

Abstract

A method for the investigation of the influence of protecting groups on the anomeric equilibrium in the sialic acid glycosides has been developed on the basis of the equilibration of O-sialyl hydroxylamines by reversible homolytic scission of the glycosidic bond following the dictates of the Fischer–Ingold persistent radical effect. It is found that a trans-fused 4O,5N-oxazolidinone group stabilizes the equatorial glycoside, i.e., reduces the anomeric effect, when compared to the 4O,5N-diacetyl protected systems. This effect is discussed in terms of the powerful electron-withdrawing nature of the oxazolidinone system, which in turn is a function of its strong dipole moment in the mean plane of the pyranose ring system. The new equilibration method displays a small solvent effect and is most pronounced in less polar media consistent with the anomeric effect in general. The unusual (for anomeric radicals) poor kinetic selectivity of anomeric sialyl radicals is discussed in terms of the planar π-type structure of these radicals and of competing 1,3-diaxial interactions in the diastereomeric transition states for trapping on the α- and β-faces of the radical.
Introduction
The influence of protecting groups on the reactivity and selectivity of glycosyl donors is a widely recognized and exploited phenomenon.1−23 The corresponding influence of protecting groups on the anomeric equilibrium, while evident for many years,24,25 is less widely studied and exploited. Recent examples of the latter include the recognition that 2N,3O-oxazolidinones both facilitate anomerization in the 2-amino-2-deoxyhexopyranosides and stabilize the axial over the equatorial anomer,26−34 with similar effects being apparent in hexopyranosides carrying a 2,3-O-cyclic carbonate group,32,35 and the realization that cyclic 4,6-O-acetals modulate the anomeric effect in the hexopyranoses.14,36,37 Our interest in the use of the N-acetyl-4O,5N-oxazolidinone protected sialyl donors,38,39 and their N-desacetyl counterparts,40,41 which have emerged as some of the most powerful and equatorially selective systems for the preparation of sialyl O-glycosides,42−58 as well as their C-59,60 and S-counterparts,61 prompted us to examine the effect of these systems on the anomeric equilibrium of sialyl glycosides. The magnitude of the anomeric effect is typically assessed either by mutarotation of anomeric hemiacetals or by the Brønsted or Lewis acid-mediated equilibration of glycosides, and such methods have been used to determine the position of the anomeric equilibrium in both N-acetyl neuraminic acid itself and its methyl glycoside,62−66 leading to the conclusion that the axial glycoside is significantly more strongly favored in the sialic acids (Figure 1) than in glucose and that mutarotation of the hemiacetal takes place via a ring-opening mechanism.65,66
Figure 1.

Anomeric equilibrium in N-acetylneuraminic acid.
The attempted synthesis of a N-acetyl-4O,5N-protected sialyl hemiacetal 2 by hydrolysis of the corresponding thioglycoside 3 resulted in the formation of a complex mixture of the two anomers of 2 and of the acyclic form 4, and its hydrate 5, as determined by NMR spectroscopy, in which the latter two species predominated (Figure 2). The complexity of the spectra together with the predominance of the open-chain forms revealed the strain placed on the pyranose forms by the presence of the trans-fused oxazolidinone ring, even if no distortion of the pyranose ring is evident from X-ray structures,38,50 and precluded use of the standard methods in this investigation.
Figure 2.

Compounds employed in and arising from the mutarotation of an oxazolidinone-protected sialose derivative.
Accordingly, we turned our attention to the use of neutral methods for the equilibration of sialic acid glycosides such as would not involve the intermediacy of the anomeric hemiacetal and would be compatible with the readily cleaved38 activated oxazolidinone ring. Precedent for the equilibration of anomeric stereochemistry through the reversible homolytic cleavage and reformation of anomeric C–H,67,68 C–Co,69 and C–Te bonds,70 suggested that the answer might lie in radical reactions. The need to approximate as closely as possible the properties of O-glycosidic bonds suggested the synthesis and equilibration of O-glycosyl derivatives of sterically hindered N,N-disubstituted hydroxylamines by the Fischer–Ingold persistent radical effect.71−73 Albeit less substituted than the systems required for the radical equilibration process, glycosyl hydroxylamines are found in nature in the enediyne antibiotics calicheamicin γ1I674 and esperamicin 7,75 and simple hydroxylamines have been employed as glycosidic bond surrogates in neoglycoconjugates (Figure 3).76−81 Moreover, the distinct conformational preferences of the O-glycosyl hydroxylamine linkage have been advanced as a design element used by nature to assist in the binding of calicheamicin to the minor groove of DNA.82O-Glycosyl hydroxylamines have been identified as metabolites of various drugs,83 including glucosides and glucuronides of the antioxidant 2,2,6,6-tetramethylpiperidin-1-ol.84
Figure 3.

Natural products featuring the O-glycosyl hydroxylamine moiety.
In a preliminary communication we established the concept with sialyl glycosides of 2,2,4,4-tetramethylpiperidin-1-ol in a single solvent.85 In this article we report in full on the synthesis of the O-sialyl glycosides of two further hydroxylamines of differing steric bulk and conduct equilibration studies in three solvents spanning a broad range of polarities. The influence of the sialyl protecting groups, solvent polarity, and hydroxylamine steric bulk on the thermal equilibration reactions of the sialyl hydroxylamines are presented and afford insight into the influence of the trans-fused oxazolidinone group in sialic acid chemistry. The lack of kinetic selectivity of sialyl anomeric radicals is discussed from the viewpoint of their conformation, which is based on literature electron spin resonance data of cognate radicals.
Results
Despite the presence of O-glycosyl hydroxylamines in various enediyne antibiotics and their use in neoglycoconjugates, literature methods for their synthesis are limited to the glycosylation of (i) various N-acylated hydroxylamines, e.g., N-hydroxyphthalimide and N-carboethoxy hydroxylamine,76,80,86−91 (ii) oximes,92,93 and (iii) nitrones,94−96 each with subsequent manipulation of the functionality at nitrogen. These methods are, however, not suitable for the synthesis of the sterically hindered O-glycosyl N,N-dialkylhydroxylamines required for this investigation, and we therefore turned to the design of alternative routes. A brief investigation of the direct glycosylation of N-hydroxy-2,2,6,6-tetramethylpiperidine with efficient sialyl donors such as developed previously in our laboratory39 was unfruitful, perhaps for reasons of steric hindrance, and we turned therefore to radical reactions and their relative indifference to steric constraints.97 Previous workers have generated sialyl anomeric radicals for the purposes of C-sialoside formation by the action of stannyl radicals on sialyl chlorides,98,99 but we have preferred simple photolysis of a readily available S-sialyl xanthate ester 8.100 We note in passing that attempted photochemical equilibration of xanthate 8 resulted, in a process reminiscent of the Barton–Achmatowicz101,102 “game of bridge reaction”,103 only in the known104 elimination product 9 (Scheme 1).
Scheme 1. Photochemical Elimination Xanthate 8.

Fortunately, 254 nm photolysis of 8 through Pyrex in a Rayonnet photoreactor in the presence of the stable nitroxyl radicals TEMPO (10), TMIO (11),105 and SG1 (12)106 resulted in the formation of the desired O-glycosyl hydroxylamines 14–16 as reported in Scheme 2. Attempted application of this method to DBNO 13(107) resulted only in the formation of the glycal 9. That the photolysis of 8 can be interrupted with appropriate radical traps before the formation of glycal 9 indicates that it takes place via a two-step process giving an initial radical pair followed by disproportionation (Scheme 1). This observation, which is critical to the success of the project, differs from the original conclusion of the Barton and Porter laboratories regarding the photoelimination of thiobenzoate esters. Those workers preferred a concerted elimination from a photochemically excited state of the thiocarbonyl ester on the basis of (i) the observation of a short-lived transient on irradiation of the thiobenzoate, and (ii) the inability to trap radical intermediates with (unspecified) radical traps.101 We also note, however, and more in line with the results observed here, that the photolysis of S-acyl xanthate esters is known to proceed with homolytic scission of the S-acyl bond resulting in acyl radical formation.108
Scheme 2. Synthesis of Sterically Hindered O-Glycosyl Hydroxylamines.

Returning to the synthesis of the O-glycosyl hydroxylamines, when the photolysis of 8 was conducted in the presence of TEMPO (10) the product 14 was obtained as an approximately 1:2 mixture, favoring the isomer (β-) with the axial hydroxylamino residue, in 69% yield together with 10% of the glycal. With TMIO (11), the adduct 15 was obtained in 72% yield as an approximate 1:2.2 α:β mixture. Photolysis in dichloroethane in the presence of racemic SG1 (12) gave only the glycal 9, but the anomeric radical could be intercepted in 45% yield when neat SG1 (12 equiv) was used as solvent. Finally, photolysis in the presence of di(t-butyl)nitroxide in dichloroethane afforded only the glycal 9. Given the use of racemic SG1, there are four potential diastereomers of 16, but the product was isolated as a mixture of two predominant but inseparable isomers, the major one of which we tentatively assign as an equatorial (α-) adduct and the minor as an axial (β-) adduct without commenting on the configuration at the stereogenic center adjacent to the phosphoryl group. The anomeric stereochemistry in each of the adducts 14–16 is assigned on the basis of the 3J heteronuclear coupling constant between the anomeric carboxyl carbon and the axial methylene proton at C3 in the pyranose ring, which is diagnostic in the sialic acid glycosides.109−113 These assignments are also supported by NOE measurements for the axial (β-) anomer of 14 and subsequent members of the same series (vide infra), which reveal the spatial proximity of the axial H6 in the pyranose ring and one or more of the methyl singlets in the hydroxylamine moiety. Resubmission of 14 to the photolysis conditions did not result in any change in the anomeric ratio consistent with the lack of a suitable chromophore, and indicating the products 14–16 to be kinetic mixtures. Rate constants for the trapping of alkyl radicals by nitroxyl radicals, while high (106 to 109 s–1), are nevertheless below the diffusion controlled limit for stabilized alkyl radicals and are dependent on steric bulk in both the radical and the trap.114−117 The stereoselectivities observed in the formation of 14–16 therefore must factor in the inherent face selectivity of the intermediate radical 18 (Figure 4) and the steric bulk of the trap, which according to Bowry and Ingold, follows the trend DBNO > TEMPO > TMIO on the basis of kinetic trapping data (SG1 not being included in the study).114 We conclude therefore that radical 18 shows a modest axial selectivity that can be overridden by the use of the presumably more bulky trap SG1. This modest axial selectivity is consistent with the work of Paulsen and Matschulat, who recorded an axial/equatorial trapping preference of 1.8:1 for the reaction of 18 with allyltributylstannane at 60 °C in THF,99 but not with the work of Nagy and Bednarski, who reported an approximate axial/equatorial ratio of 1:1 for the same reaction conducted at room temperature under photochemical conditions.98
Figure 4.

Predicted structure of the intermediate radical 18 and literature structures of the model radicals 19–23 with key ESR parameters (hyperfine splitting constants a in mT).
We depict radical 18 as a close to planar π-radical with extensive delocalization onto the carboxyl oxygen resulting in two distinct conformers about the exocyclic C1–C2 bond (Figure 4) on the basis of literature ESR data for simple model radicals. Thus, ESR studies of the tri-O-acetyl-2-deoxy-1-glucosyl radical 19 show it to adopt a chair conformation with an out of plane bend of ∼6° for the anomeric carbon consistent with a radical that is 90% sp2 hybridized.118,119 The methoxycarbonylmethyl radical 20 is a planar π-radical with extensive delocalization onto the carboxyl oxygen and a barrier to rotation about the CH2–CO2Me bond of ∼11 kcal mol–1,120 and the methoxy(methoxycarbonyl)methyl radical 21 displays ESR parameters that closely mirror those of 20, including the hyperfine splitting aHδ indicative of coupling to the ester methyl hydrogens, which is diagnostic of extensive delocalization.121 Finally, the close model radicals 22 and 23 also show ESR spectral parameters consistent with those for 20, including hyperfine splitting by the ester methyl hydrogens and two distinct rotamers about the C5–CO2Me bond.119,122 The two radicals 22 and 23, isomeric at the β-position, adopt different conformations so as in each case to maximize the overlap of the β-acetoxy group with the singly filled orbital, a phenomenon known as quasi-homoanomeric stabilization.119,122 It follows that when the β-position is unsubstituted as in radicals 18 and 19, and the constraint of the quasi-homoanomeric interaction absent, the chair conformation will be retained.
Presumably, it is the close to π-type, approximately planar structure of radical 18 and the presence of the anomeric carboxyl group that accounts for the low kinetic stereoselectivities in its reactions with radical traps, which contrast with the high axial selectivity seen in the chemistry of simple per acetylated glycosyl radicals, including 19, and their 1-alkoxy derivatives.119,123−127 Thus, trapping of radical 18 along the axial direction, while favored on stereoelectronic grounds and leading directly to the chair conformation of the product, involves a 1,3-diaxial interaction between the axial H4 and H6 and the partially formed C2–O bond, while equatorial trapping results in 1,3-diaxial interaction of H4 and H6 with the fully formed C2-CO2Me bond albeit while placing the incoming bulky hydroxylamine in the sterically more accessible equatorial position (Figure 5). This latter mode only begins to predominate with the most bulky nitroxide trap, SG1 (12) and the formation of 16 (Scheme 2). A similar rationale has been previously invoked for exo–endo-selectivity, with competing steric interactions between a full covalent bond to a substituent at the radical center and partially formed covalent bond to an incoming trap for the bicyclic radical 24.128
Figure 5.

Competing transition states for the trapping of radical 18 showing 1,3-diaxial interactions with H4 and H6, and structure of the radical 24.
With a series of glycosyl hydroxylamines in hand, and with a view to examining the influence of protecting groups on the anomeric equilibrium, a set of standard protecting group manipulations were applied to 14 and 15, as illustrated in Scheme 3, giving a number of derivatives for subsequent equilibration studies.
Scheme 3. Preparation of Variously Protected Sialyl Hydroxylamines.

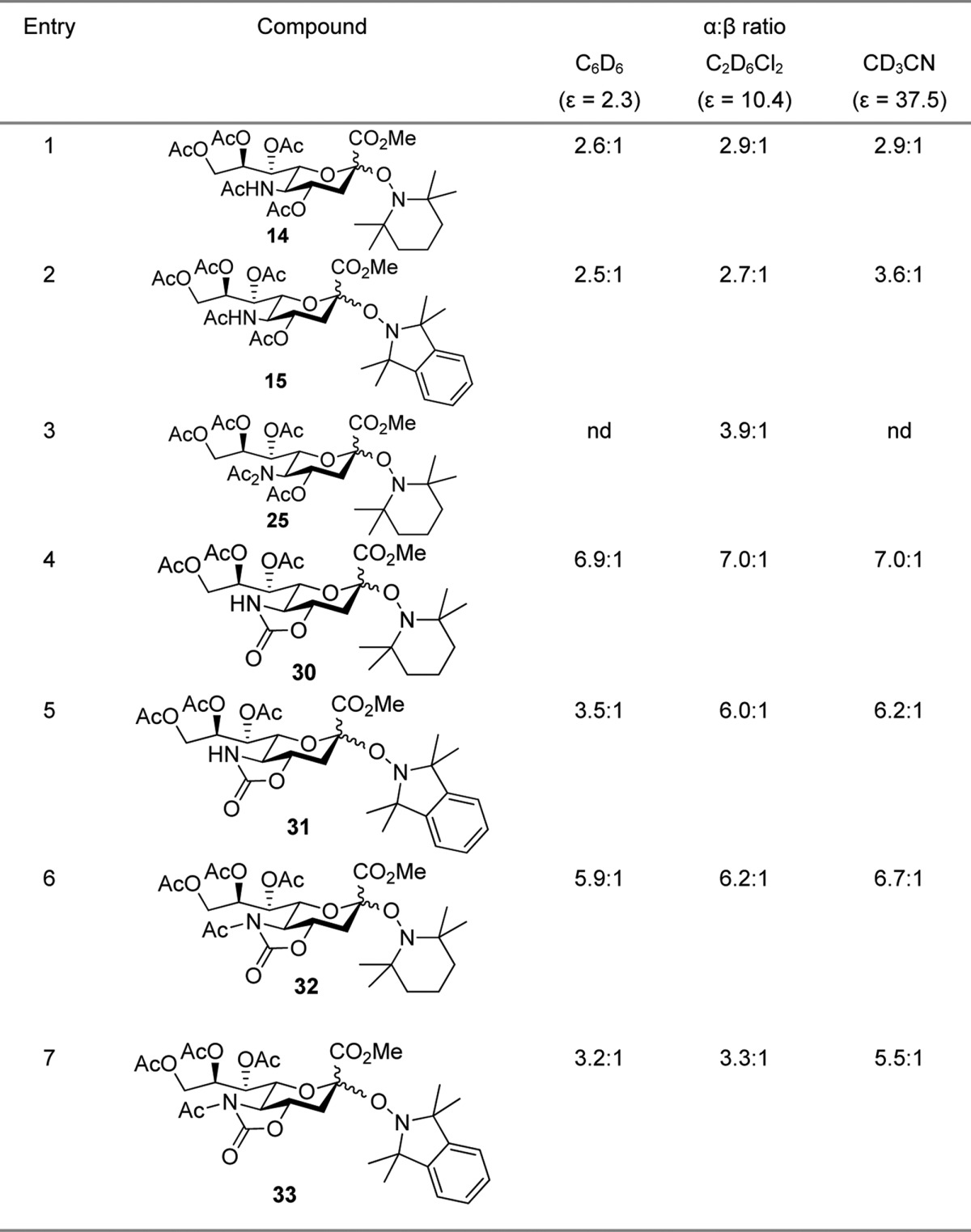
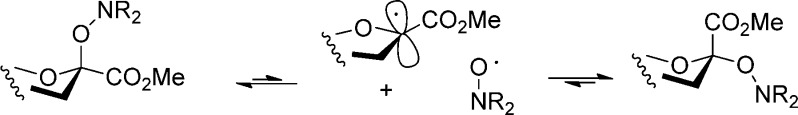
A series of equilibration reactions were then conducted by heating individually (0.05–0.1 M) solutions of substances 14, 15, and 30–33 to 90 °C in each of deuteriobenzene, deuterio-1,2-dichloroethane, and deuterioacetonitrile, with periodic monitoring by NMR spectroscopy until no further change was observed (Table 1). Attempted equilibration of the SG1 derivative 16 resulted only in the formation of the elimination product 9, for which reason, coupled with the problem of extra diastereomers arising from the stereogenic center in the aglycone, the SG1 series was not pursued further. A further compound, the N,N-diacetyl TEMPO glycoside 25 was also subjected to equilibration in deuterio-1,2-dichloroethane (Table 1); however, further work with this compound was not conducted in other solvents, nor was the TMIO analogue prepared. The mechanism of the equilibration process involves reversible homolytic scission of the glycosyl-hydroxylamine C–O bond to an anomeric radical and a persistent aminoxyl radical (Scheme 4). It is implicit in the Fischer–Ingold radical effect71−73,129 that the recombination is intermolecular rather than intramolecular within the confines of the initial radical pair. The intermolecular nature of the process enables its use in combinatorial library generation,130 and for the present purposes it was readily demonstrated by a simple crossover experiment. Thus, heating of 15 in the presence of an equimolar amount of TEMPO in 1,2-dichloroethane to 90 °C for nine days, after which no further change was observed, resulted in the formation of an approximately 1:2 mixture of 14 and 15, both as a mixture of anomers favoring the α-anomers. The predominace of the TMIO sialosides 15 over the TEMPO sialosides 14 in this equilibrated mixture indicates the greater thermodynamic stability of 15 over 14, which is consistent with the less hindered, more tied back nature114 and therefore less persistent nature of the TMIO radical 11 in comparison to the TEMPO radical 10.
Table 1. Solvent and Substituent Effects on the Equilibration of O-Sialyl Hydroxylaminesa.
ε = dielectric constant in Debye units.
Scheme 4. Equilibration by Means of the Fischer–Ingold Persistent Radical Effect.
Successful achievement of the equilibration process required rigorous exclusion of oxygen, which was achieved by copious sparring with argon, and possibly moisture, as with some batches of deuterioacetonitrile a further type of byproduct exemplified by 34 was observed (Scheme 5). While this thermal oxidative decarboxylation process of a sialic acid glycoside is certainly interesting in view of other sialic acid decarboxylation processes, both oxidative131−133 and reductive,126 we have not pursed it further at present, especially as it could be avoided by degassing and use of fresh deuterioacetonitrile.
Scheme 5. Unexpected Decarboxylative Lactone Formation during Equilibration.

Several trends are evident in the data presented in Table 1. First, as compared to sialic acid itself (Figure 1) and to simple methyl sialosides62−66 the O-sialyl hydroxylamines studied all show an inverted preference for the equatorial over the axial glycosides. Second, there is a solvent effect on the position of the anomeric equilibrium with the equatorial (α-) anomers generally being more highly favored in the higher polarity solvents. Third, the magnitude of this solvent effect is on the whole greater in the TMIO series (Table 1, entries 2, 5, and 7) than in the TEMPO series (Table 1, entries 1, 4, and 6). Fourth, the anomeric ratio at equilibrium is a function of the protecting groups employed with the same trends being seen for the both the TEMPO and TMIO series of compounds.
The general preference of the equatorial over the axial glycosides seen in Table 1, which contrasts sharply with the strong anomeric effect in simple sialosides, reflects the significant steric bulk of the hindered hydroxylamine aglycones and the attendant destabilization of the axial glycosides through classical 1,3-diaxial interactions. This reversal of the anomeric preference between simple sialosides and those of hindered hydroxylamines does not detract from the validity of comparisons within the later series of compounds that form the core of this article. The observed solvent effect, which is consistent with the well-known25 general trend whereby the anomeric effect is greatest in less polar solvents, simply reflects the more polar nature of equatorial versus axial glycosides. The greater solvent dependence of the TMIO series as compared to the TEMPO series of compounds presumably arises from the more polar nature of the TMIO group relative to the TEMPO group, which is a function of the presence of the electron-withdrawing arene in the TMIO moiety.134 The most consistent trend, which holds for both the TEMPO and TMIO series of compounds and for all three solvents assayed, involves the change in anomeric ratio as a function of protecting groups at the O4 and N5 positions. Thus, there is generally a greater preference for the equatorial glycoside, or a smaller anomeric effect, in the 4O,5N-oxazolidinone series (Table 1, entries 4 and 5), than in the N-acetyl-4O,5N-oxazolidinone series (Table 1, entries 6 and 7), which in turn show a greater equatorial preference than the 4O,5N-diacetyl series (Table 1, entries 1 and 2). Although less data is available, the 4O,5N,5N-triacetyl compounds (Table 1, entry 3) appear to fall between the diacetyl and the N-acetyloxazolidinones. This trend is summarized in Figure 6.
Figure 6.

Decreasing preference for the axial (β-) sialoside as a function of protecting group.
Turning to the reasons underlying the general greater preference for the equatorial glycosides (Figure 6) in the oxazolidinone series, we discount steric arguments based on a smaller steric clash between the aglycone and the O4 protecting group in the oxazolidinones because the usual exoanomeric effect25,135,136 conformation about the glycosidic bond will orient the aglycone toward the rear of the molecule and away from O4 (Figure 7). Indeed, the typical exoanomeric effect conformation with an N–O–anomeric C–ring oxygen torsion angle of ∼-60° is evident in the X-ray crystal structure of a calicheamicin derivative,74,137 in the X-ray structures of a simple O-glycosyl hydroxylamine,138 and of various O-glycosyl oximes and hydroxyimides,93,139−141 albeit a molecular mechanics-based computational paper appears to suggest a N–O–anomeric C–ring oxygen torsion angle of ∼170°.82
Figure 7.
Newman projection about the hydroxylamine N–O bond showing the typical exoanomeric effect conformation of the O-glycosyl hydroxylamines.74,93,137−141
Rather we suggest that the influence of the oxazolidinone on the anomeric effect is rooted in the highly polar nature of the oxazolidinone system with its strong dipole moment aligned with the axis of the carbonyl bond (Figure 8).142−144 Thus, as illustrated in Figure 9, the oxazolidinone moiety exhibits a powerful electron-withdrawing effect in the mean plane of the pyranose ring due to the alignment of the carbonyl dipole with the polar C4–O4 and C5–N5 bonds. This is to be contrasted with the 4O,5N-diacetyl system in which, assuming the standard minimum energy conformations of the ester and amide groups145−147 with the latter apparent from the 180° H5–C5–N5–NH torsion angle with 3JNH,H5 = 10 Hz, the two carbonyl dipoles are oriented in opposite directions and approximately perpendicular to the C4–O4 and C5–N5 bonds resulting overall in a smaller electron-withdrawing effect. The electron-withdrawing effect of the N-acetyloxazolidinone system, while still powerful because of the alignment of the oxazolidinone carbonyl with the C4–O4 and C5–N5 bonds in the mean plane of the pyranose ring, is moderated by the orientation of the acetyl group, which, as is clear from the available X-ray crystal structures38,50 and literature data with simple models,144 is oriented so as to minimize the overall dipole (Figure 8) at least in weakly polar environments.
Figure 8.

Dipole moments (Debye units) of open chain and cyclic carbonyl functionalities in nonpolar solvents.
Figure 9.

Orientations of the key O4 and N5 protecting group carbonyl dipole moments in the O-sialyl hydroxylamines (side chains omitted for clarity).
If the anomeric effect is understood in terms of donation of electron density from a lone pair (n) on the ring oxygen into the synperiplanar C–O σ* orbital of the axial glycosidic bond,25,148 the influence of the oxazolidinone can be attributed to the lowering of the energy of the lone pair due to the presence of the strongly electron-withdrawing group, which in turn reduces the n–σ* interaction and so diminishes the anomeric effect. In valence bond terms this equates to the destabilization of the double bond–no bond resonance form owing to the increased energy of the oxocarbenium ion arising from the presence of the strongly electron-withdrawing protecting group (Figure 10). It can also be argued in view of the high dipole moment of the oxazolidinone group that there is a dipolar component to the stabilization of the equatorial glycosides. Thus, in the axial glycosides the vectors of the oxazolidinone dipole and the anomeric carbon oxygen bond dipole subtend an angle of approximately 90°, whereas in the equatorial glycoside the angle subtended is approximately 60°, suggesting that the equatorial glycosides will be more highly stabilized in less polar media, as is the case (Table 1).
Figure 10.

Influence of strongly electron-withdrawing protecting groups on the anomeric effect.
Conclusions
A new method of equilibration of sialyl anomeric C–O bonds has been developed on the basis of the Fischer–Ingold persistent radical effect. This method enables the investigation of protecting groups on the anomeric equilibrium and is compatible with acid and base sensitive functionality. The trans-fused oxazolidinone moiety is found by this method to stabilize equatorial glycosides over their axial counterparts, i.e., to reduce the magnitude of the anomeric effect. This effect, which adds to a series of recent observations on the influence of cyclic protecting groups on the anomeric effect,14,36 is a manifestation of the strongly electron-withdrawing nature of the trans-fused oxazolidinone, (and by extrapolation trans-fused cyclic carbonates), which is a direct consequence of the highly dipolar nature of such heterocycles. The strongly electron-withdrawing nature of these groups manifest by the present study more than likely also plays an important role in the high kinetic selectivity observed in the trans-fused oxazolidinone (and cyclic carbonate)-directed α-sialoside synthesis methods.38−40,45,149 The unusually poor kinetic diastereoselectivity of anomeric radicals in the sialic acid series is a function of the π-type planar nature of these radicals coupled with the existence of competing 1,3-diaxial interactions in the diastereomeric transition states for the formation of both anomers.
Acknowledgments
We thank the NIH (GM62160) for generous support of this work.
Supporting Information Available
Full experimental details and copies of 1H and 13C NMR spectra of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fraser-Reid B.; Lopez C. Top. Curr. Chem. 2011, 301, 1–9. [DOI] [PubMed] [Google Scholar]
- Gomez A. M. Top. Curr. Chem. 2011, 301, 31–68. [DOI] [PubMed] [Google Scholar]
- Shiao T. C.; Roy R. Top. Curr. Chem. 2011, 301, 69–108. [DOI] [PubMed] [Google Scholar]
- Kim K. S.; Suk D.-H. Top. Curr. Chem. 2011, 301, 109–140. [DOI] [PubMed] [Google Scholar]
- Aubry S.; Sasaki K.; Sharma I.; Crich D. Top. Curr. Chem. 2011, 301, 141–188. [DOI] [PubMed] [Google Scholar]
- Premathilake H. D.; Demchenko A. V. Top. Curr. Chem. 2011, 301, 189–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-Y.; Wong C.-H. Top. Curr. Chem. 2011, 301, 223–252. [DOI] [PubMed] [Google Scholar]
- Codée J. D. C.; Christina A. E.; Walvoort M. T. C.; Overkleeft H. S.; van der Marel G. A. Top. Curr. Chem. 2011, 301, 253–290. [DOI] [PubMed] [Google Scholar]
- Crich D. Acc. Chem. Res. 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- Huang M.; Retailleau P.; Bohé L.; Crich D. J. Am. Chem. Soc. 2012, 134, 14746–14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Garrett G. E.; Birlirakis N.; Bohé L.; Pratt D. A.; Crich D. Nat. Chem. 2012, 4, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasomanee J. P.; Demchenko A. V. J. Am. Chem. Soc. 2012, 134, 20097–20102. [DOI] [PubMed] [Google Scholar]
- Jensen H. H.; Nordstrom M.; Bols M. J. Am. Chem. Soc. 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- Moumé-Pymbock M.; Furukawa T.; Mondal S.; Crich D. J. Am. Chem. Soc. 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen C. M.; Nordstrom L. U.; Bols M. J. Am. Chem. Soc. 2007, 129, 9222–9235. [DOI] [PubMed] [Google Scholar]
- Gylling Frihed T.; Walvoort M. T. C.; Codée J. D. C.; van der Marel G. A.; Bols M.; Pedersen C. M. J. Org. Chem. 2013, 78, 2191–2205. [DOI] [PubMed] [Google Scholar]
- Okada Y.; Mukae T.; Okajimia K.; Taira M.; Fujita M.; Yamada H. Org. Lett. 2007, 9, 1573–1576. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Woerpel K. A. Org. Biomol. Chem. 2006, 4, 1195–1201. [DOI] [PubMed] [Google Scholar]
- Satoh H.; Manabe S. Chem. Soc. Rev. 2013, 42, 4297–4309. [DOI] [PubMed] [Google Scholar]
- Ranade S. C.; Demchenko A. V. J. Carbohydr. Chem. 2013, 32, 1–43. [Google Scholar]
- Walvoort M. T. C.; van der Marel G. A.; Overkleeft H. S.; Codée J. D. C. Chem. Sci. 2013, 4, 897–906. [Google Scholar]
- Ngoje G.; Li Z. Org. Biomol. Chem. 2013, 11, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Liu Q.-W.; Bin H.-C.; Yang J.-S. Org. Lett. 2013, 15, 3974–3977. [DOI] [PubMed] [Google Scholar]
- Lemieux R. U. In Molecular Rearrangements, Part 2; De Mayo P., Ed.; Wiley: New York, 1964; pp 709–769. [Google Scholar]
- Kirby A. J.The Anomeric Effect and Related Stereoelectronic Effects at Oxygen; Springer-Verlag: Berlin, 1983. [Google Scholar]
- Crich D.; Vinod A. U. J. Org. Chem. 2005, 70, 1291–1296. [DOI] [PubMed] [Google Scholar]
- Boysen M.; Gemma E.; Lahmann M.; Oscarson S. Chem. Commun. 2005, 3044–3046. [DOI] [PubMed] [Google Scholar]
- Olsson J. D. M.; Eriksson L.; Lahmann M.; Oscarson S. J. Org. Chem. 2008, 73, 7181–7188. [DOI] [PubMed] [Google Scholar]
- Satoh H.; Manabe S.; Ito Y.; Luthi H. P.; Laino T.; Hutter J. J. Am. Chem. Soc. 2011, 133, 5610–5619. [DOI] [PubMed] [Google Scholar]
- Manabe S.; Ishii K.; Ito Y. J. Am. Chem. Soc. 2006, 128, 10666–10667. [DOI] [PubMed] [Google Scholar]
- Geng Y.; Zhang L.-H.; Ye X.-S. Chem. Commun. 2008, 597–599. [DOI] [PubMed] [Google Scholar]
- Manabe S.; Ito Y. Tetrahedron Lett. 2009, 50, 4827–4829. [Google Scholar]
- Manabe S.; Ishii K.; Ito Y. Eur. J. Org. Chem. 2011, 497–516. [Google Scholar]
- Manabe S.; Ishii K.; Satoh H.; Ito Y. Tetrahedron 2011, 67, 9966–9974. [Google Scholar]
- Geng Y.; Qin Q.; Ye X.-S. J. Org. Chem. 2012, 77, 5255–5270. [DOI] [PubMed] [Google Scholar]
- Sharma I.; Bohé L.; Crich D. Carbohydr. Res. 2012, 357, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yule J. E.; Wong T. C.; Gandhi S. S.; Qiu D.; Riopel M. A.; Koganty R. R. Tetrahedron Lett. 1995, 36, 6839–6842. [Google Scholar]
- Crich D.; Li W. J. Org. Chem. 2007, 72, 2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D.; Li W. J. Org. Chem. 2007, 72, 7794–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Nishiura Y.; Takahashi T. J. Am. Chem. Soc. 2006, 128, 7124–7125. [DOI] [PubMed] [Google Scholar]
- Farris M. D.; De Meo C. Tetrahedron Lett. 2007, 48, 1225–1227. [Google Scholar]
- Crich D.; Wu B. Org. Lett. 2008, 10, 4033–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navuluri C.; Crich D. Angew. Chem., Int. Ed. 2013, 52, 11549–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Tateno Y.; Nishiura Y.; Takahashi T. Org. Lett. 2008, 10, 5597–5600. [DOI] [PubMed] [Google Scholar]
- Hsu C.-H.; Chu K.-C.; Lin Y.-S.; Han J.-L.; Peng Y.-S.; Ren C.-T.; Wu C.-Y.; Wong C.-H. Chem.—Eur. J. 2010, 16, 1754–1760. [DOI] [PubMed] [Google Scholar]
- Hanashima S.; Sato K.-I.; Ito Y.; Yamaguchi Y. Eur. J. Org. Chem. 2009, 4215–4220. [Google Scholar]
- Gu Z.-y.; Zhang J.-x.; Xing G.-w. Chem.—Asian J. 2012, 7, 1524–1528. [DOI] [PubMed] [Google Scholar]
- Wang Y.-J.; Jia J.; Gu Z.-Y.; Liang F.-F.; Li R.-C.; Huang M.-H.; Xu C.-S.; Zhang J.-X.; Men Y.; Xing G.-W. Carbohydr. Res. 2011, 346, 1271–1276. [DOI] [PubMed] [Google Scholar]
- Wang C.-H.; Li S.-T.; Lin T.-L.; Cheng Y.-Y.; Sun T.-H.; Wang J.-T.; Cheng T.-J. R.; Mong K. K. T.; Wong C.-H.; Wu C.-Y. Angew. Chem., Int. Ed. 2013, 52, 9157–9161. [DOI] [PubMed] [Google Scholar]
- Liang F.-F.; Chen L.; Xing G.-W. Synlett 2009, 425–428. [Google Scholar]
- Harris B. N.; Patel P. P.; Gobble C. P.; Stark M. J.; De Meo C. Eur. J. Org. Chem. 2011, 4023–4027. [Google Scholar]
- Tanaka H.; Nishiura Y.; Takahashi T. J. Org. Chem. 2009, 74, 4383–4386. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Nishiura Y.; Takahashi T. J. Am. Chem. Soc. 2008, 130, 17244–17245. [DOI] [PubMed] [Google Scholar]
- Xing G.-x.; Chen L.; Liang F.-f. Eur. J. Org. Chem. 2009, 5963–5970. [Google Scholar]
- Sun B.; Jiang H. Tetrahedron Lett. 2011, 52, 6035–6038. [Google Scholar]
- Chu K.-C.; Ren C.-T.; Lu C.-P.; Hsu C.-H.; Sun T.-H.; Han J.-L.; Pal B.; Chao T.-A.; Lin Y.-F.; Wu S.-H.; Wong C.-H.; Wu C.-Y. Angew. Chem., Int. Ed. 2011, 50, 9391–9395. [DOI] [PubMed] [Google Scholar]
- Chuang H.-Y.; Ren C.-T.; Chao C.-A.; Wu C.-Y.; Shivatare S. S.; Cheng T.-J. R.; Wu C.-Y.; Wong C.-H. J. Am. Chem. Soc. 2013, 135, 11140–11150. [DOI] [PubMed] [Google Scholar]
- Boltje T. J.; Heise T.; Rutjes F. P. T.; van Delft F. L. Eur. J. Org. Chem. 2013, 5257–5261. [Google Scholar]
- Noel A.; Delpech B.; Crich D. Org. Lett. 2012, 14, 1342–1345. [DOI] [PubMed] [Google Scholar]
- Gu Z.-y.; Zhang X.-t.; Zhang J.-x.; Xing G.-w. Org. Biomol. Chem. 2013, 11, 5017–5022. [DOI] [PubMed] [Google Scholar]
- Noel A.; Delpech B.; Crich D. Org. Lett. 2012, 14, 4138–4141. [DOI] [PubMed] [Google Scholar]
- Kuhn R.; Lutz P.; Macdonald D. L. Chem. Ber. 1966, 99, 611–617. [DOI] [PubMed] [Google Scholar]
- Yu P. K.; Ledeen R. J. Biol. Chem. 1969, 244, 1306–1313. [PubMed] [Google Scholar]
- Wilson J. C.; Angus D. I.; von Itzstein M. J. Am. Chem. Soc. 1995, 117, 4214–4217. [Google Scholar]
- Klepach T.; Carmichael I.; Serianni A. S. J. Am. Chem. Soc. 2008, 130, 11892–11900. [DOI] [PubMed] [Google Scholar]
- Chan J.; Sandhu G.; Bennet A. J. Org. Biomol. Chem. 2011, 9, 4818–4822. [DOI] [PubMed] [Google Scholar]
- Akhlaq M. S.; Schuchmann H. P.; Von Sonntag C. Int. J. Radiat. Biol. 1987, 5, 91–102. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Roberts B. P. J. Chem. Soc., Chem Commun. 1998, 1145–1146. [Google Scholar]
- Yu G. X.; Tyler D. R.; Branchaud B. P. J. Org. Chem. 2001, 66, 5687–5691. [DOI] [PubMed] [Google Scholar]
- Yamago S.; Miyazoe H.; Yoshida J.-i. Tetrahedron Lett. 1999, 40, 2339–2342. [Google Scholar]
- Studer A. Chem.—Eur. J. 2001, 7, 1159–1164. [DOI] [PubMed] [Google Scholar]
- Fischer H. Chem. Rev. 2001, 101, 3581–3610. [DOI] [PubMed] [Google Scholar]
- Tebben L.; Studer A. Angew. Chem., Int. Ed. 2011, 50, 5034–5068. [DOI] [PubMed] [Google Scholar]
- Lee M. D.; Dunne T. S.; Chang C. C.; Ellestad G. A.; Siegel M. M.; Morton G. O.; McGahren W. J.; Borders D. B. J. Am. Chem. Soc. 1987, 109, 3466–3468. [Google Scholar]
- Golik J.; Dubay G.; Groenewold G.; Kawaguchi H.; Konishi M.; Krishnan B.; Ohkuma H.; Saitoh K.; Doyle T. W. J. Am. Chem. Soc. 1987, 109, 3462–3464. [DOI] [PubMed] [Google Scholar]
- Cao F.; Tropper F. D.; Roy R. Tetrahedron 1995, 51, 6679–6686. [Google Scholar]
- Rodriguez E. C.; Marcaurelle L. A.; Bertozzi C. R. J. Org. Chem. 1998, 63, 7134–7135. [DOI] [PubMed] [Google Scholar]
- Chabre Y. M.; Roy R. Adv. Carbohydr. Chem. Biochem. 2010, 63, 165–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik G.; Ferry A.; Guinchard X.; Cresteil T.; Crich D. Chem.—Eur. J. 2013, 19, 2168–2179. [DOI] [PubMed] [Google Scholar]
- Hudak J. E.; Yu H. H.; Bertozzi C. R. J. Am. Chem. Soc. 2011, 133, 16127–16135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgault J. P.; Trabbic K. R.; Shi M.; Andreana P. R. Org. Biomol. Chem. 2014, 12, 1699–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S.; Gange D.; Gupta V.; Kahne D. J. Am. Chem. Soc. 1994, 116, 3197–3206. [Google Scholar]
- Shaffer C. L.; Gunduz M.; Ryder T. F.; O’Connell T. N. Drug Metab. Dispos. 2010, 38, 292–301. [DOI] [PubMed] [Google Scholar]
- Li F.; Pang X.; Krausz K. W.; Jiang C.; Chen C.-S.; Cook J. A.; Krishna M. C.; Mitchell J. B.; Gonzalez F. J.; Patterson A. D. J. Proteome Res. 2013, 12, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kancharla P. K.; Navuluri C.; Crich D. Angew. Chem., Int. Ed. 2012, 51, 11105–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochowski E.; Jurczak J. Carbohydr. Res. 1976, 50, c15–c16. [Google Scholar]
- Nicolaou K. C.; Groneberg R. D. J. Am. Chem. Soc. 1990, 112, 4085–4086. [Google Scholar]
- Nicolaou K. C.; Groneberg R. D.; Miyazaki T.; Stylianides N. A.; Schulze T. J.; Stahl W. J. Am. Chem. Soc. 1990, 112, 8193–8195. [Google Scholar]
- Halcomb R. L.; Wittman M. D.; Olson S. H.; Danishefsky S. J.; Golik J.; Wong H.; Vyas D. J. Am. Chem. Soc. 1991, 113, 5080–5082. [Google Scholar]
- Kim S.-H.; Augeri D.; Yang D.; Kahne D. J. Am. Chem. Soc. 1994, 116, 1766–1775. [Google Scholar]
- Yang D.; Kim S.-H.; Kahne D. J. Am. Chem. Soc. 1991, 113, 4715–4716. [Google Scholar]
- Kuryanov V. O.; Chupakhina T. a.; Shapovalova A. A.; Katsev A. M.; Chirva V. Y. Russ. J. Bioorg. Chem. 2011, 37, 231–239. [Google Scholar]
- Yu J.; Sun J.; Yu B. Org. Lett. 2012, 14, 4022–4025. [DOI] [PubMed] [Google Scholar]
- Bamhaoud T.; Lancelin J.-M.; Beau J.-M. J. Chem. Soc., Chem. Commun. 1992, 1494–1496. [Google Scholar]
- Da Silva E.; Prandi J.; Beau J.-M. J. Chem. Soc., Chem Commun. 1994, 2127–2128. [Google Scholar]
- Moutel S.; Prandi J. J. Chem. Soc., Perkin Trans. 1 2001, 305–315. [Google Scholar]
- Barton D. H. R.; Parekh S. I.. Half a Century of Free Radical Chemistry; Cambridge University Press: Cambridge, 1993. [Google Scholar]
- Nagy J. O.; Bednarski M. D. Tetrahedron Lett. 1991, 32, 3953–3956. [Google Scholar]
- Paulsen H.; Matschulat P. Liebigs Ann. Chem. 1991, 487–495. [Google Scholar]
- Martichonok V.; Whitesides G. M. J. Org. Chem. 1996, 61, 1702–1706. [DOI] [PubMed] [Google Scholar]
- Barton D. H. R.; Bolton M.; Magnus P. D.; West P. J.; Porter G.; Wirz J. J. Chem. Soc., Chem. Commun. 1972, 632–633. [Google Scholar]
- Achmatowicz S.; Barton D. H. R.; Magnus P. D.; Poulton G. A.; West P. J. J. Chem. Soc., Chem. Commun. 1971, 1014–1015. [DOI] [PubMed] [Google Scholar]
- Barton D. H. R.Some Recollections of Gap Jumping; American Chemical Society: Washington, D.C., 1991. [Google Scholar]
- Horn E. J.; Gervay-Hague J. J. Org. Chem. 2009, 74, 4357–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths P. G.; Moad G.; Rizzardo E.; Solomon D. H. Aust. J. Chem. 1983, 36, 397–401. [Google Scholar]
- Guerret O.; Couturier J.-L.; Le Mercier C. U.S. Patent Appl 2004/0077873 A1.
- Cardellini L.; Greci L.; Tosi G. Synth. Commun. 1992, 22, 201–207. [Google Scholar]
- Barton D. H. R.; George M. V.; Tomoeda M. J. Chem. Soc. 1962, 1967–1974. [Google Scholar]
- Czarniecki M. F.; Thornton E. R. J. Am. Chem. Soc. 1977, 99, 8273–8279. [Google Scholar]
- Hori H.; Nakajima T.; Nishida Y.; Ohrui H.; Meguro H. Tetrahedron Lett. 1988, 29, 6317–6320. [Google Scholar]
- Haverkamp J.; Spoormaker T.; Dorland L.; Vliegenthart J. F. G.; Schauer R. J. Am. Chem. Soc. 1979, 101, 4851–4853. [Google Scholar]
- Prytulla S.; Lauterwein J.; Klessinger M.; Thiem J. Carbohydr. Res. 1991, 215, 345–349. [Google Scholar]
- Kancharla P. K.; Crich D. J. Am. Chem. Soc. 2013, 135, 18999–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowry V. W.; Ingold K. U. J. Am. Chem. Soc. 1992, 114, 4992–4996. [Google Scholar]
- Chateauneuf J.; Lusztyk J.; Ingold K. U. J. Org. Chem. 1988, 53, 1629–1632. [Google Scholar]
- Beckwith A. L. J.; Bowry V. W.; Moad G. J. Org. Chem. 1988, 53, 1632–1641. [Google Scholar]
- Arends I. W. C. E.; Mulder P.; Clark K. B.; Wayner D. D. M. J. Phys. Chem. 1995, 99, 8182–8189. [Google Scholar]
- Korth H. G.; Sustmann R.; Giese B.; Rueckert B.; Groeninger K. S. Chem. Ber. 1990, 123, 1891–1898. [Google Scholar]
- Praly J. P. Adv. Carbohydr. Chem. Biochem. 2001, 56, 65–151. [DOI] [PubMed] [Google Scholar]
- Lung-min W.; Fischer H. Helv. Chim. Acta 1983, 66, 138–147. [Google Scholar]
- Korth H.-G.; Sustmann R.; Merényi R.; Viehe H. G. J. Chem. Soc., Perkin Trans. 2 1983, 67–74. [Google Scholar]
- Korth H. G.; Praly J. P.; Somsak L.; Sustmann R. Chem. Ber. 1990, 123, 1155–1160. [Google Scholar]
- Adlington R. M.; Baldwin J. E.; Basak A.; Kozyrod R. P. J. Chem. Soc., Chem. Commun. 1983, 944–945. [Google Scholar]
- Baumberger F.; Vasella A. Helv. Chim. Acta 1983, 66, 2210–2222. [Google Scholar]
- Giese B.; Dupuis J. Angew. Chem., Int. Ed. 1983, 22, 622–623. [Google Scholar]
- Crich D.; Ritchie T. J. J. Chem. Soc., Chem. Commun. 1988, 1461–1463. [Google Scholar]
- Kahne D.; Yang D.; Lim J. J.; Miller R.; Paguaga E. J. Am. Chem. Soc. 1988, 110, 8716–8717. [Google Scholar]
- Crich D.; Natarajan S. J. Org. Chem. 1995, 60, 6237–6241. [Google Scholar]
- Griller D.; Ingold K. U. Acc. Chem. Res. 1976, 9, 13–19. [Google Scholar]
- Crich D.; Grant D.; Bowers A. A. J. Am. Chem. Soc. 2007, 129, 12106–12107. [DOI] [PubMed] [Google Scholar]
- Potter J. J.; von Itzstein M. Carbohydr. Res. 1996, 282, 181–187. [Google Scholar]
- Horn E. J.; Saludes J. P.; Gervay-Hague J. Carbohydr. Res. 2008, 343, 936–940. [DOI] [PubMed] [Google Scholar]
- Shie J.-J.; Fang J.-M.; Lai P.-T.; Wen W.-H.; Wang S.-Y.; Cheng Y.-S. E.; Tsai K.-C.; Yang A.-S.; Wong C.-H. J. Am. Chem. Soc. 2011, 133, 17959–17965. [DOI] [PubMed] [Google Scholar]
- Bagryanskaya E. G.; Marque S. R. A.; Tsentalovich Y. P. J. Org. Chem. 2012, 77, 4996–5005. [DOI] [PubMed] [Google Scholar]
- Rao V. S. R.; Qasba P. K.; Balaji P. V.; Chandrasekaran R.. Conformation of Carbohydrates; Harwood Academic Publishers: Amsterdam, 1998. [Google Scholar]
- Grindley T. B. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid B., Tatsuta K., Thiem J., Eds.; Springer: Berlin, 2001; Vol. 1, pp 3–51. [Google Scholar]
- Lee M. D.; Dunne T. S.; Chang C. C.; Siegel M. M.; Morton G. O.; Ellestad G. O.; McGahren W. J.; Borders D. B. J. Am. Chem. Soc. 1992, 114, 985–997. [Google Scholar]
- Duléry V.; Renaudet O.; Philouze C.; Dumy P. Carbohydr. Res. 2007, 342, 894–900. [DOI] [PubMed] [Google Scholar]
- Renaudet O.; Dumy P.; Philouze C.; Averbuch-Pouchot M.-T. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2001, C57, 649–650. [DOI] [PubMed] [Google Scholar]
- Renaudet O.; Philouze C.; Bailly C.; Durif A.; Dumy P. J. Chem. Crystallogr. 2011, 41, 204–208. [Google Scholar]
- Tian R.; Haoriqinbatu; Liu H.; Wang X.; Yang Y. Acta Crystallogr., Sect. E: Struct. Rep. Online 2012, E68, o635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan A. L.Tables of Experimental Dipole Moments; Freeman: San Francisco, 1963. [Google Scholar]
- Lee C. M.; Kumler W. D. J. Am. Chem. Soc. 1961, 83, 4596–4600. [Google Scholar]
- Lee C. M.; Kumler W. D. J. Am. Chem. Soc. 1962, 84, 571–578. [Google Scholar]
- Gonzalez-Outeiriño J.; Nasser R.; Anderson J. E. J. Org. Chem. 2005, 70, 2486–2493. [DOI] [PubMed] [Google Scholar]
- Schweitzer W. B.; Dunitz J. D. Helv. Chim. Acta 1982, 65, 1547–1554. [Google Scholar]
- Fowler P.; Bernet B.; Vasella A. Helv. Chim. Acta 1996, 79, 269–287. [Google Scholar]
- Deslongchamps P.Stereoelectronic Effects in Organic Chemistry; Pergamon: Oxford, 1983. [Google Scholar]
- Crich D.; Navuluri C. Angew. Chem., Int. Ed. 2010, 49, 3049–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.