, DMSO-d6) δ 7.54–7.35 (m, 4H), 7.16 (m, 4H), 6.92 (dd, J = 8.9, 1.8 Hz, 1H), 4.08 (s, 2H), 2.60 (q, J = 7.5 Hz, 2H), 2.34 (s, 3H), 1.16 (t, J = 7.5 Hz, 3H). LRMS [ES]+: m/z = 359.2 (M + H)+.
l. 4-[(6-Bromo-2-hydroxynaphthalen-1-yl)methyl]-5-phenyl-2,3-dihydro-1H-pyrazol-3-one (13)
The crude product was purified by medium pressure chromatography using a gradient MeOH/EtOAc solvent system (1–10% MeOH) yielding 16 mg (0.045 mmol, 58%). 1H NMR (300 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.53 (d, J = 8.8 Hz, 1H), 7.43 (m, 2H), 7.35 (m, 3H), 7.18 (d, J = 8.8 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 4.10 (s, 2H), 2.32 (s, 3H). LRMS [ES]+: m/z = 394.93 (M + H)+.
m. 4-[(6-Bromo-2-hydroxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (14)
The crude product was purified by medium pressure chromatography using a gradient MeOH/EtOAc solvent system (1–10% MeOH) yielding 45 mg (0.110 mmol, 62%). 1H NMR (500 MHz, DMSO-d6) δ 7.89 (d, J = 2.1 Hz, 1H), 7.52 (d, J = 8.8 Hz, 1H), 7.46 (s, 1H), 7.33 (d, J = 7.8 Hz, 2H), 7.13 (m, 4H), 4.06 (s, 2H), 2.31 (s, 3H). LRMS [ES]+: m/z = 409.10 (M + H)+.
n. 4-[(7-Bbromo-2-hydroxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (15)
The crude product was purified by medium pressure chromatography using a gradient MeOH/EtOAc solvent system (1–10% MeOH) yielding 16 mg (0.110 mmol, 52%). 1H NMR (300 MHz, DMSO-d6) δ 7.54–7.35 (m, 4H), 7.16 (m, 4H), 6.92 (dd, J = 8.9, 1.8 Hz, 1H), 4.08 (s, 2H), 2.34 (s, 3H). LRMS [ES]+: m/z = 409.10 (M + H)+.
o. 4-[(3-Bromo-2-hydroxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (16)
The crude product was purified by medium pressure chromatography using a gradient MeOH/EtOAc solvent system (1–10% MeOH) yielding 20 mg (0.110 mmol, 65%). 1H NMR (300 MHz, DMSO-d6) δ 8.01 (s, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.51–7.33 (m, 4H), 7.15 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 9.0 Hz, 1H), 6.87 (t, J = 7.4 Hz, 1H), 4.08 (s, 2H), 2.44 (s, 3H). LRMS [ES]+: m/z = 409.1 (M + H)+.
p. 4-[(2-Hydroxy-3-methoxynaphthalen-1-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (17)
The crude product was purified by medium pressure chromatography using a gradient MeOH/EtOAc solvent system (1–10% MeOH) yielding 25 mg (0.069 mmol, 48%). 1H NMR (500 MHz, chloroform-d) δ 7.58–7.53 (m, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.19–7.12 (m, 1H), 7.00–6.91 (m, 2H), 6.81 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 4.18 (s, 2H), 2.54 (s, 3H). LRMS [ES]+: m/z = 361.2 (M + H)+.
q. 4-[(6-Bromo-2-hydroxynaphthalen-1-yl)methyl]-5-(4-methoxyphenyl)-2,3-dihydro-1H-pyrazol-3-one (18)
The crude product was purified by medium pressure chromatography using a gradient MeOH/EtOAc solvent system (1–10% MeOH) yielding 15 mg (0.035 mmol, 48%). 1H NMR (300 MHz, DMSO-d6) δ 7.92 (d, J = 2.1 Hz, 1H), 7.59–7.43 (m, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.14 (dd, J = 19.4, 8.9 Hz, 2H), 6.93 (d, J = 8.3 Hz, 2H), 4.06 (s, 2H), 3.78 (s, 3H). LRMS [ES]+: m/z = 427.1 (M + H)+.
General Procedure (IV) for Preparation of 2-Halogenated 6-Hydroxy-Quinoline Carbaldehydes
The corresponding 6-hydroxy-2(1H)-quinolinone (0.6 mmol) was taken up in trifluoroacetic acid (1 mL). To this solution was added hexamethylenetetramine (1.2 mmol), and the resulting mixture was then heated at 100 °C for 2 h. The reaction was allowed to cool, methanol (10 mL) was added, and the solvent was evaporated to afford a dark residue. Water (15 mL) was added, and the resulting precipitate was filtered and dried. The precipitate was dissolved in DMF (0.8 mL), the solution cooled to 0 °C, and phosphorus oxyhalide (POCl3 or POBr3) (1.8 mmol) was added dropwise. The light gray colored suspension was then stirred at room temp overnight. Ice-cold water (15 mL) was added, and the precipitate was filtered and dried under a vacuum to yield the corresponding aldehyde as a pure product. Synthesis of 6-hydroxyquinoline-5-carbaldehyde (60) has been reported previously by using Reimer-Teimann conditions.31
a. 2-Chloro-6-hydroxyquinoline-5-carbaldehyde (61)
Yield 80 mg (0.38 mmol, 63%). 1H NMR (500 MHz, DMSO-d6) δ 11.06 (s, 1H), 8.23 (d, J = 8.8 Hz, 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 9.3, 1H), 7.36 (d, J = 9.3, 1H). LRMS [ES]+: m/z = 208.2 (M + H)+.
b. 2-Bromo-6-hydroxyquinoline-5-carbaldehyde (62)
POBr3 (3 mmol) was added dropwise to 2,6-dihydroxy-quinaldehyde. The suspension was heated to 50 °C and stirred for 18 h. Ice-cold water (15 mL) was then added, and resulting light brown precipitate was filtered and dried under a vacuum to afford the pure product. Yield 60 mg (0.23 mmol, 45%). 1H NMR (500 MHz, DMSO-d6) δ 11.16 (s, 1H), 8.70 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.42 (d, J = 9.3, 1H), 7.39 (d, J = 9.3, 1H). LRMS [ES]+: M + H+ 253.1 (M + H)+.
c. 2-Chloro-6-hydroxy-4-methylquinoline-5-carbaldehyde (63)
Yield 18 mg (0.08 mmol, 66%). 1H NMR (500 MHz, DMSO-d6) δ 12.71 (s, 1H), 10.74 (s, 1H), 8.07 (d, J = 9.3 Hz, 1H), 7.58 (s, 1H), 7.48 (d, J = 9.3, 1H), 2.69 (s, 3H). LRMS [ES]+: m/z = 222.1 (M + H)+.
Preparation of Quinoline-Based Pyrazolone Compounds
Compounds were prepared using General Procedure (II) Knoevenagel condensation as described above.
a. 2-(4-Methylbenzoyl)-3H-chromeno[6,5-b]pyridin-3-on (46)
Yield 28 mg (0.093 mmol, 54%). 1H NMR (500 MHz, chloroform-d) δ 9.06–9.01 (m, 1H), 8.81 (s, 1H), 8.62–8.56 (m, 1H), 8.38 (d, J = 9.3 Hz, 1H), 7.86–7.75 (m, 2H), 7.64 (dd, J = 8.6, 4.2 Hz, 1H), 7.34–7.24 (m, 2H), 2.45 (s, 3H). LRMS [ES]+: m/z = 316.1 (M + H)+.
b. 8-Chloro-2-(4-methylbenzoyl)-3H-chromeno[6,5-b]pyridin-3-one (47)
Yield 65 mg (0.019 mmol, 80%). 1H NMR (500 MHz, chloroform-d) δ 8.72 (d, J = 0.6 Hz, 1H), 8.55–8.49 (m, 1H), 8.29 (dd, J = 9.3, 0.8 Hz, 1H), 7.85–7.76 (m, 2H), 7.63 (d, J = 8.8 Hz, 1H), 7.34–7.20 (m, 2H), 2.45 (s, 3H). LRMS [ES]+: m/z = 350.0 (M + H)+.
c. 8-Bromo-2-(4-methylbenzoyl)-3H-chromeno[6,5-b]pyridin-3-one (48)
Yield 65 mg (0.171 mmol, 72%). 1H NMR (500 MHz, chloroform-d) δ 8.72 (d, J = 0.7 Hz, 1H), 8.44–8.38 (m, 1H), 8.30 (dd, J = 9.3, 0.7 Hz, 1H), 7.85–7.72 (m, 2H), 7.31 (ddt, J = 7.1, 1.3, 0.7 Hz, 2H), 7.25–7.19 (m, 1H), 2.45 (s, 3H). LRMS [ES]+: m/z = 396.0 (M + H)+.
d. 8-Chloro-10-methyl-2-(4-methylbenzoyl)-3H-chromeno[6,5-b]pyridin-3-one (49)
Yield 55 mg (0.15 mmol, 67%). 1H NMR (500 MHz, chloroform-d) δ 9.17 (s, 1H), 8.29 (d, J = 9.3 Hz, 1H), 7.80 (dd, J = 23.1, 8.5 Hz, 3H), 7.43 (s, 1H), 7.31 (d, J = 7.9 Hz, 2H), 2.97 (s, 3H), 2.45 (s, 3H). LRMS [ES]+: m/z = 364.1 (M + H)+.
e. 8-Chloro-2-[4-(propan-2-yl)benzoyl]-3H-chromeno[6,5-b]pyridin-3-one (50)
Yield 25 mg (0.066 mmol, 55%). 1H NMR (500 MHz, chloroform-d) δ 8.71 (s, 1H), 8.29 (dd, J = 9.3, 0.8 Hz, 1H), 7.89–7.83 (m, 2H), 7.80 (d, J = 9.3 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.39–7.33 (m, 2H), 3.00 (p, J = 6.9 Hz, 1H), 1.29 (d, J = 6.9 Hz, 6H). LRMS [ES]+: m/z = 378.1 (M + H)+.
General Procedure (V) for Preparation of Quinoline-Based Pyrazolones
General procedure (III) for naphthalene pyrazolones was followed. The compounds were obtained as white precipitates, which were purified by medium pressure chromatography using a gradient MeOH/DCM solvent system (1–10% MeOH) unless otherwise noted.
a. 4-[(6-Hydroxyquinolin-5-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (19)
Yield 4 mg (0.012 mmol, 48%). 1H NMR (500 MHz, D2O) δ 11.60 (s, 1H), 8.56 (m, 2H), 8.04 (s, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.34 (dd, J = 17.7, 8.5 Hz, 4H), 7.17 (d, J = 7.7 Hz, 1H), 4.11 (s, 3H), 2.32 (s, 3H). LRMS [ES]+: m/z = 332.1 (M + H)+.
b. 4-[(2-Chloro-6-hydroxyquinolin-5-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (20)
Yield 20 mg (0.054 mmol, 65%). 1H NMR (500 MHz, DMSO-d6) δ 11.57 (s, 1H), 10.25 (s, 1H), 8.08 (d, J = 8.7 Hz, 1H), 7.58 (d, J = 9.0 Hz, 1H), 7.36–7.30 (m, 4H), 7.22 (d, J = 9.0 Hz, 1H), 7.15 (d, J = 8.7 Hz, 1H), 4.09 (s, 2H), 2.31 (s, 3H). LRMS [ES]+: m/z = 366.1 (M + H)+.
c. 4-[(2-Bromo-6-hydroxyquinolin-5-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (21)
Yield 15 mg (0.036 mmol, 49%). 1H NMR (300 MHz, DMSO-d6) δ 7.96 (d, J = 8.9 Hz, 1H), 7.59 (d, J = 9.0 Hz, 1H), 7.32 (dd, J = 8.5, 5.2 Hz, 4H), 7.16 (d, J = 7.8 Hz, 1H), 4.08 (s, 2H), 2.31 (s, 3H). LRMS [ES]+: m/z = 411.1 (M + H)+.
d. 4-[(2-Chloro-6-hydroxy-4-methylquinolin-5-yl)methyl]-5-(4-methylphenyl)-2,3-dihydro-1H-pyrazol-3-one (22)
The crude product was purified by medium pressure chromatography using a gradient EtOAc/hexanes solvent system (10–25% EtOAc). Yield 12 mg (0.031 mmol, 59%). 1H NMR (500 MHz, DMSO-d6) δ 11.54 (s, 1H), 8.97 (s, 1H), 7.57 (d, J = 8.9 Hz, 1H), 7.28 (d, J = 9.0 Hz, 2H), 7.20 (d, J = 7.7 Hz, 2H), 7.08 (d, J = 8.9 Hz, 1H), 4.28 (s, 2H), 2.72 (s, 3H), 2.28 (s, 3H). LRMS [ES]+: m/z = 380.1 (M + H)+.
e. 4-[(2-Chloro-6-hydroxyquinolin-5-yl)methyl]-5-[4-(propan-2-yl)phenyl]-2,3-dihydro-1H-pyrazol-3-one (23)
Yield 8 mg (0.020 mmol, 52%). 1H NMR (500 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.03 (d, J = 9.0 Hz, 1H), 7.56 (d, J = 9.0 Hz, 1H), 7.31 (m, 3H), 7.18 (m, 3H), 4.07 (s, 2H), 2.87 (p, J = 6.9 Hz, 1H), 1.20 (d, J = 6.9 Hz, 6H). LRMS [ES]+: m/z = 394.1 (M + H)+.
General Procedure (IV) for Preparation of Isoxazolone Derivatives
2-(4-Methylbenzoyl)-3H-benzo[f]chromen-3-one (0.06 mmol) was taken in DMF (0.2 mL) to which was added the corresponding hydroxylamine (0.12 mmol), and the reaction mixture was stirred at 60 °C for 18 h. The solvent was evaporated, and the residue was dissolved in EtOAc (5 mL). The organic layer was then washed with water, dried with Na2SO4, and reduced to dryness to yield an oily product. The oil was further purified by medium pressure chromatography using gradient EtOAc/hexanes solvent system (0–50% EtOAc) to afford the pure product.
a. 4-[(6-Bromo-2-hydroxynaphthalen-1-yl)methyl]-3-(4-methylphenyl)-2,5-dihydro-1,2-oxazol-5-one (24)
Yield 13 mg (0.031 mmol, 50%) of 1:1 mixture of imine and enamine tautomers. 1H NMR (300 MHz, DMSO-d6) δ 9.88 (s, 1H), 8.04–7.85 (m, 4H), 7.65 (d, J = 8.9 Hz, 1H), 7.53–7.42 (m, 2H), 7.24 (dd, J = 21.9, 8.4 Hz, 3H), 7.05–6.91 (m, 1H), 4.65 (dd, J = 8.6, 5.0 Hz, 1H), 2.35 (s, 3H). LRMS [ES]+: m/z = 412.1 (M + H)+.
b. 4-[(6-Bromo-2-hydroxynaphthalen-1-yl)methyl]-2-methyl-3-(4-methylphenyl)-2,5-dihydro-1,2-oxazol-5-one (25)
Pyridine (1.5 equiv) was added to the reaction mixture as methyl hydroxylamine was purchased as a hydrochloride salt. Yield 15 mg (0.035 mmol, 47%). 1H NMR (300 MHz, DMSO-d6) δ 9.74 (s, 1H), 7.93 (d, J = 2.1 Hz, 1H), 7.54 (dd, J = 9.0, 2.3 Hz, 2H), 7.37 (dd, J = 9.1, 2.2 Hz, 1H), 7.28–7.15 (m, 4H), 7.03 (d, J = 8.9 Hz, 1H), 3.92 (s, 2H), 2.97 (s, 3H), 2.33 (s, 3H). LRMS [ES]+: m/z = 424.0.
c. 2-Benzyl-4-[(6-bromo-2-hydroxynaphthalen-1-yl)methyl]-3-(4-methylphenyl)-2,5-dihydro-1,2-oxazol-5-one (26)
Pyridine (1.5 equiv) was added to the reaction mixture as benzyl hydroxylamine was purchased as a hydrochloride salt. Yield 13 mg (0.018 mmol, 41%). 1H NMR (500 MHz, D2O) δ 9.73 (s, 1H), 7.94 (d, J = 2.1 Hz, 1H), 7.52 (m, 4H), 7.40–7.22 (m, 6H), 7.14–7.07 (m, 2H), 7.04 (d, J = 8.9 Hz, 1H), 3.90 (s, 2H), 2.51 (s, 2H), 2.36 (s, 3H). LRMS [ES]+: m/z = 500.1 (M + H)+.
Acknowledgments
The research was funded by the National Cancer Institute (CA-129132). The authors would like to thank Prof. Bill Atkins and the Department of Medicinal Chemistry, University of Washington, for providing access to their NMR facility and resources and Dr. Eric Foss (Fred Hutchinson Cancer Research Center) for help with statistical analysis.
Glossary
Abbreviations
- SIRT
sirtuin
- STD NMR
saturation transfer difference nuclear magnetic resonance
- HMTA
hexamethylenetetramine
Author Present Address
# (J.P.) Kineta Inc., Seattle, WA 98109.
Author Present Address
⊥ (A.D.S.) Syntrix Biosystems, Auburn, WA 98001.
Author Contributions
J.A.S. and A.B. conceived this project and designed the experiments, S.S.M., J.P., A.T, and A.S. contributed to compound design and carried out the syntheses, T.K.L., Y.L., and V.L. carried out the biochemical enzyme and cell-based assays, and S.S.M. and M.S. conceived and carried out the STD NMR experiments. S.S.M., A.B., and J.A.S. wrote the manuscript.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
References
- Heltweg B.; Gatbonton T.; Schuler A. D.; Posakony J.; Li H.; Goehle S.; Kollipara R.; Depinho R. A.; Gu Y.; Simon J. A.; Bedalov A. Antitumor activity of a small-molecule inhibitor of human silent inform ation regulator 2 enzymes. Cancer Res. 2006, 66, 4368–4377. [DOI] [PubMed] [Google Scholar]
- Aljada A.; Dong L.; Mousa S. A. Sirtuin-targeting drugs: Mechanisms of action and potential therapeutic applications. Curr. Opin. Investig. Drugs 2010, 11, 1158–1168. [PubMed] [Google Scholar]
- Sanchez-Fidalgo S.; Villegas I.; Sanchez-Hidalgo M.; de la Lastra C. A. Sirtuin modulators: mechanisms and potential clinical implications. Curr. Med. Chem. 2012, 19, 2414–2441. [DOI] [PubMed] [Google Scholar]
- Roth M.; Chen W. Y. Sorting out functions of sirtuins in cancer. Oncogene 2014, 33, 1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S. S.; Leko V.; Simon J. A.; Bedalov A. Sirtuin modulators. Handb. Exp. Pharmacol. 2011, 206, 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur J. A.; Sinclair D. A. Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discovery 2006, 5, 493–506. [DOI] [PubMed] [Google Scholar]
- Burnett C.; Valentini S.; Cabreiro F.; Goss M.; Somogyvari M.; Piper M. D.; Hoddinott M.; Sutphin G. L.; Leko V.; McElwee J. J.; Vazquez-Manrique R. P.; Orfila A. M.; Ackerman D.; Au C.; Vinti G.; Riesen M.; Howard K.; Neri C.; Bedalov A.; Kaeberlein M.; Soti C.; Partridge L.; Gems D. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 2011, 477, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M.; McDonagh T.; Heltweg B.; Hixon J.; Westman E. A.; Caldwell S. D.; Napper A.; Curtis R.; DiStefano P. S.; Fields S.; Bedalov A.; Kennedy B. K. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [DOI] [PubMed] [Google Scholar]
- Borra M. T.; Smith B. C.; Denu J. M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [DOI] [PubMed] [Google Scholar]
- Lain S.; Hollick J. J.; Campbell J.; Staples O. D.; Higgins M.; Aoubala M.; McCarthy A.; Appleyard V.; Murray K. E.; Baker L.; Thompson A.; Mathers J.; Holland S. J.; Stark M. J.; Pass G.; Woods J.; Lane D. P.; Westwood N. J. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara E.; Mai A.; Calvanese V.; Altucci L.; Lopez-Nieva P.; Martinez-Chantar M. L.; Varela-Rey M.; Rotili D.; Nebbioso A.; Ropero S.; Montoya G.; Oyarzabal J.; Velasco S.; Serrano M.; Witt M.; Villar-Garea A.; Imhof A.; Mato J. M.; Esteller M.; Fraga M. F. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791. [DOI] [PubMed] [Google Scholar]
- Solomon J. M.; Pasupuleti R.; Xu L.; McDonagh T.; Curtis R.; DiStefano P. S.; Huber L. J. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 2006, 26, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amengual J. E.; Clark-Garvey S.; Kalac M.; Scotto L.; Marchi E.; Neylon E.; Johannet P.; Wei Y.; Zain J.; O’Connor O. A. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood 2013, 122, 2104–2113. [DOI] [PubMed] [Google Scholar]
- Huber J. L.; McBurney M. W.; Distefano P. S.; McDonagh T. SIRT1-independent mechanisms of the putative sirtuin enzyme activators SRT1720 and SRT2183. Future Med. Chem. 2010, 2, 1751–1759. [DOI] [PubMed] [Google Scholar]
- Carafa V.; Nebbioso A.; Altucci L. Sirtuins and disease: the road ahead. Front. Pharmacol. 2012, 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholec M.; Bleasdale J. E.; Chrunyk B.; Cunningham D.; Flynn D.; Garofalo R. S.; Griffith D.; Griffor M.; Loulakis P.; Pabst B.; Qiu X.; Stockman B.; Thanabal V.; Varghese A.; Ward J.; Withka J.; Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Nikolaev A. Y.; Imai S.; Chen D.; Su F.; Shiloh A.; Guarente L.; Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [DOI] [PubMed] [Google Scholar]
- North B. J.; Marshall B. L.; Borra M. T.; Denu J. M.; Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [DOI] [PubMed] [Google Scholar]
- Medda F.; Russell R. J.; Higgins M.; McCarthy A. R.; Campbell J.; Slawin A. M.; Lane D. P.; Lain S.; Westwood N. J. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J. Med. Chem. 2009, 52, 2673–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posakony J.; Hirao M.; Stevens S.; Simon J. A.; Bedalov A. Inhibitors of Sir2: evaluation of splitomicin analogues. J. Med. Chem. 2004, 47, 2635–2644. [DOI] [PubMed] [Google Scholar]
- Neugebauer R. C.; Uchiechowska U.; Meier R.; Hruby H.; Valkov V.; Verdin E.; Sippl W.; Jung M. Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J. Med. Chem. 2008, 51, 1203–1213. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Khan M. N.; Sawada H.; Imai E.; Itoh Y.; Yamatsuta K.; Tokuda N.; Takeuchi J.; Seko T.; Nakagawa H.; Miyata N. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [DOI] [PubMed] [Google Scholar]
- Angulo J.; Nieto P. M. STD-NMR: application to transient interactions between biomolecules-a quantitative approach. Eur. Biophys. J. 2011, 40, 1357–1369. [DOI] [PubMed] [Google Scholar]
- Rotili D.; Carafa V.; Tarantino D.; Botta G.; Nebbioso A.; Altucci L.; Mai A. Simplification of the tetracyclic SIRT1-selective inhibitor MC2141: coumarin- and pyrimidine-based SIRT1/2 inhibitors with different selectivity profile. Bioorg. Med. Chem. 2011, 19, 3659–3668. [DOI] [PubMed] [Google Scholar]
- Peck B.; Chen C. Y.; Ho K. K.; Di Fruscia P.; Myatt S. S.; Coombes R. C.; Fuchter M. J.; Hsiao C. D.; Lam E. W. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer. Ther. 2010, 9, 844–855. [DOI] [PubMed] [Google Scholar]
- Rotili D.; Tarantino D.; Nebbioso A.; Paolini C.; Huidobro C.; Lara E.; Mellini P.; Lenoci A.; Pezzi R.; Botta G.; Lahtela-Kakkonen M.; Poso A.; Steinkuhler C.; Gallinari P.; De Maria R.; Fraga M.; Esteller M.; Altucci L.; Mai A. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J. Med. Chem. 2012, 55, 10937–10947. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [DOI] [PubMed] [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992, 2, 661–665. [DOI] [PubMed] [Google Scholar]
- Garcia O.; Nicolas E.; Albericio F. o-Formylation of electron-rich phenols with dichloromethyl methyl ether and TiCl4. Tetrahedron Lett. 2003, 44, 4961–4963. [Google Scholar]
- Utley J. H. P.; Rozenberger G. G. Electroorganic reactions. Part 56: Anodic oxidation of 2-methyl- and 2-benzylnaphthalenes: factors influencing competing pathways. Tetrahedron 2002, 58, 5251–5265. [Google Scholar]
- Wynberg H.; Meijer E. W. The Reimer-Teimann reaction. Org. React. 1982, 28, 1–36. [Google Scholar]