

Background: The role of the circadian protein Per1 in the regulation of sodium reabsorption in the distal convoluted tubule (DCT) is unknown.
Results: Per1 transcriptionally regulates the sodium transporter NCC and the WNK kinase cascade.
Conclusion: Per1 regulates sodium reabsorption in the DCT through NCC and the WNK cascade.
Significance: These data demonstrate a role for Per1 in the regulation of renal sodium transporters.
Keywords: Circadian Clock, Gene Expression, Kidney, Renal Physiology, Sodium Transport, Transcription
Abstract
It has been well established that blood pressure and renal function undergo circadian fluctuations. We have demonstrated that the circadian protein Per1 regulates multiple genes involved in sodium transport in the collecting duct of the kidney. However, the role of Per1 in other parts of the nephron has not been investigated. The distal convoluted tubule (DCT) plays a critical role in renal sodium reabsorption. Sodium is reabsorbed in this segment through the actions of the NaCl co-transporter (NCC), which is regulated by the with-no-lysine kinases (WNKs). The goal of this study was to test if Per1 regulates sodium transport in the DCT through modulation of NCC and the WNK kinases, WNK1 and WNK4. Pharmacological blockade of nuclear Per1 entry resulted in decreased mRNA expression of NCC and WNK1 but increased expression of WNK4 in the renal cortex of mice. These findings were confirmed by using Per1 siRNA and pharmacological blockade of Per1 nuclear entry in mDCT15 cells, a model of the mouse distal convoluted tubule. Transcriptional regulation was demonstrated by changes in short lived heterogeneous nuclear RNA. Chromatin immunoprecipitation experiments demonstrated interaction of Per1 and CLOCK with the promoters of NCC, WNK1, and WNK4. This interaction was modulated by blockade of Per1 nuclear entry. Importantly, NCC protein expression and NCC activity, as measured by thiazide-sensitive, chloride-dependent 22Na uptake, were decreased upon pharmacological inhibition of Per1 nuclear entry. Taken together, these data demonstrate a role for Per1 in the transcriptional regulation of NCC, WNK1, and WNK4.
Introduction
The circadian clock regulates a multitude of physiological functions, such as blood pressure (BP),2 metabolism, the immune response, sleep-wake cycles, and renal function (reviewed in Refs. 1 and 2). In terms of renal function, sodium excretion, glomerular filtration rate, renal blood flow, and plasma aldosterone levels exhibit rhythmic fluctuations (reviewed in Ref. 3). The molecular mechanism of the circadian clock consists of four core proteins, which interact with one another to regulate expression of circadian target genes (4). The four core proteins are CLOCK, Bmal1, Period (Per1–3), and Cryptochrome (Cry1 and -2). CLOCK and Bmal1 heterodimerize and interact with E-box response elements to transcriptionally up-regulate circadian target genes, which include Per and Cry. Per and Cry interact and then repress the transcriptional activity of CLOCK and Bmal1 (5). In addition to transcriptional regulation, the circadian clock also undergoes post-translational modifications through the phosphorylation of the Per proteins by the circadian kinases casein kinase 1 isoforms δ/ϵ (CK1δ/ϵ). Phosphorylation by CK1δ/ϵ allows Per1 nuclear entry (6, 7).
We have shown previously that Per1 regulates the basal and aldosterone-mediated expression of αENaC, the regulated subunit of the renal epithelial sodium channel (ENaC) (7–10). We also recently demonstrated that Per1 coordinately regulates the expression of multiple genes that are involved in the regulation of renal sodium reabsorption in collecting duct cells (8). This includes the positive regulation of Fxyd5, a positive regulator of the Na,K-ATPase (11), and the negative regulation of endothelin-1, a powerful inhibitor of ENaC (12). It was initially presumed that Per1 behaved primarily as a repressor of CLOCK and Bmal1 activity; however, increasing evidence suggests that Per1 can activate gene expression through an unknown mechanism, in a manner that appears to be gene- and tissue-specific (8–10, 13–15). We have recently demonstrated that a putative mechanism for Per1 action may occur through repression of the circadian repressor Cry2 (16). Taken together, these data predict that loss of Per1 should result in decreased renal sodium reabsorption, with subsequent decreased plasma volume and decreased BP. Consequently, we have shown previously that Per1 KO mice exhibit significantly lower BP (8) and increased urinary sodium levels and urine volume (9) compared with WT controls. Consistent with these results, we have shown recently that mice with reduced levels of Per1 have a defect in renal sodium retention and decreased plasma aldosterone (17).
We have shown that Per1 plays an extensive role in the regulation of ENaC and multiple other genes that contribute to renal sodium reabsorption in the collecting duct. However, the potential role of Per1 in other parts of the nephron has not been investigated. The distal convoluted tubule (DCT), which precedes the collecting duct, plays a critical role in sodium reabsorption in the renal cortex. Sodium is reabsorbed in this nephron segment through the actions of the NaCl co-transporter (NCC). NCC is post-translationally regulated through the actions of the with-no-lysine kinases (WNKs), including WNK1 and WNK4 (18). WNK1 increases NCC activity through activation of Ste20/SPS-1-related proline/alanine-rich kinase (19, 20), a downstream kinase of the WNKs; at baseline, WNK4 appears to inhibit NCC activity (21). The goal of this study was to test whether Per1 contributes to the regulation of sodium reabsorption in the DCT through transcriptional modulation of NCC and the WNK kinases. Pharmacological blockade of nuclear Per1 entry resulted in decreased mRNA expression of NCC and WNK1 and increased expression of WNK4 mRNA in the renal cortex of mice. To investigate this potential regulation further, a recently generated cell model of the mouse DCT (mDCT15 cells) was employed (22–24). Using several independent methods, we demonstrate a novel role for Per1 in the transcriptional regulation of NCC, WNK1, and WNK4. Importantly, NCC function, as measured by thiazide-sensitive, chloride-dependent 22Na uptake, was decreased upon pharmacological inhibition of Per1 nuclear entry. Stable knockdown of WNK4 prevented this effect. Taken together, these data demonstrate a role for Per1 in the coordinate regulation of sodium reabsorption in mouse DCT cells.
EXPERIMENTAL PROCEDURES
Animals
All animal use protocols were approved by the University of Florida and North Florida/South Georgia Veterans Affairs Institutional Animal Care and Use Committees in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. WT and Per1 KO mice (129/sv) mice were originally provided by Dr. David Weaver (University of Massachusetts) (25). WT and Per1 heterozygote (het) mice were bred in house by University of Florida Animal Care Services staff and maintained on a normal 12-h light/dark cycle. For in vivo PF670462 (CK1δ/ϵ inhibitor) experiments, weight-matched male WT 129/sv mice were given either vehicle (20% hydroxypropyl β-cyclodextrin) or 30 mg/kg PF670462 subcutaneously every 12 h for 2.5 days starting at noon and sacrificed at midnight 12 h after the last injection. Mice were anesthetized by inhalant isoflurane, and tissues were collected and snap-frozen in liquid nitrogen. The dose and time of PF670462 administration have been published previously and described by others (26). Kidneys were later dissected, and cortex was removed for protein or RNA isolation.
Cell Culture
mDCT15 cells were maintained in DMEM/F-12, 5% heat-inactivated FBS, and 1% penicillin-streptomycin-neomycin at 37 °C. For RNA silencing experiments, non-target siRNA (Dharmacon) and a previously characterized individual siRNA for Per1 (9) were used according to the manufacturer's instructions and as described previously (7, 16). Cells were transfected using Dharmafect 1 (Dharmacon). Casein kinase 1δ/ϵ inhibitor experiments were performed as described previously (7, 16). For dose and time course, mDCT15 cells were treated with either 0.1, 1, 10, or 100 μm casein kinase 1δ/ϵ inhibitor PF670462 or vehicle (water) for 24, 48, or 72 h (and as described previously (7)). For nuclear entry experiments, mDCT15 cells were treated with 10 μm PF670462 or vehicle for 48 h.
Assessment of NCC Function
The determination of NCC activity in mDCT15 cells was performed as described previously (22). Briefly, mDCT15 cells were seeded in 12-well plates and prepared as described. mDCT15 cells were treated with either 0.1, 1, 10, or 100 μm casein kinase 1δ/ϵ inhibitor PF670462 or vehicle (water) for 48 h prior to uptake. Thirty minutes before uptake, 0.1 mm metolazone was added to the medium. The medium was then changed to a 22Na+-containing medium (140 mm NaCl, 1 mm CaCl, 1 mm MgCl, 5 mm HEPES/Tris, pH 7.4, 1 mm amiloride, 0.1 mm bumetanide, 0.1 mm benzamil, 1 mm ouabain, and 1 μCi/ml 22Na+) with or without thiazide (0.1 mm metolazone) and incubated for 20 min. Tracer uptake was then stopped via washes with ice-cold wash buffer. Cells were subsequently lysed with 0.1% SDS. Radioactivity was measured via liquid scintillation, and protein concentrations of the lysates were determined (BCA protein assay, Pierce). Uptakes were normalized to nmol/mg. Thiazide-sensitive uptake was given by the difference of the uptakes with and without thiazide.
RNA Isolation and Quantitative Real-time PCR
Total RNA was isolated and DNase I-treated using Direct-ZolTM RNA Miniprep (Zymo Research) as per the manufacturer's instructions. DNase I-treated RNA (2 μg) samples were used as template for reverse transcription with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The resulting cDNAs (20 ng) were then used as a template in quantitative real-time PCR (qPCR) reactions (Applied Biosystems) to evaluate changes in Per1, NCC, WNK1, WNK4, and actin mRNA levels. Cycle threshold (Ct) values were normalized against β-actin, and relative quantification was performed using the ΔΔCt method (27). -Fold change values were calculated as the change in mRNA expression levels relative to the control. TaqMan primer/probe sets were purchased from Applied Biosystems.
Membrane Protein Analysis, Nuclear Protein Isolation, and Western Blot Analysis
Membrane protein was isolated using the Cell Surface Protein Isolation Kit (Pierce) according to the manufacturer's instructions. Nuclear extracts were isolated using the NE-PER kit (Pierce) according to the manufacturer's instructions. Protein concentrations were then quantified by BCA assay (Pierce). Proteins were separated on a 4–20% Tris-HCl Ready Gel (Bio-Rad) and transferred to a PVDF membrane. The membrane was blocked with 2% nonfat dry milk in TBS-S (TBS plus 0.05% RodeoTM Saddle Soap) (USB Corp.) and incubated overnight at 4 °C with anti-Per1 (1:500) (Pierce), anti-NCC (1:500) (described previously (22)), or anti-β-actin (1:500) (Santa Cruz Biotechnology, Inc.) antibodies. β-Actin was used as a loading control. The membrane was washed with 2% nonfat dry milk in TBS-S for 15 min and then incubated with horseradish peroxidase conjugate anti-rabbit secondary antibody and incubated in 2% nonfat dry milk in TBS-S for 1 h at 4 °C. After incubation, the blot was washed with TBS-S for 15 min. Detection was performed using Novex® ECL chemiluminescent substrate reagents (Invitrogen). Densitometry was performed using ImageJ (National Institutes of Health).
Chromatin Immunoprecipitation (ChIP)
mDCT15 cells were grown to ∼80% confluence and then treated with vehicle (water) or 10 μm PF670462 for 48 h. ChIP was performed using the ChIP-ITTM Express Enzymatic Kit (Active Motif) according to the manufacturer's instructions and as described previously (28). Chromatin concentrations were calculated, and equal amounts of vehicle-treated and 10 μm PF670462-treated chromatin were used per pull-down. Pull-downs were performed using 3 μg of either anti-Per1 (Pierce), anti-CLOCK (Pierce), anti-Pol II (Santa Cruz Biotechnology), or rabbit IgG as a negative control (Bethyl). Complexes were incubated overnight at 4 °C with end-over-end rotation. Immunoprecipitated DNA was amplified by end point PCR. Band intensities were quantitated using densitometry, which was performed using ImageJ. Bands were relativized to the relevant vehicle or PF670462-treated 10% input. Putative E-boxes in the Slc12a3 (NCC), Wnk1, and Wnk4 promoters were analyzed using ALGGEN-PROMO (29, 30). For each gene, three primer sets were designed as shown in Table 1. Putative E-box sequences are listed in Table 2.
TABLE 1.
Primers used
| Primer set | Forward | Reverse |
|---|---|---|
| Slc12a3 | ||
| 1 | 5′-GGCAGTGAGTTAGCACAA | 5′-CAGTTAGAGTCTCCCGTGTC |
| 2 | 5′-AGACAGATGCCAGGGTCT | 5′-TAAAGGAGTGGGACTGGG |
| 3 | 5′-CCTTCCCTGGTTGTCCAC | 5′-GAGGACTCTTGTCACTGTCC |
| Wnk1 | ||
| 1 | 5′-GTCAGCTCAAGGGCATTT | 5′-CCCCCTTTTCAGAGAGATG |
| 2 | 5′-AGATGGGAGGCAGAACTA | 5′-TGCATGCCTTTAATTCCAGC |
| 3 | 5′-TGCCTCTACCTCCCAAAC | 5′-GCAGCTGTTGACAACTTC |
| Wnk4 | ||
| 1 | 5′-TGTATGACGGCAGTTATA | 5′-GGTTATGCTTTTCAGTTG |
| 2 | 5′-AGCGTCAACAGCCTAAATAT | 5′-CTCCTGGAATGGTACCTATA |
| 3 | 5′-CACTGTCGGCCTGTTACT | 5′-CCTGTCCCCAAGCTATGT |
TABLE 2.
Putative E-box sequences
| Primer set | Predicted E-box sequence | |
|---|---|---|
| Slc12a3 | ||
| 1 | CACCTG | |
| 2 | CACGTG | |
| 3 | CAGCTG | |
| Wnk1 | ||
| 1 | CAGCTG | |
| 2 | CACCTG CACCTG | |
| 3 | CACCTG | |
| Wnk4 | ||
| 1 | CACCTG | |
| 2 | CACCTG | |
| 3 | CACCTG | |
Heterogeneous Nuclear RNA (hnRNA) Analysis
hnRNA analysis was performed as described previously (9). Primers were designed to amplify regions spanning the intron/exon boundary of the Wnk1, kidney-specific WNK1 (KS-Wnk1), Gadph, and Wnk4 genes. GAPDH was used as a cDNA loading control (sequence previously published (9)). Sequences and exon numbers are shown in Table 3. Total RNA was isolated from vehicle- and CKinh-treated cells, DNase I-treated, and converted to cDNA as described above. PCRs were performed using 40 ng of cDNA as template and the following cycling parameters. Reactions were heated to 95 °C for 15 min to activate the Taq polymerase. 35 amplification cycles were performed using the following parameters: 95 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, followed by a final 10-min extension at 72 °C.
TABLE 3.
hnRNA primer sequences and exon numbers
| hnRNA | Spanning exon | Forward | Reverse |
|---|---|---|---|
| NCC | 2 | 5′-TAGCATTGTACCCAGGAA | 5′-CATCAGTCAGCTCACGAC |
| Wnk1 | 1 | 5′-CAGTCTACAAAGGTCTGG | 5′-CAAGCTCCACAATCTCCC |
| KS-Wnk1 | 4a | 5′-TGTGTATGTGCTGGGTCC | 5′-TTGCTACTAGTAGTTTCTTCTG |
| Wnk4 | 2 | 5′-TTCTGGCTAGCGGTTTCA | 5′-GAGCATCTCAACTTCCTC |
Statistical Analysis
All data are represented as means ± S.E. Statistical analyses were performed using GraphPad Prism version 6. All graphs/plots were made with GraphPad Prism version 6. An unpaired Student's t test was used to compare differences between control and treated groups. For the 22Na+ uptake experiments, statistical analysis was performed using the SigmaPlot software package (Systat, San Jose, CA). Data were analyzed for statistical significance using analysis of variance (Holm-Sidak) or Mann-Whitney rank sum where appropriate. All p values less than 0.05 were considered significant.
RESULTS
Inhibition of CK1δ/ϵ in Vivo Results in Decreased mRNA Expression of NCC and WNK1 but Increased Expression of WNK4
Per1 must be phosphorylated by CK1δ/ϵ in order to enter the nucleus (6). Previously, we have shown that pharmacological inhibition of CK1δ/ϵ recapitulates the effects of Per1 knockdown, including decreased αENaC mRNA and protein expression and decreased ENaC channel activity (7, 16). Therefore, to determine if Per1 potentially regulated genes involved in sodium reabsorption in the renal cortex in vivo, WT mice were treated with vehicle or PF670462 (CK1δ/ϵ inhibitor) as described previously by others (26). Kidneys were harvested, cortex was dissected, and mRNA levels of NCC, WNK1, and WNK4 were assessed by qPCR. CK1δ/ϵ inhibition resulted in significantly decreased levels of NCC (Fig. 1A) and WNK1 (Fig. 1B). Conversely, CK1δ/ϵ inhibition resulted in significantly increased levels of WNK4 (Fig. 1C). To assess if PF670462 treatment in vivo resulted in decreased nuclear Per1 protein levels, Western blot analysis was performed on nuclear fractions obtained from the mice described above. As shown, nuclear Per1 levels were significantly decreased by more than 50% (Fig. 1D; quantified in Fig. 1E).
FIGURE 1.

Pharmacological blockade of Per1 nuclear entry in vivo results in decreased NCC and WNK1 expression and increased WNK4 expression in the renal cortex. Weight-matched male WT 129/sv mice were given either vehicle (20% hydroxypropyl β-cyclodextrin) or 30 mg/kg PF670462 subcutaneously every 12 h for 2.5 days starting at noon and euthanized at midnight 12 h after the last injection, as published and described previously (26). Kidneys were harvested, cortex was dissected, and NCC (A), WNK1 (B), or WNK4 (C) mRNA expression was measured by qPCR. Nuclear protein fractions were isolated from the renal cortex using the NE-PER kit (Pierce) and analyzed by Western blot using an anti-Per1 antibody (Pierce/Thermo) (D). Data were quantified using densitometry (ImageJ) (E). *, p < 0.05 compared with WT, n = 4 animals/treatment group. Values are represented as the mean ± S.E. (error bars).
Per1 Knockdown in Vitro or in Vivo or Pharmacological Inhibition of Per1 Nuclear Entry in mDCT15 Cells Results in Decreased mRNA Expression of NCC and WNK1 and Increased Expression of WNK4
To further investigate our in vivo findings, Per1 was knocked down using siRNA in a model of the distal convoluted tubule, mDCT15 cells. Total mRNA levels of NCC, WNK1, and WNK4 were measured by qPCR. As expected, Per1 knockdown resulted in significantly decreased mRNA expression of Per1 (Fig. 2A), as we have shown previously (7, 8, 10, 16). Consistent with our in vivo findings, Per1 knockdown resulted in significantly decreased expression of NCC (Fig. 2B) and WNK1 (Fig. 2C) but increased expression of WNK4 (Fig. 2D).
FIGURE 2.

Per1 knockdown results in decreased NCC and WNK1 expression and increased WNK4 expression in mDCT15 cells in vitro. mDCT15 cells were treated with non-target or Per1 siRNA for 48 h. qPCR was used to evaluate changes in Per1 (A), NCC (B), WNK1 (C), or WNK4 (D) mRNA expression in Per1 siRNA versus non-target siRNA control (n = 3). *, p < 0.05; **, p < 0.01. Values are represented as the mean ± S.E. (error bars).
To further assess the role of Per1 in the regulation of NCC, WNK1, and WNK4 in vivo, mRNA levels of NCC, WNK1, and WNK4 were assessed by qPCR in WT and Per1 het mice euthanized at midnight (Fig. 3). We have previously demonstrated that Per1 het mice have a ∼50% reduction of Per1 in the renal cortex (16). Consistent with our in vitro results and our in vivo CK1δ/ϵ inhibitor results, mRNA expression of NCC and WNK1 were both significantly decreased in Per1 het mice when compared with WT (Fig. 3, A and B). Conversely, WNK4 mRNA expression was significantly increased (Fig. 3C).
FIGURE 3.

Reduced Per1 expression in vivo results in decreased NCC and WNK1 expression and increased WNK4 expression in kidney cortex. Dissected cortex was harvested from WT 129/sv and Per1 het mice euthanized at midnight. qPCR was used to evaluate changes in NCC (A), WNK1 (B), or WNK4 (C) mRNA expression in WT versus Per1 het (n = 3). *, p < 0.05; **, p < 0.01. Values are represented as the mean ± S.E. (error bars).
To further assess the regulation of NCC, WNK1, and WNK4 mRNA expression by blockade of Per1 nuclear entry, mDCT15 cells were treated with CK1δ/ϵ inhibitor, and NCC, WNK1, and WNK4 mRNA expression was determined by qPCR. After 48 h of CK1δ/ϵ inhibition, treatment with 10 μm PF670462 resulted in significant reduction of NCC and WNK1 mRNA (Fig. 4, A and B), whereas WNK4 was significantly up-regulated after 24, 48, or 72 h of 10 μm CK1δ/ϵ inhibition (Fig. 4C). The goal of this experiment was to determine the lowest effective dose, as described previously (7). Although the data points for WNK4 at the 100 μm dose were difficult to interpret, it was clear that 10 μm was the lowest effective dose. Thus, the 10 μm dose was used for subsequent experiments.
FIGURE 4.

Pharmacological blockade of Per1 nuclear entry results in decreased NCC and WNK1 expression and increased WNK4 expression in vitro. mDCT15 cells were treated with the casein kinase 1δ/ϵ inhibitor PF670462 with either 0.1, 1, 10, or 100 μm for 24, 48, or 72 h. qPCR was used to evaluate changes in NCC (A), WNK1 (B), or WNK4 (C) expression in PF670462-treated cells versus vehicle (water)-treated cells (n = 3). *, p < 0.05; **, p < 0.01. Values are represented as the mean ± S.E. (error bars).
To confirm that CK1δ/ϵ inhibition decreased Per1 nuclear entry, mDCT15 cells were treated with vehicle or 10 μm PF670462 for 48 h, and nuclear extracts were collected with Western blotting performed to assess nuclear Per1 levels. As we have shown previously in other cell types (7, 16), nuclear Per1 levels were significantly decreased with PF670462 treatment (Fig. 5A; quantified in Fig. 5B). It should be noted that nuclear Per1 was detected at a molecular mass of just less than 50 kDa, which has been consistently demonstrated in Western blots and pull-down experiments in vivo in the liver and the kidney and in vitro in many different cell types (7, 8, 10, 16).
FIGURE 5.
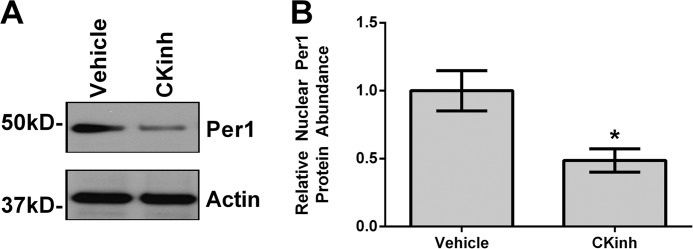
Pharmacological blockade of Per1 nuclear entry results in decreased nuclear Per1 in vitro. A, nuclear extracts were collected from mDCT15 cells treated with 10 μm casein kinase 1δ/ϵ inhibitor PF670462 or water for 48 h. Western blot analysis was performed using anti-Per1 or anti-β-actin antibodies as a loading control. Data are representative of three independent experiments. B, densitometry analysis was used to quantitate the level of Per1 in A (n = 3). *, p < 0.05. Values are represented as the mean ± S.E. (error bars).
Pharmacological Inhibition of Per1 Nuclear Entry Results in Decreased Transcription of NCC and WNK1 and Increased Transcription of WNK4 and KS-WNK1
Measurement of the short lived hnRNA is a measure of transcriptional activity (31, 32). Therefore, to assess if the effect of CK1δ/ϵ inhibition and Per1 knockdown on the inhibition on NCC, WNK1, KS-WNK1, and WNK4 was transcriptional, hnRNA levels were assessed by PCR amplification of intron-exon junctions using cDNA templates from mDCT15 cells treated with vehicle or 10 μm PF670462 for 48 h or non-target and Per1 siRNA for 48 h. KS-WNK1 is a kinase-dead splice variant, unique to the kidney, that inhibits the activity of WNK1 (33). Both Per1 siRNA-mediated knockdown and CK1δ/ϵ inhibition led to significantly decreased hnRNA expression of both NCC (Fig. 6A) and WNK1 (Fig. 6B) but increased expression of KS-WNK1 (Fig. 6C) and WNK4 (Fig. 6D).
FIGURE 6.

Pharmacological blockade of Per1 nuclear entry results in decreased transcription of NCC and WNK1 and increased transcription of KS-WNK1 and WNK4. Primers were designed to amplify regions of hnRNA spanning an intron/exon boundary for NCC (A), WNK1 (B), KS-WNK1 (C), and WNK4 (D). GAPDH was used as a cDNA loading control. The arrows are representative of primer location. Bands were quantitated using ImageJ densitometry. Signal strength was normalized to the relevant vehicle or CKinh-treated GAPDH control (n = 3; *, p < 0.05). Values are represented as the mean ± S.E. (error bars).
Pharmacological Inhibition of Per1 Nuclear Entry Results in Increased Transcription of KS-WNK1 in Vivo
To assess if KS-WNK1 is up-regulated in vivo, hnRNA levels were assessed in WT versus Per1 het mice and in vehicle-treated versus CK1δ/ϵ-treated mice (Fig. 7). Significantly increased hnRNA expression of KS-WNK1 was observed in Per1 het mice compared with WT and in CK1δ/ϵ-treated mice compared with vehicle-treated controls, confirming our in vitro findings.
FIGURE 7.

Reduction of Per1 in vivo results in increased transcription of KS-WNK1. WT, Per1 het, vehicle-treated, and PF670462-treated mice were used as described in the legends to Figs. 1 and 3. Primers were designed to amplify regions of hnRNA spanning an intron/exon boundary of KS-WNK1. GAPDH was used as a cDNA loading control. Arrows, primer location. Bands were quantitated using ImageJ densitometry. Signal strength was normalized to the relevant GAPDH control (n = 3; *, p < 0.05). Values are represented as the mean ± S.E. (error bars).
Pharmacological Inhibition of Per1 Nuclear Entry Results in Decreased Binding of Per1 and CLOCK to the Promoters of Slc12a3 and Wnk1 but Increased Binding to the Wnk4 Promoter
As discussed above, circadian regulation of target genes is mediated through interaction of the core circadian proteins with E-box response elements in target gene promoters. To assess if Per1 and CLOCK interact with the endogenous promoters of Slc12a3, Wnk1, and Wnk4 and if CK1δ/ϵ inhibition could modulate this interaction, ChIP was performed in mDCT15 cells treated with either vehicle or 10 μm PF670462 for 48 h. Three primer sets per gene were designed to amplify short regions containing putative E-box sites within approximately the first 10 kb upstream of the transcription start site (Figs. 8A, 9A, and 10A). Loading was demonstrated by performing PCR using 10% of the input as template (Figs. 8B, 9B, and 10B). IgG was used as a negative control (Figs. 8F, 9F, and 10F). CK1δ/ϵ inhibition led to significantly decreased binding of RNA Pol II at all three sites in the Slc12a3 promoter (Fig. 8C), indicative of decreased transcription, further corroborating our mRNA and hnRNA data. CLOCK and Per1 were detected at all three NCC promoter regions, and these interactions were significantly decreased with CK1δ/ϵ inhibition (Fig. 8, D and E). On the Wnk1 promoter, CK1δ/ϵ inhibition led to significantly decreased RNA Pol II binding at all three promoter regions (Fig. 9C). As with Slc12a3, CLOCK and Per1 were detected on all three promoter regions, and binding was significantly decreased upon CK1δ/ϵ inhibition (Fig. 9, D and E). On the Wnk4 promoter, RNA Pol II has increased binding upon CK1δ/ϵ inhibition in all three promoter regions, indicative of increased transcription, in agreement with the results of the hnRNA assay (Fig. 10C). CK1δ/ϵ inhibition led to increased CLOCK and Per1 interaction with the second and third promoter regions but no apparent interaction with the promoter region amplified by the first primer set (Fig. 10, D and E).
FIGURE 8.

Pharmacological blockade of Per1 nuclear entry leads to decreased occupancy of CLOCK and Per1 on the Slc12a3 promoter in mDCT15 cells. Chromatin immunoprecipitation experiments were performed using mDCT15 cells treated with either vehicle (water) or 10 μm PF670462 for 48 h. End point PCR was performed using primer sets flanking three putative E-boxes in the Slc12a3 promoter (A, not drawn to scale). B, input control. Chromatin immunoprecipitations were performed using anti-Pol II (Santa Cruz Biotechnology) (C), anti-CLOCK (Pierce) (D), anti-Per1 (Pierce) (E), or rabbit IgG (Bethyl) (negative control) (F) antibodies. Bands were quantitated using densitometry, which was performed using ImageJ. Signal strength was normalized to the relevant vehicle- or CKinh-treated input control (n = 3; *, p < 0.05; **, p < 0.01). Values are represented as the mean ± S.E. (error bars).
FIGURE 9.

Pharmacological blockade of Per1 nuclear entry leads to decreased occupancy of CLOCK and Per1 on the Wnk1 promoter in mDCT15 cells. Chromatin immunoprecipitation experiments were performed using mDCT15 cells treated with either vehicle (water) or 10 μm PF670462 for 48 h. End point PCR was performed using primer sets flanking three putative E-boxes in the WNK1 promoter (A, not drawn to scale). B, input control. Chromatin immunoprecipitations were performed using anti-Pol II (Santa Cruz Biotechnology) (C), anti-CLOCK (Pierce) (D), anti-Per1 (Pierce) (E), or rabbit IgG (Bethyl) (negative control) (F) antibodies. Bands were quantitated using densitometry, which was performed using ImageJ. Signal strength was normalized to the relevant vehicle or CKinh treated input control (n = 3; *, p < 0.05; **, p < 0.01). Values are represented as the mean ± S.E. (error bars).
FIGURE 10.

Pharmacological blockade of Per1 nuclear entry leads to increased occupancy of CLOCK and Per1 on the Wnk4 promoter in mDCT15 cells. Chromatin immunoprecipitation experiments were performed using mDCT15 cells treated with either vehicle (water) or 10 μm PF670462 for 48 h. End point PCR was performed using primer sets flanking three putative E-boxes in the WNK4 promoter (A, not drawn to scale). B, input control. Chromatin immunoprecipitations were performed using anti-Pol II (Santa Cruz Biotechnology) (C), anti-CLOCK (Pierce) (D), anti-Per1 (Pierce) (E), or rabbit IgG (Bethyl) (negative control) (F) antibodies. Bands were quantitated using densitometry, which was performed using ImageJ. Signal strength was normalized to the relevant vehicle- or CKinh-treated input control (n = 3; *, p < 0.05; **, p < 0.01). Values are represented as the mean ± S.E. (error bars).
Pharmacological Blockade of Per1 Nuclear Entry in Vitro and in Vivo Results in Increased mRNA Levels of Cry1/2 and Per2 in Kidney Cortex and mDCT15 Cells
Recent evidence from our laboratory and others has demonstrated that Per1 may act on target gene expression through modulation of the circadian repressors Cry1/2 and Per2 (15, 16, 34). We have demonstrated that inhibition of Per1 results in up-regulation of Cry1/2 mRNA and protein levels and that Per1 and Cry2 mediate opposing actions on target gene expression in the kidney, liver, and adrenal glands in vitro and in vivo (16, 17). However, this has not been investigated in the context of CK1δ/ϵ inhibition in vitro in mDCT15 cells or in in vivo in kidney cortex. Therefore, mRNA levels were assessed by qPCR for Clock, Bmal1, Cry1, Cry2, and Per2, in either mDCT15 cells (Fig. 11) or WT animals treated with CKinh (Fig. 12). Pharmacological blockade of Per1 nuclear entry in vitro did not affect Clock or Bmal1 mRNA (Fig. 11, A and B). Consistent with our previous observations, however, CKinh treatment resulted in significantly increased mRNA levels of Cry1, Cry2, and Per2 (Fig. 11, C–E). These results were corroborated in vivo with CKinh treatment (Fig. 12). These confirm our previously reported results that we have shown for Cry2 both in vitro and in vivo in kidney, liver, and the adrenal gland (17) and that others have shown previously for Per2 and Cry1 (34).
FIGURE 11.

Pharmacological blockade of Per1 nuclear entry results in increased Cry1, Cry2, and Per2 expression in vitro. mDCT15 cells were treated with the casein kinase 1δ/ϵ inhibitor PF670462 (10 μm for 48 h). qPCR was used to evaluate changes in Clock (A), Bmal1 (B), Cry1 (C), Cry2 (D), or Per2 (E) expression in PF670462-treated cells versus vehicle (water)-treated cells (n = 3; *, p < 0.05; **, p < 0.01). Values are represented as the mean ± S.E. (error bars).
FIGURE 12.

Pharmacological blockade of Per1 nuclear entry results in increased Cry1, Cry2, and Per2 expression in vivo. Weight-matched male WT 129/sv mice were given either vehicle (20% hydroxypropyl β-cyclodextrin) or 30 mg/kg PF670462 subcutaneously as described in the legend to Fig. 1. Kidneys were harvested, cortex was dissected, and qPCR was used to evaluate changes in Clock (A), Bmal1 (B), Cry1 (C), Cry2 (D), or Per2 (E) expression (n = 4; *, p < 0.05 compared with WT). Values are represented as the mean ± S.E. (error bars).
Pharmacological Inhibition of Per1 Nuclear Entry Results in Decreased Membrane and Intracellular Protein Expression of NCC
To test whether the effects of CK1δ/ϵ inhibition on NCC expression extended to the level of protein, plasma membrane and intracellular fractions were isolated from mDCT15 cells treated with vehicle or 10 μm PF670462 for 48 h. Western blot analysis was performed to determine changes in NCC protein levels from both intracellular and membrane fractions. CKinh treatment led to a significant decrease of NCC at the intracellular level and a ∼40% decrease at the membrane level (Fig. 13). Decreases at both the intracellular and membrane level are consistent with our hypothesis that regulation of NCC by Per1 occurs at the transcriptional level.
FIGURE 13.

Pharmacological blockade of Per1 nuclear entry results in decreased membrane and intracellular protein expression of NCC in mDCT15 cells. Membrane and intracellular extracts were collected from mDCT15 cells treated with 10 μm casein kinase 1δ/ϵ inhibitor PF670462 or water for 48 h. Western blot analysis was performed using anti-NCC or anti-β-actin antibodies as a loading control of cell lysate. Data are representative of three independent experiments. Densitometry analysis was used to quantitate the level of NCC (n = 3; *, p < 0.05). Values are represented as the mean ± S.E. (error bars).
Pharmacological Inhibition of Per1 Nuclear Entry Results in Decreased NCC Activity
Having demonstrated that pharmacological inhibition of Per1 nuclear entry affects expression of NCC, WNK1, and WNK4, NCC activity was measured with and without CK1δ/ϵ inhibition to assess for an effect on NCC function. The mDCT15 cells were treated with vehicle or 1, 10, or 100 μm PF670462 for 24 or 48 h prior to measurement of NCC activity by thiazide-sensitive 22Na+ uptake. At 24 h, no significant change was seen in NCC activity in the CK1δ/ϵ inhibition groups (Fig. 14A). However, at 48 h, NCC activity declined by 32 ± 7% in the 10 μm group and by 39 ± 6% in the 100 μm group (Fig. 14B, n = 4, p < 0.05 compared with vehicle for both). This significant decrease parallels the decrease in NCC membrane protein levels (Fig. 13). Interestingly, a similar treatment in mDCT15 cells with stable knockdown of WNK4 expression (generated as described previously (22)) showed a sharply attenuated decline in NCC activity with CK1δ/ϵ inhibition (non-significant at 10 μm, 8 ± 2% for 100 μm, n = 4, p < 0.05, Fig. 14C).
FIGURE 14.

Pharmacological blockade of Per1 nuclear entry results in decreased NCC activity in mDCT15 cells. NCC activity was measured in mDCT15 cells (A and B) or mDCT15 cells with WNK4 expression stably reduced by 70 ± 3% (C). Cells were treated with either vehicle (water) or 1, 10, or 100 μm PF670462 for 24 h (A) or 48 h (B and C) prior to measurement of thiazide-sensitive 22Na+ uptake. Uptakes were normalized to nmol/mg. Thiazide-sensitive uptake was calculated as the difference between the uptakes with and without thiazide. n = 4; *, p < 0.05. Error bars, S.E.
DISCUSSION
The results of the present study demonstrate a potential role for Per1 in the transcriptional regulation of NCC and the WNK cascade. We demonstrated that pharmacological blockade of Per1 nuclear entry or decreased Per1 in vivo and in vitro results in decreased mRNA expression of NCC and WNK1 but increased expression of WNK4 and KS-WNK1. Furthermore, ChIP and hnRNA assays performed in the mouse distal convoluted tubule cell line, mDCT15, consistently demonstrated that Per1 acts on the NCC/WNK pathway via a transcriptional mechanism and that this regulation extends to the level of protein. Importantly, we show for the first time that NCC activity was decreased upon pharmacological inhibition of Per1 nuclear entry, an effect that appeared to be partially dependent on WNK4. Based on these data, we propose a model for Per1 action on the NCC and WNK pathway in which Per1 promotes increased sodium reabsorption in the distal convoluted tubule, through transcriptional regulation of NCC, WNK1, KS-WNK1, and WNK4 (Fig. 15). This apparent coordinate regulation of pathway genes by Per1 suggests a role for Per1 in driving renal epithelial sodium reabsorption in DCT cells; this coordinate regulation is entirely consistent with our previously proposed model for Per1 action on ENaC in CCD cells (8).
FIGURE 15.

A model for Per1 action on NCC and the WNK cascade in mDCT15 cells. Per1, presumably through interactions with other circadian clock proteins, appears to coordinately regulate the NCC/WNK pathway via positive regulation of NCC and WNK1 and negative regulation of WNK4 and KS-WNK1.
We also demonstrated that CK1δ/ϵ inhibition led to increased transcription of KS-WNK1, as measured by increased amplification of the intron-exon sequence of the KS-WNK1-specific Exon4A, which is the first exon in KS-WNK1 and replaces the first four exons of WNK1 (33). However, further evidence is required to understand the role of Per1 in the regulation of KS-WNK1. The WNK kinase pathway includes many other factors in addition to WNK1, WNK4, and KS-WNK1. These include WNK3, which promotes sodium reabsorption by activating NCC and inhibiting WNK4. STE20/SPS-1-related proline/alanine-rich kinase and oxidative stress-responsive kinase 1 were determined to be downstream targets of the WNK cascade and are presumably responsible for the phosphorylation of NCC itself (reviewed in Refs. 21 and 35). It was recently demonstrated that the levels of phosphorylated oxidative stress-responsive kinase 1, Ste20/SPS-1-related proline/alanine-rich kinase, and NCC exhibited circadian fluctuations in mice (36), a result that is consistent with our observations demonstrating regulation of this pathway by circadian clock proteins. Interestingly, a microarray study showed that NCC, WNK1, and WNK4 did not appear to oscillate with a circadian pattern (37). More work is needed to determine if the Per1-mediated regulation of NCC and the WNK kinases is independent of time. Consistent with our previous observations in the liver and kidney (16), knockdown or inhibition of Per1 in the present study resulted in increased mRNA expression of Cry2. As well, Cry1 and Per2 mRNA expression was increased following knockdown or inhibition of Per1, but CLOCK and Bmal1 levels did not appear to be affected. Thus, the mechanism of Per1 action probably involves the regulation of other circadian repressors.
Mounting evidence has demonstrated the critical importance of the circadian clock for the regulation of BP control and renal sodium handling (reviewed in Refs. 1, 3, and 38). CLOCK KO mice are relatively hypotensive and display dysregulated urinary sodium excretion and mild diabetes insipidus (37, 39). Cry1/2 KO mice display salt-sensitive hypertension partially due to increased aldosterone levels (40). This result was linked to increased expression of 3β-dehydrogenase isomerase, a rate-limiting enzyme in the aldosterone synthesis pathway. Per2 KO mice exhibit decreased 24-h diastolic BP, increased heart rate, and a reduction in diurnal dipping (41). Bmal1 KO mice have reduced BP in the active phase, leading to a lack of circadian BP variation (42). We have demonstrated that Per1 KO animals exhibit significantly lower BP than WT controls (8). It is unknown if the BP phenotype of these animals is solely due to modulation of renal sodium retention; however, recently, we have shown that mice with reduced levels of Per1 have a defect in renal sodium retention (17). It is likely that a combination of factors contribute to the BP phenotype of these animals, including the heart (43), sympathetic activity (44, 45), the vasculature (46), and/or nitric oxide (47). However, consistent with our findings that Per1 is important for the regulation of sodium reabsorption in the kidney, we have recently demonstrated that reduced expression of Per1 in mice is associated with decreased renal sodium reabsorption (17). Available data in humans are consistent with a role for Per1 in BP regulation as well; a recent report demonstrated that Per1 was overexpressed in the renal medulla of human hypertensive patients compared with normotensive control patients (48).
Along with our previous data demonstrating the regulation of ENaC and sodium transport genes in collecting duct cells and the kidney (8–10), Per1 appears to regulate gene expression in a cell model of the distal convoluted tubule. It will be interesting to see if Per1 and the circadian clock regulate gene expression in other parts of the kidney. It has been demonstrated that the sodium hydrogen exchanger NHE3 exhibits circadian oscillations in the mouse renal medulla and is regulated by Bmal1/CLOCK (49, 50) (51, 52). Taken together, the data presented here demonstrate a novel role for Per1 in the regulation of sodium reabsorption in distal convoluted tubule cells via transcriptional regulation of NCC and several kinase genes from the WNK cascade.
Acknowledgment
We thank Dr. Brian D. Cain for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK085193 and DK098460 (to M.L.G.), NIHK08 DK081728 (to B. K.), and NIHR01 DK085097 (to R.S.H.). This work was also supported by the American Society of Nephrology Foundation for Kidney Research (to M. L. G.), American Heart Association Predoctoral Fellowship 13PRE16910096 (to J. R.), and the Research Service, Atlanta Veterans Affairs Medical Center (to R. S. H.).
- BP
- blood pressure
- CK1δ/ϵ
- casein kinase 1 δ/ϵ
- CKinh
- casein kinase 1δ/ϵ inhibitor
- ENaC
- epithelial sodium channel
- DCT
- distal convoluted tubule
- NCC
- NaCl co-transporter
- WNK
- with-no-lysine kinase
- KS-WNK1
- kidney-specific WNK1
- hnRNA
- heterogeneous nuclear RNA
- het
- heterozygote
- qPCR
- quantitative real-time PCR
- Pol
- polymerase.
REFERENCES
- 1. Richards J., Gumz M. L. (2013) Mechanism of the circadian clock in physiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R1053–R1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richards J., Gumz M. L. (2012) Advances in understanding the peripheral circadian clocks. FASEB J. 26, 3602–3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stow L. R., Gumz M. L. (2011) The circadian clock in the kidney. J. Am. Soc. Nephrol. 22, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dibner C., Schibler U., Albrecht U. (2010) The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 72, 517–549 [DOI] [PubMed] [Google Scholar]
- 5. Albrecht U., Eichele G. (2003) The mammalian circadian clock. Curr. Opin. Genet. Dev. 13, 271–277 [DOI] [PubMed] [Google Scholar]
- 6. Lee H. M., Chen R., Kim H., Etchegaray J. P., Weaver D. R., Lee C. (2011) The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc. Natl. Acad. Sci. U.S.A. 108, 16451–16456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richards J., Greenlee M. M., Jeffers L. A., Cheng K. Y., Guo L., Eaton D. C., Gumz M. L. (2012) Inhibition of αENaC expression and ENaC activity following blockade of the circadian clock-regulatory kinases CK1δ/ϵ. Am. J. Physiol. 303, F918–F927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stow L. R., Richards J., Cheng K. Y., Lynch I. J., Jeffers L. A., Greenlee M. M., Cain B. D., Wingo C. S., Gumz M. L. (2012) The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension 59, 1151–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gumz M. L., Stow L. R., Lynch I. J., Greenlee M. M., Rudin A., Cain B. D., Weaver D. R., Wingo C. S. (2009) The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J. Clin. Invest. 119, 2423–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gumz M. L., Cheng K. Y., Lynch I. J., Stow L. R., Greenlee M. M., Cain B. D., Wingo C. S. (2010) Regulation of αENaC expression by the circadian clock protein Period 1 in mpkCCD(c14) cells. Biochim. Biophys. Acta 1799, 622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lubarski I., Pihakaski-Maunsbach K., Karlish S. J., Maunsbach A. B., Garty H. (2005) Interaction with the Na,K-ATPase and tissue distribution of FXYD5 (related to ion channel). J. Biol. Chem. 280, 37717–37724 [DOI] [PubMed] [Google Scholar]
- 12. Bugaj V., Pochynyuk O., Mironova E., Vandewalle A., Medina J. L., Stockand J. D. (2008) Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am. J. Physiol. 295, F1063–F1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bose S., Boockfor F. R. (2010) Episodes of prolactin gene expression in GH3 cells are dependent on selective promoter binding of multiple circadian elements. Endocrinology 151, 2287–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen R., Schirmer A., Lee Y., Lee H., Kumar V., Yoo S. H., Takahashi J. S., Lee C. (2009) Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol. Cell 36, 417–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oster H., Baeriswyl S., Van Der Horst G. T., Albrecht U. (2003) Loss of circadian rhythmicity in aging mPer1−/−mCry2−/− mutant mice. Genes Dev. 17, 1366–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards J., All S., Skopis G., Cheng K. Y., Compton B., Srialluri N., Stow L., Jeffers L. A., Gumz M. L. (2013) Opposing actions of Per1 and Cry2 in the regulation of Per1 target gene expression in the liver and kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R735–R747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richards J., Cheng K. Y., All S., Skopis G., Jeffers L., Lynch I. J., Wingo C. S., Gumz M. L. (2013) A role for the circadian clock protein Per1 in the regulation of aldosterone levels and renal Na+ retention. Am. J. Physiol. 305, F1697–F1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang C. L., Zhu X., Wang Z., Subramanya A. R., Ellison D. H. (2005) Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J. Clin. Invest. 115, 1379–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moriguchi T., Urushiyama S., Hisamoto N., Iemura S., Uchida S., Natsume T., Matsumoto K., Shibuya H. (2005) WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 280, 42685–42693 [DOI] [PubMed] [Google Scholar]
- 20. Zagórska A., Pozo-Guisado E., Boudeau J., Vitari A. C., Rafiqi F. H., Thastrup J., Deak M., Campbell D. G., Morrice N. A., Prescott A. R., Alessi D. R. (2007) Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J. Cell Biol. 176, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gamba G. (2012) Regulation of the renal Na+-Cl− cotransporter by phosphorylation and ubiquitylation. Am. J. Physiol. 303, F1573–F1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ko B., Mistry A. C., Hanson L., Mallick R., Cooke L. L., Hack B. K., Cunningham P., Hoover R. S. (2012) A new model of the distal convoluted tubule. Am. J. Physiol. 303, F700–F710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chávez-Canales M., Arroyo J. P., Ko B., Vázquez N., Bautista R., Castañeda-Bueno M., Bobadilla N. A., Hoover R. S., Gamba G. (2013) Insulin increases the functional activity of the renal NaCl cotransporter. J. Hypertens. 31, 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ko B., Mistry A. C., Hanson L., Mallick R., Wynne B. M., Thai T. L., Bailey J. L., Klein J. D., Hoover R. S. (2013) Aldosterone acutely stimulates NCC activity via a SPAK-mediated pathway. Am. J. Physiol. 305, F645–F652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bae K., Jin X., Maywood E. S., Hastings M. H., Reppert S. M., Weaver D. R. (2001) Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron 30, 525–536 [DOI] [PubMed] [Google Scholar]
- 26. Perreau-Lenz S., Vengeliene V., Noori H. R., Merlo-Pich E. V., Corsi M. A., Corti C., Spanagel R. (2012) Inhibition of the casein-kinase-1-ϵ/δ prevents relapse-like alcohol drinking. Neuropsychopharmacology 37, 2121–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔ C(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 28. Richards J., Jeffers L. A., All S. C., Cheng K. Y., Gumz M. L. (2013) Role of Per1 and the mineralocorticoid receptor in the coordinate regulation of αENaC in renal cortical collecting duct cells. Front. Physiol. 4, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Messeguer X., Escudero R., Farré D., Núñez O., Martínez J., Albà M. M. (2002) PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 [DOI] [PubMed] [Google Scholar]
- 30. Farré D., Roset R., Huerta M., Adsuara J. E., Roselló L., Albà M. M., Messeguer X. (2003) Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lipson K. E., Baserga R. (1989) Transcriptional activity of the human thymidine kinase gene determined by a method using the polymerase chain reaction and an intron-specific probe. Proc. Natl. Acad. Sci. U.S.A. 86, 9774–9777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen H., Kilberg M. S. (2006) Alignment of the transcription start site coincides with increased transcriptional activity from the human asparagine synthetase gene following amino acid deprivation of HepG2 cells. J. Nutr. 136, 2463–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Subramanya A. R., Yang C. L., Zhu X., Ellison D. H. (2006) Dominant-negative regulation of WNK1 by its kidney-specific kinase-defective isoform. Am. J. Physiol. 290, F619–F624 [DOI] [PubMed] [Google Scholar]
- 34. Padmanabhan K., Robles M. S., Westerling T., Weitz C. J. (2012) Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science 337, 599–602 [DOI] [PubMed] [Google Scholar]
- 35. Ko B., Hoover R. S. (2009) Molecular physiology of the thiazide-sensitive sodium-chloride cotransporter. Curr. Opin. Nephrol. Hypertens. 18, 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Susa K., Sohara E., Isobe K., Chiga M., Rai T., Sasaki S., Uchida S. (2012) WNK-OSR1/SPAK-NCC signal cascade has circadian rhythm dependent on aldosterone. Biochem. Biophys. Res. Commun. 427, 743–747 [DOI] [PubMed] [Google Scholar]
- 37. Zuber A. M., Centeno G., Pradervand S., Nikolaeva S., Maquelin L., Cardinaux L., Bonny O., Firsov D. (2009) Molecular clock is involved in predictive circadian adjustment of renal function. Proc. Natl. Acad. Sci. U.S.A. 106, 16523–16528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonny O., Firsov D. (2013) Circadian regulation of renal function and potential role in hypertension. Curr. Opin. Nephrol. Hypertens. 22, 439–444 [DOI] [PubMed] [Google Scholar]
- 39. Nikolaeva S., Pradervand S., Centeno G., Zavadova V., Tokonami N., Maillard M., Bonny O., Firsov D. (2012) The circadian clock modulates renal sodium handling. J. Am. Soc. Nephrol. 23, 1019–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doi M., Takahashi Y., Komatsu R., Yamazaki F., Yamada H., Haraguchi S., Emoto N., Okuno Y., Tsujimoto G., Kanematsu A., Ogawa O., Todo T., Tsutsui K., van der Horst G. T., Okamura H. (2010) Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat. Med. 16, 67–74 [DOI] [PubMed] [Google Scholar]
- 41. Vukolic A., Antic V., Van Vliet B. N., Yang Z., Albrecht U., Montani J. P. (2010) Role of mutation of the circadian clock gene Per2 in cardiovascular circadian rhythms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R627–R634 [DOI] [PubMed] [Google Scholar]
- 42. Curtis A. M., Cheng Y., Kapoor S., Reilly D., Price T. S., Fitzgerald G. A. (2007) Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc. Natl. Acad. Sci. U.S.A. 104, 3450–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Durgan D. J., Young M. E. (2010) The cardiomyocyte circadian clock: emerging roles in health and disease. Circ. Res. 106, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sheward W. J., Naylor E., Knowles-Barley S., Armstrong J. D., Brooker G. A., Seckl J. R., Turek F. W., Holmes M. C., Zee P. C., Harmar A. J. (2010) Circadian control of mouse heart rate and blood pressure by the suprachiasmatic nuclei: behavioral effects are more significant than direct outputs. PLoS One 5, e9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki J., Ogawa M., Tamura N., Maejima Y., Takayama K., Maemura K., Honda K., Hirata Y., Nagai R., Isobe M. (2010) A critical role of sympathetic nerve regulation for the treatment of impaired daily rhythm in hypertensive Dahl rats. Hypertens. Res. 33, 1060–1065 [DOI] [PubMed] [Google Scholar]
- 46. Anea C. B., Zhang M., Stepp D. W., Simkins G. B., Reed G., Fulton D. J., Rudic R. D. (2009) Vascular disease in mice with a dysfunctional circadian clock. Circulation 119, 1510–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kunieda T., Minamino T., Miura K., Katsuno T., Tateno K., Miyauchi H., Kaneko S., Bradfield C. A., FitzGerald G. A., Komuro I. (2008) Reduced nitric oxide causes age-associated impairment of circadian rhythmicity. Circ. Res. 102, 607–614 [DOI] [PubMed] [Google Scholar]
- 48. Marques F. Z., Campain A. E., Tomaszewski M., Zukowska-Szczechowska E., Yang Y. H., Charchar F. J., Morris B. J. (2011) Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension 58, 1093–1098 [DOI] [PubMed] [Google Scholar]
- 49. Nishinaga H., Komatsu R., Doi M., Fustin J. M., Yamada H., Okura R., Yamaguchi Y., Matsuo M., Emoto N., Okamura H. (2009) Circadian expression of the Na+/H+ exchanger NHE3 in the mouse renal medulla. Biomed. Res. 30, 87–93 [DOI] [PubMed] [Google Scholar]
- 50. Saifur Rohman M., Emoto N., Nonaka H., Okura R., Nishimura M., Yagita K., van der Horst G. T., Matsuo M., Okamura H., Yokoyama M. (2005) Circadian clock genes directly regulate expression of the Na+/H+ exchanger NHE3 in the kidney. Kidney Int. 67, 1410–1419 [DOI] [PubMed] [Google Scholar]
- 51. Bhutta H. Y., Deelman T. E., Ashley S. W., Rhoads D. B., Tavakkoli A. (2013) Disrupted circadian rhythmicity of the intestinal glucose transporter SGLT1 in Zucker diabetic fatty rats. Dig. Dis. Sci. 58, 1537–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Balakrishnan A., Stearns A. T., Ashley S. W., Rhoads D. B., Tavakkolizadeh A. (2012) PER1 modulates SGLT1 transcription in vitro independent of E-box status. Dig. Dis. Sci. 57, 1525–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]