

Abstract
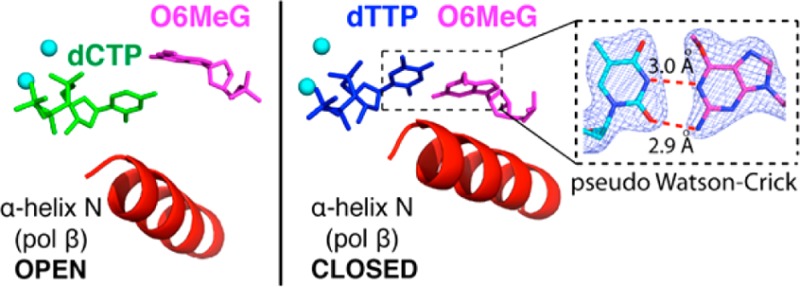
Human DNA polymerase β (polβ) inserts, albeit slowly, T opposite the carcinogenic lesion O6-methylguanine (O6MeG) ∼30-fold more frequently than C. To gain insight into this promutagenic process, we solved four ternary structures of polβ with an incoming dCTP or dTTP analogue base-paired with O6MeG in the presence of active-site Mg2+ or Mn2+. The Mg2+-bound structures show that both the O6MeG·dCTP/dTTP–Mg2+ complexes adopt an open protein conformation, staggered base pair, and one active-site metal ion. The Mn2+-bound structures reveal that, whereas the O6Me·dCTP–Mn2+ complex assumes the similar altered conformation, the O6MeG·dTTP–Mn2+ complex adopts a catalytically competent state with a closed protein conformation and pseudo-Watson–Crick base pair. On the basis of these observations, we conclude that polβ slows nucleotide incorporation opposite O6MeG by inducing an altered conformation suboptimal for catalysis and promotes mutagenic replication by allowing Watson–Crick-mode for O6MeG·T but not for O6MeG·C in the enzyme active site. The O6MeG·dTTP–Mn2+ ternary structure, which represents the first structure of mismatched polβ ternary complex with a closed protein conformation and coplanar base pair, the first structure of pseudo-Watson–Crick O6MeG·T formed in the active site of a DNA polymerase, and a rare, if not the first, example of metal-dependent conformational activation of a DNA polymerase, indicate that catalytic metal-ion coordination is utilized as a kinetic checkpoint by polβ and is crucial for the conformational activation of polβ. Overall, our structural studies not only explain the promutagenic polβ catalysis across O6MeG but also provide new insights into the replication fidelity of polβ.
Introduction
Although O6-methylguanine (O6MeG) is a minor component of methylated DNA lesions produced by various endogenous (e.g., S-adenosylmethionine) and exogenous (e.g., N-methyl-N-nitrosourea) alkylating agents,1−5 it is a highly mutagenic lesion. The genotoxic O6MeG lesion is also generated by anticancer methylating agents such as temozolomide and is believed to be responsible for the cytotoxicity of various methylating anticancer agents. O6MeG is directly repaired in an error-free manner by a sacrificial protein called methylguanine methyltransferase (MGMT).6 If not repaired by MGMT, the persistent O6MeG in templating DNA causes G to A transition mutations.2,7 Since MGMT activity is impaired in many cancer cells, the treatment of such cells with methylating anticancer agents can promote the formation of O6MeG·T mismatch, which can trigger a futile cytotoxic repair by the mismatch repair system (MMR).8,9 Cells deficient in MMR are resistant to the cytotoxicity induced by temozolomide-mediated methylation. In MMR-deficient cells thymine in O6MeG·T mismatch can be removed by thymine DNA glycosylase and methyl-CpG-binding domain protein 4,10−13 and the resulting abasic sites can be further processed by downstream base-excision repair (BER) proteins such as DNA polymerase β (polβ).14,15 Therefore, elucidating the mechanism of the replication across O6MeG by polβ could potentially further our understanding of the mutagenicity and the cytotoxicity of O6MeG.
The X-family DNA polymerase polβ is a short-nucleotide-gap filling BER enzyme16 and has been shown to replicate across O6MeG in vitro.14,15 Polβ is mutated and overexpressed in many cancer cells17 and has been implicated to play a role in resistance to various anticancer agents such as cisplatin, bleomycin, and methylating agents.18,19 Inhibition of polβ has been shown to sensitize temozolomide activity,19−21 implicating polβ’s potential role in the repair of temozolomide-induced DNA lesions.
The O6MeG mutagenicity mainly results from the preferential incorporation of T opposite templating O6MeG by DNA polymerases.1,22−24 Although thermodynamic and NMR studies on duplex DNA indicate that O6MeG·C base pair is more stable than O6MeG·T base pair (Figure 1),25−29 many DNA polymerases preferentially insert T over C opposite O6MeG.24,30−33 For example, the Y-family DNA polymerase polι and replicative DNA polymerases such as T7 DNA polymerase and Bacillus stearothermophilus DNA polymerase I fragment (BF) incorporate T opposite O6MeG with insertion efficiency ∼10-fold greater than that for C. In addition, the X-family DNA polymerase polβ inserts T opposite O6MeG ∼30-fold more efficiently than C in vitro, while the rate of replication across the lesion is decreased ∼100-fold.15
Figure 1.

Structures of (A) O6MeG·C base pair and (B) O6MeG·T base pair.
Currently, although structures of various DNA polymerases in complex with O6MeG-containing DNA have provided important insights into the mutagenic potential of O6MeG,22,30,33 the structural basis underlying the observed preferential misincorporation of T opposite O6MeG by several DNA polymerases remains elusive. For example, X-ray structures of BF have shown that structural differences among BF ternary complexes bearing the newly incorporated O6MeG·dCTP and O6MeG·dTTP base pairs are not prominent, with both O6MeG·C and O6MeG·T forming isosteric Watson–Crick-type base pairings in the confines of the BF active site.30 To gain deeper insight into the mutagenic replication across O6MeG conducted by several DNA polymerases, we solved X-ray structures of O6MeG-containing DNA bound to polβ, which highly inaccurately replicates across O6MeG. Herein, we report five X-ray structures of polβ bound to O6MeG-containing DNA, representing varying stages of nucleotide insertion opposite O6MeG; a binary structure with a single-nucleotide gap opposite O6MeG and four ternary structures with an incoming dCTP or dTTP analogue paired with O6MeG in the presence of active-site Mg2+ or Mn2+. In addition, to evaluate the effects of the active-site metal ion on the polβ catalysis, we have determined steady-state kinetic parameters for the insertion of dCTP/dTTP opposite templating O6MeG by the enzyme in the presence of Mg2+ or Mn2+. Our X-ray structures reveal that polβ slows nucleotide incorporation opposite O6MeG by inducing an altered conformation suboptimal for catalysis and that polβ discriminates O6MeG·T against O6MeG·C in the nascent base-pair binding pocket. Our structural studies not only provide the basis for the promutagenic replication across O6MeG by polβ but also provide new insights into the replication fidelity of polβ.
Experimental Section
DNA Sequences Used for X-ray Crystallographic Studies
All oligonucleotides used for crystallographic studies were purchased from Midland Certified Reagent Company (Midland, TX). The DNA sequence for template DNA is 5′-CCGAC(O6MeG)TCGCATCAGC-3′. The DNA sequence for upstream primer is 5′-GCTGATGCGA-3′, and the sequence for the downstream primer is 5′-phosphate/GTCGG-3′.34
Cocrystallization of polβ:DNA Binary and Ternary Complexes
Polβ was expressed and purified as described previously.35 Polβ binary complex with a single-nucleotide gap opposite templating O6MeG was prepared using the same conditions described previously.35 Polβ ternary complex was prepared by adding nonhydrolyzable dCMPNPP or dTMPNPP (5.0 mM, Jena Biosciences) to the mixture of the polβ gapped binary complex. Polβ ternary complex crystals with nonhydrolyzable dCMPNPP or dTMPNPP opposite templating O6MeG were grown over 2–4 weeks in a buffer solution containing 50 mM imidazole, pH 7.5, 14%–23% PEG3400, and 350 mM NaOAc.35 The polβ binary and ternary complex crystals were cryo-protected with 12% ethylene glycol and flash-frozen in liquid nitrogen. Diffraction data were collected at the beamline 5.0.3 at the Advanced Light Source, Lawrence Berkeley National Laboratory and were processed using the HK-2000 program. The polβ gapped binary complex structure and the ternary complex structures were solved by molecular replacement36 using published binary (PDB ID 1BPX) and ternary (PDB ID 1BPY) structures as the search models, respectively.37 The model building and structure refinement were conducted using COOT,38 Phenix,39 and MolProbity,40 and all the crystallographic figures were generated using PyMOL.
Steady-State Kinetics of Nucleotide Incorporation Opposite Templating O6MeG by polβ
Steady-state kinetic parameters for nucleotide incorporation opposite O6MeG by polβ were determined as described.41 Oligonucleotides used for kinetic assays (primer, 5′-FAM/CTGCAGCTGATGCG-3′; downstream primer, 5′-phosphate/CGTACGGATCCCCGGGTAC-3′; and template, 5′-GTACCCGGGGATCCGTACG (O6MeG)CGCATCAGCGCAG-3′) were purchased from Midland Certified Reagent Company.
DNA substrate containing a single-nucleotide gap opposite templating O6MeG was prepared by annealing the template oligonucleotide with the upstream and the downstream primers at 95 °C for 3 min followed by slow cooling to room temperature. Polymerase activities were determined using the reaction mixture containing 50 mM Tris-HCl pH 7.4, 100 mM KCl, 5 mM MgCl2 or MnCl2, 80 nM single-nucleotide gapped DNA, and varying concentrations of incoming nucleotide. The phosphoryl transfer reactions were initiated by adding polβ and stopped by adding 95% formamide solution containing 20 mM EDTA, 45 mM Tris-borate, 0.1% bromophenol blue, and 0.1% xylene cyanol. The polymerase reaction mixtures were separated on 18–20% denaturing polyacrylamide gels, and the product formation was analyzed using a PhosphorImager (Molecular Dynamics). The efficiency and the relative efficiency of nucleotide incorporation opposite templating O6MeG by polβ were calculated as kcat/Km and f = (kcat/Km) [dC or dT:O6MeG]/(kcat/Km) [dC:dG], respectively.
Results
Kinetic Studies
Using steady-state kinetic methods,41 we determined kinetic parameters for nucleotide incorporation opposite O6MeG by polβ (Table 1). In the presence of Mg2+, nucleotide insertion efficiency for T opposite O6MeG is ∼20-fold higher than that for C opposite O6MeG, and ∼300-fold lower than that for C opposite G. In the presence of Mn2+, the insertion efficiency for T opposite O6MeG is ∼100-fold higher than that for C opposite O6MeG, and ∼30-fold lower than that for C opposite G. Substituting Mn2+ for Mg2+ increases the C and T insertion efficiencies ∼2-fold and ∼10-fold, respectively.
Table 1. Steady-State Kinetic Parameters for Nucleotide Insertion Opposite O6MeG by polβ.
| template:dNTP (metal ion) | Km (μM) | kcat (1/s) | kcat/Km | f |
|---|---|---|---|---|
| dG:dCTP (Mg2+) | 0.6 ± 0.1 | 212.0 ± 19.9 | 3.5 × 102 | 1 |
| dG:dTTP (Mg2+) | 56.1 ± 4.6 | 2.8 ± 0.4 | 5.0 × 10–2 | 1.4 × 10–4 |
| O6MedG:dCTP (Mg2+) | 234.2 ± 24.5 | 14.5 ± 1.2 | 6.2 × 10–2 | 1.7 × 10–4 |
| O6MedG:dTTP (Mg2+) | 56.2 ± 4.7 | 62.4 ± 11.0 | 1.2 | 3.3 × 10–3 |
| O6MedG:dCTP (Mn2+) | 193.3 ± 7.6 | 20.4 ± 1.6 | 1.1 × 10–1 | 2.9 × 10–4 |
| O6MedG:dTTP (Mn2+) | 38.7 ± 4.1 | 431.8 ± 53.2 | 11.2 | 3.2 × 10–2 |
Binary Structure of polβ Bound to DNA Containing a Single-Nucleotide Gap Opposite O6MeG
We determined a binary complex structure of polβ bound to DNA containing a single-nucleotide gap opposite O6MeG (A and B of Figure 2). The O6MeG gapped binary structure was solved by molecular replacement using a published gapped structure (PDB ID 1BPX), and refined to 2.4 Å resolution (Table 2). The overall structure is similar to that of the published gapped binary structure35 (PDB ID 1BPX; RMSD = 0.65 Å), with the protein in an open conformation and a 90° kink in the DNA. Comparison of the O6MeG gapped structure with published G gapped structure35 (PDB ID 1BPX) shows a minor conformational difference in templating base and primer terminus base pair (Figure 2E). O6MeG adopts an anti base conformation (Figures 2C and 2D). Tyr271 is H-bonded to N2 of O6MeG. The α-Helix N containing Asn279 and Arg283, the minor-groove recognition motifs, is in an open conformation.
Figure 2.

Structure of polβ bound to DNA containing a single-nucleotide gap opposite templating O6MeG (PDB ID 4MF2). (A) Overall structure of the gapped pol β complex. (B) DNA sequence used for crystallization of the O6MeG gapped complex. The O6MeG·C/T ternary complex structures have dCTP or dTTP analogue opposite templating O6MeG. (C) Active-site view of the gapped structure. Protein is in an open conformation. The three aspartic acid residues as well as Tyr271, Asn279, and Arg283 are indicated. H-bonding interactions are indicated as dotted lines. An ordered water molecule is depicted as a magenta sphere. (D) A 2Fo – Fc map contoured at 1σ around O6MeG lesion. (E) Structural overlay of the templating base and primer terminus in the O6MeG gapped binary complex and published G gapped binary complex35 (PDB ID 1BPX).
Table 2. Data Collection and Refinement Statistics.
| PDB code | gapped binary (4MF2) | O6MeG·C Mg2+ ternary (4MFC) | O6MeG·T Mg2+ ternary (4MFF) | O6MeG·C Mn2+ ternary (4NY8) | O6MeG·T Mn2+ ternary (4NXZ) |
|---|---|---|---|---|---|
| Data Collection | |||||
| space group | P21 | P21 | P21 | P21 | P21 |
| Cell Constants | |||||
| a (Å) | 54.438 | 54.596 | 54.546 | 54.625 | 50.803 |
| b (Å) | 79.265 | 79.648 | 78.839 | 79.288 | 79.842 |
| c (Å) | 54.789 | 54.856 | 54.751 | 54.838 | 55.442 |
| α (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β (deg) | 105.66 | 105.86 | 105.95 | 105.97 | 107.05 |
| γ (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| resolution (Å)a | 20–2.40 (2.44–2.40) | 20–2.14 (2.18–2.14) | 20–2.56 (2.60–2.56) | 20–2.25 (2.29–2.25) | 20–2.56 (2.60–2.56) |
| ⟨I/σ⟩ | 14.8 (2.27) | 20.2 (3.38) | 11.0 (2.39) | 24.8 (4.60) | 14.9 (1.80) |
| completeness (%) | 93.8 (95.7) | 100 (100) | 99.1 (96.8) | 99.4 (96.4) | 95.0 (93.7) |
| Rmergeb (%) | 8.1 (32.2) | 9.2 (31.3) | 13.0 (47.3) | 8.0 (30.8) | 13.6 (59.4) |
| redundancy | 3.3 (3.1) | 4.5 (4.4) | 4.5 (4.1) | 5.6 (4.9) | 4.6 (3.9) |
| Refinement | |||||
| Rworkc/Rfreed (%) | 19.8/27.9 | 21.3/27.4 | 22.2/29.4 | 21.1/26.7 | 19.3/25.5 |
| unique reflections | 16418 | 25023 | 14381 | 21347 | 12667 |
| Mean B Factor (Å2) | |||||
| protein | 33.1 | 29.3 | 20.7 | 28.2 | 29.2 |
| ligand | 31.5 | 28.6 | 18.4 | 35.2 | 27.5 |
| solvent | 26.5 | 27.7 | 14.8 | 24.5 | 26.8 |
| Ramachandran Plot | |||||
| most favored (%) | 95.9 | 94.9 | 94.7 | 97.8 | 97.5 |
| add. allowed (%) | 3.8 | 4.5 | 4.3 | 2.2 | 2.5 |
| RMSD | |||||
| bond lengths (Å) | 0.011 | 0.015 | 0.011 | 0.004 | 0.004 |
| bond angles (deg) | 1.620 | 1.964 | 1.617 | 1.134 | 1.097 |
Values in parentheses are for the highest resolution shell.
Rmerge = ∑|I – ⟨I⟩|/ ∑+I where +I is the integrated intensity.
Rwork = ∑|F(obs) – F(calc)|/∑F(obs).
Rfree = ∑|F(obs) – F(calc)|/∑F(obs), calculated using 5% of the data.
Ternary Structure of polβ Inserting a dCTP Analogue Opposite O6MeG in the Presence of Mg2+
To gain structural insight into how polβ performs accurate replication across O6MeG, we determined a ternary structure of polβ incorporating a dCTP analogue opposite templating O6MeG in the presence of active-site Mg2+ (A and B of Figure 3). Nonhydrolyzable dCMPNPP (dCTP* hereafter) was used because it retains binding affinity with polβ,42 while preventing the nucleotidyl transfer catalyzed by the enzyme. The O6MeG·C–Mg2+ ternary structure was refined to 2.3 Å resolution (Figure 3A). Since all published ternary structures of polβ with base pair mismatch involve either active-site Mn2+ or mutations in the minor-groove recognition motif (Arg283Lys),42−46 our structure represents the first structure of wild-type polβ with base pair mismatch and active-site Mg2+.
Figure 3.

Ternary structure of polβ incorporating nonhydrolyzable dCTP analogue (dCTP*, shown in green) opposite templating O6MeG in the presence of Mg2+ (PDB ID 4MFC). (A) Overall structure of the O6MeG·C–Mg2+ ternary structure. (B) Structural overlay of the O6MeG·C–Mg2+ ternary complex and the O6MeG binary gapped complex. Protein in the binary structure is shown in blue. (C) Active-site view of the O6MeG·C–Mg2+ ternary structure. Protein is in an open conformation. O6MeG and dCTP* form a staggered base pair. The distance between the 3′-OH of the primer terminus and Pα of dTTP* is indicated as a red double-headed arrow. (D) A 2Fo – Fc map contoured at 1σ around O6MeG and dCTP*. (E) Close-up view of the active-site metal ion binding site. Only the nucleotide-binding metal ion is present in this structure, and the metal ion is not coordinated to Asp192. (F) Overlay of the O6MeG·C–Mg2+ ternary structure with published C·A–Mn2+ ternary structure (PDB ID 3C2L(42)). (G) Overlay of the O6MeG·C–Mg2+ ternary structure with published A·G ternary structure with Arg283Lys mutation (PDB ID 4F5P(43)).
Surprisingly, the O6MeG·C–Mg2+ ternary complex shows an open protein conformation, a staggered O6MeG·C base pair conformation. The overall structure of the O6MeG·C–Mg2+ ternary complex is almost indistinguishable from that of the O6MeG binary gapped structure (RMSD = 0.265 Å, Figure 3B), indicating that binding of dCTP* does not readily induce an open-to-closed conformational activation of the enzyme. Polβ ternary structure with an open protein conformation has only been observed with the enzyme with Arg283Lys mutation, and has not been observed with the wild-type enzyme.44,45 Published polβ ternary structures with the wild-type enzyme show a closed protein conformation for correct insertion and an intermediate protein conformation for incorrect insertion. The O6MeG·C–Mg2+ ternary structure most likely represents a ground-state conformation,47 which is suboptimal for nucleotidyl transfer reaction. Apparently, this ternary structure has not reached a catalytically competent state. The distance between the 3′-OH of the primer terminus and the Pα of dCTP* is ∼2.6 Å longer than the distance typically observed for correct insertion (6.0 vs ∼3.4 Å).35 The α-helix N-containing minor-groove recognition motifs, which typically move ∼10 Å toward a nascent base pair for correct insertion, have not moved from the positions observed in the O6MeG gapped binary complex structure with an open conformation (Figure 3B).35 Overall, the O6MeG·C–Mg2+ ternary complex does not adopt a catalytically competent conformation, which is consistent with the observed slow dCTP insertion opposite O6MeG in the presence of Mg2+ (Table 1).
The O6MeG·C–Mg2+ ternary complex structure explains why accurate replication across O6MeG is greatly inhibited by polβ (Table 1). The structure reveals that polβ slows the accurate replication across O6MeG by inducing a novel catalytically incompetent conformation. First, polβ prevents dCTP incorporation opposite O6MeG by inducing an open protein conformation rather than a closed conformation required for chemistry. Second, polβ deters the dCTP incorporation by inducing a staggered O6MeG·C base pair conformation, which lacks H-bonding and base-stacking interactions typically observed for the correct insertion (Figures 3A and 3C). The staggered base pair conformation has been observed with polβ ternary structures with base pair mismatch. Last, polβ precludes dCTP incorporation opposite O6MeG by altering the coordination state of the active-site metal ions. The O6MeG·C–Mg2+ ternary structure shows only one active-site metal ion,43 rather than the two active-site metal ions required for catalysis. Furthermore, the coordination sphere of the nucleotide-binding metal ion is only partially completed. Taken together, the combined effects of the open protein conformation, staggered base pair, and one active-site metal ion greatly distort the active-site conformation, thereby hampering the incorporation of dCTP opposite O6MeG by polβ.
The active-site structure of the O6MeG·C–Mg2+ ternary complex is very different from that of published ternary complex42 with C·A mismatch and active-site Mn2+ (Figure 3F, RMSD = 1.565 Å). These structures differ in the conformations of protein and DNA, the number of active-site metal ions, and position of incoming nucleotide. The distance between the O3 of primer terminus and the Pα of incoming nucleotide for the O6MeG·C–Mg2+ structure is 2.7 Å longer than that for the C·A–Mn2+ structure (E and F of Figure 3). In addition, the distance between the C1′ of primer terminus and the C1′ of incoming nucleotide for the O6MeG·C–Mg2+ structure is ∼4.0 Å longer than that for the C·A–Mn2+ structure. In stark contrast, the active-site structure of the O6MeG·C–Mg2+ ternary complex is very similar to that of published G·A–Mg2+ ternary complex with Arg283Lys mutation43 (RMSD = 0.274 Å, Figure 3G), which was introduced to capture polβ ternary structure with an open protein conformation. A minor structural difference between the wild-type polβ:O6MeG·C–Mg2+ and the Arg283Lys polβ:G·A–Mg2+ complexes is the presence/absence of Asn279-mediated minor-groove edge recognition of incoming nucleotide (3.0 vs 4.8 Å, Figure 3G). The structural similarity among the O6MeG gapped binary, the O6MeG·C–Mg2+ ternary, and the Arg283Lys polβ:G·A–Mg2+ ternary complexes suggests that ground-state structures of polβ ternary complex with base pair mismatch adopt open protein conformation and staggered base pair conformation, which would be suboptimal for catalysis. Polβ appears to discourage nucleotide misincorporation by preventing an open-to-closed conformational activation and inducing noncoplanar base pair conformation in the presence of a mismatched incoming nucleotide.
Ternary Structure of polβ Inserting a dTTP Analogue Opposite O6MeG in the Presence of Mg2+
To gain structural insight into the highly promutagenic replication across O6MeG by polβ, we solved a ternary structure of polβ with an incoming nonhydrolyzable dTMPNPP (dTTP* hereafter) paired with O6MeG in the presence of active-site Mg2+. The X-ray structure of the O6MeG·T–Mg2+ ternary complex was solved to 2.3 Å resolution. The overall structure of the O6MeG·T–Mg2+ ternary complex is essentially identical to that of the O6MeG·C–Mg2+ ternary complex, with assuming an open protein conformation, a staggered base pair, and one active-site metal ion (A–C of Figure 4). Therefore, the O6MeG·T–Mg2+ ternary structure most likely represents a ground-state conformation, which is suboptimal for polymerase reaction.
Figure 4.

Ternary structure of polβ incorporating a nonhydrolyzable dTTP analogue (dTTP*, shown in cyan) opposite templating O6MeG in the presence of Mg2+ (PDB ID 4MFF). (A) Overall structure of the O6MeG·T–Mg2+ ternary structure. (B) Active-site view of the O6MeG·T–Mg2+ ternary structure. Protein is in an open conformation. O6MeG and dTTP* form a staggered base pair. Ordered water-mediated H-bondings not observed in the O6MeG·C–Mg2+ ternary structure are indicated in red dotted lines. (C) Close-up view of the metal-ion-binding site. Only nucleotide-binding metal ion is present in this structure. An ordered water molecule that bridges Asp256, Asp190, and primer terminus 3′-OH replaces the catalytic metal ion observed in polβ ternary structure. (D) Overlay of the metal-ion-binding site of the O6MeG·C–Mg2+ structure (green) and the O6MeG·C–Mg2+ structure (blue). Note differences in the positions of the primer terminus 3′-OHs and ordered water molecules. The 5′ side of the primer terminus base is omitted for clarity.
Comparison of the O6MeG·C/T–Mg2+ ternary structures suggests that a water-mediated H-bond network may contribute the promutagenic replication of O6MeG by the enzyme (Figure 4D). Since both the O6MeG·C–Mg2+ and the O6MeG·T–Mg2+ ternary complexes adopt a staggered base pair, the preferential T insertion opposite O6MeG by the enzyme is unlikely, due to a difference in base pairing stabilities of their ground-state structures. Interestingly, the distance between O3′ of the primer terminus and Pα of the incoming nucleotide seen in the O6MeG·T–Mg2+ ternary structure (3.7 Å, Figure 4C) is 2.3 Å shorter than that seen in the O6MeG·C–Mg2+ ternary structure, indicating that the O6MeG·T–Mg2+ ternary complex adopts more favorable conformation for nucleotidyl transfer than the O6MeG·C–Mg2+ ternary complex. The favorable conformation of the O6MeG·T–Mg2+ ternary complex appears to be triggered by a water-mediated H-bond network present in the O6MeG·T–Mg2+ ternary structure (Figure 4D), but not in the O6MeG·C–Mg2+ ternary structure. More specifically, in the active site of the O6MeG·T–Mg2+ ternary complex, an ordered water molecule is H-bonded to Asp190, Asp256, and primer terminus 3′-OH, which is reminiscent of the catalytic metal ion’s coordination with Asp190, Asp192, Asp256, primer terminus 3′-OH, and Pα of an incoming nucleotide. This water-mediated H-bond network brings the 3′-OH of the primer terminus closer to the Pα of the incoming nucleotide with a distance comparable to that observed in ternary structures with correct insertion (3.7 vs ∼3.4 Å). To reach a catalytically competent state, the O6MeG·T–Mg2+ complex would thus require a conformational reorganization of the protein to a lesser extent than the O6MeG·C–Mg2+ complex. In other words, the O6MeG·T–Mg2+ complex will have a lower energy barrier for chemistry than the O6MeG·C–Mg2+ complex, resulting in faster T insertion opposite O6MeG relative to C insertion opposite O6MeG.
Ternary Structure of polβ Inserting a dCTP Analogue Opposite O6MeG in the Presence of Mn2+
As mentioned above, the O6MeG·C–Mg2+ ternary structure likely represent a ground state structure with a catalytically incompetent conformation. To gain insight into precatalytic state of polβ incorporating dCTP opposite O6MeG, we determined ternary structure of polβ with dCTP* paired with templating O6MeG in the presence of Mn2+. The use of Mn2+ has been shown to enhance the binding affinity of the incoming mismatched nucleotide, facilitate the formation of an intermediate protein conformation during misincorporation, and significantly increase (>10-fold) the rate of misincorporation.35,37,41,42,46 The O6MeG·C–Mn2+ ternary structure was refined to 2.25 Å (Figure 5).
Figure 5.

Ternary structure of polβ incorporating dCTP* opposite templating O6MeG in the presence of Mn2+ (PDB ID 4NY8). (A) Overall structure of the O6MeG·C–Mn2+ ternary structure. (B) Active-site view of the O6MeG·C–Mn2+ ternary structure. Protein is in an open conformation. O6MeG and dCTP* form a staggered base pair. (C) Close-up view of the metal-ion-binding site. Both the nucleotide-binding and the catalytic metal ions are present, yet the critical coordination of Asp256 to the catalytic metal ion is lacking. The O3′(primer terminus)-Pα(dCTP*) (5.0 Å) and the C1′(primer terminus)-C1′(dCTP*) (9.0 Å) distances are longer than those for correct insertion (3.4 Å and 5.0 Å, respectively). (D) Overlay of the active-site structure of the O6MeG·C–Mg2+/Mn2+ complexes (RMSD = 0.165 Å).
The O6MeG·C–Mn2+ ternary structure indicates that, even in the presence of Mn2+, polβ strongly discourages the accurate replication across O6MeG by inducing a catalytically imcompetent conformation (Figure 5A). The active-site of the O6MeG·C–Mn2+ ternary structure is significantly different from those of published polβ ternary structures with base pair mismatch and active-site Mn2+ (Figure S1in SI). Whereas published structures with C·A or A·G mismatch show a partially closed protein conformation, our O6MeG·C–Mn2+ structure shows an open protein conformation (Figure S1in SI). In addition, positions of templating base and incoming nucleotide in those complexes are quite different. Interestingly, the overall structure of the O6MeG·C–Mn2+ ternary complex is essentially identical to that of the O6MeG·C–Mg2+ ternary complex with an open protein conformation (RMSD = 0.165 Å, Figure 5D). Like the O6MeG·C–Mg2+ ternary complex, the O6MeG·C–Mn2+ ternary complex adopts an open protein conformation and staggered base pair, indicating that the substitution of Mn2+ for Mg2+ does not significantly facilitate the open-to-closed conformational activation of the O6MeG·C ternary complex (Figure 5A). The only notable difference between the O6MeG·C–Mg2+/Mn2+ complexes is the absence and the presence of the catalytic metal ion, respectively (Figure 5B and C). The structural similarity between the O6MeG·C–Mg2+/Mn2+ complexes is consistent with our kinetic data showing only a modest (∼2-fold) increase in insertion efficiency by the metal-ion substitution (Table 1). The distance between the O3′ of primer terminus and the Pα of incoming nucleotide in the O6MeG·C–Mn2+ structure is ∼1.6 Å longer than that observed in polβ structure with correct insertion. In addition, the catalytic metal ion is not coordinated to catalytic Asp256 (4.5 Å) and is weakly coordinated to the primer terminus 3′-OH (3.1 Å) (Figure 5C). Molecular dynamics studies47 have suggested that the reaction pathway for polβ-catalyzed misincorporation involves an open-to-closed conformational change of protein and proton transfer from primer O3′-H to Asp256. Recent computational and structural studies44 with Asp256Glu polβ support that proton transfer from primer O3′ to nearby Asp256 is important for catalysis. The formation of open protein conformation, the lack of the coordination of Asp256 to the catalytic metal ion, and the longer O3′-Pα and O3′–Mn2+ distances observed in the O6MeG·C–Mn2+ ternary structure thus suggest that this structure most likely represents a conformational intermediate that requires a further conformational adjustment of the active site to reach a catalytically competent state. Overall, both the O6MeG·C–Mg2+/Mn2+ ternary structures with open protein conformation explain the inefficient incorporation of dCTP opposite O6MeG by the enzyme (Table 1).
Ternary Structure of polβ Inserting a dTTP Analogue Opposite O6MeG in the Presence of Mn2+
To gain insight into the precatalytic state of polβ performing the mutagenic replication across O6MeG, we determined a ternary structure of polβ with dTTP* paired with templating O6MeG in the presence of Mn2+. The O6MeG·T–Mn2+ ternary structure was solved to 2.56 Å (Figure 6).
Figure 6.

Ternary structure of polβ incorporating dTTP* opposite templating O6MeG in the presence of Mn2+ (PDB ID 4NXZ). (A) Overall structure of the O6MeG·T–Mn2+ ternary complex. (B) Active-site view of the O6MeG·T–Mn2+ ternary structure. Protein is in a closed conformation. O6MeG and dTTP* form coplanar Watson–Crick-type base pair. (C) H-bonding interactions and geometry of O6MeG·dTTP* base pair. A 2Fo – Fc map is contoured at 1σ around O6MeG and dTTP*. (D) Close-up view of the active-site metal ion binding site. Both the nucleotide-binding and the catalytic metal ions are present. The distance between the 3′-OH of the primer terminus and Pα of dTTP* is comparable to that for correct insertion (∼3.4 Å). The C1′(primer terminus)–C1′(dTTP*) distance is similar to that observed for correct insertion (∼5.0 Å). (E) Overlay of the active-site structure of the O6MeG·T–Mn2+ ternary complex (shown in blue) with that of published G·A–Mn2+ ternary complex55 (PDB ID 4LVS, shown in yellow green, RMSD = 0.655 Å). (F) Overlay of the active-site structure of the O6MeG·T–Mn2+ ternary complex (shown in blue) with that of published A·U–Mg2+ ternary complex41 (PDB ID 2FMS, shown in cyan, RMSD = 0.270 Å).
Remarkably, unlike the O6MeG·C–Mn2+ ternary complex, the O6MeG·T–Mn2+ ternary complex shows a catalytically competent state with a closed protein conformation, Watson–Crick-like base pair, and the two active-site metal ions (A and B of Figure 6), which have not been observed in any published polβ structures with base pair mismatch. The overall structure of the O6MeG·T–Mn2+ ternary complex is essentially identical to that of published A·U–Mg2+ ternary complex41 (PDB ID 2FMS, RMSD = 0.270 Å, Figure 6F and Figure S2 in SI). The O6MeG·T–Mn2+ ternary structure shows the signature conformational reorganization of a closed polβ conformation, where α-helix N shifts ∼10 Å toward a nascent base pair (Figure 6B). O6MeG forms coplanar pseudo-Watson–Crick base pairing with dTTP* by forming two H-bonds; N1 and N2 of O6MeG are H-bonded to N3 and O2 of dTTP*, respectively (Figure 6C). Unlike the O6MeG·T–Mg2+ and the O6MeG·C–Mn2+ ternary structures, the O6MeG·T–Mn2+ ternary structure shows completion of the coordination spheres of the both metal ions (Figure 6D). The distance between Pα of dTTP* and O3′ of primer terminus is 3.7 Å, which is comparable to that for correct insertion (∼3.4 Å).
Whereas the O6MeG·T–Mn2+ ternary complex and recently published G·A–Mn2+ ternary complex55 are found to be similar in overall structure (PDB ID 4LVS, RMSD = 0.655 Å), the active-site conformations of protein and DNA in both complexes are quite different (Figure 6E and Figure S3 in SI). Our O6MeG·T–Mn2+ structure shows a ∼2 Å shift of both α-helix N and the template strand bases and a ∼4 Å shift of the phosphate backbone of template strand from their positions observed in the G·A–Mn2+ structure. Published G·A–Mn2+ mismatched structure shows that the nascent dG·dATP base pair forms a buckled conformation (κ angle = ∼140°) and lacks the minor-groove edge interactions with Asn279 and Arg283. In addition, the primer terminus 3′-OH is not coordinated to the catalytic metal ion (4.8 Å), is distant from Pα of the incoming nucleotide (4.7 Å), and is suboptimally positioned for in-line nucleophilic attack on the Pα. In stark contrast, our O6MeG·T–Mn2+ mismatched structure shows protein and DNA conformations that are nearly indistinguishable from those observed in published A·U–Mg2+ matched structure41 (PDB ID 2FMS, RMSD = 0.270 Å, Figure 6F). The structural differences between the G·A–Mn2+ and the O6MeG·T–Mn2+ complexes suggest that polβ allows coplanar conformation only when the base pair can adopt Watson–Crick-mode conformation in the nascent base pair binding pocket.
Structural comparison of the O6MeG·C–Mg2+/Mn2+ and the O6MeG·T–Mg2+/Mn2+ complexes provides insights into the observed kinetic differences among the O6MeG complexes (Table 1). In the case of the O6MeG·C complexes, substituting Mn2+ for Mg2+ induces only a modest conformational change such as binding of the catalytic metal ion (Figure 5D). On the contrary, substituting Mn2+ for Mg2+ in the O6MeG·T ternary complex induces an open-to-closed conformational transition of protein, staggered-to-coplanar conformational change of base pair, and the completion of the coordination spheres of two metal ions. The difference in the degree of conformational change among the O6MeG complexes indicates that the substitution of the active-site metal ion has a greater effect on the O6MeG·T ternary complexes than the O6MeG·C ternary complexes, which is consistent with our kinetic studies showing that substituting Mn2+ for Mg2+ increases insertion efficiency for dTTP and dCTP opposite O6MeG by ∼10-fold and ∼2-fold, respectively (Table 1).
The analysis of the crystallographic thermal B-factors of the active sites of the O6MeG ternary structures and published A·U–Mg2+ ternary structure also provides insights into the observed kinetic differences (Figure 7). The crystallographic temperature factors have been used to analyze the active sites of various enzymes48 including polβ.34,49 The B-factors analysis of the active sites of published polβ ternary structures shows that the nascent and the primer terminus base pairs of ternary complex with a matched base pair (e.g., A·U (PDB ID 2FMS, 2.0 Å resolution),41a A·T (3LK9, 2.5 Å),41b G·C (2FMP, 1.7 Å),41a oxoG·C (1MQ3, 2.8 Å)34) are ordered with an average B-factor range of ∼20–30 Å2, while those of ternary complex with a mismatched base pair (e.g., C·A (3C2L, 2.6 Å),42 G·A (3C2M, 2.2 Å),42 G·A (4LVS, 2.0 Å)55) are disordered with an average B-factor range of ∼40–60 Å2, suggesting that the low mobility of the nascent and the primer terminus base pairs is preferred for the formation of a catalytically optimal conformation. In the case of our O6MeG·C/T–Mg2+ structures, the nascent and the primer terminus base pairs and the nucleotide-binding metal ion in the O6MeG·C–Mg2+ complex are more fluctuating than those in the O6MeG·T–Mg2+ complex (Figures 7A and 7B and Table 2), implying that the active site of the O6MeG·T–Mg2+ complex is more ordered and thus more favorable for catalysis than that of the O6MeG·C–Mg2+ complex, which is consistent with the observed higher efficiency for dTTP insertion than dCTP insertion opposite O6MeG (Table 1). The B-factors analysis also indicates that the templating O6MeG and the catalytic metal ion of the O6MeG·T–Mn2+ complex are more resolved than those of the O6MeG·C–Mn2+ complex (Figures 7C and 7D), which would attribute to the higher insertion efficiency for dTTP over dCTP opposite O6MeG. Interestingly, whereas the O6MeG·T–Mn2+ complex and published A·U–Mg2+ complex are structurally very similar, the catalytic metal ion in the O6MeG·T–Mn2+ complex is more fluctuating than that in the A·U–Mg2+ complex (Figures 7D and 7E), which partially explains ∼30-fold lower insertion efficiency for the O6MeG·T–Mn2+ complex than that for the G·C–Mg2+ complex (Table 1).
Figure 7.

The B-factors analysis of the nascent and the primer terminus base pairs of the O6MeG ternary structures and published A·U–Mg2+ ternary structure41a (PDB ID 2FMS). (A) The O6MeG·C–Mg2+ ternary structure. (B) The O6MeG·T–Mg2+ ternary structure. (C) The O6MeG·C–Mn2+ ternary structure. (D) The O6MeG·T–Mn2+ ternary structure. (E) Published A·U–Mg2+ ternary structure.41a
The structural differences between the O6MeG·T–Mn2+ and the O6MeG·C–Mn2+ complexes strongly indicate that the O6MeG·T complex has higher accessibility to the catalytically competent state than the O6MeG·C complex, which is consistent with the preferential insertion of T over C opposite O6MeG by polβ. The O6MeG·T–Mn2+ ternary structure is consistent with the ∼100-fold higher insertion efficiency for T over C opposite O6MeG in the presence of Mn2+ (Table 1). We conclude that the O6MeG·T–Mn2+ ternary structure represents a precatalytic state competent for nucleotidyl transfer.47
Discussion
Our structural studies provide important insights into the slow, yet highly promutagenic replication across O6MeG by polβ.14,15 The O6MeG·C/T–Mg2+ ternary structures with the open protein conformation, staggered base pair, and one active-site metal ion suggest that polβ slows nucleotide incorporation opposite O6MeG by inducing an altered conformation incompetent for catalysis. The striking conformational difference between the O6MeG·T–Mn2+ ternary complex (a closed protein conformation and coplanar Watson–Crick-mode base pair) and the O6MeG·C–Mn2+ ternary complex (an open protein conformation and staggered base pair) explains the preferential insertion of dTTP over dCTP opposite O6MeG during polβ catalysis. In addition to these, our studies provide insights into the replication fidelity mechanism of polβ.
Polβ Discriminates O6MeG·T against O6MeG·C in the Nascent Base-Pair Binding Pocket
Our O6MeG·C/T–Mn2+ ternary structures indicate that polβ allows coplanar O6MeG·T, but not coplanar O6MeG·C, in the enzyme active site, thereby promoting the mutagenic replication across O6MeG (Figure 8). These structures also indicate that polβ allows only a Watson–Crick-mode base pair in the nascent base-pair binding pocket, and strongly discourages non-Watson–Crick-mode base pairs (e.g., wobble O6MeG·C, one H-bonded O6MeG·T; Figure 1) in the binding pocket.
Figure 8.
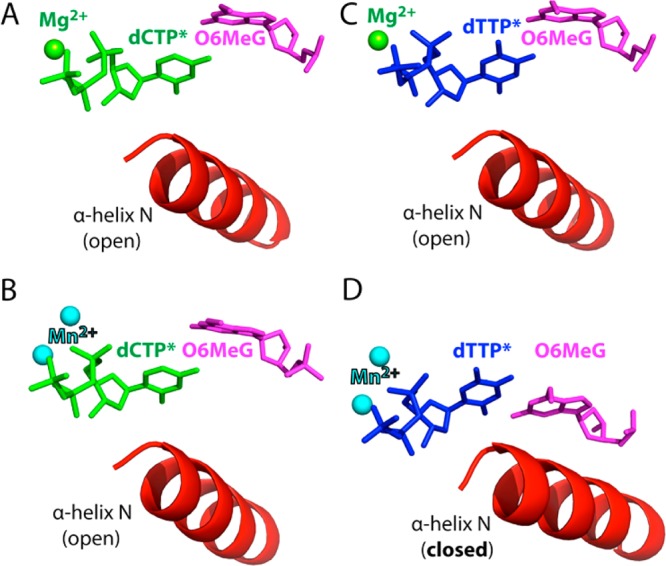
Effect of the active-site metal ion on the conformational activation of polβ. (A) The O6MeG·C–Mg2+ ternary structure with the nucleotide-binding metal ion. (B) The O6MeG·C–Mn2+ ternary structure with the two active-site metal ions. (C) The O6MeG·T–Mg2+ ternary structure with the nucleotide-binding metal ion. (D) The O6MeG·T–Mn2+ ternary structure with the two metal ions. The complex adopts a closed protein conformation and pseudo-Watson–Crick base pair.
As described above, the O6MeG·T–Mn2+ ternary structure shows coplanar Watson–Crick-like base pair and a closed protein conformation (Figure 8D), whereas the O6MeG·C–Mn2+ ternary structure shows staggered base pair and an open protein conformation (Figure 8B). Published polβ ternary structures with correct insertion show coplanar base pair and a closed protein conformation, whereas structures with base pair mismatch (e.g., T·C, A·C) show a staggered base pair and an intermediate protein conformation. Our O6MeG·T–Mn2+ ternary structure, which represents the first polβ mismatched ternary structure with coplanar base pair and a closed protein conformation, thus suggests that the closed polβ conformation is allowed only when a base pair can form coplanar Watson–Crick-type pairing in the enzyme active site. Whereas O6MeG·T can form two H-bonds via pseudo-Watson–Crick base pairing, O6MeG·C cannot readily form Watson–Crick-like pairing at physiological pH (Figure 1). Polβ appears to suppress the formation of coplanar O6MeG·C base pair in the nascent base-pair binding pocket,47,50 which is in contrast with a high fidelity DNA polymerase BF that allows relaxed isosteric Watson–Crick-mode for both O6MeG·C and O6MeG·T in its active site (Figure S4 in SI). The difference in O6MeG·C/T base pairing modes in the polβ and BF structures may result from more strict base-pair geometry constraints of polβ relative to those of BF; duplex DNAs in the active site of polβ and BF have been shown to adopt B-form and A-form, respectively.16,30 In addition, X-ray structures of BF with A·C mismatch have shown that BF induces Watson–Crick dATP·dC base pair in the presence of Mn2+,30 which is in contrast to polβ inducing staggered dATP·dC base pair in the presence of Mn2+.51 All together, polβ appears to be highly efficient at discriminating between Watson–Crick and non-Watson–Crick-mode base pairs in the binding step, allowing Watson–Crick-mode O6MeG·T base pair, but not wobble O6MeG·C base pair, in the nascent base-pair binding pocket.
Implications of polβ Ternary Complex with an Open Protein Conformation and the Nucleotide-Binding Metal Ion
The Polβ:DNA:dNTP ternary complex with an open protein conformation has been suggested to form at the initial stages of open-to-closed conformational transition of the enzyme, yet capturing such complex has been difficult.52 Our O6MeG·C/T–Mg2+ ternary structures with an open protein conformation and the nucleotide-binding metal ion may represent a close approximation of a conformational intermediate captured prior to open-to-closed conformational change, thereby providing insights into the enzyme’s conformational transition.
First, these structures indicate that binding of the nucleotide-binding metal ion occurs prior to that of the catalytic metal ion, which is consistent with kinetic studies43,53 that indicate a fast nucleotide-binding metal ion followed by a slow catalytic-ion-induced conformational transition. Second, our results illustrate that binding of an incoming nucleotide is not sufficient to trigger the open-to-closed conformational transition (A and C of Figure 8).16 Third, the structural differences between the O6MeG·T–Mg2+ ternary structure (an open protein conformation and the nucleotide-binding metal ion) and the O6MeG·T–Mn2+ ternary structure (a closed protein conformation and the two metal ions) suggest that binding of the catalytic metal ion is important for the open-to-closed conformational activation. Lastly, the observation of the open protein conformation for both O6MeG·C/T–Mg2+ structures implies that the formation of the closed protein conformation is discouraged when a base pair does not adopt a coplanar Watson–Crick geometry in the enzyme active site,34,54 which could provide a kinetic checkpoint prior to catalysis.
The observation of the polβ ternary complexes with an open protein conformation and base pair mismatch supports an induced-fit mechanism,35 whereas a closed protein conformation, which is the optimal conformation for nucleotidyl transfer reaction, is readily accessible for correct insertion but not for incorrect insertion. The open protein conformation would also facilitate diffusion of an incorrect nucleotide from the active site and thus lower binding affinity of the incorrect nucleotide, which will enhance replication fidelity of the enzyme.55
Polβ May Utilize Coordination State of the Catalytic Metal Ion As a Kinetic Checkpoint to Prevent Misincorporation
Large variation in the catalytic metal-ion coordination state among our O6MeG·C/T–Mg2+ and O6MeG·C/T–Mn2+ ternary structures and previous polβ ternary structures35,43,55 suggests that polβ utilizes the catalytic metal-ion coordination to deter nucleotide misincorporation (Figure S5 in SI). The coordination state of the catalytic metal ion appears to greatly affect conformations of polβ–DNA complexes. In polβ ternary structures with correct insertion, the catalytic metal ion is typically coordinated to three Asp residues, Pα oxygen of incoming nucleotide, and the 3′-OH of primer terminus. These matched ternary structures adopt a closed protein conformation and coplanar base pair. In published polβ–Mn2+ ternary structures with C·A and A·G mismatches,42 the two active-site metal ions are observed, yet primer terminus 3′-OH is not liganded to the catalytic metal ion. These mismatched ternary structures show an intermediate protein conformation and staggered base pair. The O6MeG·C/T–Mg2+ ternary structures with an open protein conformation and staggered base pair show only the nucleotide-binding metal-ion coordination. The O6MeG·C–Mn2+ ternary structure with an open protein conformation and staggered base pair shows the presence of the two active-site metal ions, yet Asp256 is not liganded to the catalytic metal ion. Lastly, the O6MeG·T–Mn2+ ternary structure shows completion of the coordination sphere of the catalytic metal ion, and adopts the closed protein conformation and coplanar base pair. Taken together, the observed large variation in the catalytic metal-ion coordination state among polβ structures suggests that the coordination state of the catalytic metal ion dictates the conformation of the polβ–DNA complex, that the completion of the coordination sphere of the catalytic metal ion is crucial for the conformational activation of the enzyme,43,52,56 and that the completion of the catalytic metal-ion coordination is achieved in the presence of only Watson–Crick-mode base pair in the nascent base-pair binding pocket. The observation of only the nucleotide-binding metal ion in the O6MeG·C/T–Mg2+ ternary structures with an open protein conformation supports that polβ deters the coordination of the catalytic metal ion for non-Watson–Crick base pair. We conclude that polβ may use the catalytic metal-ion coordination as a kinetic checkpoint to increase its replication fidelity.
Implication of the O6MeG·T–Mn2+ Ternary Complex with Pseudo-Watson–Crick Base Pair and a Closed Protein Conformation
The O6MeG·T–Mn2+ ternary structure represents the first polβ structure with coplanar mismatched base pair, which is in contrast to published mismatched polβ structures with staggered base pair. The observation of the coplanar mismatched O6MeG·T base pair in the nascent base pair binding pocket suggests that some mismatched base pairs, for example G·T base pair which comprises 60% of the base substitution mutations produced by polβ,57 could also form the similar coplanar conformation during DNA replication by polβ, and that mismatched base pairs with coplanar conformation would be preferentially formed over mismatched base pairs with staggered conformation during polβ catalysis.
The O6MeG·T–Mn2+ ternary structure also represents the first structure of pseudo-Watson–Crick O6MeG·T base pair formed in the nascent base-pair binding pocket of a DNA polymerase. Whereas the pseudo-Watson–Crick O6MeG·T base pair has been observed in an X-ray structure of duplex DNA,29 NMR studies have indicated the formation of one H-bonded O6MeG·T base pair rather than the two H-bonded pseudo-Watson–Crick base pair (Figure 1).26,28 In the nascent base-pair binding pocket of BF, O6MeG·T forms an isosteric Watson–Crick O6MeG·T base pair (Figure S4 in SI).30 Our observation of pseudo-Watson–Crick O6MeG·T suggests that some DNA polymerases with the base-pair geometry constraints similar to those of polβ, for example polλ, may also induce pseudo-Watson–Crick O6MeG·T base pair conformation during replication across O6MeG.
Effect of Mn2+ on Conformational Reorganization of DNA Polymerase
The polβ:O6MeG·T–Mn2+ ternary structure represents, to our knowledge, the first example of a DNA polymerase structure with a drastic Mn2+-induced conformational transition of protein and nascent base pair. The X-family DNA polymerase polλ does not undergo a conformational transition during nucleotide incorporation.58 The Y-family DNA polymerase Dpo459 and the B-family DNA polymerase RB69pol60 structures with active-site Mg2+ show that the protein conformations of ternary complexes with base pair mismatch vs match are almost the same, so substituting Mn2+ for Mg2+ is unlikely to induce an open-to-closed conformational activation of those enzymes.59 Published BF-A·C mismatched structures with the active-site Mg2+ vs Mn2+ show a wobble-to-Watson–Crick conformational change of base pair, yet conformational change of protein is not prominent.51 Interestingly, BF:T·G–Mg2+ ternary structure shows wobble T·G base pair and an “ajar” protein conformation.61 It would be interesting to know whether substitution of the active-site metal ion will induce Watson–Crick-mode T·G base pair and an ajar-to-closed conformational change of protein. Taken together, polβ is a rare DNA polymerase that can induce a drastic metal-dependent conformational change in both protein and base pair during nucleotide misincorporation.
Effects of Mn2+ on Replication Fidelity of DNA Polymerase
In vitro studies with various DNA polymerases have shown that Mn2+ is highly promutagenic.62 Substituting Mn2+ for Mg2+ increases misincorporation rate and reduces replication fidelity of several DNA polymerases, such as polβ,42 Dpo4,59 polι,64 polλ,65Escherichia coli DNA polymerase I,63 and T7 DNA polymerase.63 Although several DNA polymerase structures with base pair mismatch have been reported,42,59,61 the structural basis for Mn2+-promoted replication infidelity of DNA polymerase is poorly understood due in significant part to the scarcity of mismatched DNA polymerase structures with Mg2+/Mn2+ and wild-type active site. For example, Dpo4 structure59 with T·G mismatch lacks the active-site Mn2+, and published polβ structures42,55 with mismatch either lack active-site Mg2+ or have Arg283Lys mutation.43 BF structures with A·C–Mg2+/Mn2+ lack the primer terminus 3′-OH and the catalytic metal ion.61 Our polβ structures with Mg2+/Mn2+ and wild-type active site thus provide new insight into the Mn2+-promoted replication infidelity. Whereas the O6MeG·C/T–Mg2+ ternary complexes contain only the nucleotide-binding metal ion (Figure 7 and Figure S5 in SI), the O6MeG·C/T–Mn2+ ternary complexes contain both the catalytic and the nucleotide-binding metal ions, indicating that substituting Mn2+ for Mg2+ promotes binding and coordination of the catalytic metal ion during misincorporation. The coordination of the catalytic metal ion in the active site of DNA polymerase has been suggested to lower the pKa of the 3′-OH of primer terminus,66 place the 3′-OH of primer terminus in an optimal position for in-line nucleophilic attack on the Pα of incoming nucleotide, and promote proton transfer from the 3′-OH of primer terminus to nearby catalytic carboxylate44,47,67a or water molecule,67b thereby lowering the activation energy barrier for nucleotidyl transfer and facilating the chemical reaction.47 The catalytic metal ion may sense the presence of abnormal substrates in the nascent base pair binding pocket and play an important role in deterring nucleotide misincorporation by preventing its proper coordination,52,68 which has been suggested to be the rate-limiting step of the nucleotidyl transfer.52,53,59 DNA polymerases probably utilize the catalytic Mg2+, which is highly sensitive to the presence of active-site mutations, base pair mismatch, and suboptimal substrates,69 to increase substrate specificity and replication fidelity. The replacement of Mg2+ with Mn2+, which is more tolerant of active-site distortions and abnormal substrates than Mg2+,69 could stimulate the binding and the subsequent coordination of the catalytic metal ion during nucleotide misincorporation, thereby faciliating the incorporation of otherwise unfavorable substrates and decreasing replication fidelity of DNA polymerase.
Conclusion
In summary, we have reported the first structures of wild-type polβ ternary complex with an open protein conformation and one active-site metal ion (the O6MeG·C/T–Mg2+ complex), polβ ternary complex with base pair mismatch and a closed protein conformation (the O6MeG·T–Mn2+ complex), pseudo-Watson–Crick O6MeG·T base pair formed in the nascent base-pair binding pocket of a DNA polymerase, and a metal-dependent conformational activation of a DNA polymerase. Our studies presented here provide structural basis for the polβ catalysis across the carcinogenic O6MeG lesion. Our results indicate that polβ slows noncomplementary nucleotide incorporation by inducing an alternate conformation suboptimal for chemistry, and that polβ promotes mutagenic replication by allowing Watson–Crick-mode for O6MeG·T, but not for O6MeG·C, in the nascent base-pair binding pocket. Our studies also suggest that polβ increases its replication fidelity by utiziling the catalytic metal-ion coordination state as a kinetic checkpoint prior to catalysis, and that the completion of the catalytic-metal ion coordination is crucial for the open-to-closed conformational activation of the enzyme.
Acknowledgments
This work was supported by grants from a start-up fund from the College of Pharmacy at the University of Texas at Austin and the National Institute of Health Grant ES23101. Instrumentation and technical assistance for this work were provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health and National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Supporting Information Available
Five figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Green C. L.; Loechler E. L.; Fowler K. W.; Essigmann J. M. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loechler E. L.; Green C. L.; Essigmann J. M. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 6271–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L.; Prakash S.; Prakash L. Mol. Cell. Biol. 2000, 20, 8001–8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveless A. Nature 1969, 223, 206–207. [DOI] [PubMed] [Google Scholar]
- Dumenco L. L.; Allay E.; Norton K.; Gerson S. L. Science 1993, 259, 219–222. [DOI] [PubMed] [Google Scholar]
- Nakatsuru Y.; Matsukuma S.; Nemoto N.; Sugano H.; Sekiguchi M.; Ishikawa T. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 6468–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eadie J. S.; Conrad M.; Toorchen D.; Topal M. D. Nature 1984, 308, 201–203. [DOI] [PubMed] [Google Scholar]
- Bush Z. M.; Longtine J. A.; Cunningham T.; Schiff D.; Jane J. A.; Vance M. L.; Thorner M. O.; Laws E. R.; Lopes M. B. S. J. Clin. Endocrinol. Metab. 2010, 95, E280–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D.; Calvo J. A.; Samson L. D. Nat. Rev. Cancer 2012, 12, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjolund A. B.; Senejani A. G.; Sweasy J. B. Mutat. Res. 2013, 743–744, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lari I. U.; Day R. S.; Dobler K.; Paterson M. C. Nucleic Acids Res. 2001, 29, 2409–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S.; Xu J.; Sannai M.; Moore R.; Caretti E. Cell 2011, 146, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibghat-Ullah; Day R. S. Biochemistry 1992, 31, 7998–8008. [DOI] [PubMed] [Google Scholar]
- Reha-Krantz L. J.; Nonay R. L.; Day R. S.; Wilson S. H. J. Biol. Chem. 1996, 271, 20088–20095. [DOI] [PubMed] [Google Scholar]
- Singh J.; Su L.; Snow E. T. J. Biol. Chem. 1996, 271, 28391–28398. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Wilson S. H. Chem. Rev. 2006, 106, 361–382. [DOI] [PubMed] [Google Scholar]
- Murphy D. L.; Jaeger J.; Sweasy J. B. J. Am. Chem. Soc. 2011, 133, 6279–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi R. N.; Almeida K. H.; Fornsaglio J. L.; Schamus S.; Sobol R. W. Cancer Res. 2005, 65, 6394–6400. [DOI] [PubMed] [Google Scholar]
- Trivedi R. N.; Wang X.-H.; Jelezcova E.; Goellner E. M.; Tang J.-B.; Sobol R. W. Mol. Pharmacol. 2008, 74, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal A. S.; Banerjee S.; Aneja R.; Sarkar F. H.; Ostrov D. A.; Narayan S. PLoS ONE 2011, 6, e16691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal A. S.; Banerjee S.; Panda H.; Bulkin C. D.; Izumi T.; Sarkar F. H.; Ostrov D. A.; Narayan S. Mol. Cancer Res. 2009, 7, 1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-Y.; Chowdhury G.; Zang H.; Angel K. C.; Vu C. C.; Peterson L. A.; Guengerich F. P. J. Biol. Chem. 2006, 281, 38244–38256. [DOI] [PubMed] [Google Scholar]
- Eoff R. L.; Angel K. C.; Egli M.; Guengerich F. P. J. Biol. Chem. 2007, 282, 13573–13584. [DOI] [PubMed] [Google Scholar]
- Woodside A. M.; Guengerich F. P. Biochemistry 2002, 41, 1027–1038. [DOI] [PubMed] [Google Scholar]
- Gaffney B. L.; Jones R. A. Biochemistry 1989, 28, 5881–5889. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Shapiro L.; Kozlowski S. A.; Gaffney B. L.; Jones R. A. Biochemistry 1986, 25, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Ginell S. L.; Kuzmich S.; Jones R. A.; Berman H. M. Biochemistry 1990, 29, 10461–10465. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Shapiro L.; Kozlowski S. A. J. Mol. Biol. 1986, 188, 677–692. [DOI] [PubMed] [Google Scholar]
- Leonard G. A.; Thomson J.; Watson W. P.; Brown T. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 9573–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. J.; Forsberg L. J.; Beese L. S. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 19701–19706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside A. M.; Guengerich F. P. Biochemistry 2002, 41, 1039–1050. [DOI] [PubMed] [Google Scholar]
- Singer B.; Chavez F.; Goodman M. F.; Essigmann J. M.; Dosanjh M. K. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 8271–8274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eoff R. L.; Irimia A.; Egli M.; Guengerich F. P. J. Biol. Chem. 2007, 282, 1456–1467. [DOI] [PubMed] [Google Scholar]
- Batra V. K.; Shock D. D.; Beard W. A.; McKenna C. E.; Wilson S. H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sawaya M. R.; Prasad R.; Wilson S. H.; Kraut J.; Pelletier H. Biochemistry 1997, 36, 11205–11215. [DOI] [PubMed] [Google Scholar]; b Koag M.; Min K.; Lee S. J. Biol. Chem. 2014, 289, 6289–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Acta Crystallogr., Sect. D 2010, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- Pelletier H.; Sawaya M. R.; Wolfle W.; Wilson S. H.; Kraut J. Biochemistry 1996, 35, 12742–12761. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr., Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. Acta Crystallogr., Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B.; Snoeyink J.; Richardson J. S.; Richardson D. C. Nucleic Acids Res. 2007, 35, W375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Batra V.; Beard W.; Shock D.; Krahn J.; Pedersen L.; Wilson S. H. Structure 2006, 14, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Surya Prakash G. K.; Zibinsky M.; Upton T. G.; Kashemirov B. A.; McKenna C. E.; Oertell K.; Goodman M. F.; Batra V. K.; Pedersen L. C.; Beard W. A.; Shock D. D.; Wilson S. H.; Olah G. A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15693–15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra V. K.; Beard W. A.; Shock D. D.; Pedersen L. C.; Wilson S. H. Mol. Cell 2008, 30, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Wilson S. H. Structure 2012, 20, 1829–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra V. K.; Perera L.; Lin P.; Shock D. D.; Beard W. A.; Pedersen L. C.; Pedersen L. G.; Wilson S. H. J. Am. Chem. Soc. 2013, 135, 8078–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Wilson S. H. Nucleic Acids Res. 2013, 41, 1848–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn J.; Beard W.; Wilson S. Structure 2004, 12, 1823–1832. [DOI] [PubMed] [Google Scholar]
- Lin P.; Batra V. K.; Pedersen L. C.; Beard W. A.; Wilson S. H.; Pedersen L. G. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5670–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yuan Z.; Zhao J.; Wang J. Protein Eng. 2003, 16, 109–114. [DOI] [PubMed] [Google Scholar]; b Radivojac R.; Obradovic Z.; Vucetic S.; Brown C. J.; Lawson J. D.; Dunker A. K. Protein Sci. 2004, 13, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Parthasarathy S.; Murthy M. R. N. Protein Sci. 1997, 6, 2561–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bartlett G. J.; Porter C. T.; Borkakoti N.; Thornton J. M. J. Mol. Biol. 2002, 324, 105–121. [DOI] [PubMed] [Google Scholar]; e Qian M.; Haser R.; Buisson G.; Duee E.; Payan F. Biochemistry 1994, 33, 6284–6294. [DOI] [PubMed] [Google Scholar]; f Wood Z. A.; Poole L. B.; Karplus P. A. Science 2003, 300, 650–653. [DOI] [PubMed] [Google Scholar]
- Batra V. K.; Beard W. A.; Shock D. D.; Pedersen L. C.; Wilson S. H. Structure 2005, 1225–1233. [DOI] [PubMed] [Google Scholar]
- Christian T. D.; Romano L. J.; Rueda D. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21109–21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Hellinga H. W.; Beese L. S. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 17644–17648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Arora K.; Beard W. A.; Wilson S. H.; Schlick T. J. Am. Chem. Soc. 2004, 126, 8441–8453. [DOI] [PubMed] [Google Scholar]
- Zhong X.; Patel S. S.; Tsai M.-D. J. Am. Chem. Soc. 1998, 120, 235–236. [Google Scholar]
- Batra V. K.; Beard W. A.; Hou E. W.; Pedersen L. C.; Prasad R.; Wilson S. H. Nat. Struct. Mol. Biol. 2010, 17, 889–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Shock D. D.; Wilson S. H. Cell 2013, 154, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby T. W.; DeRose E. F.; Cavanaugh N. A.; Beard W. A.; Shock D. D.; Mueller G. A.; Wilson S. H.; London R. E. Nucleic Acids Res. 2012, 40, 2974–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheroff W. P.; Jung H. K.; Beard W. A.; Wilson S. H.; Kunkel T. A. J. Biol. Chem. 1999, 274, 3642–3650. [DOI] [PubMed] [Google Scholar]
- Bebenek K.; Pedersen L. C.; Kunkel T. A. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1862–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman A.; Ling H.; Woodgate R.; Yang W. EMBO J. 2005, 24, 2957–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Wang J.; Konisberg W. H. J. Am. Chem. Soc. 2013, 135, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. Y.; Beese L. S. J. Biol. Chem. 2011, 286, 19758–19767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman R. A.; Mildvan A. S.; Loeb A. A. Biochemistry 1985, 24, 5810–5817. [DOI] [PubMed] [Google Scholar]
- Tabor S.; Richardson C. C. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E. G.; Woodgate R. J. Biol. Chem. 2007, 282, 24689–24696. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz M.; Bebenek K.; Krahn J. M.; Pedersen L. C.; Kunkel T. A. DNA Repair 2007, 6, 1333–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abashkin Y. G.; Erickson J. W.; Burt S. K. J. Phys. Chem. B 2001, 105, 287–292. [Google Scholar]
- a Cisneros G. A.; Perera L.; Garcia-Diaz M.; Bebenek K.; Kunkel T. A.; Pedersen L. G. DNA Repair 2008, 7, 1824. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nakamura T.; Zhao Y.; Yamagata Y.; Hua Y.-j.; Yang W. Nature 2012, 487, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kunkel T. A.; Loeb L. A. J. Biol. Chem. 1979, 254, 5718–5725. [PubMed] [Google Scholar]; b Shah A. M.; Li S. X.; Anderson K. S.; Sweasy J. B. J. Biol. Chem. 2001, 276, 10824–10831. [DOI] [PubMed] [Google Scholar]
- a Villani G.; Tanguy Le Gac N.; Wasungu L.; Burnouf D.; Fuchs R. P.; Boehmer P. E. Nucleic Acids Res. 2002, 30, 3323–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vipond I. B.; Moon B. J.; Halford S. E. Biochemistry 1996, 35, 1712–1721. [DOI] [PubMed] [Google Scholar]; c Junop M. S.; Haniford D. B. EMBO J. 1996, 15, 2547–2555. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.