

Abstract
Genetic vaccines are emerging as a powerful modality to induce T-cell responses to target tumor associated antigens (TAA). Viral or plasmid DNA or RNA vectors harbor an expression cassette encoding the antigen of choice delivered in vivo by vaccination. In this context, immunizations with minigenes containing selected, highly antigenic, T-cell epitopes of TAAs may have several advantages relative to full-length proteins. The objective of this study was to identify an optimal scaffold for minigene construction. We generated a number of minigenes containing epitopes from the carcinoembryonic antigen (CEA) model TAA and utilized muscle DNA electro-gene-transfer (DNA-EGT) to vaccinate HLA-A*0201 (HHD) and CEA/HHD double transgenic mice. The components utilized to construct the minigenes included CD8+ T cell epitopes and (or) anchor modified analogs that were selected on the basis of their predicted binding to HLA-*A0201, their uniqueness in the human proteome, and the likelihood of cancer cell natural processing and presentation via MHC-I. Other candidate components comparatively tested included: helper CD4+ T-cell epitopes, flanking regions for optimal epitope processing (including both proteasome-dependent and furin-dependent polypeptide processing mechanisms), and immunoenhancing moieties. Through a series of comparative studies and iterations we have identified an optimal minigene scaffold comprising the following elements: human tissue plasminogen activator (TPA) signal peptide, T-cell epitopes connected by furin sensitive linkers, and the E. Coli enterotoxin B subunit. The selected epitope modified minigenes (EMM) delivered by DNA-EGT were able to break immune tolerance in CEA/HHD mice and induce a strong immune response against all epitopes tested, independently of their relative positions within the scaffold. Furthermore, the optimized EMMs delivered via DNA-EGT were more immunogenic and exerted more powerful antitumor effects in a B16-CEA/HHD metastatic melanoma model than a DNA vector encoding the full-length protein or a mixture of the same peptides injected subcutaneously. Our data may shed light on the optimal design of a universal vehicle for epitope-targeted, genetic cancer vaccines.
Keywords: cancer vaccine, epitope prediction, minigene
Introductıon
The development of therapeutic cancer vaccines is based on the notion that the induction of immune responses against self antigens, particularly those either mutated or overexpressed in tumors vs. the corresponding normal tissues, may attenuate cancer growth and metastasis.1 In particular, since cytotoxic T lymphocytes (CTL) have been shown to play a crucial role in tumor surveillance,2,3 cancer vaccines are being designed to elicit strong and durable T-cell responses against selected tumor-associated antigens (TAAs).4 CTLs recognize short peptides derived from intracellularly synthesized proteins that are presented on the surface of target cells in association with Major Histocompatibility Complex class I (MHC-I) molecules. CTL epitopes have been identified in multiple clinically relevant TAAs defining the so called “cancer antigenome.”5 In order to break tolerance and potentiate immune responses against self-antigens, several efforts have been spent over the years to generate anchor-modified analog peptides. By virtue of their higher affinity for MHC class I, analogs are able to bind to the MHC complex with a longer half-life thus soliciting a more efficient priming of T cells which, once primed, are capable of subsequent recognition of the wild-type epitopes on the surface of target cells, including cancer cells.6,7
Electro-gene-transfer (EGT) of plasmid DNA in vivo is an efficient and safe methodology that results in greater DNA uptake per cell and enhanced protein expression.8,9 DNA-EGT results in long-term immune responses against target antigens in a variety of species10,11 and can be repeatedly administered to boost immune responses as required for the maintenance of antitumor immunity.12-14 In addition to increased gene expression, DNA-EGT is believed to enhance the immune response through stimulating local secretion of inflammatory chemokines and cytokines, the recruitment of antigen presenting cells (APCs) to the EGT site and by promoting the trafficking of APCs to the draining lymph nodes. Indeed, the addition of in vivo EGT has been associated consistently with an enhancement of cell-mediated and humoral immune responses in small and large animals,11 supporting its use in human clinical trials. Our group has shown that DNA-EGT is able to induce high levels of cell-mediated immunity (CMI) to a variety of TAAs in small and large animal species upon injection of plasmids expressing codon-usage optimized variants of full-length or truncated forms of TAAs, including those derived from carcinoembryonic antigen (CEA), the HER2/neu oncogene, telomerase reverse transcriptase (hTERT) and matrix metalloproteinase 11 (MMP11).14-20
It has been surmised that immunizations with minigenes containing select, minimal T-cell epitopes may have several advantages as compared with full-length or even truncated proteins.21 A technical advantage of smaller minigenes is their compatibility with commonly used delivery agents, including plasmid DNA vectors. Also, full-length proteins may have unknown, non-desirable and potentially even toxic biological activity in contrast to minigenes that deliver only specific, immunologically relevant targeting information. Immunization with the entire gene may lead to the processing of immunodominant epitopes that may be highly competitive for binding to MHC, although some of these epitopes may be ineffective due to immune tolerance mechanisms and thymic ablation of the corresponding T-cell clones.22 In contrast, minigenes can be designed to contain only a select number of non-immunodominant epitopes with reduced frequency of negative thymic selection. Furthermore, polyepitope DNA vaccines can be constructed to contain epitope analogs to increase the chances of breaking immune tolerance and epitopes can be spaced by suitable linkers conducive to efficient processing.
Numerous approaches have been previously described for the generation of minigenes targeting TAAs engineered for exploitation as cancer vaccines.23 However, despite intensive studies and various strategic approaches, the design of an optimal minigene that maximizes epitope-specific immune responses has so far remained elusive. Here, we have undertaken a systematic effort to identify an optimal scaffold for epitope-modified minigene (EMM) constructs.
We have recently described an efficient T-cell epitope in silico prediction approach based on 3 criteria: 1) binding to 1 out of 5 common MHC class I alleles; 2) uniqueness to the antigen of interest; and 3) increased likelihood of natural processing. We characterized 225 candidate T-cell epitopes (wild-type and fixed-anchor analogs) selected within CEA, HER2/neu and hTERT by high-throughput stable binding to MHC using the iTopia epitope discovery assay.24 On the basis of these results, we concluded that the combination of in silico prediction and a biochemical binding/stability assay represents an accurate prediction of novel TAA-derived epitopes. Indeed this was later confirmed by further validation of Human Leukocyte Antigen (HLA)-A*0201 restricted fragments in HLA-A*0201 (HHD) transgenic mice.
In the present study, we generate and functionally characterize a series of EMMs targeting human CEA as a model antigen. Human CEA is one of the most well-studied TAAs over the past 20 y.25-27 Its aberrant expression has been long correlated with many cancer types. Furthermore several therapeutic strategies have been developed against CEA and brought into advanced clinical trials.28
Here, by comparatively assessing distinct approaches to EMM design both in vitro and by iterative immunization studies upon DNA-EGT in HHD/CEA double transgenic mice in vivo, we define an optimal minigene construct exhibiting strong immunogenicity and therapeutic efficacy.
Results
Selection of CEA/HLA-A*0201 restricted epitopes
By means of an in silico prediction algorithm previously developed by our group,24 we selected 17 CEA epitopes restricted to HLA-A*0201 (Table 1). In addition to the predicted MHC-I allele HLA-A2 binding affinity, primary epitope selection was also based on increased susceptibility to proteolytic processing and the uniqueness of the target antigen in the human genome. A further criterion was the possibility of designing fixed-anchor modified epitope analogs. The algorithm selected nonamers (CEA.569, 589, 687, 691) or decamers (CEA.100, 307, 411, 589, 682, 690) predicted to bind to MHC-I pocket and identified by the position of the first residue occurring within the CEA protein primary sequence. CEA.589 was selected in 2 versions, both as nonamer and decamer (referred as 58910mer), as both forms were predicted to exhibit MHC-I pocket binding capabilities. In 6 epitopes (CEA.100, 307, 411, 589, 58910mer, and 682), wild-type residues at position 9 or 10 were replaced with valine. CEA.690 was modified by replacing isoleucine with leucine in position 2. CEA.691, 569 and 687 were left without modifications.
Table 1. Wild-type and anchor modified HLA-A*0201 restricted CEA peptides used in this study.
| Peptide | Peptide name | % Peptide Binding vs. Positive Control | Affinity Value (EC50) | Off Rate T1/2 | ||
|---|---|---|---|---|---|---|
| # | Sequence | |||||
| 1 | VLYGPDDPTV | CEA.411V10 | 55 | 5.50E-06 | 14 | |
| 2 | GLMIGVLVGV | CEA.690L2 | 62 | 1.70E-07 | 80 | |
| 3 | VLYGPDTPIV | CEA.589V10 | 66 | 4.70E-06 | 16 | |
| 4 | GLSAGATVGV | CEA.682V10 | 63 | 6.30E-06 | 14 | |
| 5 | GLNRTTVTTV | CEA.307V10 | 47 | 6.60E-06 | 2.2 | |
| 6 | VLYGPDTPV | CEA.589V9 | 74 | 4.40E-06 | 25 | |
| 7 | IIYPNASLLV | CEA.100V10 | 70 | 4.70E-06 | 3.3 | |
| 8 | VLYGPDDPTI | CEA.411 | 69 | 3.20E-06 | 4.5 | |
| 9 | GIMIGVLVGV | CEA.690 | 46 | 2.50E-07 | 18 | |
| 10 | VLYGPDTPII | CEA.58910mer | 76 | 2.90E-06 | 3 | |
| 11 | GLSAGATVGI | CEA.682 | 78 | 2.50E-06 | 1.9 | |
| 12 | GLNRTTVTTI | CEA.307 | 67 | 1.20E-05 | 1.5 | |
| 13 | VLYGPDTPI | CEA.5899mer | 79 | ND | 5.5 | |
| 14 | IIYPNASLLI | CEA.100 | 73 | 3.00E-06 | 1.7 | |
| 15 | IMIGVLVGV | CEA.691 | 74 | 1.20E-07 | 41 | |
| 16 | YVCGIQNSV | CEA.569 | 56 | 3.90E-06 | 6.5 | |
| 17 | ATVGIMIGV | CEA.687 | 89 | 5.00E-06 | 3.8 | |
| 18 | YLSGANLNL | CEA.605 (cap1) | 77 | 1.60E-06 | 21 | |
Peptides were selected in silico by a prediction algorithm. Modified residues are underlined and indicated in bold. The iTopia assay was used to evaluate candidate peptide binding and complex stability using microtiter plates coated with MHC-I monomers. The percentage of peptide binding relative to the positive control, Affinity (EC50) and Off-rate (half-life, T1/2) are indicated in the columns. ND = not determined.
To determine their biochemical properties, wild-type peptides and corresponding analogs were synthesized and characterized for MHC binding and complex stability using the iTopia Epitope Discovery System. Peptides were incubated in duplicate in MHC-coated wells (refer to Methods) and peptides exceeding the threshold of 30% of the assay positive control were classified as binders. As shown in Table 1, according to this standard, all peptides were able to bind HLA-A2 and the percentage of the positive control is reported. Interestingly, most of the wild-type and anchor-modified peptides showed high affinity to HLA-A*0201 exhibiting a comparable half-maximal effective concentration (EC50) to the CAP-1 peptide (CEA.605) used as positive control29 (Fig. 1A). Although the majority of the analogs displayed a lower EC50, and thus had higher affinity for HLA-A2 than the wild-type epitopes from which they were derived, no improvement in binding affinity was observed for either CEA.690L2 or CEA.307V10, the latter of which actually displayed lower affinity than its wild-type counterpart.

Figure 1. Affinity and stability of HLA complexes formed with the in silico selected CEA-native peptides and analogs. (A and B) The iTopia assay was used to evaluate carcinoembryonic antigen (CEA) peptide binding and HLA complex stability. Peptides (designated by the position of the first amino acid in the CEA protein) were assayed using 11 μM peptide incubated for 18 h at 21 °C with β2M, anti-MHC mAb, and plate-bound MHC heavy chain. Binding of 10 native peptides and 7 analogs were tested, including nonomers (9mers) and decamers (10mers). Analogs are labeled by the substitution and position in the peptide sequence. Peptide variants corresponding to 9mers and 10mers beginning at the same amino acid position are indicated, with peptide lengths different from 9 (10mers) indicated. CAP-1 (CEA.605) was also evaluated as a positive control. (A) Binding affinity of CEA peptides and their analogs to the HLA-A*0201 allelic variant expressed as half-maximal effective concentration (EC50) reported in molarity. Each peptide was tested in duplicate on the same plate. This experiment has been performed twice with similar results. ND = not determined. (B) Complex stability was measured by replacing the assay buffer with fresh peptide-free buffer and incubating at 37 °C. Binding measurements were performed at 8 time points over an 8 h incubation period to calculate estimated HLA:peptide complex stability. The half-life, or T1/2, is interpreted as the time required for the relative binding to diminish by 50% and is expressed in hours on the y-axis. Black and white dots indicate the affinity and the stability of wild-type and analog peptides, respectively.
The binding stability of the peptide-MHC complex was also evaluated over time. Despite the relatively high affinity of candidate epitopes, MHC complex stability varied considerably with 10/17 peptides (59%) forming stable complexes, here arbitrarily defined as a half-life (T1/2) > 4 h (Fig. 1B). An improved stability of peptide analogs compared with native sequences was observed, in particular for CEA.411V10, 589V9, 589V10, 682V10 and 690L2 (3.1, 4.5, 5.3, 7.4 and 4.4-fold, respectively). These data were further confirmed in a cell-based T2 binding assay (data not shown). Due to poor in vitro binding features, CEA.100 and its analog were excluded from subsequent analyses. We also decided to exclude CEA.690 and CEA.690L2 considering its high sequence similarity with the already clinically validated, stable CEA.691 epitope.30
To verify the biologic relevance of the epitope modification suggested by our algorithm, we next set out to test the impact on their immunogenicity in vivo, and further, assess their cross-reactivity with wild-type epitopes. To this end, HHD mice were immunized with a mixture containing the candidate CD8+ T cell peptide immunogen along with hepatitis B virus (HBV)-core128 helper peptide and immunostimulatory synthetic oligonucleotides (CpG) in Incomplete Freund’s Adjuvant (IFA). Mice were given a second injection 15 d later with the same mix of components. Two weeks later, peripheral blood mononuclear cells (PBMCs) and splenocytes were recovered for immunological assays, including intracellular staining for IFNγ release upon stimulation with wild-type or analog peptides. As shown in Figure 2, immunization with any of the designed analogs induced a cross-reactive response (Fig. 2A), eliciting IFNγ release from PBMCs upon secondary stimulation with wild-type epitope. Furthermore, analogs were much more immunogenic than their wild-type native peptides in activating PBMCs (Fig. 2A) and splenocytes (Fig. 2B). In particular, CEA.589V10 was highly potent, in sharp contrast to its native counterpart not modified at position 10 with valine, exhibiting a 310-fold increase in immunogenicity. Similar results were obtained for CEA.682, while lower immunogenicity was measured for CEA.411V10, albeit significantly higher than the corresponding wild-type epitope, and comparable to that of CEA.691 which was not modified by the algorithm. Overall, the enhancement of immune response for peptide analogs relative to the corresponding wild-type peptides ranged from 11.8- to 310-fold (Fig. 2B). Specifically, the fold increase in the immunogenicity of peptide analogs were 11.8, 310, 12.2, 105, and 16-fold for CEA.411V10, 589V10, 589V9, 682V10, and 307V10, respectively, compared with wild-type counterparts. Importantly, these immunogenicity data correlate with binding data (Fig. 1), thus confirming that binding affinity and off-rate are important parameters to predict the immunogenic potential of a defined epitope.

Figure 2. Immunogenicity of selected epitopes. (A–C) HHD mice were immunized with the indicated carcinoembryonic antigen (CEA) peptides (shown on the y-axis) by subcutaneous injection of 100 μg of the wild-type peptide or analog in a mixture with 140 μg HBV-T helper epitope58 and 50 μg CpG-ODN in Incomplete Freund Adjuvant (IFA). (A) Peripheral blood mononuclear cells (PBMCs) from immunized HHD mice were stimulated in vitro with the indicated peptide and analyzed for CD8+ T cell interferon-γ (IFNγ) secretion by intracellular staining and flow cytometry 2 to 3 wk after the last treatment. Mice immunized with analogs were analyzed both for cell-mediated immunity (CMI) elicited against the analog itself (black bar) and against the corresponding wild-type epitope (lightly shaded bar). Each sample was obtained by pooling PBMCs from 4–5 HHD mice and analyzed in duplicate. (B) Splenocytes from peptide immunized HHD mice were stimulated in vitro with pooled peptides and analyzed for CD8+ T cell IFNγ secretion by intracellular staining of cell-surface stained splenocytes and flow cytometry. Each black dot represents a single mouse; the empty dot represents the geometric mean of the group. The dashed horizontal lines represents a cut-off of 0.1% CD8+IFNγ+ cells chosen to indicate biologically relevant immunogenicity. Statistical analysis was performed by Student's t test; *P-value < 0.05. (C) Cytotoxic T lymphocyte (CTL) effectors isolated from spleens of immunized HHD mice (n = 3 per group) 5 d after final vaccination with the indicated CEA peptides were stimulated in vitro with peptide pools and recombinant human IL-2. On day 5, these in vitro stimulated cells were used as CTL effector cells, and the CTL activity was determined by a 6 h 51Cr-release cytotoxicity assay using the indicated cell lines as targets. Effector (E) cells were incubated with target (T) cells at E:T ratio = 100. Each bar represents the lysis obtained with effectors from each group vaccinated with the indicated peptide. Colo205 (CEA−) human colon adenocarcinoma cells served as a negative control. The assay was run in triplicate.
To verify whether elicited effectors are indeed capable of lysing human cancer cells, we used as targets different human HLA-A*0201+ colon cancer cells, including SW480, Colo705, and Colo201 which express CEA, and Colo205 that is negative for CEA. These results demonstrate that cells from mice immunized with CEA.411V10, 589V9, 589V10, 682V10, and 691 peptides were able to efficiently lyse only CEA positive colon cancer cells, although with some variability (Fig. 2C). In a different experimental setting (data not shown), CEA.569- and CEA.687-specific effectors were also able to recognize CEA+ cells whereas poor lytic activity was observed by CEA.307 and CEA.307V10 primed CTLs. Importantly, these data show that most of the epitopes predicted by the algorithm are immunogenic and effectively presented in complex with MHC-I on malignant cells.
In light of these results, we decided to utilize the best performing wild-type and anchor-modified epitopes in the construction of optimized minigene constructs: CEA.691, CEA.411V10, CEA.589V10, and CEA.682V10.
Defining the best scaffold for EMMs design
To define a universal scaffold for the construction of an immunogenic and therapeutically effective minigene, several EMM variants were designed and constructed for functional characterization. A first series was based on proteasome-dependent epitope processing. The ubiquitin degradation pathway is an efficient endogenous polyepitope processing mechanism and ubiquitin fusion has been shown to improve the induction of CD8+ T-cell response to genetic epitope vaccines by targeting the polypeptide for rapid degradation by the proteasome.31,32 Thus, 2 EMMs were designed to covalently linking the same string of CEA epitopes to a mutant form of ubiquitin (G76V, 37) encoded in the vector and fused to the CEA peptide via the flexible linker peptide VGKGGSGG (Fig. 3A and B). The design also included the use of 3 spacer sequences, AAY33 (Fig. 3A), LRA or RLRA (Fig. 3B) designed to ensure efficient epitope processing by the proteasome.

Figure 3. Epitope modified minigene design. (A–F) To determine the optimum epitope modified minigene (EMM) scaffold, candidate DNA vectors were constructed for the expression of immunogenic carcinoembryonic antigen (CEA) polyepitopes. The helper epitope from Tetanus toxin (p30) is encoded on the majority of the constructs as indicated. (A–B) Ubiquitin (ubi)-fused proteasome-dependent EMM variations in which either AAY (A), or LRA, or RLRA (B) were utilized as a processing spacer between epitopes. (C–F) Furin-dependent EMM variations in which REKR was the selected sequence for furin-specific cleavage and epitope processing. The leader peptide from the secretory protein tissue plasminogen activator (TPA) was used for guiding the polyepitope into the endoplasmic reticulum. (C) Minimal furin-dependent EMM with the p30 helper epitope. (D) The membrane-translocating sequence (MTS) from the HIV-1 derived Tat gene at the C-terminus and the p30 helper epitope. (E) Fusion of the p30 helper epitope and heat-labile toxin B (LTB) subunit of E. coli to the polyepitope C-terminus. (F) LTB fusion only.
A second series (Fig. 3C–F) was based on the proteasome-independent mechanism of proprotein processing operated by furin in the trans-Golgi compartment of the secretory pathway.34 For this reason, the leader peptide from the secretory protein tissue plasminogen activator (TPA) was included in all these particular EMMs in order to ensure translocation of the nascent protein into the endoplasmic reticulum (ER).35 The leader peptide was followed by the string of the previously selected four epitopes CEA.411V10, CEA.691, CEA.589V10, and CEA.682V10 to comprise a polyepitope. Between each epitope, a furin-specific cleavage site (REKR) from the human immunodeficiency virus type-1 (HIV-1) glycoprotein gp120/gp41 was inserted.36 The first minigene (TPA-CEA-Furin-p30, Figure 3C) bears the p30 helper epitope from Tetanus toxin in addition to the above-described elements.37 In a second furin-specific minigene construct, the membrane translocating sequence (MTS) from the HIV-1 derived Tat gene at the C-terminus (TPA-CEA-Furin-p30-MTS, Figure 3D) was included for its ability to deliver exogenous antigens into the intracellular compartments where processing into MHC-binding peptides occurs. To assess if fusion of the polypeptide to the heat-labile toxin B subunit of E. coli (LTB) is able to increase cell-mediated immune responses after DNA-EGT, as previously observed by our group for several antigens such as CEA, hTERT and MMP-11,38-41 2 other LTB-containing minigenes were constructed, with or without the p30 helper epitope (TPA-CEA-Furin-p30-LTB, Figure 3E and TPA-CEA-Furin-LTB, Figure 3E and 3F, respectively).
Next, in order to assay our candidate constructs specifically designed to elicit targeted immunity, these EMMs were used to vaccinate HHD mice by DNA-EGT. Mice received 4 weekly injections and CMI was analyzed 2 wk after the last boost. PMBCs from the immunized animals were challenged in vitro using the pool of modified or native peptides, and CMI was measured by intracellular staining for IFNγ. As shown in Figure 4, we found that all the minigenes tested elicited a strong CD8+ T cell immune response among PBMCs from EMM-immunized HHD mice. Importantly, the observed CMI was cross-reactive with wild-type epitopes. Of note, the fusion with LTB resulted in significantly higher levels of CMI.
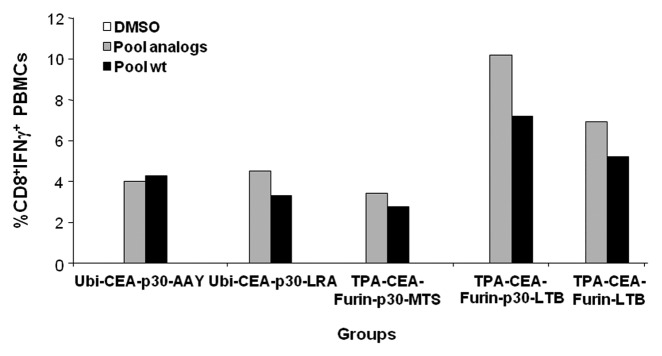
Figure 4. Cross-reactive cell-mediated immunity in epitope modified minigene immunized HHD mice. HHD mice (n = 5 per group) were immunized by DNA electro-gene transfer with candidate epitope modified minigenes (EMMs; refer to Figure 3) receiving 4 weekly subcutaneous injections of 50 μg plasmid DNA followed by electroporation. Two weeks after the 4th injection, peripheral blood mononuclear cells (PBMCs) from mice within the indicated group were combined and challenged in vitro using the pool of analogs or wild-type (wt) epitopes. Cell-mediated immunity was measured by CD8+ T cell IFNγ secretion by intracellular staining of cell-surface stained PBMCs and flow cytometry. The experiment was performed in duplicate and average values are reported.
To assess the contribution of each epitope to the overall immunogenicity of the polyepitope containing EMMs, immunized mice were euthanized and the immune response against individual wild-type epitopes was measured by intracellular staining of splenocytes for IFNγ. As shown in Figure 5, epitope- and scaffold-specific differences in the percentage of CD8+IFNγ+ lymphocytes were observed. In response to immunization with EMMs acting through the ubiquitin degradation pathway with the AAY spacer, the immune response against CEA.682 was consistently relatively high, followed by the epitopes CEA.691 and CEA.589. However, CEA.411V10 component of the EMM was non-immunogenic, as no response was observed upon stimulation with the wild-type CEA.4ll epitope (Fig. 5A). Use of the LRA spacer gave rise to an effective immune response only against CEA.682 epitope (Fig. 5B) but not against the others, thus indicating that LRA spacer sequence is not an efficacious spacer element. Similar results were obtained with the RLRA spacer constructs (data not shown).

Figure 5. Immunologic analysis of splenocytes from epitope modified minigene immunized HHD mice. (A–E) HHD mice (n = 5 to 6 per group) were immunized by DNA electro-gene transfer with candidate epitope modified minigenes (EMMs; refer to Figure 3) receiving 4 weekly subcutaneous injections of 50 μg plasmid DNA followed by electroporation. Two weeks later, mice were sacrificed and splenocytes were stimulated in vitro with the individual wild-type epitope as indicated. CD8+ T cell interferon-γ (IFNγ) secretion was determined by intracellular staining of cell-surface stained splenocytes and flow cytometry. In each panel, the CD8+ response is reported corresponding to the EMM construct used for immunization: Ubi-CEA-p30-AAY (A) Ubi-CEA-p30-LRA (B), TPA-CEA-furin-p30-MTS (C), TPA-CEA-Furin-p30-LTB (D), and TPA-CEA-Furin-LTB (E). Black dots represent the immune response per each single mouse; empty dots represent the geometric mean of the group.
Overall the proteasome-independent, furin cleavage-dependent EMMs gave a similar pattern of immunogenicity (Fig. 5C–E), revealing a strong CD8+IFNγ+ immune response against CEA.682 and CEA.691, followed by a good response to CEA.589, but again an ineffective response against CEA.411. Interestingly, the fusion with LTB alone (TPA-CEA-furin-LTB) in the absence of both the MTS leader peptide and the p30 helper peptide (Fig. 5E) appeared to stimulate the highest level of immune response in this assay.
In conclusion, taken together these data show that furin-based EMMs are more efficacious than proteasome-dependent EMMs, and importantly, that MTS and p30 are not essential components of the candidate EMM scaffold.
Lack of immunogenicity of CEA.411V10 is position-independent and is due to epitope competition
The results from the previous section indicate that proteasome-independent, furin-dependent EMMs have the best configuration for processing and presentation of the chosen epitopes. However, regardless of the processing context CEA.411V10 was poorly immunogenic as compared with the other 3 epitopes. There are 2 possible explanations that could account for this observation. In the first scenario, CEA.411V10 processing may be affected by a positional effect since this epitope was placed in the first position in the construction of all candidate EMM variants. Thus, it could not be excluded that the processing of the first epitope was inefficient due to intrinsic features of polypeptide folding and poor exposure of the cleavage site between the first and the second epitope to furin. A second possibility accounting for the reduced immunogenic potential of CEA.411V10 is that of epitope competition. Within the antigen presenting cell, co-expression and processing of the 4 epitopes with differing binding affinity for the MHC class I pocket may impair the binding, presentation, and the resultant immunogenicity of the weakest epitope.
To address the first point, a new furin-LTB-based EMM (TPA-CEA-Furin-Inverted-LTB) was designed in which the CEA.411V10 and CEA.682V10 positions were switched (Fig. 6A, upper panels). Upon immunization in HHD mice, the immunogenicity of the new construct was virtually identical to that of the original orientation (Fig. 6A, compare the right and left lower panels). Although neither construct elicited a strong response against the CEA.411 epitope, CEA.682V10 remained highly immunogenic when placed in the first position, suggesting that the poor immune response to CEA.411V10 was not due to a positional effect.

Figure 6. Epitope immunogenicity is position independent and affected by competition. (A) To analyze the effects of the epitope position within the polyepitope, HHD mice (n = 7 to 8 per group) were immunized with TPA-CEA-furin-LTB (left panels) or the version in which 411V10 and 682V10 were inverted (right panels) by electro-gene transfer. Splenocytes from peptide immunized mice were stimulated in vitro with the indicated epitopes three weeks after the last boost and analyzed for CD8+ T cell interferon-γ (IFNγ) secretion by intracellular staining of cell-surface stained splenocytes and flow cytometry. (B) To analyze the effects of MHC-binding pocket epitope competition, HHD mice (n = 3 to 4 per group) received 2 injections of either each peptide alone (left) or admixed together (right), both with CpG-ODN in Incomplete Freund Adjuvant. Three weeks later, the immune response of in vitro epitope-stimulated splenocytes was measured by CD8+ T cell interferon-γ (IFNγ) secretion via intracellular staining of cell-surface stained splenocytes and flow cytometry. Black dots represent the immune response for each mouse; empty dots represent the geometric mean of the group.
In order to test the second hypothesis, we performed peptide immunizations using either individual peptides or a mixture of the 4 and measured CMI by intracellular staining for IFNγ in response subsequent stimulation with each individual epitope. As shown in Figure 6B, mixed peptide vaccinations with CEA.682 all maintain comparable immunogenicity to single injection whereas CEA.691 and CEA.589 are slightly less immunogenic when injected as mixtures. In sharp contrast, CEA.411 is dramatically affected by co-injection. These data confirm that the intrinsic affinity to MHC in conjunction with epitope competition in the context of a polyepitope construct may affect the performance of a selected epitope candidate.
The optimized EMM is more immunogenic than the peptide mixture and exerts a potent therapeutic effect
In order to compare the immunogenicity of our optimized EMM with that of other platforms, we performed parallel anti-CEA vaccinations in tolerant HHD/CEA mice using three different immunogens, including the best performing EMM minigene (TPA-CEA-furin-LTB), the mixture of the same CEA peptides encoded by the minigene plus a helper epitope, and the full-length codon optimized cDNA coding for CEA fused to LTB. Mice receiving either full-length CEA or EMM were immunized by EGT and received 4 weekly DNA injections, whereas mice immunized by peptide mixture received two injections of peptides spaced by a 14-d interval. In either case, the CMI was measured 2 wk later against wild-type peptides as assessed by intracellular staining of splenocytes for IFNγ. Results (Fig. 7A) show that for each epitope, the highest level of CMI was achieved using the EMM construct, followed by peptide immunizations. In particular, CEA.682 immunogenicity was approximately 13-fold higher with EMM as compared with the full-length cDNA and 27-fold and 2-fold higher for CEA.691 and CEA.589, respectively. For this latter epitope, vaccination with EMM was the only approach capable to overcome the 0.1% response threshold, and was thus considered of sufficient strength to break immune tolerance.
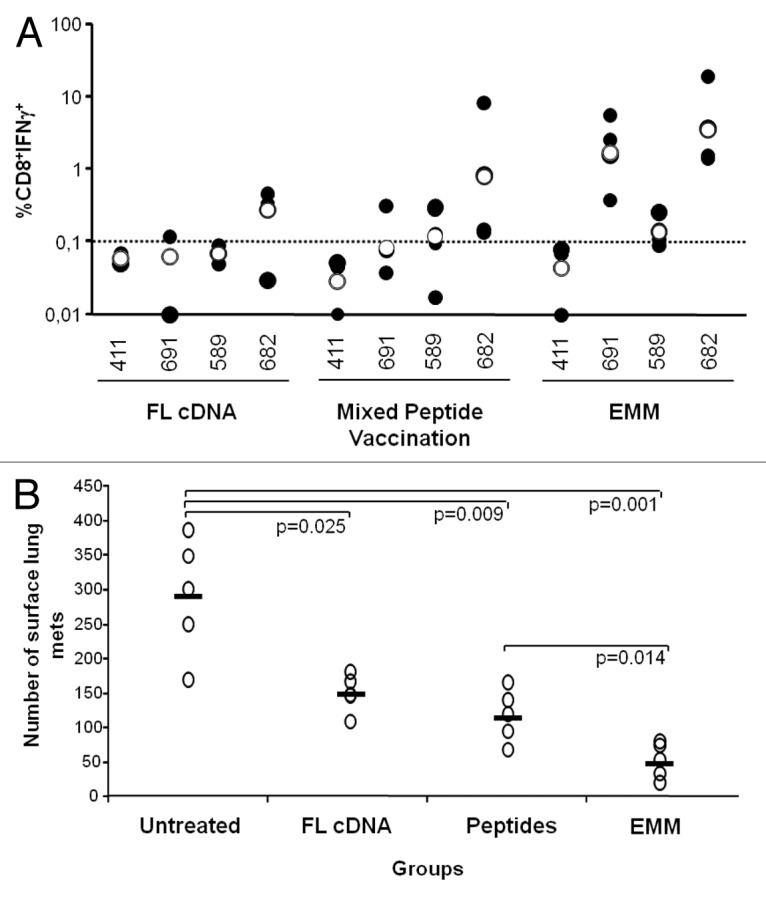
Figure 7. Comparison of full-length cDNA, peptides and epitope-modified minigenes targeting carcinoembryonic antigen. (A) To assay the relative immunogenicity of the epitope-modified minigene (EMM) vehicle, 2 groups (n =3-4) of HHD/CEA mice were vaccinated with 4 weekly DNA immunizations via electro-gene transfer (EGT) using full-length carcinoembryonic antigen (CEA) cDNA or the TPA-CEA-furin-LTB version of the EMM construct (refer to Figure 3). A third group (n = 4) received 2 injections of peptides (with CpG-ODN in Incomplete Freunds’ Adjuvant) spaced by 14 d interval. The immune response was analyzed 2 wk later by stimulating splenocytes from immunized mice with the indicated epitope and measuring CD8+ T cell interferon-γ (IFNγ) secretion via immunofluorescent staining of splenocytes and flow cytometry. Black dots represent the individual immune response of each mouse and empty dots represent the geometric mean of the group. Due to the low number of mice available, no statistical difference was observed among the specific immune responses as measured by Student’s t test. (B) To assay the anticancer effects of EMM immunization, vaccinated mice were challenged i.v. with 5 × 104 B16-HHD/CEA cells. Fourteen days later, lungs were explanted and surface metastases enumerated by means of an optical microscope. Empty dots represent the number of metastases (mets) per each single mouse lung and the bar represent the geometric mean of the group. Statistical analyses were done by Student's 2-tailed t test; P values are shown.
On the basis of these results we were encouraged to test the antitumor efficacy of the most immunogenic EMM in a prophylactic model. To this end, 2 groups of 5 HHD/CEA mice were first immunized in parallel by 4 weekly minigene vaccinations or with 2 bi-weekly injections of the peptide mixture. Two weeks after the end of the immunization schedule mice were challenged with B16-HHD/CEA cells injected i.v. Two weeks later, the mice were euthanized and the number of lung metastases was determined. As shown in Figure 7B, the full-length CEA cDNA was able to confer a significant antitumor effect, as previously observed in this model.42 However, TPA-CEA-furin-LTB vaccination exhibited a stronger anticancer activity in this aggressive model. Peptide mixture vaccination was also capable of exerting a potent anti-tumor effect, albeit to a lesser extent than TPA-CEA-furin-LTB.
Taken altogether, these data indicate that EMMs are a potent platform for the development of novel epitope-based cancer vaccines.
Discussion
T lymphocytes are a critical cellular component of immunity and play a crucial role in the eradication of cancer cells in mammals. The activation of cytotoxic (CD8+) and helper (CD4+) subsets of T lymphocytes is integral to cellular immunity, including immune responses targeting neoplastic cells.
CTLs and their T-cell receptors (TCR) recognize small peptides derived from intracellular antigens and presented by MHC-I molecules on the cell surface via the endogenous antigen processing and presentation pathway.43,44 Peptides for human CD8+ T-cell epitopes range in length from 7 to 14 amino acids, although they are typically 9–10 amino acids long. TCR recognition of the peptide-MHC class I molecule complexes on the cell surface triggers the cytolytic activity of CTLs, resulting in the death of cells presenting the stimulating peptide-MHC class I complexes.45
MHC class I restricted epitope vaccines have been shown to confer immune protection in animal models. Epitope-based vaccines offer a number of advantages compared with vaccines based on full-length TAAs. For example, peptide vaccines can induce effective immune responses to subdominant epitopes when there is tolerance to a dominant epitope. Furthermore, anchor-modified or heteroclitic peptide analogs can be constructed with the ability to break tolerance and (or) further increase immunogenicity relative to native peptides.46 Finally, the use of peptides as immunogens also minimizes safety risks potentially associated with the use of intact proteins.
In this study, we identified an efficient scaffold for the expression of minigenes encoding T-cell epitopes. To accomplish this aim, we first identified a set of immunogenic peptides within CEA and generated analogs selected on the basis of their class I MHC binding properties, specifically the HLA-A*0201 allelic variant. Prediction and selection of HLA-epitopes relied on specific algorithms that rank potential sequences (within a given protein, in our study CEA) on the basis of their binding properties to the MHC-I epitope-binding pocket. This ranking is particularly relevant for large antigens and can result in long lists of putative epitopes with differing relative scores and affinities. However, higher binding affinity does not necessarily translate into increased immunogenicity in vivo. For this reason, mice transgenic for HLA constitute a powerful means to measure candidate epitope immunogenicity in the native context of human major histocompatibility complexes, an approach conducive to prospective evaluation of vaccination strategies including the identification of novel or enhanced epitopes.24
The peptides and analogs described herein were selected on the basis of their ability to elicit a maximal tumor-specific immune response, as well as for their minimal potential for eliciting off-target autoimmunity.24 Specifically, the CEA protein was analyzed using an algorithm that ranked protein fragments based on various factors impacting immunogenicity, including CEA epitope binding affinity for HLA-A*0201, similarity of candidate epitopes to fragments of other human proteins, and amenability to immunogenic enhancement. The program introduced single amino acid substitutions in the synthesis of analogs inducing consistently increased biochemical affinity and stability (Fig. 1; Table 1). Of particular importance and as shown in Figure 2, these parameters were predictive of the peptide immunogenic efficacy in HHD mice in vivo as well as epitope processing and presentation efficiency by human colon cancer cells. CEA peptide-primed HHD effector T cells were in fact able to lyse only HLA-A2+/CEA+ target cells, albeit to varying degrees. However, it remained to be determined in a DNA vaccine setting how the relative cytotoxicity of polyepitope primed effector cells may be impacted by the CEA epitope expression level or other factors linked to processing machinery.
To address this question, we selected the optimized CEA.411V10, 589v10, 682, and 691 epitopes as the antigenic components of the EMM immunization construct and considered the vaccine scaffold structural elements. Previous studies have explored minigene vaccines comprising multiple contiguous minimal murine CTL epitopes47 or contiguous dominant HLA-A*0201 and HLA-A*11-restricted epitopes from the polymerase, envelope, and core proteins of hepatitis B virus and HIV, together with the PADRE (pan-DR epitope) universal T-cell epitope and an endoplasmic reticulum-translocating signal sequence.48 However, processing of individual epitopes in these minigenes is not assured due to the non-specific nature of proteasomal processing.49 The approach of inserting AAY spacers between the epitopes and the use of ubiquitin as a protein-targeting sequence was previously tested in the context of minigenes containing CTL epitopes derived from MPT64 and 38 kDa proteins of Mycobacterium tubercolosis.32 Pitcovski et al. (2006) assayed a melanoma DNA immunotherapy encoding a multi-epitope polypeptide having 3 repeats of 4 modified melanoma antigens linked by 5 spacer elements that signal proteasomal cleavage and fused to the E.coli LTB enterotoxin as adjuvant.50 In a separate study,51 an oral DNA minigene vaccine was also evaluated containing the HIV tat translocation peptide and a spacer (AAA) followed by an HLA-A2-restricted CEA T cell epitope, all inserted into a pCMV vector including an ER signal peptide.51 In another study, Lu et al.,52 described minigenes having multiple CTL epitopes joined via furin-sensitive linkers and containing the HIV-1 tat sequence.
In order to define a “universal” minigene structure, we have systematically explored most of these components in 2 minigenes categories: proteasome-dependent EMMs and furin-dependent EMMs. In the first category, the epitopes were expressed as a polypeptide fused to ubiquitin and spaced by proteasome-sensitive linkers. The second class of minigenes comprised translated immunogenic polypeptides addressed to the secretory pathway (via a TPA leader sequence), followed by furin-mediated processing in the trans-Golgi. As shown in Figures 4 and 5, our comparative approach revealed stronger immune responses using furin-based EMMs. In addition, fusion with LTB further enhanced the elicited response whereas other elements, such as the p30 helper epitope and MTS sequence were not found to be necessary. We have previously shown that LTB sequence contains CD4+ specific epitopes,38,53 therefore it is likely that LTB-fusion stimulates CD4+ T helper cells sufficiently to achieve optimal immune response. Lastly, we demonstrate that the relative position of the epitope within the minigene does not influence its immunogenicity. On the other hand, some epitopes, like CEA.411V10, suffer competition with other epitopes both when delivered in the format of a minigene or as a peptide mixture (Fig. 5). Observations that this epitope is poorly immunogenic when co-administered together with other CEA epitopes despite single peptide vaccination efficacy (Fig. 6) evinces the occurrence of epitope competition and may represent a potential limitation to our approach. However, combinations of short immunogenic peptides or, alternatively, synthetic long-peptide vaccines are currently being evaluated in clinical trials. One example is the Eastern Cooperative Oncology Group Phase II Trial E1696,54 in which a mixture of peptides containing multiple epitopes derived from MART-1, gp100, and tyrosinase was administered in patients with metastatic nonresectable melanoma. In another approach, cancer patients are vaccinated in Phase II and III studies with multiple tumor-associated peptides (TUMAPs) isolated from tumor specimens and identified by mass spectrometry.55 It would be of interest to evaluate whether epitope competition also occurs in patients in these trials.
Finally, the best performing EMM (TPA-CEA-furin-LTB) was also evaluated in a tumor challenge study, using the previously established and aggressive B16-CEA/HHD metastatic model in tolerant HHD/CEA transgenic mice. B16-CEA/HHD cells have been previously shown to be recognized by HHD effector T cells stimulated with CEA vaccines.42 Immunization of recipient mice with our minigene vaccine elicited significant immunogenic protection, thus demonstrating the anticancer therapeutic efficacy of our strategy (refer to Figure 7). It remains to be seen, however, how the immune response against each single epitope within the EMM construct contributes to the overall antitumor effect.
In conclusion, we have discovered a universal strategy applicable to the design of vaccine minigenes comprising either predicted or experimentally identified epitopes for delivery via DNA-EGT. Our results provide rationale for further studies, including testing this approach in combination with other treatment modalities, such as peptide vaccines or other genetic immunotherapy vectors.
Materials and Methods
Transgenic mice and cell lines
HLA-A*0201 (HHD) transgenic mice were bred at Charles River Laboratories and were kindly provided by Dr Lemonnier (Pasteur Institute). These mice are transgenic for the HHD complex (human β2-microglobulin fused to HLA-A*0201 α1 and α2 domain, H-2Db α3 domain) and are devoid of H-A2b and murine β2-microglobulin.56 For this reason the immune response elicited in these mice is specifically restricted to human HLA-A*0201, making this line a suitable model for epitope identification and optimization. HHD/CEA hybrid mice have been obtained by breeding transgenic mice homozygous for CEA with HHD mice. These mice express human CEA antigen presented exclusively by human HLA-A*0201 and represent a unique in vivo animal model to predict and study human immune response of a human CEA–based vaccine.42 Six to 8-wk-old HHD and HHD/CEA mice were used in this study. At the end of the treatment period and before necropsy, mice were euthanized by compressed CO2 gas as indicated in the AVMA (American Veterinary Medical Association) Panel on Euthanasia and according to the United Kingdom CO-ordinating Committee of Cancer Research (UKCCCR) guidelines.57 The experiments were conducted according to EU Directive EC86/609 on the protection of animals used for experimental and other scientific purposes, which was ratified by Italian Legislation with DL no. 116/92 on 19 February, 1992.
SW-480, Colo205, Colo705, and Colo201 cells were obtained from American Type Culture Collection (ATCC). B16-HHD/CEA cells were generated by transfecting the murine melanoma cell line B16-F10 (ATCC, cat. 6475) sequentially with 2 plasmids, one encoding CEA (pcDNA3-CEA) and the other HHD (pcDNA3-Hygro-HHD). Cells were maintained in culture at 10% CO2 in Dulbelco’s modified Eagle’s medium (DMEM), 10% FCS with 1% Pen/Strep, 800 µg/mL G418 and 400 µg/mL hygromycin (Invitrogen).
Peptides
Lyophilized CEA peptides were purchased from Jerini (JPT) and resuspended in DMSO at 40 mg/mL. Pools of peptides of 15 amino acids overlapping by 11 residues were assembled as previously described.15 Peptides and pools were stored at −80 °C.
In silico prediction of T-cell epitopes, fixed-anchor analogs and in vitro MHC binding assays
Seventeen candidate epitopes were selected in silico from CEA on the basis of 3 criteria: (1) predicted binding to MHC-I alleles HLA-A*0201; (2) uniqueness in the human genome; and (3) increased likelihood of natural processing, as previously described.24 Peptide MHC binding and complex stability was characterized using the iTopiaTM Epitope Discovery System (Beckman Coulter), as previously described.24 Briefly, the assay utilizes avidin-coated microtiter plates containing biotinylated MHC-I monomers loaded with β2-microglobulin (β2M) and placeholder peptides. The monomer-coated plates, assay buffers, FITC-conjugated anti-HLA-ABC monoclonal antibody (mAb) B9.12.1, β2M and allele-matched positive control peptides were obtained as part of the iTopia kit. Monomer-coated plates were first denatured, releasing the placeholder peptide and leaving only the MHC heavy chain bound to the plate. Test peptides were then introduced under optimal folding conditions, along with the anti-HLA-ABC-FITC monoclonal tracer antibody. Peptides were first evaluated in the initial binding assay (11 μM peptide incubated for 18 h at 21 °C with β2M, anti-MHC mAb and plate-bound MHC heavy chain). Irrespective of the initial binding, peptides were also screened in the stability assay to determine MHC complex half-life (T1/2; time taken for 50% reduction in binding at 37 °C after removing peptide from the assay buffer). Briefly, peptides were incubated overnight at 21 °C in MHC-coated wells. Assay buffer was replaced with fresh buffer containing no peptide. Plates were then incubated at 37 °C and read at multiple time points over a 24 h period. Dissociation rates were calculated by GraphPad Prism software using a single-phase exponential decay equation fitted to data recorded during the first 8 h, as recommended by the manufacturer and for consistency with earlier studies using this assay. All assay plates were read using a Cytofluor II fluorometer (PerSeptive Biosystems).
Immunizations
Mice were injected with 50 μg of plasmid DNA in a 50 μL volume into the quadriceps followed by electroporation, as previously described.15 Peptide vaccination was performed by subcutaneous injection of a mixture of 100 μg peptide, 140 μg HBV-T Helper epitope58 and 50 μg CpG-ODN (Sigma) in Incomplete Freund’s Adjuvant (IFA) per mouse. The immune response was evaluated 2 to 3 wk after the final treatment.
Immune response
The detection of peripheral immune response was measured by intracellular staining for IFNγ as previously described.59 Briefly, PBMC or splenocytes harvested from immunized mice (or controls) were resuspended in 0.6 mL RPMI, 10% FCS and incubated with the indicated pool of peptides (5 μg/mL final concentration of each peptide) and brefeldin A (1 μg/mL; BD PharMingen) at 37 °C for 12–16 h. Cells were then washed and stained with CD3, CD4, and CD8 surface antibodies (BD PharMingen). After washing, cells were fixed, permeabilized and incubated with the IFNγ-FITC antibodies (BD PharMingen), fixed with formaldehyde 1% in PBS and analyzed on a FACSCalibur flow cytometer, using CellQuest software (Becton Dickinson). Dimethyl sulfoxide (DMSO) and staphylococcal enterotoxin B (SEB, Sigma) at 10 μg/mL were used as internal negative and positive control of the assay, respectively.
In vitro cytotoxicity assays
Assays were performed according to standard protocols.60 Briefly, lymphocytes were isolated from harvested spleen of 3 mice per group 5 d after the final vaccination. Cells were resuspended to 2 × 106 cells/mL and were stimulated with CEA peptide pools along with 20 IU/mL recombinant human IL-2 (Sigma). Five days later, these in vitro stimulated cells were used as CTL effector cells, and the CTL activity was determined by a standard 6 h 51Cr-release cytotoxicity assay using the indicated cell lines as targets. 51Cr labeled cells were then added to wells for 6 h at an effector to target cell ratio = 100. Specific lysis was calculated as (experimental 51Cr release − spontaneous 51Cr release)/(maximal 51Cr release − spontaneous 51Cr release) × 100.
Tumor challenge
HHD/CEA mice were injected i.v. with 5 × 104 of B16-HHD/CEA cells. This dose of tumor cells is lethal in 100% of mice within 4 to 6 wk after transplant if left untreated. Three weeks after challenge, surface lung metastases were enumerated via optical microscope (Leica).
Statistical analysis
Statistical analyses were performed by Student's 2-tailed t test. The data are presented as means ± SD. A P-value < 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
LA is employed at Takis Biotech. AF and AB are employees of Merck and Co., Inc. NLM is employed at Johnson and Johnson.
Acknowledgments
This work was supported in part by the grant AIRC IG 10507. We thank Cinzia Roffi for editorial assistance.
Glossary
Abbreviations:
- APC
antigen presenting cell
- CEA
carcinoembryonic antigen
- CTL
cytotoxic T lymphocyte
- CMI
cell-mediated immunity
- DNA-EGT
DNA electro-gene transfer
- EC50
half-maximal effective concentration
- EMM
epitope-modified minigene
- IFNγ
interferon-γ
- LTB
heat-labile toxin B subunit
- MTS
membrane translocating sequence
- PBMC
peripheral blood mononuclear cell
- TAA
tumor-associated antigen
- TCR
T-cell receptor
- TPA
tissue plasminogen activator
Citation: Aurisicchio L, Fridman A, Bagchi A, Scarselli E, La Monica N, Ciliberto G. A novel minigene scaffold for therapeutic cancer vaccines . OncoImmunology 2014; 3:e27529; 10.4161/onci.27529
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/27529
REFERENCES
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–4. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ascierto PA, Capone M, Urba WJ, Bifulco CB, Botti G, Lugli A, Marincola FM, Ciliberto G, Galon J, Fox BA. The additional facet of immunoscore: immunoprofiling as a possible predictive tool for cancer treatment. J Transl Med. 2013;11:54. doi: 10.1186/1479-5876-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palucka K, Ueno H, Banchereau J. Recent developments in cancer vaccines. J Immunol. 2011;186:1325–31. doi: 10.4049/jimmunol.0902539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J. 2013;32:194–203. doi: 10.1038/emboj.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, Zhang RH, Scheinberg DA. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia. 2006;20:2025–33. doi: 10.1038/sj.leu.2404380. [DOI] [PubMed] [Google Scholar]
- 7.Krug LM, Dao T, Brown AB, Maslak P, Travis W, Bekele S, Korontsvit T, Zakhaleva V, Wolchok J, Yuan J, et al. WT1 peptide vaccinations induce CD4 and CD8 T cell immune responses in patients with mesothelioma and non-small cell lung cancer. Cancer Immunol Immunother. 2010;59:1467–79. doi: 10.1007/s00262-010-0871-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.André F, Mir LM. DNA electrotransfer: its principles and an updated review of its therapeutic applications. Gene Ther. 2004;11(Suppl 1):S33–42. doi: 10.1038/sj.gt.3302367. [DOI] [PubMed] [Google Scholar]
- 9.Aurisicchio L, Ciliberto G. Genetic cancer vaccines: current status and perspectives. Expert Opin Biol Ther. 2012;12:1043–58. doi: 10.1517/14712598.2012.689279. [DOI] [PubMed] [Google Scholar]
- 10.Cappelletti M, Zampaglione I, Rizzuto G, Ciliberto G, La Monica N, Fattori E. Gene electro-transfer improves transduction by modifying the fate of intramuscular DNA. J Gene Med. 2003;5:324–32. doi: 10.1002/jgm.352. [DOI] [PubMed] [Google Scholar]
- 11.Capone S, Zampaglione I, Vitelli A, Pezzanera M, Kierstead L, Burns J, Ruggeri L, Arcuri M, Cappelletti M, Meola A, et al. Modulation of the immune response induced by gene electrotransfer of a hepatitis C virus DNA vaccine in nonhuman primates. J Immunol. 2006;177:7462–71. doi: 10.4049/jimmunol.177.10.7462. [DOI] [PubMed] [Google Scholar]
- 12.Aurisicchio L, Peruzzi D, Conforti A, Dharmapuri S, Biondo A, Giampaoli S, Fridman A, Bagchi A, Winkelmann CT, Gibson R, et al. Treatment of mammary carcinomas in HER-2 transgenic mice through combination of genetic vaccine and an agonist of Toll-like receptor 9. Clin Cancer Res. 2009;15:1575–84. doi: 10.1158/1078-0432.CCR-08-2628. [DOI] [PubMed] [Google Scholar]
- 13.Peruzzi D, Mesiti G, Ciliberto G, La Monica N, Aurisicchio L. Telomerase and HER-2/neu as targets of genetic cancer vaccines in dogs. Vaccine. 2010;28:1201–8. doi: 10.1016/j.vaccine.2009.11.031. [DOI] [PubMed] [Google Scholar]
- 14.Gavazza A, Lubas G, Fridman A, Peruzzi D, Impellizeri JA, Luberto L, Marra E, Roscilli G, Ciliberto G, Aurisicchio L. Safety and efficacy of a genetic vaccine targeting telomerase plus chemotherapy for the therapy of canine B-cell lymphoma. Hum Gene Ther. 2013;24:728–38. doi: 10.1089/hum.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mennuni C, Calvaruso F, Facciabene A, Aurisicchio L, Storto M, Scarselli E, Ciliberto G, La Monica N. Efficient induction of T-cell responses to carcinoembryonic antigen by a heterologous prime-boost regimen using DNA and adenovirus vectors carrying a codon usage optimized cDNA. Int J Cancer. 2005;117:444–55. doi: 10.1002/ijc.21188. [DOI] [PubMed] [Google Scholar]
- 16.Aurisicchio L, Mennuni C, Giannetti P, Calvaruso F, Nuzzo M, Cipriani B, Palombo F, Monaci P, Ciliberto G, La Monica N. Immunogenicity and safety of a DNA prime/adenovirus boost vaccine against rhesus CEA in nonhuman primates. Int J Cancer. 2007;120:2290–300. doi: 10.1002/ijc.22555. [DOI] [PubMed] [Google Scholar]
- 17.Aurisicchio L, Peruzzi D, Conforti A, Dharmapuri S, Biondo A, Giampaoli S, Fridman A, Bagchi A, Winkelmann CT, Gibson R, et al. Treatment of mammary carcinomas in HER-2 transgenic mice through combination of genetic vaccine and an agonist of Toll-like receptor 9. Clin Cancer Res. 2009;15:1575–84. doi: 10.1158/1078-0432.CCR-08-2628. [DOI] [PubMed] [Google Scholar]
- 18.Fattori E, Aurisicchio L, Zampaglione I, Arcuri M, Cappelletti M, Cipriani B, Mennuni C, Calvaruso F, Nuzzo M, Ciliberto G, et al. ErbB2 genetic cancer vaccine in nonhuman primates: relevance of single nucleotide polymorphisms. Hum Gene Ther. 2009;20:253–65. doi: 10.1089/hum.2008.153. [DOI] [PubMed] [Google Scholar]
- 19.Dharmapuri S, Peruzzi D, Mennuni C, Calvaruso F, Giampaoli S, Barbato G, Kandimalla ER, Agrawal S, Scarselli E, Mesiti G, et al. Coadministration of telomerase genetic vaccine and a novel TLR9 agonist in nonhuman primates. Mol Ther. 2009;17:1804–13. doi: 10.1038/mt.2009.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peruzzi D, Mori F, Conforti A, Lazzaro D, De Rinaldis E, Ciliberto G, La Monica N, Aurisicchio L. MMP11: a novel target antigen for cancer immunotherapy. Clin Cancer Res. 2009;15:4104–13. doi: 10.1158/1078-0432.CCR-08-3226. [DOI] [PubMed] [Google Scholar]
- 21.Ishioka GY, Fikes J, Hermanson G, Livingston B, Crimi C, Qin M, del Guercio MF, Oseroff C, Dahlberg C, Alexander J, et al. Utilization of MHC class I transgenic mice for development of minigene DNA vaccines encoding multiple HLA-restricted CTL epitopes. J Immunol. 1999;162:3915–25. [PubMed] [Google Scholar]
- 22.Kazansky DB. Intrathymic selection: new insight into tumor immunology. Adv Exp Med Biol. 2007;601:133–44. doi: 10.1007/978-0-387-72005-0_14. [DOI] [PubMed] [Google Scholar]
- 23.Bei R, Scardino A. TAA polyepitope DNA-based vaccines: a potential tool for cancer therapy. J Biomed Biotechnol. 2010;2010:102758. doi: 10.1155/2010/102758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fridman A, Finnefrock AC, Peruzzi D, Pak I, La Monica N, Bagchi A, Casimiro DR, Ciliberto G, Aurisicchio L. An efficient T-cell epitope discovery strategy using in silico prediction and the iTopia assay platform. Oncoimmunology. 2012;1:1258–70. doi: 10.4161/onci.21355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shively JE, Beatty JD. CEA-related antigens: molecular biology and clinical significance. Crit Rev Oncol Hematol. 1985;2:355–99. doi: 10.1016/S1040-8428(85)80008-1. [DOI] [PubMed] [Google Scholar]
- 26.Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal. 1991;5:344–66. doi: 10.1002/jcla.1860050510. [DOI] [PubMed] [Google Scholar]
- 27.Kuespert K, Pils S, Hauck CR. CEACAMs: their role in physiology and pathophysiology. Curr Opin Cell Biol. 2006;18:565–71. doi: 10.1016/j.ceb.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gameiro SR, Jammeh ML, Hodge JW. Cancer vaccines targeting carcinoembryonic antigen: state-of-the-art and future promise. Expert Rev Vaccines. 2013;12:617–29. doi: 10.1586/erv.13.40. [DOI] [PubMed] [Google Scholar]
- 29.Weihrauch MR, Ansén S, Jurkiewicz E, Geisen C, Xia Z, Anderson KS, Gracien E, Schmidt M, Wittig B, Diehl V, et al. Phase I/II combined chemoimmunotherapy with carcinoembryonic antigen-derived HLA-A2-restricted CAP-1 peptide and irinotecan, 5-fluorouracil, and leucovorin in patients with primary metastatic colorectal cancer. Clin Cancer Res. 2005;11:5993–6001. doi: 10.1158/1078-0432.CCR-05-0018. [DOI] [PubMed] [Google Scholar]
- 30.Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. 2009;15:169–80. doi: 10.1158/1078-0432.CCR-08-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stack JH, Whitney M, Rodems SM, Pollok BA. A ubiquitin-based tagging system for controlled modulation of protein stability. Nat Biotechnol. 2000;18:1298–302. doi: 10.1038/82422. [DOI] [PubMed] [Google Scholar]
- 32.Wang QM, Sun SH, Hu ZL, Zhou FJ, Yin M, Xiao CJ, Zhang JC. Epitope DNA vaccines against tuberculosis: spacers and ubiquitin modulates cellular immune responses elicited by epitope DNA vaccine. Scand J Immunol. 2004;60:219–25. doi: 10.1111/j.0300-9475.2004.01442.x. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez F, An LL, Harkins S, Zhang J, Yokoyama M, Widera G, Fuller JT, Kincaid C, Campbell IL, Whitton JL. DNA immunization with minigenes: low frequency of memory cytotoxic T lymphocytes and inefficient antiviral protection are rectified by ubiquitination. J Virol. 1998;72:5174–81. doi: 10.1128/jvi.72.6.5174-5181.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina F, Ramos M, Iborra S, de León P, Rodríguez-Castro M, Del Val M. Furin-processed antigens targeted to the secretory route elicit functional TAP1-/-CD8+ T lymphocytes in vivo. J Immunol. 2009;183:4639–47. doi: 10.4049/jimmunol.0901356. [DOI] [PubMed] [Google Scholar]
- 35.Köhne C, Johnson A, Tom S, Peers DH, Gehant RL, Hotaling TA, Brousseau D, Ryll T, Fox JA, Chamow SM, et al. Secretion of glycosylation site mutants can be rescued by the signal/pro sequence of tissue plasminogen activator. J Cell Biochem. 1999;75:446–61. doi: 10.1002/(SICI)1097-4644(19991201)75:3<446::AID-JCB10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 36.Hallenberger S, Moulard M, Sordel M, Klenk HD, Garten W. The role of eukaryotic subtilisin-like endoproteases for the activation of human immunodeficiency virus glycoproteins in natural host cells. J Virol. 1997;71:1036–45. doi: 10.1128/jvi.71.2.1036-1045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valmori D, Pessi A, Bianchi E, Corradin G. Use of human universally antigenic tetanus toxin T cell epitopes as carriers for human vaccination. J Immunol. 1992;149:717–21. [PubMed] [Google Scholar]
- 38.Facciabene A, Aurisicchio L, Elia L, Palombo F, Mennuni C, Ciliberto G, La Monica N. Vectors encoding carcinoembryonic antigen fused to the B subunit of heat-labile enterotoxin elicit antigen-specific immune responses and antitumor effects. Vaccine. 2007;26:47–58. doi: 10.1016/j.vaccine.2007.10.060. [DOI] [PubMed] [Google Scholar]
- 39.Mennuni C, Ugel S, Mori F, Cipriani B, Iezzi M, Pannellini T, Lazzaro D, Ciliberto G, La Monica N, Zanovello P, et al. Preventive vaccination with telomerase controls tumor growth in genetically engineered and carcinogen-induced mouse models of cancer. Cancer Res. 2008;68:9865–74. doi: 10.1158/0008-5472.CAN-08-1603. [DOI] [PubMed] [Google Scholar]
- 40.Conforti A, Cipriani B, Peruzzi D, Dharmapuri S, Kandimalla ER, Agrawal S, Mori F, Ciliberto G, La Monica N, Aurisicchio L. A TLR9 agonist enhances therapeutic effects of telomerase genetic vaccine. Vaccine. 2010;28:3522–30. doi: 10.1016/j.vaccine.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 41.Peruzzi D, Mori F, Conforti A, Lazzaro D, De Rinaldis E, Ciliberto G, La Monica N, Aurisicchio L. MMP11: a novel target antigen for cancer immunotherapy. Clin Cancer Res. 2009;15:4104–13. doi: 10.1158/1078-0432.CCR-08-3226. [DOI] [PubMed] [Google Scholar]
- 42.Conforti A, Peruzzi D, Giannetti P, Biondo A, Ciliberto G, La Monica N, Aurisicchio L. A novel mouse model for evaluation and prediction of HLA-A2-restricted CEA cancer vaccine responses. J Immunother. 2009;32:744–54. doi: 10.1097/CJI.0b013e3181aee1b6. [DOI] [PubMed] [Google Scholar]
- 43.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–99. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 44.Pamer E, Cresswell P. Mechanisms of MHC class I--restricted antigen processing. Annu Rev Immunol. 1998;16:323–58. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 45.Kägi D, Vignaux F, Ledermann B, Bürki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–30. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 46.Lazoura E, Apostolopoulos V. Insights into peptide-based vaccine design for cancer immunotherapy. Curr Med Chem. 2005;12:1481–94. doi: 10.2174/0929867054039017. [DOI] [PubMed] [Google Scholar]
- 47.Thomson SA, Sherritt MA, Medveczky J, Elliott SL, Moss DJ, Fernando GJ, Brown LE, Suhrbier A. Delivery of multiple CD8 cytotoxic T cell epitopes by DNA vaccination. J Immunol. 1998;160:1717–23. [PubMed] [Google Scholar]
- 48.Ishioka GY, Fikes J, Hermanson G, Livingston B, Crimi C, Qin M, del Guercio MF, Oseroff C, Dahlberg C, Alexander J, et al. Utilization of MHC class I transgenic mice for development of minigene DNA vaccines encoding multiple HLA-restricted CTL epitopes. J Immunol. 1999;162:3915–25. [PubMed] [Google Scholar]
- 49.Cortez-Gonzalez X, Sidney J, Adotevi O, Sette A, Millard F, Lemonnier F, Langlade-Demoyen P, Zanetti M. Immunogenic HLA-B7-restricted peptides of hTRT. Int Immunol. 2006;18:1707–18. doi: 10.1093/intimm/dxl105. [DOI] [PubMed] [Google Scholar]
- 50.Pitcovski J, Bazak Z, Wasserman E, Elias O, Levy A, Peretz T, Fingerut E, Frankenburg S. Heat labile enterotoxin of E. coli: a potential adjuvant for transcutaneous cancer immunotherapy. Vaccine. 2006;24:636–43. doi: 10.1016/j.vaccine.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 51.Zhou H, Luo Y, Mizutani M, Mizutani N, Becker JC, Primus FJ, Xiang R, Reisfeld RA. A novel transgenic mouse model for immunological evaluation of carcinoembryonic antigen-based DNA minigene vaccines. J Clin Invest. 2004;113:1792–8. doi: 10.1172/JCI21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu J, Higashimoto Y, Appella E, Celis E. Multiepitope Trojan antigen peptide vaccines for the induction of antitumor CTL and Th immune responses. J Immunol. 2004;172:4575–82. doi: 10.4049/jimmunol.172.7.4575. [DOI] [PubMed] [Google Scholar]
- 53.Aurisicchio L, Peruzzi D, Koo G, Wei WZ, La Monica N, Ciliberto G. Immunogenicity and therapeutic efficacy of a dual component Genetic Cancer Vaccine Co-Targeting CEA and HER2/neu in preclinical models. Hum Gene Ther. 2013 doi: 10.1089/hum.2013.103. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirkwood JM, Lee S, Moschos SJ, Albertini MR, Michalak JC, Sander C, Whiteside T, Butterfield LH, Weiner L. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/-granulocyte-monocyte colony-stimulating factor and/or IFN-alpha2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin Cancer Res. 2009;15:1443–51. doi: 10.1158/1078-0432.CCR-08-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dengjel J, Nastke MD, Gouttefangeas C, Gitsioudis G, Schoor O, Altenberend F, Müller M, Krämer B, Missiou A, Sauter M, et al. Unexpected abundance of HLA class II presented peptides in primary renal cell carcinomas. Clin Cancer Res. 2006;12:4163–70. doi: 10.1158/1078-0432.CCR-05-2470. [DOI] [PubMed] [Google Scholar]
- 56.Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Pérarnau B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J Exp Med. 1997;185:2043–51. doi: 10.1084/jem.185.12.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.CO-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the Welfare of Animals in Experimental Neoplasia. Second Edition, vol. 77. London, WC2A 3PX: Br J Cancer, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Livingston BD, Crimi C, Fikes J, Chesnut RW, Sidney J, Sette A. Immunization with the HBV core 18-27 epitope elicits CTL responses in humans expressing different HLA-A2 supertype molecules. Hum Immunol. 1999;60:1013–7. doi: 10.1016/S0198-8859(99)00103-2. [DOI] [PubMed] [Google Scholar]
- 59.Giannetti P, Facciabene A, La Monica N, Aurisicchio L. Individual mouse analysis of the cellular immune response to tumor antigens in peripheral blood by intracellular staining for cytokines. J Immunol Methods. 2006;316:84–96. doi: 10.1016/j.jim.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Lu J, Higashimoto Y, Appella E, Celis E. Multiepitope Trojan antigen peptide vaccines for the induction of antitumor CTL and Th immune responses. J Immunol. 2004;172:4575–82. doi: 10.4049/jimmunol.172.7.4575. [DOI] [PubMed] [Google Scholar]