

Abstract
Background
Gene microarray technology provides the ability to study the regulation of thousands of genes simultaneously, but its potential is limited without an estimate of the statistical significance of the observed changes in gene expression. Due to the large number of genes being tested and the comparatively small number of array replicates (e.g., N = 3), standard statistical methods such as the Student's t-test fail to produce reliable results. Two other statistical approaches commonly used to improve significance estimates are a penalized t-test and a Z-test using intensity-dependent variance estimates.
Results
The performance of these approaches is compared using a dataset of 23 replicates, and a new implementation of the Z-test is introduced that pools together variance estimates of genes with similar minimum intensity. Significance estimates based on 3 replicate arrays are calculated using each statistical technique, and their accuracy is evaluated by comparing them to a reliable estimate based on the remaining 20 replicates. The reproducibility of each test statistic is evaluated by applying it to multiple, independent sets of 3 replicate arrays. Two implementations of a Z-test using intensity-dependent variance produce more reproducible results than two implementations of a penalized t-test. Furthermore, the minimum intensity-based Z-statistic demonstrates higher accuracy and higher or equal precision than all other statistical techniques tested.
Conclusion
An intensity-based variance estimation technique provides one simple, effective approach that can improve p-value estimates for differentially regulated genes derived from replicated microarray datasets. Implementations of the Z-test algorithms are available at http://vessels.bwh.harvard.edu/software/papers/bmcg2004.
Background
Biologists can now use microarray technology to determine the expression levels of tens of thousands of genes simultaneously, in less time than it previously took to measure the expression level of a single gene. Currently, cDNA and oligo microarrays can measure a sizeable fraction of all mRNA species in cell or tissue samples. The richness of the resulting data is opening up a new era of systems biology that promises to reveal the complex inner workings of cellular machinery [1]. However, there remains the challenge of processing the microarray data from array images into a format that best facilitates the discovery of new biological insights. In fact, applying improved computational tools to previously published microarray data has led to the discovery of new biology (e.g. [2]). We strongly believe, as do others, that the quality of the data processing steps is critical to the overall success of a microarray experiment [3].
A typical data processing pipeline consists of several steps. (See [4] for a review, and see [5,6] for a review of microarray processing software.) First, image analysis software locates the arrayed spots in the scanned image, quantifies the foreground and background brightness of each spot, and notes any irregularities in spot morphology. The background intensity value is then subtracted from the foreground intensity value. The background-subtracted intensity data from each array must then be normalized, or rescaled, to remove artifactual differences in signal brightness due, for example, to different labeling efficiencies that produced arrays of different overall intensity. Normalization techniques are often based on the assumption that a large number of spots will have similar expression levels between conditions. Curve-fitting techniques, such as a locally weighted regression, are used to equalize expression values between arrays, or between array channels for two-color arrays [7,8]. After this normalization, the intensity values can be used by a variety of algorithms for detecting differences in expression between the measured biological conditions. This processing is applied whether two samples are compared directly or a "reference sample" experimental design is used. In a reference sample design, the same reference RNA sample is hybridized to one channel of all arrays, and the other channel is hybridized with each individual experimental sample. This design is often used when multiple biological conditions are being investigated and it becomes impractical to perform every pairwise combination of conditions directly [4,9].
Accurately detecting differentially regulated genes
Given a list of normalized intensity values across various biological conditions, the next step is to determine which genes are differentially regulated among the conditions being studied. In the early days of microarray experimentation, an emphasis was placed on analyzing the data using exploratory data mining techniques, such as hierarchical clustering [10] and self-organizing maps [11]. Clustering algorithms measure the similarity between observed gene regulation patterns across the various conditions, and assemble clusters such that similarly regulated genes are grouped together. The resulting clusters produce an effective overview of the data, showing which of the many possible patterns of regulation are actually present in the data. Since these patterns are somewhat robust, a few erroneous spots are unlikely to change them dramatically. For a researcher who is simply interested in the overall pattern of the data, performing replicate arrays to reduce the number of errors is not particularly efficient. Many researchers choose instead to explore a greater number of experimental conditions.
Increasingly, microarrays are being used in a different context; researchers want to know with high confidence which specific genes are regulated across a small number of experimental conditions (e.g., treatment vs. control, or mutant vs. wildtype). To answer this question, it becomes extremely important to use an accurate method to rank individual genes by their probability of truly being regulated, especially since this information may be used to plan more labor-intensive experiments around biological questions raised by a small number of such putatively regulated genes. In the absence of replicate arrays, the reliability of the data can be estimated (e.g. [12,13]), but such "single slide" methods require a model of the expected noise characteristics of the system, a property that can potentially change between datasets. Performing replicate arrays can significantly improve predictions of differentially regulated genes, thereby decreasing the false positive (false detection) rate and false negative rate [9,14,15]. Using replicate arrays allows the calculation of more accurate significance estimates (p-values) that will aid in the interpretation of a list of "top regulated genes," which are commonly ranked by ratio alone.
Here we address the problem of accurately detecting genes that are significantly differentially regulated between a pair of biological conditions, given microarray datasets with a small number of replicates (e.g. N = 3 arrays). If the number of replicates were very large (e.g., hundreds), the task would be relatively easy; since the ratio of expression levels between the two conditions would be well estimated by the average ratio or median ratio, the genes could simply be ranked by one of these estimates. In practice, however, the number of replicate arrays is rarely greater than 3, and estimates of average expression ratios are not always sufficiently accurate to predict which genes are truly regulated. The variation of a measured expression ratio is critical in determining whether the observed ratio is due to random measurement fluctuations or to a true difference between the quantities being measured. Genes with larger measured expression ratios between conditions are more likely to be truly regulated, while genes whose ratios have a high measured variance are less likely to be truly regulated. This idea can be expressed mathematically as a test statistic where the numerator contains an estimate of the size of the effect, i.e. the ratio of gene expression intensities between conditions, and the denominator includes an estimate of the variance, i.e. the standard deviation of the ratio. A variety of such statistical tests have been applied to microarray data (reviewed in [4,16]); the challenge is to choose the numerator and denominator of the test statistic such that it makes the best use of all available data in order to get the most accurate determination of which genes are most likely to be regulated.
Comparing statistical tests used to find differentially regulated genes
The familiar Student's t-test (hereafter, "standard t-test") is the most straightforward method of calculating whether there is a significant difference in expression levels between conditions for each gene. Suppose that mRNAs from two biological conditions, "X" and "Y", are hybridized to a small number of replicate arrays (N two-color arrays or 2N one-color arrays). Mavg, the average logged ratio of expression levels between conditions X and Y, and its sample standard deviation, σM, are given by the standard formulas (see Methods). A standard t-statistic is calculated as 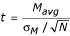 . From this formula, it is clear that a large t-statistic (and the corresponding highly significant p-value) can occur because of either a large Mavg (high ratio) or a small σM (low noise). Although the standard t-statistic (or derivates thereof based on permutation [17] or Bayesian analysis [18]) can produce acceptable results for larger numbers of replicates (e.g., N = 8), the results are less than satisfactory when applied to a small number of microarray replicates (e.g., N = 3, Fig. 1). Fig. 1a shows data from an experiment that was repeated 6 times on two-color arrays. The six arrays were split into two random groups of three arrays, and the t-statistic described above was calculated for each gene in each group of three. The t-statistics from the two groups are graphed against each other in Fig. 1a. Although the two groups contain replicate arrays from the same experimental conditions, the t-statistic is clearly not reproducible between the groups. Fig. 1b and 1c demonstrate that Mavg, the numerator of the t-statistic, is more reproducible between the two groups, while 1/σM, representing the denominator of the t-statistic, is not reproducible. This example highlights the major shortcoming of the t-statistic: due to random chance, the replicate ratios can occasionally be extremely similar, producing an artificially low σM and high t values. False positives stemming from this effect prevent the standard t-statistic from serving as a reliable or useful test of which genes are truly regulated.
. From this formula, it is clear that a large t-statistic (and the corresponding highly significant p-value) can occur because of either a large Mavg (high ratio) or a small σM (low noise). Although the standard t-statistic (or derivates thereof based on permutation [17] or Bayesian analysis [18]) can produce acceptable results for larger numbers of replicates (e.g., N = 8), the results are less than satisfactory when applied to a small number of microarray replicates (e.g., N = 3, Fig. 1). Fig. 1a shows data from an experiment that was repeated 6 times on two-color arrays. The six arrays were split into two random groups of three arrays, and the t-statistic described above was calculated for each gene in each group of three. The t-statistics from the two groups are graphed against each other in Fig. 1a. Although the two groups contain replicate arrays from the same experimental conditions, the t-statistic is clearly not reproducible between the groups. Fig. 1b and 1c demonstrate that Mavg, the numerator of the t-statistic, is more reproducible between the two groups, while 1/σM, representing the denominator of the t-statistic, is not reproducible. This example highlights the major shortcoming of the t-statistic: due to random chance, the replicate ratios can occasionally be extremely similar, producing an artificially low σM and high t values. False positives stemming from this effect prevent the standard t-statistic from serving as a reliable or useful test of which genes are truly regulated.
Figure 1.

Evaluating the reproducibility of t-statistics between spots using a standard t-test. Two subsets of Dataset 4 each contain three replicate arrays derived from identical biological experiments. (a) Comparison of t-statistics for each subset. Values greater than ± 500 are not shown. (b) Comparison of average logged ratios Mavg, which is the numerator of the t-statistic. (c) Comparison of the inverse of the standard deviation σM, which is in the denominator of the t-statistic. Values greater than 150 are not shown.
To overcome this limitation, various modifications to the t-statistic have been proposed. First, a "penalized" t-statistic (also called a "moderated" or "regulated" t-statistic) can be used, where a constant value is added to the denominator. Tusher et al. use a penalized t-statistic of the form 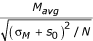 [19]. The addition of the constant s0 prevents the denominator from becoming small for low σM, reducing the false positive rate of genes with unusually low σM. Choosing too large an s0, however, effectively makes the denominator a constant, removing useful information about the variability of genes. Estimating the optimal s0 for a particular dataset can be based on minimizing the coefficient of variation of the absolute t-statistic values ("SAM") [19], minimizing false positive and false negative estimates obtained through permutation ("SAMroc") [16], or simply choosing s0 as the 90th percentile of the σM values [20]. These studies have demonstrated that when ranking genes from a microarray dataset, a penalized t-statistic can perform better than a standard t-statistic in terms of decreasing the false positive and false negative rate [4,16,18-21], but it also has the potential disadvantage of showing bias against genes of high intensity [16].
[19]. The addition of the constant s0 prevents the denominator from becoming small for low σM, reducing the false positive rate of genes with unusually low σM. Choosing too large an s0, however, effectively makes the denominator a constant, removing useful information about the variability of genes. Estimating the optimal s0 for a particular dataset can be based on minimizing the coefficient of variation of the absolute t-statistic values ("SAM") [19], minimizing false positive and false negative estimates obtained through permutation ("SAMroc") [16], or simply choosing s0 as the 90th percentile of the σM values [20]. These studies have demonstrated that when ranking genes from a microarray dataset, a penalized t-statistic can perform better than a standard t-statistic in terms of decreasing the false positive and false negative rate [4,16,18-21], but it also has the potential disadvantage of showing bias against genes of high intensity [16].
An alternative to using a penalized t-statistic is obtaining a more precise estimate of the standard deviation σM. Such an estimate should be less susceptible to a chance concordance of measurements of M that occasionally produces an extremely low σM and a high t-statistic. For this purpose, knowledge of the relationships between the data points can be used to improve the estimate. Namely, the variance values, or σM2, for one spot can be pooled, or smoothed, with the σM2 values of spots that are likely to have similar variances. The variance of microarray data has often been observed to be a function of the spot intensity [12,15,21-30], raising the possibility that the variances of individual spots can be pooled with those of spots of similar intensity to produce a more precise estimate of the standard deviation. Several studies have taken into account this intensity-dependent heteroscedasticity. For example, Rocke et al. [27] and Newton et al. [13] have presented models of measurement error in microarrays that can explicitly take into account higher variance at lower expression levels. More general approaches to variance pooling have been implemented in a variety of ways, using loess-based curve fits [15], robust nonparametric spline fits [28] and sliding windows for calculating either local averages [26,29,30] or interquartile ranges [24]. These more reliable estimates of the standard deviation can be used directly to calculate Z-statistics, which are calculated according to the same formula as the standard t-statistic, but correspond to lower p-values [26,31].
Strategies for pooling standard deviations
The studies cited above use methods that pool spots together based on their average intensity or logged intensity. For example, consider one set of replicate spots with an average intensity of 128 (27) in one channel and 16384 (214) in the other channel compared to a set of replicate spots with an average intensity of 1024 (210) in one channel and 2048 (211) in the other channel (Fig. 2). Since both of these sets of spots have the same average log2 intensity of 10.5, the standard deviations of their ratios would be presumed to be similar and would be pooled together using the pooling methods described above. However, these spots may actually be expected to have quite different standard deviations; we have noted that many ratios with high variances result from spots that have a medium or high intensity in one channel and a very low intensity in the other (data not shown). Thus, the ratios for the first spot are expected to be more variable because of the very low intensities (~100) in one channel. In this study, we test the hypothesis that if spots are pooled together with other spots of similar minimum intensity over both channels (Imin), rather than average intensity over both channels (Iavg), then a larger proportion of the high-variance spots will be grouped together, resulting in a tighter fit of the pooled standard deviation curve to the actual variance and generating more accurate estimates of the standard deviation.
Figure 2.

Motivation for pooling standard deviations by minimum intensity. A hypothetical noise distribution is given with higher noise at low intensities. Two sets of replicate spots (N = 3 arrays) that have the same average intensity are shown. However, example 1 produces a higher standard deviation of the logged ratio compared to example 2, because example 1 contains very low intensity measurements that fall into the noisiest range of the intensity scale. In this case, the minimum intensity would differentiate between these two examples while the average intensity would not.
This study expands upon previous work on intensity-dependent variance estimation for microarray data by introducing a new metric, Imin, for pooling standard deviations. We evaluate the performance of the Iavg and Imin metrics by explicitly comparing the reproducibility and accuracy of the Z-statistics calculated using these two metrics. We also compare the performance of the Z-statistics to the performance of other statistical techniques in current use, the standard and penalized t-tests. Finally, we extend our technique for pooling standard deviations to two-color microarray data from a reference sample experimental design.
Results
Datasets
The analyses in this study were performed on five different datasets. Datasets 1–4 use the direct comparison experimental design, i.e. labeled cDNA from two biological conditions, "X" and "Y," were co-hybridized onto a single array. Each dataset was generated from a different biological experiment using two-color Agilent cDNA arrays. For Datasets 1–3, the biological experiment, RNA processing and array hybridization were repeated three times. Dataset 4 contains 23 replicate arrays (see Methods). Dataset 5 uses a reference sample design, where RNA from each experimental condition is co-hybridized on an array with a standardized reference RNA sample. Dataset 5 contains three replicates arrays for each experimental condition.
Average logged intensity (Iavg) vs. minimum logged intensity (Imin) pooling metric
We demonstrate our technique of pooling standard deviations using the three arrays in Dataset 1 as a representative example of a "direct comparison" dataset. For each spot, we calculate the average logged ratio Mavg and the standard deviation of the logged ratio σM, across the three replicates. The spots are then sorted by either average intensity (Iavg) or minimum logged intensity (Imin) before pooling. Fig. 3a and 3b show the results of pooling standard deviations for Dataset 1, using either the Iavg or Imin metrics; the measured standard deviation σM and the pooled standard deviation σM' are plotted together against either Iavg or Imin. For better comparison, the pooled standard deviation curves for σM'(Iavg) and σM'(Imin) are both plotted together on Fig. 3c against their respective intensity metric, Iavg or Imin. Fig. 3 is based on data produced using the Agilent Feature Extraction software Version A.6.1.1 to quantify spot intensities in the original microarray image. This entire analysis was repeated on Datasets 2 and 3, as well as using two additional image processing techniques: SPOT Processing [32] and a combination of Agilent foreground and SPOT background values (see Methods).
Figure 3.

Two methods of pooling standard deviations of M: sorting by Iavg or by Imin. The standard deviation (σM) is pooled by taking the moving average of the variance (σM2). (a) Measured (σM, gray) and pooled (σM'(Iavg), black) standard deviation of the logged ratio M, plotted against Iavg. For spots with σM' > σM, the average residual error is 0.28; for spots with σM' < σM, the average residual error is 0.31. (b) Measured (σM, gray) and pooled (σM'(Imin), black) standard deviation of M, plotted against Imin. For spots with σM' > σM, the average residual error is 0.28; for spots with σX' < σX, the average residual error is 0.31. (c) Pooled standard deviation of M (σM') plotted against the intensity metric used for pooling, Iavg or Imin. Data are from Dataset 3.
We evaluated the tightness of the Iavg-pooled vs. Imin-pooled standard deviation curve fits to the measured standard deviations. Figs. 4a and 4b plot both measured (σM) and pooled (σM') standard deviations against either the Iavg or Imin pooling metric, analogous to Fig. 3a and 3b but using an especially noisy three-array subset of Dataset 4 that includes a population of extremely high variance spots. Instead of pooling together spots with similar variance, the Iavg metric combines the high-variance spots with the lower-variance spots. In contrast, the Imin metric pushes the high-variance spots to the left end of the curve, apart from the less noisy spots. This effect is reflected in the lower mean residual errors between σM and σM' for the Imin metric, calculated for Datasets 1–3 and six independent three-array subsets of Dataset 4 (see Table 1). For all of the datasets processed with Agilent Feature Extraction software only, the mean residual errors from using the Imin pooling metric are always less than or equal to the corresponding mean residual errors from using the Iavg pooling metric. This observation is most striking for Dataset 4 subset #3, which corresponds to the data in Fig. 4. The tighter fit that is obtained using the Imin metric is also reflected in the improved accuracy of the final Z statistic calculated using σM'(Imin), which is demonstrated in Fig. 5 and discussed below. The trend in residual values is not present when datasets are processed with the SPOT technique or with Agilent foreground and SPOT background.
Figure 4.

Comparison of pooled standard deviation curves using Iavg or Imin pooling metrics. The pooling algorithms are applied to a noisy three-array subset of Dataset 4. (a) Measured (σM, gray) and pooled (σM'(Iavg), black) standard deviation of M, plotted against Iavg. For spots with σM' > σM, the average residual error is 0.45; for spots with σM' < σM, the average residual error is 0.49. (b) Measured (σM, gray) and pooled (σM'(Imin), black) standard deviation of M, plotted against Imin. For spots with σM' > σM, the average residual error is 0.24; for spots with σM' < σM, the average residual error is 0.23.
Table 1.
Mean residual errors for spots with σM' > σM and σM' < σM, using I avg or I min pooling metric.
| Agilent Feature Extraction | SPOT Processing | Agilent FG + SPOT BG | ||||||||||
| σM2' > σM2 | σM2' < σM2 | σM2' > σM2 | σM2' < σM2 | σM2' > σM2 | σM2' < σM2 | |||||||
| Iavg |
Imin | Iavg | Imin | Iavg | Imin | Iavg | Imin | Iavg | Imin | Iavg | Imin | |
| Dataset 1 | 0.28 | 0.28 | 0.31 | 0.31 | 0.19 | 0.20 | 0.22 | 0.25 | 0.19 | 0.20 | 0.22 | 0.25 |
| Dataset 2 | 0.24 | 0.23 | 0.25 | 0.25 | 0.15 | 0.15 | 0.16 | 0.17 | 0.15 | 0.15 | 0.16 | 0.17 |
| Dataset 3 | 0.20 | 0.18 | 0.21 | 0.18 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 |
| Dataset 4 #1 | 0.15 | 0.14 | 0.17 | 0.15 | NA | NA | NA | NA | NA | NA | NA | NA |
| Dataset 4 #2 | 0.20 | 0.17 | 0.23 | 0.19 | NA | NA | NA | NA | NA | NA | NA | NA |
| Dataset 4 #3 | 0.45 | 0.24 | 0.49 | 0.23 | NA | NA | NA | NA | NA | NA | NA | NA |
| Dataset 4 #4 | 0.21 | 0.20 | 0.23 | 0.21 | NA | NA | NA | NA | NA | NA | NA | NA |
| Dataset 4 #5 | 0.25 | 0.19 | 0.28 | 0.20 | NA | NA | NA | NA | NA | NA | NA | NA |
| Dataset 4 #6 | 0.18 | 0.17 | 0.21 | 0.18 | NA | NA | NA | NA | NA | NA | NA | NA |
Data is given for three datasets using different image processing techniques (Agilent Feature Extraction, SPOT Image Processing and Agilent foreground combined with SPOT background), and for 6 independent three-array subsets of Dataset 4.
Figure 5.

Comparing the accuracy of different test statistics. Statistics were calculated for 3 replicate arrays from Dataset 4 and compared to the "gold standard" t-statistic for the remaining 20 arrays. The x-axis for all plots is the "gold standard" t-statistic. The y-axis shows: (a) average logged ratio Mavg, (b) standard t-statistic, (c) 90th percentile penalized t-statistic, (d) SAM penalized t-statistic, (e) Z-statistic using the Iavg pooling metric, or (f) Z-statistic using the Imin pooling metric.
Comparing the accuracy of different ranking statistics
In order to test the accuracy of the different test statistics – Mavg, the standard t-statistic, the 90th percentile penalized t-statistic, the SAM penalized t-statistic, Z(Iavg) and Z(Imin) – a subset of three arrays was randomly selected from the total set of 23 replicate arrays in Dataset 4 (see Methods). Each statistic was calculated for each gene in this set. The large number of remaining replicate arrays allowed us to calculate an approximate "gold standard" statistic, tgold, by computing the standard t-statistic over the set of 20 remaining replicates. The value of each test statistic from the three-array subset was compared to the value of the "gold standard" t-statistic, tgold, as shown in Fig. 5. The squared Pearson's linear correlation coefficient value (R2), representing the degree of concordance between the test statistic and tgold, was calculated. This analysis was repeated five additional times, selecting different subsets of experimental and "gold standard" arrays from Dataset 4 each time, and the R2 values from all six repetitions are given in Table 2. The Z-statistics and penalized t-statistics both have appreciably higher R2 values than either Mavg or the standard t-statistic. The R2 values for Z(Imin) are greater than the R2 value for any other technique across all six datasets. Note that there is less scatter for high-magnitude values when using Z(Imin) instead of Z(Iavg) (Fig. 5e and 5f respectively). Accordingly, the R2 value is higher for the Z(Imin) than the Z(Iavg) ranking metric for all three datasets, confirming that the tighter curve fits seen in Fig. 4a and 4b and Table 1 (see above) translate into improved accuracy of using the Imin pooling metric over Iavg.
Table 2.
Accuracy of each test statistic when compared to a "gold standard" t-statistic.
| Mavg |
t | d90th percentile | dSAM | Z(Iavg) | Z(Imin) | |
| Dataset 4 #1 | 0.69 | 0.09 | 0.80 | 0.80 | 0.79 | 0.83 |
| Dataset 4 #2 | 0.66 | 0.08 | 0.78 | 0.78 | 0.79 | 0.82 |
| Dataset 4 #3 | 0.54 | 0.04 | 0.79 | 0.70 | 0.77 | 0.84 |
| Dataset 4 #4 | 0.68 | 0.02 | 0.80 | 0.79 | 0.80 | 0.83 |
| Dataset 4 #5 | 0.64 | 0.06 | 0.80 | 0.74 | 0.78 | 0.84 |
| Dataset 4 #6 | 0.67 | 0.01 | 0.80 | 0.80 | 0.79 | 0.83 |
Each column contains the R2 value calculated between each experimental test statistic and the "gold standard" t-statistic for (left to right): the average logged ratio Mavg, the standard t-statistic, the 90th percentile penalized t-statistic, the SAM penalized t-statistic, the Z-statistic using the Iavg pooling metric and the Z-statistic using the Imin pooling metric. Data are from six independent three-array subsets of Dataset 4. Although Mavg is not a statistical test, it is included in this table for comparison.
Comparing the reproducibility of different ranking statistics
We also evaluated the reproducibility of these different test statistics, by constructing test datasets that split six replicate arrays from Dataset 4 into two subsets of 3 arrays (see Methods). Each test statistic – Mavg, the standard t-statistic, the 90th percentile penalized t-statistic, the SAM penalized t-statistic, Z(Iavg) and Z(Imin) – was calculated for both three-array subsets. A precise, i.e., reproducible, test statistic should produce similar values for both subsets since all of the arrays in both subsets were drawn from a pool of replicates prepared from identical biological experiments. Fig. 1a,1b and Fig. 6a,6b,6c,6d show the correlation for each test statistic between the two subsets, including a linear regression line in Fig. 6. The slope coefficient of the linear regression indicates whether overall magnitudes of the test statistics are different between the two subsets, while R2 indicates the degree of correlation on a gene-by-gene basis (Table 3). This analysis was repeated for an additional two pairs of independent three-array subsets of Dataset 4 (graphs not shown), with the slope coefficients and R2 values given in Table 3.
Figure 6.

Comparing the reproducibility of different test statistics. Two subsets of Dataset 4 each contain three replicate arrays derived from identical biological experiments. Each test statistic is calculated twice, once for each subset, and the two statistics are plotted against each other. (a) Comparison of 90th percentile penalized statistics. (b) Comparison of SAM penalized statistics. (c) Comparison of Z-statistics using Iavg pooling metric. (d) Comparison of Z-statistics using Imin pooling metric. Also see Fig. 1a for comparison of the standard t-test.
Table 3.
Reproducibility of each test statistic when used on replicate datasets.
| Mavg |
t | d90th percentile | dSAM | Z(Iavg) | Z(Imin) | ||
| R2 | Dataset 4 #1 | 0.89 | 0.00 | 0.86 | 0.87 | 0.93 | 0.94 |
| Dataset 4 #2 | 0.90 | 0.00 | 0.87 | 0.87 | 0.93 | 0.93 | |
| Dataset 4 #3 | 0.89 | 0.00 | 0.88 | 0.88 | 0.95 | 0.95 | |
| Slope | Dataset 4 #1 | 0.90 | 0.00 | 0.97 | 1.63 | 1.00 | 1.00 |
| Dataset 4 #2 | 0.93 | 0.01 | 0.96 | 0.58 | 1.02 | 1.04 | |
| Dataset 4 #3 | 1.00 | 0.07 | 0.96 | 0.74 | 1.09 | 1.08 | |
Linear regression slope coefficients and R2 coefficients are calculated between corresponding statistics from two replicate three-array subsets of Dataset 4. Columns represent (left to right): the average logged ratio Mavg, the t-statistic, the 90th percentile penalized statistic, the SAM penalized statistic, the Z-statistic using Iavg pooling metric and the Z-statistic using Imin for three different pairs of subsets. Although Mavg is not a statistical test, it is included in this table for comparison.
The R2 values for the two Z-statistics were similar to each other and consistently higher than those of the other techniques. Nonparametric measures of correlation, the Spearman Rho and Kendall Tau rank correlation coefficients, were also higher for both Z-statistics than any of the other statistics for all three pairs of subsets (data not shown). All three calculations of the slope coefficients for both Z-statistics, as well as Mavg and the 90th percentile penalized t-statistic, are close to 1, indicating that the overall magnitudes of the Z-statistics are consistent across datasets, whereas the standard t-statistic and the SAM penalized t-statistic produced test statistics whose overall magnitudes vary across the subsets.
Outlier detection
When calculating the Z-statistic, using a much smaller pooled σM' in place of a large σM has the potential to overestimate the significance of gene regulation in the case where one of the replicates is an outlier measurement and the large measured standard deviation provides a better estimate of the variability. As seen in Fig. 3a,3b,3c, there are several spots that lie far above the pooled standard deviation curve. Datasets 1 and 4 were reprocessed using an outlier detection technique (see Methods). Fig. 7a shows σM and σM' from Dataset 1 plotted against Imin, as in Fig. 3c, except that the y-axis has been rescaled to show all spots detected as outliers, which are now highlighted in black.
Figure 7.

Implementation of outlier detection. (a) Measured (σM, gray) and pooled (σM'(Imin), black curve) standard deviation of M, plotted against Imin, with the outlier spots highlighted (black points), for Dataset 3. (Compare to Fig. 3b.) (b) Scatterplot of average condition X intensity vs. average condition Y intensity for Dataset 3, with p-values indicated in color. (c) Z-statistic using Imin pooling metric vs. "gold standard" t-statistic with outliers highlighted in black, for a 3-array subset and 20-array "gold-standard" subset of Dataset 4. Outlier Z-statistics calculated using the pooled standard deviation. (d) Z-statistic using Imin pooling metric vs. "gold standard" t-statistic, with outliers highlighted in black, for the same data in (c). Outlier Z-statistics calculated using the measured standard deviation, for Dataset 4 subset #2.
The accuracy of this outlier detection technique was also evaluated by comparing the Z-statistic to tgold using Dataset 4. Fig. 7c plots Z(Imin) vs. tgold for Dataset 4 set #2. The outliers, which are highlighted, include false positive spots for which tgold is low and Z(Imin) is high, although not all such points are detected as outliers. Fig. 7d is an identical plot to Fig. 7c except that the Z-statistics for the outlier spots are calculated using the higher-valued measured standard deviation σM instead of the pooled value σM'. The outliers are now mostly clustered around the origin with the other non-significant spots. A few spots with moderately high tgold values are detected as outliers and have low corrected Z-statistics, and some potential false positives with high Z-statistic values and low tgold values are not detected as outliers.
At the end of the analysis, the outlier-corrected Z statistics are converted to p-values. To demonstrate the additional information that the p-values provide, Fig. 7b shows a scatterplot of X vs. Y for Dataset 1, with statistically significant spots colored according to their multiple-test-corrected p-values (see Methods). Spots with similar ratios may have different p-values due to their different standard deviations. In addition, after outlier detection, some spots with high ratios are not found to be significant.
Analysis of reference sample arrays
The techniques used above for a direct comparison experimental design were extended to a reference sample design (see Methods). Under a reference sample design, one can estimate either the standard deviation of the individual logged ratios comparing experimental samples to the reference sample, Mx and My, or the standard deviation of the paired differences of these logged ratios, μ = Mx-My. Under the first, or unpaired method, the Z-statistic is calculated as 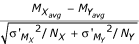 where NX and NY are the number of replicates for the given spot for condition X and condition Y, respectively. Under the second, or paired method, the Z-statistic is calculated as
where NX and NY are the number of replicates for the given spot for condition X and condition Y, respectively. Under the second, or paired method, the Z-statistic is calculated as 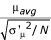 where N is the number of paired replicates for the spot. The samples used in a reference sample design may not always have been collected or processed in pairs, so we evaluated both of these methods.
where N is the number of paired replicates for the spot. The samples used in a reference sample design may not always have been collected or processed in pairs, so we evaluated both of these methods.
For each replicate in reference sample Dataset 5, the biological specimens for conditions X and Y were prepared on the same day, so a natural pairing exists for the condition X and Y arrays. These data were processed using all three image processing techniques and then analyzed using both paired and unpaired methods, and using either the Iavg or Imin pooling metric for each approach. Fig. 8a shows the measured and pooled standard deviation of the paired differences of logged ratios (σμ and σμ') plotted together against the pooling metric, Imin. Curve fits were analogously constructed using the Iavg pooling metric with the paired method (with results similar to using the Imin metric, data not shown), and using both Iavg and Imin with the unpaired method (with results similar to using the ratio method with direct comparison arrays, data not shown). The unpaired σM' and paired σμ' curves are plotted together against their the Iavg or Imin pooling metric in Fig. 8b. The paired standard deviations are lower than the unpaired standard deviations except at low intensity metric values.
Figure 8.

Methods of pooling the standard deviation for a reference sample design. The standard deviation (σ) is pooled by taking the moving average of the variance(σ2). (a) Measured (σμ, gray) and pooled (σμ' (Imin), black) standard deviation of the difference of logged ratios μ, plotted against Imin. (b) Pooled standard deviation of MX, MY and μ plotted against the intensity metric used for pooling, Iavg or Imin. Data are from Dataset 5.
Linear regression was performed between Z-statistics calculated using the paired and unpaired methods for all spots. Table 4 gives the linear regression slope coefficients when either the Iavg or Imin pooling metric was used, for Dataset 5 processed with the three different image processing techniques. For most spots, both the difference of logged ratios (μ) and number of replicates (N) are the same, except for the occasional difference between the two conditions in the number of low quality spots that are excluded from the analysis. Thus, differences in the Z-statistic primarily reflect differences in the standard deviations. The slope coefficients are all greater than 1, indicating that the paired technique produced higher Z-statistic values, due to the lower standard deviations that are produced with paired analysis.
Table 4.
Linear regression slope coefficients calculated between the corresponding Z-statistics using independent or pairwise analysis.
| Agilent Feature Extraction | SPOT | Agilent FG + SPOT BG | |
| Iavg |
1.70 | 1.68 | 1.70 |
| Imin |
1.59 | 1.61 | 1.63 |
Coefficients given for reference sample design Dataset 5. Values greater than 1 indicate higher Z-statistics with the pairwise technique. Data is shown for both pooling metrics Iavg and Imin and for three different image processing techniques. Every linear regression analysis produced an R2 value greater than 0.89 (data not shown).
The mean residual errors for spots with σ' < σ and σ' > σ were calculated when using the Iavg or Imin pooling metric in unpaired or paired analyses of Dataset 5, and are given in Table 5. For the unpaired analysis, as in the direct comparison experiments, mean residual values produced by using the Imin pooling metric are less than or equal to those produced by the Iavg pooling metric. The same trend is seen between the two pooling metrics for the paired analysis. These results are consistent regardless of the image processing technique used.
Table 5.
Mean residual errors for spots with σ2' > σ2 and σ2' < σ2, using I avg or I min pooling metric.
| Agilent Feature Extraction | SPOT Processing | Agilent FG + SPOT BG | ||||||||||
| σ2' > σ2 | σ2' < σ2 | σ2' > σ2 | σ2' < σ2 | σ2' > σ2 | σ2' < σ2 | |||||||
| Iavg |
Imin | Iavg | Imin | Iavg | Imin | Iavg | Imin | Iavg | Imin | Iavg | Imin | |
| σM2 Cond. X | 0.19 | 0.18 | 0.21 | 0.20 | 0.14 | 0.14 | 0.15 | 0.15 | 0.13 | 0.13 | 0.14 | 0.14 |
| σM2 Cond. Y | 0.22 | 0.18 | 0.25 | 0.23 | 0.16 | 0.14 | 0.17 | 0.17 | 0.15 | 0.13 | 0.16 | 0.16 |
| σμ2 | 0.19 | 0.18 | 0.22 | 0.19 | 0.13 | 0.13 | 0.14 | 0.14 | 0.12 | 0.12 | 0.12 | 0.12 |
Analysis was performed on Dataset 5 (reference sample design). Data is given for both unpaired and paired analyses, using three different image processing techniques: Agilent Feature Extraction, SPOT Image Processing, and Agilent foreground combined with SPOT background.
Discussion
Building up new knowledge about biological systems is the ultimate purpose of microarray experiments, but all such insights have to be built on a solid foundation to be accurate and useful. Proper normalization of data and accurate detection of which genes are regulated are vital to the success of downstream exploration of microarray data. Even for exploratory cluster analyses, the genes that are significantly regulated must be selected beforehand. This task of detecting these genes is a difficult statistical problem; a statistical hypothesis is made for each of tens of thousands of genes tested, but only a small number of replicate arrays are available to test those hypotheses. The statistical methods presented in this study attempt to draw as much information as possible out of a small number of array replicates to determine which genes are likely to be regulated.
It is clear that looking at the measurements of each gene in isolation can produce a test with low statistical power (e.g. using the standard t-test, Fig. 1). To improve statistical power, we can use knowledge about the relationships among the many thousands of points in the arrays. Specifically, we group together spots that have similar standard deviations and then pool together many less accurate estimates of standard deviation into a single, more accurate estimate. Our data also show that the Z-statistics are more precise than either standard or penalized t-statistics for detecting differential gene expression in microarray data. We further demonstrate that pooling standard deviations using the minimum intensity metric produces Z-statistics that are more accurate than the standard t-test, the penalized t-tests, and the average intensity-based Z-statistic.
Average combined logged intensity (Iavg) vs. minimum logged intensity (Imin) pooling metric
We evaluated two different intensity-based metrics for pooling standard deviations. There are many reports that the variance is a function of intensity, but the exact shape of this relationship could depend on many factors extrinsic to the biological experiment, such as the array technology being used, the signal-to-noise ratio of the data, the similarity between the two conditions[30], the normalization technique or the background subtraction technique. For this reason, we favor an estimation of the standard deviation using a curve-fitting technique rather than a fixed model based on previous data. Furthermore, when dealing with two-channel arrays, there are two different intensity values associated with each replicated spot. It is possible that the variation is best described as a function of the average intensities of both channels. However, our own experience and many other reports suggest that the highest variances are often seen for low intensity spots. If so, the variance may be better described as a function of the minimum intensity over all the spots.
The data presented here show that the mean residual errors are either equal or lower when using the Imin compared to the Iavg pooling metric, for every dataset using the Agilent Feature Extraction image processing technique. The subset of Dataset 4 for which this difference is most striking, #3 in Table 1, also has a population of spots with particularly high variance (see Fig. 4). The Iavg metric pools these spots together with other spots that have a much lower variance. In contrast, the Imin metric moves these spots to the low end of the x-axis, and the curve fit tracks the standard deviation of the spots much better. The noisiest spots on microarrays are often those where at least one channel is "blank", i.e. a noisy, low level of signal that presumably represents no expression. The Imin metric is better at grouping such spots together. For datasets with low background levels, there is a smaller difference in the performance of the two pooling metrics.
The trends in the mean residual errors from the unpaired reference sample analysis agree with the results from the direct comparison analyses. This similarity is to be expected, since processing each reference sample condition separately is equivalent to doing a direct comparison between each condition and reference RNA samples. Both pooling metrics generate similar mean residual error values when pooling σμ, but one dataset is not enough to make any generalizations about which pooling metric will perform best for all paired reference sample datasets. The improved performance of the Imin pooling metric is lost when using SPOT processing or combined Agilent foreground and SPOT background image processing, suggesting that these image processing techniques may be more effective at removing noise at low intensities.
The Iavg and Imin pooling techniques are reproducible to the same degree, since their R2 coefficients between Z-statistics from paired datasets (see Table 1) are similar to each other. The Imin pooling technique generates slightly more accurate results, as indicated by the greater R2 coefficients between Z(Imin) and tgold compared to those between Z(Iavg) and tgold (see Table 2). This trend holds for all six subsets of Dataset 4.
The higher accuracy of Z(Imin)
The Z-statistic calculated using the Imin pooling metric provides an improvement in accuracy over the other techniques. The t-statistic derived from datasets with 20 replicates was used as a surrogate "gold standard" since 8 or more replicates can be considered sufficient to give power to the t-statistic [17]. The t-statistic was chosen as the "gold standard" instead of the average logged ratio since the latter does not take variability into account. For each of the six permuted subsets of Datasets 4, the 90th percentile penalized t-statistic, SAM penalized t-statistic, and Z(Iavg) had similar R2 values when correlated with the "gold standard" t-statistic, although the SAM statistic did perform poorly for the noisiest subset of Dataset 4 (#3 in Table 2) with an R2 value of only 0.70. Z(Imin), however, consistently produced the highest R2 value for each of the six datasets. Since the ratios used in each of these statistics is identical, this result indicates that the standard error generated with the Imin technique produces the best correlation with the gold standard t-statistic based on 20 replicates. Although excluding spots with very low intensity could eliminate the difference in performance between the Imin and Iavg pooling metrics, this approach would make it impossible to detect low-expressed regulated genes, which may be biologically significant.
The Z-statistics from the Imin technique do not correlate perfectly with the "gold standard" t-statistic, however. Some disagreement can be expected because the Z(Imin) data was based on only three replicate arrays, which contain much less information than the 20 replicates used to calculate the "gold standard" t-statistic. Also the significance estimates calculated using the "gold standard" t-statistic may still contain some inaccuracies, even with 20 replicates. Kerr et al. found this to be true with 12 replicates, where accuracy is reduced if the error distribution for each gene is modeled separately instead of using a pooled estimate [15]. Analyzing the large (N = 20) replicate dataset using robust estimators of ratio and standard deviation may be able to create a more accurate "gold standard" to use for further testing of the Z-statistic or other statistics. Note that we do not employ an explicit permutation-based approach to estimate the false detection rates of the statistics investigated in this study, as in Ref. [16]. Rather than permute gene labels from a small set of arrays to estimate the distribution of expected test statistics, with the availability of the large (N = 23) replicate dataset described herein, we preferred to use this rich source of actual test statistics directly.
The higher reproducibility of z-statistics
The Z-statistic – calculated with either pooling the Imin or Iavg pooling metric – provides an appreciable improvement in reproducibility over the average logged ratio alone, the standard t-test and the 90th percentile and SAM penalized t-statistics. Both linear (R2) and non-parametric rank correlation coefficients were highest for the Z-statistic when comparing corresponding spots between three independent pairs of replicate datasets. Also, the standard t-statistic and SAM penalized t-statistic generate linear regression slope coefficients that vary greatly from pair to pair, indicating that their absolute magnitude is not as reproducible as the Z-statistics, whose linear regression slope coefficients are much closer to 1.
The high correlation values and near-unity slope coefficients for the Z-statistic support the hypothesis that pooling the standard deviations of spots with similar intensities provides a stable, precise estimate of the standard deviation. This assumption of a well-estimated standard deviation supports the use of the Gaussian distribution to map the Z-statistic to a p-value. Using only the measured standard deviation, one is forced to use a t-distribution with only 2 degrees of freedom to generate a p-value. This test does not have sufficient power to generate any significantly regulated points; because of the very small number of degrees of freedom, not a single spot seen in Fig. 1a is found to be significant after multiple test correction. In contrast, even after a conservative multiple test correction that makes the cutoff for statistical significance much more stringent, many spots are found significant using the Z-statistic. The penalized t-statistics do not produce a stable estimate of the standard deviation with these data, perhaps because the constant added to the denominator of the test statistic showed a large variation between replicate datasets. Therefore they cannot be mapped to a p-value in a reproducible manner.
Outlier detection
One limitation of using a pooled standard deviation is that for a spot with replicate ratios that include one or more outliers, the appropriately high measured standard deviation will be replaced by an inappropriately low pooled standard deviation. This substitution could produce a false positive result. We have sought to minimize this limitation by implementing an overlying outlier detection algorithm. (For other implementations of outlier detection, see Ref. [26,30].) The algorithm in this study uses the measured standard deviation instead of the pooled standard deviation for spots for which the pooling model may not hold. These spots are identified as ones for which residual error σ–σ' is positive and greater than twice the standard deviation of the positive residual errors.
The measured standard deviations for these outlier points are valid sample measurements of the variance process and should be used to calculate the pooled standard deviations for spots with similar intensities. These ratio measurements, however, are too widely varying for one to have the same confidence in the average ratio as one would have for other spots; thus, it is appropriate to substitute the measured standard deviation for the pooled standard deviation in these cases. Fig. 7c,7d, which highlight outlier spots on a plot of the Z(Imin) vs. the "gold standard" t-statistic for Dataset 4b, show that this outlier detection technique correctly detects many of the presumably false positive spots that have a high Z-statistic and low tgold value. The plots also show some false positive spots that are not detected through this algorithm, as well as a few spots that become false negatives after outlier detection. Other, more complex outlier detection algorithms may perform better, and should be explored. A simple modification to the current algorithm, using local instead of global estimates of the standard deviation of the residual error, may improve outlier detection. Alternative implementations include modifying the pooling window shape to give more weight to a spot's measured standard deviation or that of its nearest neighbors by intensity. Strictly speaking, the p-values for outlier spots should be calculated using a t-distribution instead of a Gaussian distribution since the measured standard deviation is being used. We have shown, however, that with 3 replicates, no spots in our datasets can be found statistically significant using the t-test and strict multiple test correction. In order to preserve detection of spots, we continue to use the Gaussian distribution to convert outlier Z-statistics to p-values, which may slightly increase the false positive rate for spots detected as outliers. In practice, however, such spots are rarely found to be significantly regulated.
Unpaired vs. paired analysis for reference sample experiments
Finally, we have extended our algorithms to apply to data from a reference sample experimental design. This design gives one the flexibility to compare many different conditions to one another, but the trade-off is a loss in precision. In theory, using a reference sample design instead of a direct comparison design should increase the variance by a factor of 2. This increase has in fact been observed in practice [33].
The paired analysis method can reduce the measured variation in a reference sample design. The linear regression slope coefficients in Table 1 indicate that the Z-statistic values using the paired analysis are higher than the unpaired Z-statistic values. Thus, the paired difference of logged ratios, μ, is less variable than the independent logged ratios, MX and MY. This observation suggests that the effects of biological or analytical variation from replicate to replicate can be reduced if comparisons are made between paired samples. Whether this reduction is due to using paired biological samples or paired array processing dates [34] is still an open question, and probably will be context-dependent. Although it may not always be practical, it would be beneficial for investigators to design reference sample experiments to be performed in parallel whenever possible to take advantage of the lower standard deviations produced by paired analysis.
Finding the optimal statistical test
Several areas remain for further refinement of our implementation of pooling-based statistical analysis of microarray data. Currently, the standard deviation is pooled using a simple moving rectangular window of 501 spots, but other window sizes and shapes may improve performance slightly. More generally, we have not explicitly compared the moving average estimator with the spline-fit or loess-based techniques to estimate the standard deviation used in other studies (see Background). While we expect performance to be similar, further testing may reveal an advantage.
Following Ref. [35], we do not try to estimate the dye-specific bias of individual spots or genes (i.e., dye-gene interaction) in order to preserve degrees of freedom needed to estimate the variance. Informally we noted that dye bias in some spots produced high measured variances that caused those spots to be considered non-significant outliers. A post-hoc test to warn of potential dye bias of individual spots may be appropriate for small numbers of array replicates (e.g. N = 3), especially if the experimental design is unbalanced (i.e., the number of dye-swapped and unswapped arrays is not equal).
Note that this study only considered statistics of the general form (ratio) / (standard deviation). ANOVA models that consider the variance as intensity-dependent, as seen in Ref. [15,25], can be seen as an extension of this concept. An ANOVA framework, however, also allows for a more complicated experimental model that can incorporate normalization and multiple biological conditions. Pooling standard deviations as a function of minimum intensity instead of average intensity may benefit such models. Permutation tests can also be used to detect regulated genes, and are known to be robust to outliers but can have low power for small N. Xu et al. found a permutation test to be equally or less accurate than parametric methods in ranking genes [36]. Bayesian analysis can also be applied to microarray data [13,20,21], and may be useful in this context to draw more information out of the distribution of intensities and ratios in the data.
In this study, data is first normalized, and then detection of regulated genes is performed in a separate step. In contrast, other approaches incorporate normalization and statistical inference into a unified model [29,35]. Furthermore, the options for normalizing the data are numerous, including algorithms based on local regression (loess) [7], splines [37], a constant shift [15], or more exotic transforms that tend to remove the intensity dependence of the variance [38]. Increased attention to the low-level details of scanning and image processing may also improve accuracy [22,33,39], while at the same time potentially changing the intensity dependence of the variance. It remains to be seen how the techniques used for normalization or variance-stabilizing transforms will impact the accuracy and precision of regulated gene detection. In addition, we are concerned that some of these transforms may create a systematic bias for or against genes of low intensity (e.g., [40]).
Test performance can depend on data characteristics
Although many datasets have a variance that is intensity-dependent [12,15,21-26], some studies have analyzed datasets whose variance characteristics are not strongly intensity-dependent (e.g., [35]). In general, we have experienced that microarray datasets with a low background relative to signal, loess-based normalization, and conservative background subtraction (e.g. SPOT Image Processing) produce standard deviations that are not strongly intensity-dependent. In this context, the differences between the Imin and Iavg metrics disappear. In fact, for data with unusually low noise, the standard deviations is nearly constant across all spots and all of the statistical tests considered in this paper, even simply the average logged ratio, tend to converge. This observation is not unexpected; as the standard deviations converge to the same value, the denominator of the test statistics will become constant, leaving the test statistics simply proportional to the ratio. We would recommend finding a normalization [7,29,33,37] and background subtraction technique [22,32,39] that produces low, intensity-independent standard deviations. Applying variance stabilizing transforms may eliminate the intensity dependence of the standard deviation [38], but might also reduce statistical power or bias the test toward spots of certain intensities. It cannot be predicted in advance whether all intensity dependence of the variation will be removed, so we continue to use the more robust statistic Z(Imin) for all of our datasets. Furthermore, in situations where changing the background subtraction or normalization technique is not possible because the original data is not available, using a more robust statistic like Z(Imin) will be advantageous.
While the pooling techniques described herein can compensate for intensity-dependent variation, this intensity dependence can be minimized or exaggerated by different normalization techniques and background subtraction techniques. These techniques may have subtle effects on the power to detect regulated genes at different intensities, perhaps creating bias for or against detection of low-expressed genes. For this reason, until the most sensitive and unbiased normalization and background subtraction methods are optimized for each microarray system, we would encourage creators of microarray data archives to preserve unnormalized intensity and background data, and the original image data when possible.
Of the many useful tests used to detect regulated genes from a small number of microarray replicates, we see the intensity-based variance estimation and Z-statistic described in this study to be a good combination of simplicity, robustness, precision, and accuracy. This technique allows meaningful p-values to be added to a list of regulated genes. With this assessment of statistical significance, an investigator can proceed to focus on genes that are most likely to be regulated.
Methods
Data acquisition
For Datasets 1–3, microarrays were prepared essentially according to the manufacturer's instructions [41]. Briefly, 20 μg of total RNA were direct-labeled with Cy-3 and Cy-5, and labeled cDNAs were hybridized overnight to Agilent Human 1 cDNA arrays (G4100a, Agilent Technologies, Palo Alto, CA) containing 16,142 features representing approximately 10,500 unique genes. After washing, the microarrays were scanned in an Agilent model G2505A microarray scanner.
Dataset 3 contains 3 replicate two-color arrays with condition X in the Cy-5 channel and condition Y in the Cy-3 channel. Dataset 1 contains 3 replicates from another experiment, including one dye-swapped array; i.e. condition X in the Cy-3 channel and condition Y in the Cy-5 channel. Dataset 2 contains 3 replicate arrays without dye-swap, but each array was hybridized with a different amount of RNA, 5, 10 or 20 μg.
Dataset 4 consists of 23 replicate Agilent cDNA arrays from the Alliance for Cellular Signaling. The files MAE030201N00.txt to MAE030223N00.txt were downloaded from http://www.signaling-gateway.org/data/micro/cgi-bin/microcond.cgi. These arrays correspond to the conditions "B-cell + SIMDM exposure = 0 minutes" vs. "Spleen". Four additional arrays are available for this condition (numbered MAE02070xN00.txt), but these arrays appeared to be slightly different from the other 23 arrays (using hierarchical clustering, data not shown) and were excluded from further analysis. The B-cell RNA was derived from 23 preparations, each from a different set of mice, while the spleen RNA was drawn from a single large pool (Rebecca Hart, Alliance for Cellular Signaling at the California Institute of Technology, Pasadena, CA, USA, personal communication).
Reference sample Dataset 5 is a set of 6 microarrays generated in our laboratory. Each of the arrays contains a reference RNA sample in the Cy-3 channel. Three have condition "X" samples in the Cy-5 channel and the other three have condition "Y" samples in the Cy-5 channel. Since corresponding biological specimens for conditions X and Y were prepared together for each replicate, a natural pairing exists for the condition X and Y arrays.
Computer techniques
Statistical modules were programmed in Perl v5.8. Microsoft Visual Basic 6.0 was used to integrate the image processing and statistical modules.
Image processing
For Datasets 1–3 array images were processed using Agilent Feature Extraction software version A.6.1.1. The Feature Extraction Software provides normalized Cy-3 and Cy-5 channel intensity values for each spot on an array (in the gProcessedSignal and rProcessedSignal fields of the output files). The default settings were used for all options. Quality control algorithms in the software detect unusual (poor quality) spots; spots were excluded from analysis that contained a nonzero value any of the following fields: IsSaturated, IsFeatNonUnifOL, IsBGNonUnifOL, IsFeatPopnOL, IsBGPopnOL, IsManualFlag. For a detailed description of the Agilent Feature Extraction software and the algorithms it uses, see the Agilent Feature Extraction Version 6.1 Users' Manual. Briefly, Agilent Feature Extraction determines the foreground value for each channel based on the pixel values in a fixed-size circle centered on each spot. The median of pixel values in a concentric ring around the circle, with an excluded region between the outer boundary of the circle and the inner boundary of the ring, gives the spot background value. The raw spot value is calculated as its foreground value less its background value. A surrogate raw value is assigned when the foreground value does not exceed the background value by two standard deviations of the spot's background pixel values. Intensity-based normalization between channels using a linear regression and a lowess curve-fit technique is then applied to remove any systematic dye incorporation biases.
Images were also processed using SPOT (CSIRO, New South Wales, Australia)[32], an R-based implementation which uses seeded region growing to determine the foreground pixels for each spot and morphological opening to determine the background value for each spot. The raw spot values, foreground less the background values, are normalized between channels using an intensity-based Loess implementation in R available in the maNorm function of the marrayNorm package of the open-source Bioconductor software http://www.bioconductor.org. We considered three image processing techniques: Agilent Feature Extraction output alone, SPOT output alone with maNorm-based normalization and Agilent foreground (gMedianSignal and rMedianSignal columns) less SPOT background (morphG and morphR columns) with maNorm-based normalization.
Pooled standard deviations – direct comparison design
Three replicate arrays were processed for each direct comparison experiment. To map intensities from different replicates onto similar scales without altering the absolute ratio values, we multiplied the intensity values on each array by a constant such that mean square error between the intensities of that array and the intensities of the first replicate array was minimized. The multiplicative factor for array j is given by 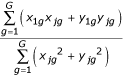 , where G is the total number of spots and x and y are intensities for condition X and condition Y. Then, for each spot, the mean and sample (measured) standard deviation (σ) across array replicates were calculated for the logged ratio M = X-Y, where X and Y are log2(x) and log2(y). The sample standard deviation of M, σM, is calculated as
, where G is the total number of spots and x and y are intensities for condition X and condition Y. Then, for each spot, the mean and sample (measured) standard deviation (σ) across array replicates were calculated for the logged ratio M = X-Y, where X and Y are log2(x) and log2(y). The sample standard deviation of M, σM, is calculated as 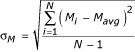 . A replicate spot for which either channel was flagged as poor quality was excluded from these calculations. Spots for which there were less than two replicates of good quality were discarded from analysis.
. A replicate spot for which either channel was flagged as poor quality was excluded from these calculations. Spots for which there were less than two replicates of good quality were discarded from analysis.
The pooled logged ratio standard deviation, σ'M, was calculated by sorting all the spots by the average logged intensity 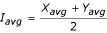 or the minimum logged intensity Imin across both channels of all replicates and then taking the square root of the moving average of the variance σM2 with a window of 501 spots. We averaged the variance instead of the standard deviation, since averaging the standard deviation directly will produce a negatively biased (~13%) estimate for N = 3 [42]. The Z-statistic was then calculated as
or the minimum logged intensity Imin across both channels of all replicates and then taking the square root of the moving average of the variance σM2 with a window of 501 spots. We averaged the variance instead of the standard deviation, since averaging the standard deviation directly will produce a negatively biased (~13%) estimate for N = 3 [42]. The Z-statistic was then calculated as 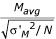 . Note that Iavg and M as defined above are equivalent to the symbols Ā and M, respectively, as used in other studies [17]. The common "M-A plot" would be called an "M-I plot" using the notation of this study.
. Note that Iavg and M as defined above are equivalent to the symbols Ā and M, respectively, as used in other studies [17]. The common "M-A plot" would be called an "M-I plot" using the notation of this study.
Pooled standard deviations – reference sample design
Three pairs of arrays were processed for each reference sample experiment. For the unpaired analysis, the arrays within a given condition were linearly normalized to each other, in order to map intensities from different replicates onto similar scales without altering the absolute ratio values (as described above). For each condition, the mean Mavg and sample standard deviation σM of the logged ratio were calculated for each feature. The pooled standard deviation of the logged ratio, σ'M, was calculated by sorting all the spots by the average intensity, Iavg, or the minimum intensity, Imin, across both channels of all replicates for the condition and then taking the square root of the moving average of the variance σM2, with a window of 501 spots, centered on the given spot. The Z-statistic was calculated as where NX and NY are the number of replicates for the given spot for condition X and condition Y, respectively.
For the paired reference sample analysis, the intensity vectors were all linearly normalized to the vector for the first replicate array of condition X to put all intensity values from both conditions on the same scale without changing the value of the ratios. Then the paired difference of logged ratios μ = MX - MY for each pair of replicates was computed. The mean and sample standard deviation of μ was then calculated across replicates. The pooled standard deviation of μ, σ'μ, was calculated by sorting all the spots by the average intensity Iavg or the minimum intensity Imin across both channels of all replicates for both conditions, and then taking the square root of the moving average of the variance σμ2, with a window of 501 spots. The Z-statistic was calculated as where N is the number of paired replicates for the spot.
To compare Z-statistic values between the paired and unpaired methods, the linear regression slope coefficient with intercept set to 0 was calculated between corresponding Z-statistics from the two methods.
Calculation of p-values
For a Z-statistic Z, the two-tailed p-value is given by 1 - 2Φ(|Z|), where Φ is the cumulative distribution function for the zero-mean, unit-variance Gaussian. The p-value is corrected for multiple tests using Sidak's formula, p' = 1-(1-p)L, where L is the total number of spots being examined. Note that we did not find it necessary to use more sophisticated means of controlling the error rate [43,44], as we are primarily concerned with ranking regulated genes and not in establishing firm statistical cutoffs.
Calculation of standard t-statistics and penalized t-statistics
Standard t-statistics for direct comparison arrays were calculated with the formula 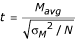 . The two-tailed p-value was calculated using a t distribution with N-1 degrees of freedom. In a penalty-based technique, a constant penalty s0 is included in the denominator of the t-statistic. The new statistic, d, is given by . Two different methods of choosing s0 were used: setting s0 to equal the 90th percentile of the actual standard deviations and the significance analysis of microarrays (SAM) technique, which chooses s such that the coefficient of variation of d is minimized. The SAM technique was implemented using software developed at Stanford University Labs [19,45]. This software imputes missing logged ratio values before calculating s0, and this feature cannot be disabled. The K-nearest-neighbor technique was selected for imputation.
. The two-tailed p-value was calculated using a t distribution with N-1 degrees of freedom. In a penalty-based technique, a constant penalty s0 is included in the denominator of the t-statistic. The new statistic, d, is given by . Two different methods of choosing s0 were used: setting s0 to equal the 90th percentile of the actual standard deviations and the significance analysis of microarrays (SAM) technique, which chooses s such that the coefficient of variation of d is minimized. The SAM technique was implemented using software developed at Stanford University Labs [19,45]. This software imputes missing logged ratio values before calculating s0, and this feature cannot be disabled. The K-nearest-neighbor technique was selected for imputation.
Outlier detection
When outlier detection was enabled, Z-statistics were calculated using the measured standard deviation instead of the pooled standard deviation for outlier spots. Outliers were determined by calculating σε, the standard deviation of the residual error ε = σ - σ' for spots with σ > σ'. Spots for which ε > 2σε were treated as outliers, similar to [26]. The measured standard deviations for the outlier points were considered to be valid sample measurements of the variance process and were not excluded from the calculation of the pooled standard deviations for spots with similar intensities.
Comparison of Z-statistic and penalty-based statistics
In order to test the reproducibility of different test statistics (Fig. 6), two sets of three arrays were randomly selected from the 23 replicate arrays in Dataset 4. For both of these subsets, we calculated the several different test statistics described above. For each gene, the value of each of the test statistics from one 3-array subset was compared to the corresponding value from the other subset, using the squared Pearson's linear correlation coefficient, R2, and two non-parametric, rank-based correlation coefficients, Spearman Rho and Kendall Tau, which were calculated using JMP (SAS Inc., Cary, NC). This entire process was repeated twice with the remaining arrays in Dataset 4, yielding a total of three independent comparisons. In total, six non-overlapping sets of three arrays – 18 arrays in all – were drawn from the original pool of 23 arrays, leaving 5 arrays that were not used in this analysis. As the sets are non-overlapping, each comparison is based on independent data.
In order to evaluate the accuracy of the different test statistics, we compared these statistics to an approximate "gold standard" measure (see Fig. 5). 3 arrays were randomly selected from the 23 arrays in Dataset 4; the other 20 were used to calculate "gold standard" t-statistics to which the results from the n = 3 dataset could be compared. The R2 value and the linear regression slope coefficient with intercept set to 0 were calculated between the corresponding experimental statistic and "gold standard" t-statistic for each gene. Only spots for which there were at least 15 replicates in the "gold standard" set of arrays were used. This process was repeated on a total of 6 random subsets.
List of abbreviations used
Xavg: Average logged intensity in channel X
Yavg: Average logged intensity in channel Y
Mavg: Average ratio
σM2: Variance of the average ratio
σM: Standard deviation of the average ratio
σM': Pooled standard deviation of the average ratio
Iavg: Average logged intensity
Imin: Minimum logged intensity
σM'(Iavg): Pooled standard deviation of the average ratio, pooled using Imin
σM'(Imin): Pooled standard deviation of the average ratio, pooled using Imin
μ: Paired difference of ratios in a reference sample experiment
σμ': Pooled standard deviation of the paired difference of ratios
Z(Iavg): Z-statistic calculated with standard deviation pooled using average logged intensity
Z(Imin): Z-statistic calculated with standard deviation pooled using minimum logged intensity
N: Number of replicate arrays
t: Standard t-statistic
s0: Penalty factor
d90th percentile: Penalized t-statistic calculated using 90th percentile s0
dSAM: Penalized t-statistic calculated using Significance Analysis of Microarrays
tgold: "Gold standard" t-statistic calculated using 20 replicate arrays
R: Pearson's correlation coefficient
Authors' contributions
JC and SN conceived of the study, implemented the Iavg and Imin algorithms, processed the microarray data used in the analyses, and completed the comparisons between the algorithms described herein. JC and GGC performed some of the microarray experiments analyzed in this study. MAG and GGC participated in the design and coordination of the study. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We would like to thank John Aach, Adnan Derti, and Yonatan Grad for a critical review of this manuscript, and Sandrine Dudoit for helpful discussions. We are grateful to the Alliance for Cellular Signaling for publicly releasing one of the microarray datasets used in this study. This work was supported by the Leet and Patterson Trust (GG-C) and NIH P50-HL56985 (MAG).
Contributor Information
Jason Comander, Email: jcomand@mit.edu.
Sripriya Natarajan, Email: nataraja@mit.edu.
Michael A Gimbrone, Jr, Email: mgimbrone@rics.bwh.harvard.edu.
Guillermo García-Cardeña, Email: ggarcia-cardena@rics.bwh.harvard.edu.
References
- Segal E, Yelensky R, Koller D. Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics. 2003;19 Suppl 1:I273–I282. doi: 10.1093/bioinformatics/btg1038. [DOI] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Tilstone Claire. Vital statistics. Nature. 2003;424:610–612. doi: 10.1038/424610a. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Yang YH, Speed T. Statistical issues in cDNA microarray data analysis. Methods Mol Biol. 2003;224:111–136. doi: 10.1385/1-59259-364-X:111. [DOI] [PubMed] [Google Scholar]
- Comander J, Weber GM, Gimbrone M. A., Jr., Garcia-Cardena G. Argus--a new database system for Web-based analysis of multiple microarray data sets. Genome Res. 2001;11:1603–1610. doi: 10.1101/gr.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudoit S, Gentleman RC, Quackenbush J. Open source software for the analysis of microarray data. Biotechniques. 2003;Suppl:45–51. [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler TB, Crosby L, Morgan KT. Normalization and analysis of DNA microarray data by self-consistency and local regression. Genome Biol. 2002;3:RESEARCH0037. doi: 10.1186/gb-2002-3-7-research0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA. Fundamentals of experimental design for cDNA microarrays. Nat Genet. 2002;32 Suppl:490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Kamat V, Dougherty ER, Bittner ML, Meltzer PS, Trent JM. Ratio statistics of gene expression levels and applications to microarray data analysis. Bioinformatics. 2002;18:1207–1215. doi: 10.1093/bioinformatics/18.9.1207. [DOI] [PubMed] [Google Scholar]
- Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui KW. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J Comput Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- Pan W, Lin J, Le CT. How many replicates of arrays are required to detect gene expression changes in microarray experiments? A mixture model approach. Genome Biol. 2002;3:research0022. doi: 10.1186/gb-2002-3-5-research0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MK, Afshari CA, Bennett L, Bushel P, Martinez J, Walker NJ, Churchill GA. Statistical Analysis of a Gene Expression Microarray Experiment with Replication. Bar Harbor, The Jackson Laboratory; 2001. http://www.jax.org/staff/churchill/labsite/research/expression/niehs.pdf [Google Scholar]
- Broberg P. Statistical methods for ranking differentially expressed genes. Genome Biol. 2003;4:R41. doi: 10.1186/gb-2003-4-6-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudoit S, Yang YH, Callow MJ, Speed T. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica Sinica. 2002;12:111–139. [Google Scholar]
- Lonnstedt I, Speed T. Replicated microarray data. Uppsala, Uppsala University; 2001. http://www.stat.berkeley.edu/users/terry/zarray/TechReport/Baypap4d.pdf [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Tibshirani R, Storey JD, Tusher V. Empirical Bayes analysis of a microarray experiment. J Am Stat Assoc. 2001;96:1151–1160. doi: 10.1198/016214501753382129. [DOI] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Colantuoni C, Henry G, Zeger S, Pevsner J. SNOMAD (Standardization and NOrmalization of MicroArray Data): web-accessible gene expression data analysis. Bioinformatics. 2002;18:1540–1541. doi: 10.1093/bioinformatics/18.11.1540. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/S0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Baggerly KA, Coombes KR, Hess KR, Stivers DN, Abruzzo LV, Zhang W. Identifying differentially expressed genes in cDNA microarray experiments. J Comput Biol. 2001;8:639–659. doi: 10.1089/106652701753307539. [DOI] [PubMed] [Google Scholar]
- Coombes KR, Highsmith WE, Krogmann TA, Baggerly KA, Stivers DN, Abruzzo LV. Identifying and quantifying sources of variation in microarray data using high-density cDNA membrane arrays. J Comput Biol. 2002;9:655–669. doi: 10.1089/106652702760277372. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32 Suppl:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Rocke DM, Durbin B. A model for measurement error for gene expression arrays. J Comput Biol. 2001;8:557–569. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- Nadon R, Shi P, Skandalis A, Woody E, Hubschle H, Susko E, Rghei N, Ramm P. Statistical inference methods for gene expression arrays. St. Catharines, Imaging Research Inc.; 2001. http://imaging.brocku.ca/PDF_files/AST_Technicalnote.pdf [Google Scholar]
- Tsodikov A, Szabo A, Jones D. Adjustments and measures of differential expression for microarray data. Bioinformatics. 2002;18:251–260. doi: 10.1093/bioinformatics/18.2.251. [DOI] [PubMed] [Google Scholar]
- Yang IV, Chen E, Hasseman JP, Liang W, Frank BC, Wang S, Sharov V, Saeed AI, White J, Li J, Lee NH, Yeatman TJ, Quackenbush J. Within the fold: assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 2002;3:research0062. doi: 10.1186/gb-2002-3-11-research0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Buckley MJ, Dudoit S, Speed T. Comparison of Methods for Image Analysis on cDNA Microarray Data, Technical report #584. 2000. http://www.stat.berkeley.edu/users/terry/zarray/TechReport/584.pdf
- Dudley AM, Aach J, Steffen MA, Church GM. Measuring absolute expression with microarrays with a calibrated reference sample and an extended signal intensity range. Proc Natl Acad Sci U S A. 2002;99:7554–7559. doi: 10.1073/pnas.112683499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ, Furukawa T, Steffen MA, Church GM, Cepko CL. Microarray analysis of the transcriptional network controlled by the photoreceptor homeobox gene Crx. Curr Biol. 2000;10:301–310. doi: 10.1016/S0960-9822(00)00379-1. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Xu R, Li X. A comparison of parametric versus permutation methods with applications to general and temporal microarray gene expression data. Bioinformatics. 2003;19:1284–1289. doi: 10.1093/bioinformatics/btg155. [DOI] [PubMed] [Google Scholar]
- Workman C, Jensen LJ, Jarmer H, Berka R, Gautier L, Nielser HB, Saxild HH, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002;3:research0048. doi: 10.1186/gb-2002-3-9-research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin B, Rocke DM. Estimation of transformation parameters for microarray data. Bioinformatics. 2003;19:1360–1367. doi: 10.1093/bioinformatics/btg178. [DOI] [PubMed] [Google Scholar]
- Qian J, Kluger Y, Yu H, Gerstein M. Identification and correction of spurious spatial correlations in microarray data. Biotechniques. 2003;35:42–4, 46, 48. doi: 10.2144/03351bm03. [DOI] [PubMed] [Google Scholar]
- Manduchi E, Grant GR, McKenzie SE, Overton GC, Surrey S, Stoeckert C. J., Jr. Generation of patterns from gene expression data by assigning confidence to differentially expressed genes. Bioinformatics. 2000;16:685–698. doi: 10.1093/bioinformatics/16.8.685. [DOI] [PubMed] [Google Scholar]
- Agilent Fluorescent Direct Label Kit Protocol Rev. 2.1. G2555-98003. Agilent Technologies; 2003. http://www.chem.agilent.com [Google Scholar]
- Sokal R, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. 3rd ed.; page 53. W.H. Freeman and Co.; 2000. p. page 53. [Google Scholar]
- Ge Y, Dudoit S, Speed T. Resampling-based multiple testing for microarray data hypothesis. Test. 2003;12:1–77. [Google Scholar]
- Dudoit S, Shaffer JP, Boldrick JC. Multiple Hypothesis Testing in Microarray Experiments. U.C. Berkeley Division of Biostatistics Working Paper Series. Working Paper 110. 2002. http://www.bepress.com/ucbbiostat/paper110
- Significance Analysis of Microarrays http://www-stat.stanford.edu/~tibs/SAM/