

Abstract
We investigated whether an uncharacterized protein from guinea pig could be the enzyme behind Kidd’s serendipitous discovery, made over 60 years ago, that guinea pig serum has cell killing ability. It has been long known that an enzyme with l-asparaginase activity is responsible for cell killing, although astonishingly, its identity remains unclear. Bacterial asparaginases with similar cell killing properties have since become a mainstay therapy of certain cancers such as acute lymphoblastic leukemia. By hydrolyzing asparagine to aspartate and ammonia, these drugs deplete the asparagine present in the blood, killing cancer cells that rely on extracellular asparagine uptake for survival. However, bacterial asparaginases can elicit an adverse immune response. We propose that replacement of bacterial enzymes with the guinea pig asparaginase responsible for serum activity, by its virtue of being more closely related to human enzymes, will be less immunogenic. To this goal, we investigated whether an uncharacterized protein from guinea pig with putative asparaginase activity, which we call gpASNase3, could be that enzyme. We examined its self-activation process (gpASNase3 requires autocleavage to become active), kinetically characterized it for asparaginase and β-aspartyl dipeptidase activity, and elucidated its crystal structure in both the uncleaved and cleaved states. This work reveals that gpASNase3 is not the enzyme responsible for the antitumor effects of guinea pig serum. It exhibits a low affinity for asparagine as measured by a high Michaelis constant, KM, in the millimolar range, in contrast to the low KM (micromolar range) required for asparaginase to be effective as an anticancer agent.
In 1953, Kidd made the serendipitous discovery that guinea pig serum can kill transplanted lymphomas in mice.1 The active agent behind this unexpected activity was revealed by Broome in 1961 to be an enzyme with l-asparaginase (ASNase) activity.2,3 Astonishingly, the precise identification of the guinea pig ASNase responsible for the anticancer effect is still unclear. In this study, we tested whether the guinea pig l-asparaginase that we refer to as gpASNase3 has the required properties to be that enzyme. In addition to characterizing its kinetic properties, we elucidated its crystal structure in its inactive and active states.
The work of Kidd and Broome suggested that ASNases may have clinical utility as anticancer agents. However, since it was found that the guinea pig serum ASNase was difficult to work with, alternative sources for ASNases with anticancer activity were sought. In 1964, an ASNase isolated from E. coli was discovered to have cell killing activity comparable to that of the ASNase present in guinea pig serum.4 Antitumor activity was also observed for an ASNase from Erwinia chrysanthemi.5 Both of these bacterial enzymes are currently FDA approved to treat acute lymphoblastic leukemia (ALL).6
ASNases catalyze the conversion of the amino acid l-asparagine into l-aspartate and ammonia (Figure 1A). Both the substrate asparagine and product aspartate are used for protein synthesis, while aspartate also participates in several biosynthetic pathways, including serving as a precursor to several amino acids and in the biosynthesis of purine bases. Despite the indispensable role of these amino acids, neither is considered an essential amino acid in humans (i.e., requiring intake through the diet), due to de novo pathways for their synthesis. In the specific case of asparagine, human cells can either acquire this amino acid from the blood, where it is present at a concentration of approximately 50 μM,7 or employ the enzyme asparagine synthetase to produce it.
Figure 1.

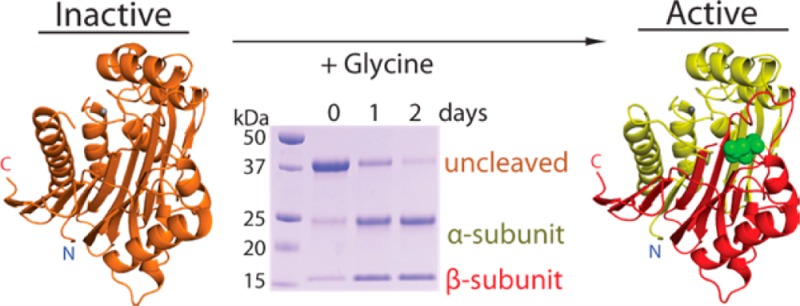
Reaction schemes, sequence alignment, and constructs of guinea pig ASNase3. (A) Reaction scheme whereby asparaginase hydrolyzes asparagine into aspartate and ammonia. (B) Sequence alignment using Clustal Omega25 of gpASNase3 (H0VQC8_CAVPO), hASNase3 (ASGL1_HUMAN), E. coli type III (IAAA_ECOLI), gpAGA (H0UZ36_CAVPO), and hAGA (ASPG_HUMAN). The hAGA signal peptide and the uncharacterized peptide at the N-terminus of gpASNase3 are denoted in green. Conserved active site residues are in red. The black dashed line separates the α- and β-subunits after autoproteolytic cleavage. The cartoon β-strands and helices in cyan and light cyan represent the secondary structural elements of the α- and β-subunits, respectively, of gpASNase3. (C) Scheme of the uncharacterized H0VQC8_CAVPO UniProt entry. We refer to gpASNase3 as the catalytic domain (gray), lacking the preceding 23 residues (green). (D) Cleavage reaction scheme. GpASNase3 is cleaved between Gly167 and Thr168 through autoproteolysis (very slow) or accelerated by glycine (see text). The freed amino group of Thr168, the first residue of the β-subunit, is required for its l-asparaginase activity.
Interestingly, some cancer types, such as ALL, have lost the ability to synthesize asparagine de novo, making them dependent on extracellular asparagine for survival.8,9 When used clinically to treat ALL, administration of ASNase depletes asparagine from the blood, and this disrupts vital growth processes and ultimately causes apoptosis.10 Not surprising due to the bacterial origin of the enzymes used as antileukemic agents, immunogenic responses are a major drawback to their clinical use, manifesting themselves as rash, edema, bronchospasm, erythema, and even systemic anaphylaxis.11 Moreover, the generated antibodies inactivate the drug.12,13 Attempting to shield the ASNase from recognition by the immune system, the E. coli enzyme was conjugated to polyethylene glycol (PEG).13−17 However, it has recently been shown that antibodies to PEG can cause silent inactivation by depleting the serum of ASNase through clearance of the PEG conjugates, rendering the treatment ineffective.10,18−20 Hence, a nonimmunogenic ASNase would greatly improve this treatment strategy. In order to be useful as an anticancer therapeutic, an ASNase must exhibit a Michaelis constant, KM for asparagine in the micromolar range, a property of the clinically used bacterial ASNases. This requirement precludes the use of human ASNases since these have been reported to have a KM for asparagine in the millimolar range.
While human ASNases can be ruled out as potential replacements for the bacterial ASNases, the still-unidentified ASNase present in guinea pig serum, due to the decreased taxonomic distance to human, would potentially be less immunogenic than the bacterial enzymes.21−23 To identify that particular enzyme, we searched the UniProt database24 for guinea pig proteins with asparaginase activity and found four such entries. One is annotated as H0UX35_CAVPO and shares 98.8% identity with human threonine aspartase (all sequence alignments performed with Clustal Omega25); a second is annotated as H0W0T5_CAVPO and has 70.4% identity with human 60 kDa lysophospholipase. The other two uncharacterized proteins, classified as protein fragments, have similarities to members of the N-terminal nucleophile (Ntn)-hydrolase superfamily: H0UZ36_CAVPO as a homologue of aspartylglucosaminidase (AGA) sharing 86.7% identity with human AGA and H0VQC8_CAVPO, which when analyzed from the first methionine in the reported sequence has 79.9% sequence identity with human ASNase3 (hASNase3), also known as hASRGL1,26,27 ALP,26 and CRASH28 (see Figure 1B). H0VQC8_CAVPO would also be classified as a type III or plant-type asparaginase given its similarity to Lupinus luteus plant asparaginase, E. coli EcAIII, and hASNase3.29−33 On the basis of the homology of H0VQC8_CAVPO to human ASNase3, in this article we refer to this enzyme as the guinea pig ASNase3 (gpASNase3).
In this study, we focus on gpASNase3. Interestingly, gpASNase3 contains an N-terminal extension of 23 amino acids that is absent in the human homologue hASNase3 (Figure 1C). This leader sequence, located prior to what would be the initiation methionine as present in hASNase3, could be part of a signal peptide for secretion into the serum, where the cell killing asparaginase from guinea pig is located.1 Note that most ASNases are not secreted. While gpASNase3 belongs to the type III enzyme class as opposed to the type II class of the clinically used ASNases (type II and III enzymes do not share any homology), a common property of the type II bacterial enzymes is periplasmic localization due to an ∼20 amino acid N-terminal signal peptide. We pondered whether the N-terminal extension, indicated by the UniProt database to be present in gpASNase3, could perform a similar translocation function.
Guinea pig ASNase3, being a member of the Ntn-hydrolase family, would be enzymatically inactive when first produced. Previously, we reported our studies of hASNase3 and its autocleavage (and hence activation) properties.32,34 Activation occurs upon autoproteolytic cleavage that results in α- and β-subunits.35 The catalytic residue for the cleavage reaction (in the context of the intact protein) and for the asparaginase activity (in the context of the cleaved protein) is the N-terminal threonine of the β-subunit. In hASNase3, and by homology in gpASNase3, activation by autocleavage into α- and β-subunits occurs between Gly167 and Thr168 (Figure 1D).27
In addition to catalyzing asparagine hydrolysis, AGA and type III l-asparaginases also function as β-aspartyl peptidases (EC 3.4.19.5).29,36,37 β-Aspartyl bond formation leads to significant structural alteration and often dysfunction and is one of the most common nonenzymatic processes by which protein damage occurs.29,36,37 Therefore, we also examined the ability of gpASNase3 to catalyze β-aspartyl peptide bond breakage.
In this study, we investigated the intrinsic cleavage (and hence activation) rate of gpASNase3 and the ability of glycine to accelerate the cleavage process, a property we previously reported for hASNase3.34 We determined that activated gpASNase3 acts both as an asparaginase and as a β-aspartyl peptidase and characterized its kinetic properties. Furthermore, we determined the first structures of gpASNase3, both in its inactive, uncleaved state and in its active, cleaved state bound to the product aspartate. In addition to increasing our mechanistic understanding of this enzyme, our analysis suggests that gpASNase3 is not the asparaginase present in guinea pig serum that is responsible for the antitumor activity observed by Kidd1 in 1953.
Materials and Methods
Cloning of Guinea Pig ASNase3
Cavia porcellus (guinea pig) asparaginase UniProt entry H0VQC8 is annotated as an uncharacterized protein fragment consisting of 332 amino acid residues (see Figure 1B,C) that is lacking its N-terminal translation initiation methionine. The first methionine in the UniProt entry is at position 24, which corresponds to the position of the translation initiation methionine in the human homologue hASNase3. For our studies, we ordered a synthetic gene (Genscript) codon optimized for expression in E. coli that began at that first methionine since we were interested in the catalytic domain of the enzyme, not in its putative secretion properties. The synthetic gene was transferred to a His6-TEV-pET14b expression vector using NdeI and BamHI restriction sites at the 5′ and 3′ ends, respectively. The expression plasmid encodes for an N-terminal hexahistidine tag followed by a TEV protease cleavage site (MGSSHHHHHHSSGGNENLYFQGH) (see Figure 2A).
Figure 2.

Glycine accelerates cleavage of gpASNase3 into its α- and β-subunits in a dose-dependent manner. (A) Scheme of the uncleaved and cleaved gpASNase3 constructs used in this study. In yellow are regions that had no clear electron density. (B) SDS–PAGE gels showing intrinsic cleavage as a function of time and the accelerated cleavage due to the addition of 10, 25, 50, 100, and 250 mM glycine at pH 6.5. (C) Quantification of the uncleaved, the α-, and the β-subunit bands of gpASNase3 using ImageJ software.57 The higher the concentration of glycine added, the greater is the decrease in uncleaved protein and the greater is the accumulation of the α- and β-subunits.
Expression and Purification
The His6-TEV-gpASNase3(1–309)-pET14b plasmid was transformed into BL21 (DE3) C41 E. coli cells for expression. A starter culture of the plasmid was grown overnight and inoculated into 6 L of 2YT medium at a ratio of 1:100. The cells were grown at 37 °C to an optical density (at 600 nm) of 0.6, and overexpression was induced with isopropyl-β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM. Growth continued at 37 °C for 6 h, at which point the cells were harvested by centrifugation, and the pellet was frozen at −20 °C. For purification, the cell pellet was thawed, resuspended in lysis buffer (25 mM Tris-HCl at pH 7.5, 200 mM KCl, 10 mM MgCl2, 10 mM imidazole, 10% glycerol, 1% Triton X100, 1 mM PMSF), and disrupted by sonication. The lysate was cleared by ultracentrifugation (33K rpm, 1 h, 4 °C), and the supernatant was loaded onto an equilibrated 5 mL HisTrap HP Ni Sepharose column (GE Healthcare), washed with buffer containing 25 mM Tris-HCl at pH 7.5, 200 mM KCl, 10 mM MgCl2, and 25 mM imidazole followed by a wash with buffer containing 25 mM Tris-HCl at pH 7.5, 200 mM KCl, 10 mM MgCl2, and 50 mM imidazole. The column was washed overnight at 4 °C with buffer containing 25 mM Tris-HCl at pH 7.5 and 100 mM KCl. The next morning, a final wash step was performed using buffer containing 25 mM Tris-HCl at pH 7.5, 200 mM KCl, and 75 mM imidazole. The uncleaved His6-TEV-gpASNase3(1–309) was unstable and prone to precipitation at pH 7.5 and was eluted in buffer that aided stabilization (20 mM CAPS at pH 10.5, 100 mM KCl, and 250 mM imidazole) with a yield of 155 mg as determined by UV absorbance at 280 nm. Two millimolar DTT was added to the eluted protein. The protein was concentrated to 10 mL (14.5 mg/mL). Five milliliters was loaded at a time onto a Superdex 200 Hi Load 26/60 gel filtration column (GE Healthcare) pre-equilibrated with 10–20 mM CAPS at pH 10.5, 100 mM KCl, and 2 mM DTT. The protein eluted as one main peak (corresponding to the monomer) with a slight shoulder at the beginning (corresponding to the dimer). The monomer peak was pooled, concentrated to ∼50 mg/mL, aliquoted, flash frozen in liquid nitrogen, and stored at −80 °C.
Glycine-Accelerated Cleavage Assay
Cleavage of His6-TEV-gpASNase3(1–309) into its α- and β-subunits was measured over several days in the absence and presence of glycine. The protein was diluted to 1.5–1.75 mg/mL in buffer in which the cleaved form was stabilized instead of the uncleaved form and was composed of 25 mM CAPS (used as an additive), 25 mM MES at pH 6.5, 100 mM KCl, and 2 mM DTT ± glycine at pH 6.5. For the comparison between cleavage of the guinea pig and human ASNase3 enzymes in the absence or presence of 250 mM glycine, the buffer contained 25 mM MES at pH 6.5, 100 mM KCl, and 2 mM DTT ± glycine. A small amount of initial precipitation occurred due to the instability of the uncleaved protein in this buffer at this pH. Samples were centrifuged at 13,000 rpm for 10 min at 4 °C to ensure that samples run on the gel contained only soluble, stable protein.
Kinetic Assay
In order to activate gpASNase3, which was purified primarily in its uncleaved form, gpASNase3 was dialyzed against buffer that contained glycine (20 mM MES at pH 6.5, 100 mM KCl, 3 mM DTT, and 400 mM glycine) for 4 days. The same kinetic assay was utilized to test for both asparaginase and isoaspartyl dipeptidase activities.36,38 It measures the production of l-aspartate through the 1:1 oxidation of reduced nicotinamide adenine dinucleotide (NADH). The conversion of NADH to NAD+ was measured spectrophotometrically as a decrease in absorbance at 340 nm at 37 °C. Glutamic-oxalacetic transaminase (Sigma G2751) and malic dehydrogenase (Sigma M2634) were helper enzymes for the coupled enzymatic reactions; ∼12 and ∼0.8 U were used, respectively. Our 2× kinetic assay buffer was composed of 100 mM Tris at pH 7.5, 400 μM α-ketoglutarate, and 400 μM NADH. The l-Asn substrate stock (Sigma-Aldrich A93003) was made fresh as a 125 mM solution in 50 mM Tris at pH 7.5. Controls were performed wherein all the components were included in the cuvette except for gpASNase3 or l-Asn. The control lacking gpASNase3 was to ensure that in the absence of enzyme the assay showed no signal. This control also confirmed the lack of prehydrolyzed l-Asn. The control lacking the substrate was to ensure the dependency of the reaction on l-Asn. Kinetic data were analyzed with SigmaPlot 2000/Enzyme Kinetics Module 1.0, from Systat Software, Inc., San Jose, California, USA, (www.sigmaplot.com). Error estimates for kcat (turnover number)/KM were calculated from the individual errors of kcat and KM using the following equation: (Δz/z) = ((Δx/x)2 + (Δy/y)2)1/2.
Crystallization
A variety of crystallization screens (Qiagen) were applied to purified His6-TEV-gpASNase(1–309). Several hits were obtained that contained buffers ranging from pH 5.0–6.5, 0.1–0.2 M salt (ammonium acetate, ammonium sulfate, lithium sulfate), and 17–25% PEG 3350–10000. Condition 22 from The Protein Complex Suite (0.15 M ammonium sulfate, 0.1 M MES at pH 6.0, and 15% PEG 4000) was optimized. For setups, 1 μL of protein (35–45 mg/mL in 20 mM CAPS at pH 10.5, 100 mM KCl, and 2 mM DTT) was mixed with 1 μL of reservoir solution (0.2 M ammonium sulfate, 0.1 M MES at pH 6.0, and 13–15% PEG 3350) on a glass coverslip and left to undergo vapor diffusion using the hanging drop method at 20 °C. Large rod-like crystals grew within 1 day. To obtain the Asp-bound gpASNase3 complex, crystals were first soaked in a solution (0.1 M MES at pH 6.0 and 15% PEG 3350) containing 2 M glycine to cleave the enzyme followed by a soak in cryoprotectant solution (0.1 M MES at pH 6.0, 15% PEG 3350, and 30% ethylene glycol) containing saturated l-aspartic acid sodium salt monohydrate (Sigma A6683).
Data Collection and Structure Solution
X-ray diffraction data were collected at the Advanced Photon Source LS-CAT beamline. Data were processed using XDS.39 The structure of uncleaved gpASNase3 was determined by molecular replacement (CCP4Molrep40) using hASNase3 with Protein Data Bank (PDB) entry 4GDV.32 The structure of uncleaved gpASNase3 was then used to determine the phases for the cleaved gpASNase3 diffraction data. The diffraction data collected from the uncleaved and cleaved gpASNase3 crystals were determined to be untwinned and twinned, respectively, using the twinning server Web site located at http://services.mbi.ucla.edu/Twinning/(41) and the program Phenix.xtriage.42 Refinement was performed with Refmac5,43 during which twinning of the cleaved gpASNase3 diffraction data was selected in the configuration settings. The sodium cations in the binding site were validated with the CheckMyMetal: Metal Binding Site Validation Server.44 Coot45 was used for model building, and figures of the structures were made using a MacPyMOL (PyMOL Molecular Graphics System, version 1.4.1, Schrödinger, LLC).
Results and Discussion
Guinea Pig ASNase3 Predominantly Purifies as the Uncleaved Protein
A synthetic gene of gpASNase3 codon optimized for expression in E. coli was made to encompass the complete 309 residue catalytic domain. The majority of gpASNase3 eluted from the nickel column as a single band of ∼35 kDa corresponding to the inactive, uncleaved precursor protein. About 25% of the eluted protein was present as active, cleaved gpASNase3 with bands occurring at ∼20 and ∼15 kDa, which correspond to the α- and β-subunits, respectively.
Glycine Accelerates Cleavage of gpASNase3 into Its Activated form
Being an Ntn-hydrolase family member, gpASNase3 requires autoproteolysis for activation, converting it from an uncleaved precursor into α- and β-subunits (Figure 2A). In gpASNase3, cleavage occurs between Gly167 and Thr168. Freeing the amino group of Thr168, the N-terminal nucleophile of the β-subunit, endows gpASNase3 with its catalytic ability to convert asparagine into aspartate and ammonia. The observation that the majority of gpASNase3 remained in its inactive, uncleaved state throughout purification is similar to what was seen for hASNase3 where intrinsic self-activation occurs very slowly.27,34 This is in contrast to what was reported for other bacterial and plant type III ASNases where autoproteolysis occurs quickly and efficiently even at low temperatures.29,31,34,46
We previously reported that glycine accelerates the rate of cleavage of hASNase3.34 To test whether the free amino acid glycine can also accelerate the cleavage of gpASNase3, we incubated the enzyme in the presence and absence of glycine and analyzed the cleavage state by SDS–PAGE. In the absence of glycine, the percent of uncleaved gpASNase3 is only reduced ∼30% over a three-day incubation period (Figure 2B,C). This showcases the slow intrinsic cleavage rate of gpASNase3. In contrast, during the same incubation time but in the presence of 250 mM glycine, gpASNase3 was cleaved almost completely. This would correspond to the glycine-stimulated cleavage rate. Glycine stimulates cleavage in a dose-dependent manner (Figure 2B,C): the higher the concentration of glycine, the greater is the decrease in uncleaved and subsequent increase in cleaved gpASNase3.
We also compared the rate of intrinsic and glycine-stimulated cleavage of gpASNase3 and hASNase3. Although a higher proportion of hASNase3 remained uncleaved after purification, the intrinsic rate was similar to that of gpASNase3 (Figure 3). Similarities between guinea pig and human ASNase3 cleavage rates were also observed in the presence of 250 mM glycine with the majority of cleavage occurring within 24 h (Figure 3). Through this study, we show that both the slow intrinsic rate of cleavage and the ability to stimulate cleavage with glycine occur in gpASNase3 and are not properties unique to hASNase3. It is possible that these properties may be common to mammalian ASNase3 enzymes. In a previous report, we speculated as to the biological implication of glycine being an activator of this type of enzymes.34
Figure 3.

Comparison of the intrinsic and glycine-stimulated cleavage rates of guinea pig and human ASNase3. (A) SDS–PAGE gels of gp and hASNase3 with and without glycine at pH 6.5. Intrinsic cleavage occurs slowly over time; however, the addition of 250 mM glycine greatly stimulates cleavage. (B) Quantification of band intensities using ImageJ software.57 Both intrinsic and glycine-stimulated cleavage rates are comparable between guinea pig and human ASNase3.
GpASNase3 Exhibits β-Aspartyl Peptidase Activity in Addition to ASNase Activity
β-Aspartyl bond formation is a spontaneous process that is a common form of protein damage, leading to significant structural changes, which can be responsible for loss of function and can alter recognition by proteases.47 In the case that these aberrant peptides cannot be repaired, β-aspartyl peptidases are able to degrade these damaged peptides. We investigated whether gpASNase3 exhibited β-aspartyl peptidase activity in addition to ASNase activity. Despite gpASNase3 having a higher turnover rate of 4 s–1 for l-Asn compared to 2 s–1 for β-l-Asp-l-Phe (see Table 1), gpASNase3 has ∼3-fold lower catalytic efficiency (defined as kcat/KM) of 1.8 mM–1 s–1 for l-Asn compared to 4.8 mM–1 s–1 for β-l-Asp-l-Phe. This is due to gpASNase3 exhibiting a higher KM value with l-Asn (2.2 mM) versus β-l-Asp-l-Phe (0.4 mM). The KM of hASNase3 for β-l-Asp-l-Phe was reported to be 0.4 mM by another group.27 According to data from Nomme et al. (manuscript submitted), who utilized the same kinetic assay and handling as those in this study, hASNase3 has a kcat of 3.2 s–1 and a KM of 2.1 mM for l-Asn hydrolysis and a kcat of 0.8 s–1 and a KM of 0.12 mM for β-l-Asp-l-Phe. This demonstrates that gpASNase3 and hASNase3 behave nearly identically in regard to their l-ASNase activity and very similarly regarding their β-aspartyl peptidase activity.
Table 1. Kinetic Parameters for gpASNase3 Hydrolysis of l-Asn and β-l-Asp-l-Phe.
| substrate | kcat (s–1) | KM (mM) | kcat/KM (mM–1 s–1) |
|---|---|---|---|
| l-Asn | 3.95 ± 0.08 | 2.24 ± 0.14 | 1.76 ± 0.15 |
| β-l-Asp-l-Phe | 1.98 ± 0.04 | 0.41 ± 0.03 | 4.83 ± 0.45 |
The implication of this kinetic characterization of gpASNase3 is that this cannot be the ASNase responsible for the cell killing property of guinea pig serum. That serum ASNase would have a KM for l-Asn in the low micromolar range like the bacterial enzymes used therapeutically. The KM of 2.2 mM for l-Asn means that gpASNase3 would most likely be unable to deplete circulating asparagine levels sufficiently to cause the death of tumor cells. The relevance for ASNase3’s low affinity for asparagine is unclear and indicates that its precise physiological role has yet to be elucidated.
Uncleaved versus Ligand-Bound, Cleaved States of gpASNase3
We determined two crystal structures of gpASNase3: one structure displayed the inactive, uncleaved enzyme (solved at 1.9 Å resolution) and the other the active, fully cleaved enzyme in complex with the product aspartate (2.2 Å). To obtain the latter, since the enzyme was primarily uncleaved after purification, full cleavage was accomplished by soaking the crystal in mother liquor containing 2 M glycine; this was followed by a soak in cryoprotectant that was saturated with l-aspartic acid sodium salt monohydrate. Data collection and refinement statistics can be found in Table 2. Both crystals were of the P3221 space group with nearly identical unit cell parameters and two molecules of gpASNase3 in the asymmetric unit. The crystal of uncleaved gpASNase3 was not twinned, while the crystal from cleaved gpASNase3 with a molecule of aspartate bound in the active site was twinned.
Table 2. Data Collection and Refinement Statistics for gpASNase3.
| uncleaved | cleaved ASP complex | |
|---|---|---|
| PDB entry | 4O47 | 4O48 |
| Data Collectiona | ||
| X-ray source | LS-CAT, ID-G | LS-CAT, ID-G |
| wavelength (Å) | 0.978 57 | 0.979 11 |
| temperature (K) | 100 | 100 |
| resolution (Å) | 30–1.9 (2.01–1.9) | 30–2.29 (2.43–2.29) |
| number of reflections | ||
| observed | 384991 (60228) | 220590 (33863) |
| unique | 83640 (13338) | 47228 (7297) |
| completeness (%) | 99.7 (99.7) | 98.8 (96.0) |
| Rsym (%) | 5.4 (49.6) | 8.2 (59.7) |
| average I/σ(I) | 14.8 (2.6) | 15.7 (3.3) |
| space group | P3221 | P3221 |
| unit cell (Å) | ||
| a = b | 114.87 | 114.58 |
| c | 138.97 | 138.59 |
| Twinning Analysis (xtriage) | ||
| ⟨I2⟩/⟨I⟩2, (untwinned: 2.0), perfect twin: 1.5) | 1.952 | 1.604 |
| ⟨|L|⟩, (untwinned: 0.500), perfect twin: 0.375) | 0.478 | 0.385 |
| ⟨L2⟩, (untwinned: 0.333), perfect twin: 0.200) | 0.310 | 0.210 |
| twin law | no twinning | -h,-k,l |
| Refinement | ||
| refinement program | REFMAC5 | REFMAC5 |
| twinning fraction | 0.4, 0.6 | |
| Rcryst (%) | 19.3 | 18.2 |
| Rfree (%) | 21.3 | 22.1 |
| resolution range (Å) | 28.7–1.90 | 29.7–2.29 |
| no. of protein molecules per asymmetric unit | 2 | 2 |
| no. of atoms | 4456 | 4443 |
| protein (protA, protB) | 2157, 2119 | 2171, 2174 |
| aspartate | 2 × 9 | |
| water molecules | 178 | 78 |
| Na+ | 2 × 1 | 2 × 1 |
| rmsd from ideal | ||
| bond lengths (Å) | 0.013 | 0.011 |
| bond angles (deg) | 1.58 | 1.50 |
| average B factor (Å2) | ||
| protein (protA, protB) | 40.5, 45.9 | 39.5, 45.3 |
| aspartate (protA, protB) | 39.2, 47.1 | |
| water molecules | 43.5 | 37.0 |
| Na+ (protA, protB) | 36.0, 35.3 | 30.7, 36.2 |
| Ramachandran plot (%) | ||
| favored regions | 96.9 | 95.7 |
| additionally allowed regions | 2.8 | 3.9 |
| outlier regions | 0.3 | 0.3 |
Data for the last shell are in parentheses.
As observed with the structures of hASNase3 (PDB entries 4GDU, 4GDT, 4GDV, 4GDW, 4HLO, and 4HLP),32,34E. coli type III ASNase (PDB entry 1T3M),33 and other Ntn-hydrolases,48 both the uncleaved and the cleaved, ligand-bound gpASNase3 exist as dimers where each protomer is formed from one α- and one β-subunit. The protomers display the conserved αββα motif consisting of two β-sheets sandwiched between α-helices (Figure 4). On one side, the α-subunit contributes α-helices 1–6, which flank the mixed β-sheet composed of strands from both the α- and β-subunits (refer to strands M, L, K, A, E, F, B, C, and D of Figure 1B). The strands are all antiparallel with the exception of C and D. The other antiparallel β-sheet is made up of strands J, I, H, and G from the β-subunit, and α-helices (7–9) from the β-subunit complete the sandwich.
Figure 4.

Structure of cleaved guinea pig ASNase3. Cartoon diagram of the cleaved gpASNase3 dimer with its ligand Asp (green) in the active site. Secondary structure elements are indicated. Despite no longer being covalently linked as a single polypeptide chain, the α- and β-subunits remain tightly associated after autocleavage.
Although the construct used was a fusion protein with an intact N-terminal His6-TEV tag, the first residue for which electron density is observed was the histidine before the starting methionine. The uncleaved structure also lacks electron density for the flexible loop region between residues 11–15 in chain A and 11–19 in chain B. In contrast to the uncleaved state, in the cleaved enzyme this N-terminal loop displayed clear electron density and was modeled. Additionally, residues spanning 154–165 (chain A) and 153–165 (chain B) are also disordered in the uncleaved structure. Hence, despite the unambiguously uncleaved state, the only residues with clear electron density preceding the cleavage site (Thr168) are Gly167 and Leu166. This disorder of the region analogous to the C-terminus of the α-chain of the cleaved enzyme was also observed with other Ntn-hydrolase enzymes including hASNase3.31−33,49 In the cleaved structure, this region (residues 156–167 in both chains A and B, i.e., this time including Gly167) also did not display clear electron density.
Consistent with E. coli type III ASNase, plant-type ASNase, and hASNase3, sodium cations were also observed in gpASNase3 to be part of a stabilizing metal binding loop that connects α-helix 2 to strand B, both of the α-subunit.31−33 The cation is coordinated by main chain carbonyl interactions with Leu55, Glu56, Asp58, Phe61, Ala63, and His65.
The dimer interface, as calculated by PDBsum50 per protomer, spans ∼1700 Å2 and involves ∼35 residues for both uncleaved and cleaved gpASNase3; 91% of these are nonpolar interactions with the remaining contributions being hydrogen bonds. There were no major differences between protomers (i.e., comparing protomer A to protomer B) within the uncleaved (root-mean-square deviation (rmsd) of 0.13 Å over 257 Cα atoms) or cleaved (rmsd of 0.21 Å over 267 Cα atoms) structures, respectively. Similarly, there were no major differences between protomers of the uncleaved and cleaved structures (rmsd of 0.19 Å over 275 Cα atoms). The latter observation demonstrates that there are no large structural changes resulting from the cleavage of gpASNase3 into its α- and β-subunits and binding of an aspartate molecule in the active site.
One region that does show a striking difference between the uncleaved and cleaved states is the motif that we refer to as the HGG loop (named for His8-Gly9-Gly10 which are conserved in plant-type ASNases). Cleavage of gpASNase3 results in a local conformational change involving the flipping of the carbonyl group of Gly9. In the uncleaved structure, the carbonyl of Gly9 forms a hydrogen bond with His8 (Figure 5A, dashed line), whereas in the cleaved structure, the carbonyl of Gly9 points away from His8, and the interaction is lost (Figure 5B). The flexibility of the HGG loop and its conformational flip between uncleaved and cleaved states most likely plays an important regulatory role in the cleavage and asparaginase reactions. Results from Nomme et al. (ref (32) and manuscript submitted) regarding hASNase3 also support this proposition.
Figure 5.

Comparison of the structures of uncleaved and cleaved guinea pig ASNase3. (A) The conserved HGG loop of uncleaved gpASNAse3 (protomer A) with the 2Fo – Fc electron density map (blue, contour level of 2σ). Enzyme activation via cleavage of the peptide bond between Gly167 and Thr168 is accompanied by a flip in the carbonyl at Gly9 (arrow), which disrupts its interaction (dashed black line) with the imidazole ring of His8. (B) The HGG motif of cleaved gpASNase3 (protomer A) with the carbonyl group (arrow) at Gly9 facing the opposite direction as clearly seen in the 2Fo – Fc electron density map (blue, contour level of 2σ). (C) The Fo – Fc omit map (gray, contoured at +3σ) present in the active site of cleaved gpASNase3 (protomer A). A molecule of Asp is modeled into it. (D) Overlay of uncleaved and cleaved gpASNase3 protomer A showing the active site residues and the ligand Asp (green). In the uncleaved structure, the Asp binding site is occupied by water molecules (not shown). The covalent bond between Gly167 and Thr168 is observed in the uncleaved structure. For cleaved gpASNase3, dashed lines indicate interactions of the Asp ligand with the active site residues. (E) Modeling of the two best fitting rotamers of Thr168 in uncleaved gpASNase3 protomer A. The 2Fo – Fc and Fo – Fc electron density maps shown were calculated for the dark gray rotamer, labeled rotamer 2. Although rotamer 2 partially fits the 2Fo – Fc electron density map (blue, contour level of 1σ), negative electron density (red, contoured at −3σ) surrounds the methyl group, indicating that too many electrons are modeled at that position. There is also positive electron density (green, contoured at +3σ) at the position occupied by the hydroxyl group of rotamer 1, indicating that more electrons should be modeled at that position. This shows that rotamer 1 (light gray) is the correct one.
An additional contrast between the uncleaved and cleaved structures is that in the former, water molecules occupy the active site, whereas in the latter the Fo – Fc omit map showed large positive density in the active site (Figure 5C) into which a molecule of Asp was modeled. The aspartic acid molecule forms hydrogen bonds with the conserved Ntn-hydrolase residues Thr168, Arg196, Asp199, Thr219, Gly220, and Gly222 (Figure 5D).
Conformation of the Catalytic Thr168
Threonine 168 belongs to a threonine catalytic triad (the others being Thr186 and Thr219) that is strictly conserved in this enzyme family. The role of Thr168 is to nucleophilically attack the asparagine side chain carbonyl C atom, a step that initiates the hydrolysis reaction. The nucleophilicity of the Thr168 side chain is increased by its α-amino group, a moiety that is freed by the cleavage reaction.51−54 In fact, Thr168 is not only required for the hydrolysis of asparagine into aspartic acid, but in addition, the hydroxyl group of its side chain is required for its own activation by initiating cleavage through nucleophilic attack on the carbonyl of Gly167.
Interestingly, Thr168 exists in different conformations in the uncleaved versus cleaved structures, with the conformation observed in the uncleaved structure deviating from that observed in both uncleaved and cleaved hASNase3. Because of the key catalytic functions of this residue, we took great care to confirm that indeed the correct rotamer of Thr168 was modeled at the active site in the uncleaved structure. For this reason, we examined all three possible rotamers of Thr168 and compared their fit to 2Fo – Fc and Fo – Fc electron density maps. The third rotamer clearly did not fit the electron density and was ruled out. The second rotamer mostly fit the 2Fo – Fc map; however, the Fo – Fc difference map showed negative density (indicating too many electrons modeled) at the position of the methyl group and positive density (indicating not enough electrons modeled) 120° away (Figure 5E, dark gray). This observation was confirmed for both protomers A and B and suggested that this rotamer is not the correct one. In contrast, in addition to clearly fitting in the 2Fo – Fc map, the first rotamer of Thr168 (Figure 5E, light gray) did not display any positive density and showed only slight negative density in the Fo – Fc difference map, and only in protomer B. This analysis validates the choice of the first rotamer for this key residue in the uncleaved structure.
The observed Thr168 rotamer of the uncleaved structure is perplexing since the critical distance between the carbonyl of Gly167 and the hydroxyl group of Thr168 is too long for cleavage to occur. The first step in the cleavage reaction would involve the Thr168 hydroxyl group attacking the carbonyl C atom of the preceding Gly167. This cleavage-incompetent conformation of Thr168 observed in the uncleaved structure could help to account for the slow intrinsic cleavage rate of gpASNase3. Moreover, this observation suggests that this region must be flexible to allow a conformational change of Thr168 and/or Gly167 that would build the cleavage competent state.
Comparison to Human ASNase3
Guinea pig and human ASNase3 are not only highly similar in sequence and kinetic properties, but as can be seen in Figure 6, they are very similar in structure as well. The main differences occur in the flexible loop regions. The cleaved gpASNase3 Asp-bound structure displays high similarity to cleaved, Asp-bound hASNase3 (PDB entry 4GDW; rmsd of 0.36 Å over 498 Cα atoms) (Figure 6A). The position of the aspartate molecule bound in the active site of cleaved guinea pig and human ASNase3 is nearly identical (Figure 6B), and the interactions between the aspartate and conserved Ntn-hydrolase active site residues Thr168, Arg196, Asp199, Thr219, Gly220, and Gly222 are the same. Additionally, the flip of the Gly9 carbonyl of the HGG loop between the uncleaved and cleaved states is consistent between human and guinea pig ASNase3.
Figure 6.
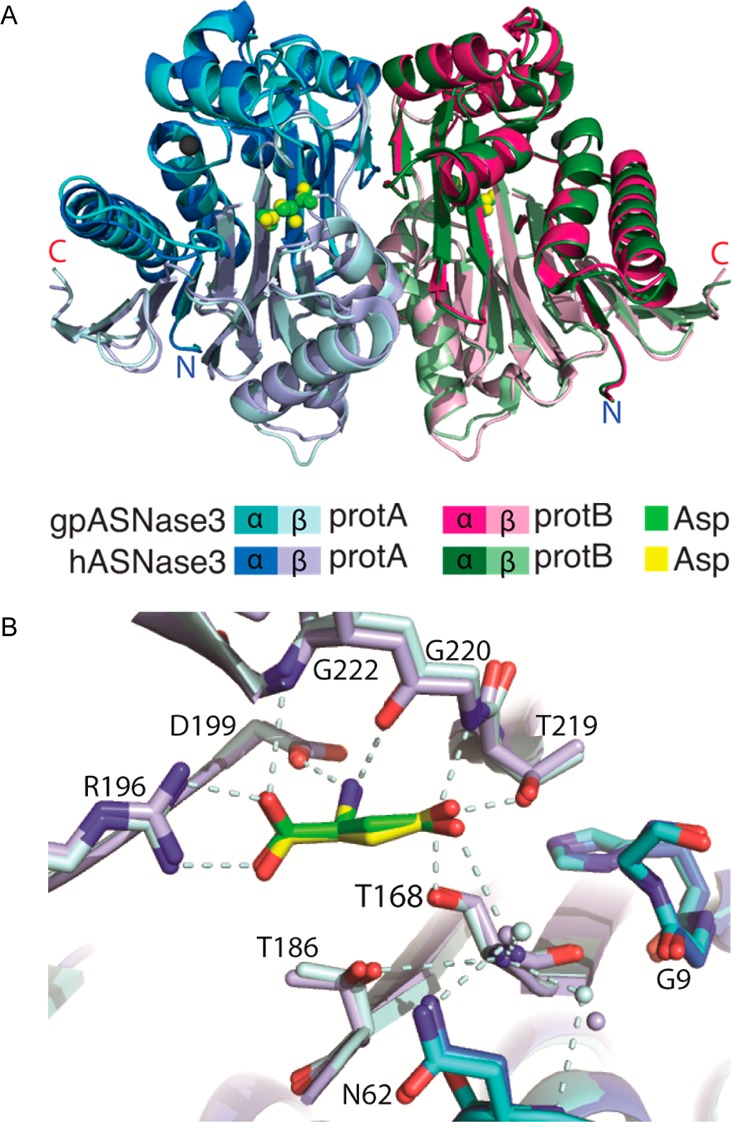
Comparison of the structures of cleaved guinea pig and human ASNase3. (A) Overlay of cartoon diagrams of the structures of cleaved gp and hASNase3 (PDB entry 4GDW) in complex with Asp. The Na+ cation present in each protomer is represented as a gray sphere. (B) Overlay of the active sites of cleaved gp and hASNase3 in complex with Asp denoted in green and yellow, respectively. The freed N-terminus at Thr168 denoting cleavage into α- and β-subunits is observed. Interactions made by conserved Ntn-hydrolase residues (dashed light cyan lines) are consistent between gp and hASNase3. The carbonyl of Gly9 in the HGG loop is also pointed away from the side chain of His8 in both gp and hASNase3 in agreement with cleavage between Gly167 and Thr168 and enzyme activation. Water molecules are shown as small spheres whose colors correspond to either h or gpASNase3.
As noted earlier, the HGG motif is conserved in plant-type asparaginases. Interestingly, the glycine of this motif that we observe to flip between the uncleaved and cleaved structures in the mammalian enzymes was observed for the E. coli enzyme to be in the same conformation regardless of cleavage (E. coli cleaved PDB entry 2ZAL;30 uncleaved attained by the T179A mutation, PDB entries 2ZAK(55) and 3C17(56)). Additionally, whereas the bacterial enzyme readily undergoes cleavage, both the human and guinea pig homologues cleave very slowly. This high conformational flexibility of the HGG motif may contribute to the slow autocleavage rate of the mammalian enzymes. The interaction between the carbonyl group of Gly9 and the imidazole ring of His8 could aid in stabilization of the uncleaved form, preventing autoproteolysis. It remains to be elucidated whether cleavage of mammalian ASNase3 into its α- and β-subunits is what triggers this conformational change at the HGG loop or whether the flipping of the carbonyl group of Gly9 makes the autocleavage reaction more favorable.
Conclusions
In this study, we determined that the uncharacterized protein fragment H0VQC8_CAVPO in the UniProt database corresponds to a type III ASNase, which we named guinea pig ASNase3. We show that like other Ntn-hydrolases, gpASNase3 is predominantly produced as an inactive, single polypeptide and requires autocatalytic activation by cleavage into α- and β-subunits that remain closely associated despite no longer being covalently linked. We have investigated the self-cleavage of gpASNase3 and found that analogously to the human enzyme, gpASNase3 has a very slow intrinsic rate of cleavage. Cleavage, and hence activation, can be stimulated by the presence of glycine. In addition to being an asparaginase, gpANase3 can also function as a β-aspartyl peptidase.
We also determined both the uncleaved (inactive) as well as the cleaved (active), aspartate-bound structures of gpASNase3. We observed that cleavage does not greatly alter the overall structure of gpASNase3 with the exception of the conformational change in the conserved HGG loop that coincides with cleavage. We also compared the gpASNase3 structure with hASNase3 and found that they are almost identical with the differences between them occurring in regions known to be highly flexible.
Most important from a translational aspect, we were able to conclusively determine that gpASNase3 is not the asparaginase responsible for the antitumor effects elicited by treatment with guinea pig serum.1 Despite the probable decrease in negative side effects related to immunogenicity, the high KM of gpASNase3 makes it unsuitable for use in anticancer therapy.
Acknowledgments
We thank Ying Su for generously providing purified hASNase3 and Dr. Julian Nomme for helpful suggestions. We thank the staff of LS-CAT for their assistance in data collection.
Glossary
Abbreviations
- ASNase
l-asparaginase
- ALL
acute lymphoblastic leukemia
- Ntn
N-terminal nucleophile
- AGA
aspartylglucosaminidase
- hASNase3
human l-asparaginase type III
- gpASNase3
guinea pig l-asparaginase type III
- PEG
poly(ethylene glycol)
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- PMSF
phenylmethanesulfonyl fluoride
- DTT
dithiothreitol
- NADH
nicotinamide adenine dinucleotide (reduced)
- rmsd
root-mean-square deviation
- PDB
Protein Data Bank
This work was supported by a National Institutes of Health grant R21 CA155424.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kidd J. G. (1953) Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. I. Course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J. Exp. Med. 98, 565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome J. D. (1961) Evidence that the L-asparaginase activity of guinea pig serum is responsible for its antilymphoma effects. Nature 191, 1114–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome J. D. (1963) Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. I. Properties of the L-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J. Exp. Med. 118, 99–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashburn L. T.; Wriston J. C. Jr. (1964) Tumor inhibitory effect of L-asparaginase from Escherichia coli. Arch. Biochem. Biophys. 105, 450–452. [DOI] [PubMed] [Google Scholar]
- Wade H. E.; Robinson H. K.; Phillips B. W. (1971) Asparaginase and glutaminase activities of bacteria. J. Gen. Microbiol. 69, 299–312. [DOI] [PubMed] [Google Scholar]
- Ertel I. J.; Nesbit M. E.; Hammond D.; Weiner J.; Sather H. (1979) Effective dose of L-asparaginase for induction of remission in previously treated children with acute lymphocytic leukemia: a report from Childrens Cancer Study Group. Cancer Res. 39, 3893–3896. [PubMed] [Google Scholar]
- Cooney D. A.; Capizzi R. L.; Handschumacher R. E. (1970) Evaluation of L-asparagine metabolism in animals and man. Cancer Res. 30, 929–935. [PubMed] [Google Scholar]
- Haley E. E.; Fischer G. A.; Welch A. D. (1961) The requirement for L-asparagine of mouse leukemia cells L5178Y in culture. Cancer Res. 21, 532–536. [PubMed] [Google Scholar]
- Neuman R. E.; McCoy T. A. (1956) Dual requirement of Walker carcinosarcoma 256 in vitro for asparagine and glutamine. Science 124, 124–125. [DOI] [PubMed] [Google Scholar]
- Muller H. J.; Boos J. (1998) Use of L-asparaginase in childhood ALL. Crit. Rev. Oncol./Hematol. 28, 97–113. [DOI] [PubMed] [Google Scholar]
- Avramis V. I.; Tiwari P. N. (2006) Asparaginase (native ASNase or pegylated ASNase) in the treatment of acute lymphoblastic leukemia. Int. J. Nanomed. 1, 241–254. [PMC free article] [PubMed] [Google Scholar]
- Ahlke E.; Nowak-Gottl U.; Schulze-Westhoff P.; Werber G.; Borste H.; Wurthwein G.; Jurgens H.; Boos J. (1997) Dose reduction of asparaginase under pharmacokinetic and pharmacodynamic control during induction therapy in children with acute lymphoblastic leukaemia. Br. J. Haematol. 96, 675–681. [DOI] [PubMed] [Google Scholar]
- Asselin B. L.; Whitin J. C.; Coppola D. J.; Rupp I. P.; Sallan S. E.; Cohen H. J. (1993) Comparative pharmacokinetic studies of three asparaginase preparations. J. Clin. Oncol. 11, 1780–1786. [DOI] [PubMed] [Google Scholar]
- Abuchowski A.; van Es T.; Palczuk N. C.; McCoy J. R.; Davis F. F. (1979) Treatment of L5178Y tumor-bearing BDF1 mice with a nonimmunogenic L-glutaminase-L-asparaginase. Cancer Treat. Rep. 63, 1127–1132. [PubMed] [Google Scholar]
- Abuchowski A.; Kazo G. M.; Verhoest C. R. Jr.; Van Es T.; Kafkewitz D.; Nucci M. L.; Viau A. T.; Davis F. F. (1984) Cancer therapy with chemically modified enzymes. I. Antitumor properties of polyethylene glycol-asparaginase conjugates. Cancer Biochem. Biophys. 7, 175–186. [PubMed] [Google Scholar]
- Pasut G.; Veronese F. M. (2009) PEG conjugates in clinical development or use as anticancer agents: an overview. Adv. Drug Delivery Rev. 61, 1177–1188. [DOI] [PubMed] [Google Scholar]
- Keating M. J.; Holmes R.; Lerner S.; Ho D. H. (1993) L-asparaginase and PEG asparaginase--past, present, and future. Leuk. Lymphoma 10(Suppl), 153–157. [DOI] [PubMed] [Google Scholar]
- Armstrong J. K.; Hempel G.; Koling S.; Chan L. S.; Fisher T.; Meiselman H. J.; Garratty G. (2007) Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 110, 103–111. [DOI] [PubMed] [Google Scholar]
- Muller H. J.; Loning L.; Horn A.; Schwabe D.; Gunkel M.; Schrappe M.; von Schutz V.; Henze G.; Casimiro da Palma J.; Ritter J.; Pinheiro J. P.; Winkelhorst M.; Boos J. (2000) Pegylated asparaginase (Oncaspar) in children with ALL: drug monitoring in reinduction according to the ALL/NHL-BFM 95 protocols. Br. J. Haematol. 110, 379–384. [DOI] [PubMed] [Google Scholar]
- Avramis V. I.; Panosyan E. H. (2005) Pharmacokinetic/pharmacodynamic relationships of asparaginase formulations: the past, the present and recommendations for the future. Clin. Pharmacokinet. 44, 367–393. [DOI] [PubMed] [Google Scholar]
- Boos J.; Werber G.; Ahlke E.; Schulze-Westhoff P.; Nowak-Gottl U.; Wurthwein G.; Verspohl E. J.; Ritter J.; Jurgens H. (1996) Monitoring of asparaginase activity and asparagine levels in children on different asparaginase preparations. Eur. J. Cancer 32A, 1544–1550. [DOI] [PubMed] [Google Scholar]
- Dellinger C. T.; Miale T. D. (1976) Comparison of anaphylactic reactions to asparaginase derived from Escherichia coli and from Erwinia cultures. Cancer 38, 1843–1846. [DOI] [PubMed] [Google Scholar]
- Muller H. J.; Beier R.; da Palma J. C.; Lanvers C.; Ahlke E.; von Schutz V.; Gunkel M.; Horn A.; Schrappe M.; Henze G.; Kranz K.; Boos J. (2002) PEG-asparaginase (Oncaspar) 2500 U/m(2) BSA in reinduction and relapse treatment in the ALL/NHL-BFM protocols. Cancer Chemother. Pharmacol. 49, 149–154. [DOI] [PubMed] [Google Scholar]
- UniProt C. (2013) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 41, D43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush L. A.; Herr J. C.; Wolkowicz M.; Sherman N. E.; Shore A.; Flickinger C. J. (2002) A novel asparaginase-like protein is a sperm autoantigen in rats. Mol. Reprod. Dev. 62, 233–247. [DOI] [PubMed] [Google Scholar]
- Cantor J. R.; Stone E. M.; Chantranupong L.; Georgiou G. (2009) The human asparaginase-like protein 1 hASRGL1 is an Ntn hydrolase with beta-aspartyl peptidase activity. Biochemistry 48, 11026–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evtimova V.; Zeillinger R.; Kaul S.; Weidle U. H. (2004) Identification of CRASH, a gene deregulated in gynecological tumors. Int. J. Oncol. 24, 33–41. [PubMed] [Google Scholar]
- Borek D.; Michalska K.; Brzezinski K.; Kisiel A.; Podkowinski J.; Bonthron D. T.; Krowarsch D.; Otlewski J.; Jaskolski M. (2004) Expression, purification and catalytic activity of Lupinus luteus asparagine beta-amidohydrolase and its Escherichia coli homolog. Eur. J. Biochem. 271, 3215–3226. [DOI] [PubMed] [Google Scholar]
- Michalska K.; Brzezinski K.; Jaskolski M. (2005) Crystal structure of isoaspartyl aminopeptidase in complex with L-aspartate. J. Biol. Chem. 280, 28484–28491. [DOI] [PubMed] [Google Scholar]
- Michalska K.; Bujacz G.; Jaskolski M. (2006) Crystal structure of plant asparaginase. J. Mol. Biol. 360, 105–116. [DOI] [PubMed] [Google Scholar]
- Nomme J.; Su Y.; Konrad M.; Lavie A. (2012) Structures of apo and product-bound human L-asparaginase: insights into the mechanism of autoproteolysis and substrate hydrolysis. Biochemistry 51, 6816–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahl A.; Pazgier M.; Hejazi M.; Lockau W.; Lubkowski J. (2004) Structure of the isoaspartyl peptidase with L-asparaginase activity from Escherichia coli. Acta Crystallogr., Sect. D 60, 1173–1176. [DOI] [PubMed] [Google Scholar]
- Su Y.; Karamitros C. S.; Nomme J.; McSorley T.; Konrad M.; Lavie A. (2013) Free glycine accelerates the autoproteolytic activation of human asparaginase. Chem. Biol. 20, 533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan J. A.; Dodson G.; Duggleby H. J.; Moody P. C.; Smith J. L.; Tomchick D. R.; Murzin A. G. (1995) A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 378, 416–419. [DOI] [PubMed] [Google Scholar]
- Hejazi M.; Piotukh K.; Mattow J.; Deutzmann R.; Volkmer-Engert R.; Lockau W. (2002) Isoaspartyl dipeptidase activity of plant-type asparaginases. Biochem. J. 364, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen R. A.; Knox T. M.; Miller C. G. (2001) Aspartic peptide hydrolases in Salmonella enterica serovar typhimurium. J. Bacteriol. 183, 3089–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez C. A.; Cai X.; Elozory A.; Liu C.; Panetta J. C.; Jeha S.; Molinelli A. R.; Relling M. V. (2013) High-throughput asparaginase activity assay in serum of children with leukemia. Int. J. Clin. Exp. Med. 6, 478–487. [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) Xds. Acta Crystallogr., Sect. D 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect. D 66, 22–25. [DOI] [PubMed] [Google Scholar]
- Padilla J. E.; Yeates T. O. (2003) A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr., Sect. D 59, 1124–1130. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Chordia M. D.; Cooper D. R.; Chruszcz M.; Muller P.; Sheldrick G. M.; Minor W. (2014) Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 9, 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and Development of Coot. Acta Crystallogr., Sect. D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek D.; Jaskolski M. (2000) Crystallization and preliminary crystallographic studies of a new L-asparaginase encoded by the Escherichia coli genome. Acta Crystallogr., Sect. D 56, 1505–1507. [DOI] [PubMed] [Google Scholar]
- Aswad D. W.; Paranandi M. V.; Schurter B. T. (2000) Isoaspartate in peptides and proteins: formation, significance, and analysis. J. Pharm. Biomed. Anal. 21, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Oinonen C.; Rouvinen J. (2000) Structural comparison of Ntn-hydrolases. Protein Sci. 9, 2329–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Guo H. C. (2010) Crystallographic snapshot of glycosylasparaginase precursor poised for autoprocessing. J. Mol. Biol. 403, 120–130. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Hutchinson E. G.; Michie A. D.; Wallace A. C.; Jones M. L.; Thornton J. M. (1997) PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci. 22, 488–490. [DOI] [PubMed] [Google Scholar]
- Duggleby H. J.; Tolley S. P.; Hill C. P.; Dodson E. J.; Dodson G.; Moody P. C. (1995) Penicillin acylase has a single-amino-acid catalytic centre. Nature 373, 264–268. [DOI] [PubMed] [Google Scholar]
- Lowe J.; Stock D.; Jap B.; Zwickl P.; Baumeister W.; Huber R. (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 268, 533–539. [DOI] [PubMed] [Google Scholar]
- Aronson N. N. Jr. (1996) Lysosomal glycosylasparaginase: a member of a family of amidases that employ a processed N-terminal threonine, serine or cysteine as a combined base-nucleophile catalyst. Glycobiology 6, 669–675. [DOI] [PubMed] [Google Scholar]
- Guan C.; Liu Y.; Shao Y.; Cui T.; Liao W.; Ewel A.; Whitaker R.; Paulus H. (1998) Characterization and functional analysis of the cis-autoproteolysis active center of glycosylasparaginase. J. Biol. Chem. 273, 9695–9702. [DOI] [PubMed] [Google Scholar]
- Michalska K.; Borek D.; Hernandez-Santoyo A.; Jaskolski M. (2008) Crystal packing of plant-type L-asparaginase from Escherichia coli. Acta Crystallogr., Sect. D 64, 309–320. [DOI] [PubMed] [Google Scholar]
- Michalska K.; Hernandez-Santoyo A.; Jaskolski M. (2008) The mechanism of autocatalytic activation of plant-type L-asparaginases. J. Biol. Chem. 283, 13388–13397. [DOI] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis, Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]