

Abstract
The Collaborative Cross (CC) is an emerging panel of recombinant inbred mouse strains. Each strain is genetically distinct but all descended from the same eight inbred founders. In 66 strains from incipient lines of the CC (pre-CC), as well as the 8 CC founders and some of their F1 offspring, we examined subsets of lymphocytes and antigen-presenting cells. We found significant variation among the founders, with even greater diversity in the pre-CC. Genome-wide association using inferred haplotypes detected highly significant loci controlling B-to-T cell ratio, CD8 T-cell numbers, CD11c and CD23 expression. Comparison of overall strain effects in the CC founders with strain effects at QTL in the pre-CC revealed sharp contrasts in the genetic architecture of two traits with significant loci: variation in CD23 can be explained largely by additive genetics at one locus, whereas variation in B-to-T ratio has a more complex etiology. For CD23, we found a strong QTL whose confidence interval contained the CD23 structural gene Fcer2a. Our data on the pre-CC demonstrate the utility of the CC for studying immunophenotypes and the value of integrating founder, CC, and F1 data. The extreme immunophenotypes observed could have pleiotropic effects in other CC experiments.
Keywords: Collaborative Cross, FcεR, QTL
1. Introduction
Understanding the genetics of the immune system has been critical in unraveling the mechanisms of the major questions in immunology of the past 50 years including antibody diversity, T cell recognition and the function of the major histocompatibility complex. Many of the major causes of immunodeficiency caused by single gene mutations have been identified and their mechanism elucidated 1.Many of the major causes of immunodeficiency caused by single gene mutations have been identified and their mechanism elucidated1. Now interest has shifted towards understanding interacting genes, and with it, complex genetics where a large number of common variants individually or through interactions each exert small but interacting effects on the lymphoid and myeloid development. Examples here include immune- mediated diseases such as asthma and type 1 diabetes 2, 3.
The mouse has been critical to progress in understanding the molecular basis of single gene diseases, and offers considerable promise for understanding diseases with more complex genetic architectures (e.g., 4 and refs therein). Here we report on the use of a new mouse genetic resource, the Collaborative Cross, as a way to probe the immune system function. The Collaborative Cross (CC) is an eight-way recombinant inbred (RI) panel conceived as a next-generation platform and community resource for systems genetics 5. The eight founders of the CC consist of five ‘classical’ laboratory strains (short names in parentheses): 129S1/SvImJ (129S1), A/J (AJ), C57BL/6J (B6), NOD/ShiLtJ (NOD), NZO/HlLtJ (NZO); and three ‘wild-derived’ strains CAST/EiJ (CAST), PWK/PhJ (PWK), WSB/EiJ (WSB). The resulting CC lines, each bred from a structured and randomized combination of these eight founders, surpass any available RI panel in genetic diversity, number of recombination events (and hence potential QTL mapping resolution), and eventually in number of available strains 6-9. From an experimental design perspective, the CC offers greater reproducibility and balance than outbred populations of similar genetic diversity. Specifically, because each CC strain is inbred, experiments on it can be replicated, and because pairs of CC strains are genetically different from each other by an approximately even degree, examining the effects of polymorphisms across a group of strains is (at its best) akin to a randomized and balanced assignment of alternative genetic treatments and their combinations. Production of the CC population was initiated in 2005 10-12 Three different cohorts of CC mice are in existence5, and our analysis focuses only on strains initiated at The Oak Ridge National Laboratory (ORNL). Analysis of incipient CC strains (pre-CC) from ORNL demonstrated balanced allele frequencies and well-distributed recombination events 13. Inbreeding of the CC lines is ongoing with up-to-date information on the status and availability of lines at http://www.csbio.unc.edu/CCstatus/ 14. Our report complements several recent studies examining different phenotypes in the pre-CC. 5, 13, 15, 16.
In our initial phenotyping of the pre-CC mice we chose to focus on the differences among well defined leukocyte subsets that could be effectively defined using flow cytometry. In all immune phenotypes measured, we found variation in the pre-CC strains exceeding that found in the founder strains. We also found striking outliers in some traits suggesting immune dysregulation in some of the pre-CC strains. Despite residual heterozygosity and the use of a very small cohort of pre-CC strains, we identified several QTLs, one of which (CD23 surface expression) we were able to localize to plausible coding mutations in a single gene of known function. The present genetic analyses add to a growing body of evidence supporting the potential power of the CC for the study of a variety of phenotypes. Our observations of extreme outlier immunological phenotypes in the pre-CC mice also have implications for other phenotyping studies performed on the pre-CC mice and completed CC strains.
Results
Founder strains have diverse resting immune phenotypes
In order to assess the contribution of each of the founder strains to the immunological diversity of the pre-CC lines, we analyzed mice from all eight CC founder strains (121 inbred mice) and from 31 of their F1 crosses (132 F1 hybrid mice). Splenocytes from all strains were analyzed using a nine-parameter flow cytometric panel consisting of antibodies staining subsets of T-cells, B-cells, and antigen-presenting cells (APCs). We established a single, thorough data analysis scheme for our flow cytometry data which was applied to every sample analyzed (description in Table 1, example gating scheme in Supplemental Figure 1.1). Our analysis scheme defined a total of 21 different phenotypes derived from our flow data (Table 1). Every batch of samples analyzed included a B6 male spleen to help with standardization. We used mean fluorescent intensity (MFI) to measure the level of protein expression on the cell surface, calibrating this measurement against a subset (61/70) of B6 mice used as controls. For this reason, although as many as 70 B6 mice are included in our analyses of non-MFI phenotypes, the 61 used as controls are necessarily excluded from phenotype data (see CD11c MFI, CD11b MFI and CD23 MFI in Table 1). We found that 19 of 21 of our measured immunophenotypes showed highly significant differences among the founder strains (see Table 1).
Table 1.
Summary of immune phenotypes and their variation among the founders. INT: inverse normal transformation; logit: is the logit function; log10: log to the base 10.
| Phenotype | Description | Measurement | Transformation for statistical analysis | ANOVA of strain effect | Number of mice per strain (129, AJ, B6, CAST, NOD, NZO, PWK, WSB, B6 controls) | |
|---|---|---|---|---|---|---|
| F statistic | P-Value | |||||
| -APCs | Total Lymphoid Cells | % of Total Cells | Logit | F7,38=4.28 | 0.00144 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| +APCs | Total Myeloid Cells | % of Total Cells | Logit | F7,48=15.1 | 3.23E-10 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| B/T | Ratio of B-cells to T-cells | CD19+ / H57+ | Log10 | F7,38=10.4 | 3.44E-05 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| CD11b Hi | Phagocytes | % of Total Cells | Logit | F7,38=3.89 | 0.00275 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| CD11b MFI | CD11b Antigen Density on Dendritic Cells | Mean Fluorescence Intensity (MFI) | Log10 | F7,38=5.39 | 2.40E-04 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| CD11c MFI | CD11c Antigen Density on Dendritic Cells | MFI | Log10 | F7,38=22.4 | 1.13E-11 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| CD11c+ | CD11c+/CD11b-Dendritic Cells | % of Total Cells | Logit | F7,38=2.01 | 0.0785 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| CD19+ | Total B-cells | % of Total Cells | Logit | F7,38=4.14 | 1.79E-03 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| CD23 MFI | CD23 antigen density on all B-cells | MFI | Log10 | F7,38=14.3 | 6.10E-09 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| CD4+ | Helper and Regulatory T-cells | % of H57+ | Logit | F7,38=8.87 | 1.96E-06 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| CD4+, CD25+ | Regulatory T-cells | % of CD4+ | Logit | F7,38=7.75 | 8.15E-06 | 6, 6, 10, 2, 6, 10, 9, 10, 58 |
| CD4+/CD8+ | Ratio of Helper T-cells to Cytotoxic T-cells | CD4+ / CD8+ | Log10 | F7,38=6.45 | 4.91E-05 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| CD8+ | Cytotoxic T-cells | % of H57+ | Logit | F7,38=5.38 | 2.42E-04 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| DCs | Classical Dendritic Cells | % of Total Cells | Logit | F7,38=16.1 | 1.31E-09 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| FoB | Follicular B-cells | % of CD19+ | INT | F7,38=9.63 | 7.88E-07 | 6, 6, 10, 2, 6, 10, 9, 10, 58 |
| Granulocytes | Granulocytes | % of Total Cells | Logit | F7,38=2.41 | 0.0381 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| H57+ | Total T-cells | % of Total Cells | Logit | F7,48=5.52 | 1.95E-04 | 6, 6, 10, 2, 6, 10, 9, 10, 62 |
| Macrophages | Macrophages | % of Total Cells | Logit | F7,38=6.49 | 4.63E-05 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| Monocytes | Monocytes | % of Total Cells | Logit | F7,38=11 | 1.60E-07 | 6, 6, 10, 2, 6, 10, 9, 10, 61 |
| MZB | Marginal Zone B-cells | % of CD19+ | INT | F7,38=18.1 | 2.56E-10 | 6, 6, 10, 2, 6, 10, 9, 10, 58 |
| TransB | Transitional B-cells | % of CD19+ | INT | F7,38=6.52 | 4.45E-05 | 6, 6, 10, 2, 6, 10, 9, 10, 58 |
In this communication we summarize the immune phenotypes, and focus on three illustrative points (while including results on all phenotypes in Supplemental Material). First, the observed phenotypes are complex with 15 independently measured principal components required to capture all of the phenotypic variation. Second, we identified QTL that contribute to immunologically important variation, especially T/B ratios, and the genetics of these are complex. Further a strong QTL for CD23 expression maps on chromosome 8 at or near Fcer2, the gene that encodes CD23. Finally examination of the parental strains that show large variations of CD23 expression show that these do not control the level of circulating IgE as had been suggested by Ford and co-workers 17.
Diversity in the pre-CC strains is greater than observed in the founders
We analyzed 66 pre-CC strains, that is, incipient CC strains that had undergone sister-brother mating for at least five generations. In many immunophenotypes analyzed, the pre-CC showed greater variability than that observed in the founders. In some cases, we found extreme phenotypes representing profound disruption in lymphocyte homeostasis (Figure 1). For example, several pre-CC strains had a notable T-cell lymphopenia, with H57+ cells constituting a mere 5-10% of the total splenocytes (Figure 1a). We also identified pre-CC strains with extremely high CD4/CD8 ratios (∼9 CD4/ 1 CD8), and interestingly, inverted CD4/CD8 ratios (Figure 1b). Such apparent immune dysregulation was not identified in the founders or any of the F1 hybrids analyzed.
Figure 1.

Immunophenotypic diversity in the CC founders and pre-CC strains. Strain means are shown embedded within the entire pre-CC dataset. Founder strains are indicated using the standard colors. Males and females are pooled. Error bars represent standard of deviation, except for CAST/EiJ where error bars represent range. A) T-cells (H57+) as a fraction of all cells in the spleen. Values are untransformed with values ranging from 0.043 (4.3%) to 0.72 (72%). B) Ratio of CD4+ to CD8+ T-cells. Data are transformed using the natural log in order to normalize the distribution and expose outliers. The untransformed ratios range from 0.50 to 8.65 CD4+ per CD8+ T-cell.
Figure 2 plots trait correlations among the 66 pre-CC, 121 inbred and 132 F1 mice and indicates that some traits were highly correlated, notably those defining T cell subsets. The percentage of B cells and the percentage of T cells, unsurprisingly, correlates strongly with the B/T cell ratio. Surprisingly, however, the number of independent traits was quite high: a principal components analysis showed that 15 uncorrelated composite traits would be required to explain 99% of the variation provided by the 21 measured phenotypes. This suggests substantial independent control of these traits (Figure 3), and is consistent with many immune traits depending on multiple interaction genetic pathways rather than a few highly influential loci.
Figure 2.

Correlations among 21 immunophenotypes among 121 inbred, 133 F1 and 66 pre-CC mice. Each block represents the correlation between two (transformed) phenotypes, with the top-left to bottom-right diagonal of black blocks depicting correlation of +1. Shading indicates strength of correlation; white diagonal lines indicate positive vs negative correlation. Correlation is based on between 276 and 319 paired observations, depending on phenotype.
Figure 3.

Principal Components Analysis (PCA) indicates that more than 99% of the variance of the 21 phenotypes can be explained by 15 statistically independent composite phenotypes (PCs).
QTL identified for lymphocyte subsets
When we performed a genome-wide scan in the pre-CC for genetic loci associated with changes in B/T ratio, we found two strong peaks on chromosome 6 centered at 23Mb and 91Mb (Figure 4; Table 2). T cell receptor beta (61Mb), Ig kappa genes (70 Mb) as well as NK receptor (129 Mb) genes map on mouse chromosome 6. In addition to significant associations noted for B/T ratios, MFI of CD23, % of T cells, % of B cells, % of CD8 cells, MFI CD11c and % of transitional B cells (see Figure S1 for scans of phenotypes without genome-wide significant QTL). The genome of each pre-CC strain is a distinct mosaic of the haplotypes from the original eight founders, and so at any given QTL the impact of genetic variation in the region can be described in terms of strain effects; that is, estimated substitution effects of the eight founder haplotypes. The strain effects at the 23Mb QTL peak for B/T ratio on chromosome 6 (Figure 5) include a strong positive effect on B/T ratio by the PWK strain haplotype and a strong negative effect by CAST. An independent pattern of strain effects is seen at the second chrosome 6 peak for B/T (at 91Mb), which includes a strong positive effect of the NZO haplotype. It is tempting to speculate on the role of WASL that maps in 24664995 on chromosome 6, and lies just outside the 1.5 LOD drop interval (but well within the more conservative bootstrap interval; Table 2). The protein is a member of the Wiskott-Aldrich syndrome family and may play a role in cyto-skeletal rearrangement during signaling. Wiskott-Aldrich syndrome protein (WASP) itself is important in T and B cell differentiation 18, 19. In the more distal region another effect is mediated by NZO encoded alleles. Although this segment is very large, it does contain skap55 that regulates binding to LFA1 in response to CCR7 20 as well as Nod1, all important in innate immunity, clearly multiple epistatic genes located in cis on the NZO background could be critical.
Figure 4.

Genome scans of pre-CC mice with significant or near-significant QTL. The x-axis plots location in the genome, y-axis gives statistical significance of association with the phenotype. Dashed horizontal lines indicate genomewide significance at the 0.05, 0.1, and 0.2 level.
Table 2.
Significant and suggestive QTL detected in the pre-CC. LogP: negative log to base 10 of the p-value (measure of statistical association).
| Phenotype | Chr | LogP 5% significance threshold | LogP at QTL peak | QTL peak location in Mb | 1.5 LOD drop interval | 95% bootstrap confidence interval | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start (Mb) | End (Mb) | Length in Mb (#Genes) | Start (Mb) | End (Mb) | Length in Mb (#Genes) | |||||
| B/T | 6 | 6.93 | 9.49 | 23.458 | 23.217313 | 23.815913 | 0.599 (14) | 9.89947 | 49.307538 | 39.408(741) |
| B/T | 6 | 6.93 | 7.33 | 91.385 | 88.058961 | 92.668757 | 4.610 (117) | 53.868818 | 116.845948 | 62.977(1118) |
| CD11c MFI | 7 | 5.96 | 8.64 | 141.616 | 141.616402 | 141.653981 | 0.038(3) | 131.296863 | 144.257850 | 12.961(466) |
| CD11c MFI | 7 | 5.96 | 8.40 | 129.749 | 136.462451 | 138.639201 | 2.18(14) | 136.462451 | 144.4206 | 7.958(366) |
| CD19+ | 6 | 6.91 | 8.25 | 23.458 | 23.303557 | 23.815913 | 0.512 (13) | 5.463471 | 51.76262 | 46.299(829) |
| CD23 MFI | 8 | 5.89 | 10.96 | 8.638 | 3.083611 | 16.833693 | 13.75(271) | 3.083611 | 22.846868 | 19.764(435) |
| CD4+ | 7 | 6.93 | 6.55 | 141.626 | 141.141539 | 142.24667 | 1.105 (99) | 136.462451 | 151.510949 | 15.048(393) |
| CD4+/CD8+ | 7 | 6.91 | 7.15 | 141.646 | 141.141539 | 142.24667 | 1.105 (99) | 136.462451 | 151.510949 | 15.048(393) |
| CD8+ | 7 | 6.91 | 7.26 | 141.646 | 141.176511 | 142.24667 | 1.070 (94) | 136.462451 | 151.510949 | 15.048(393) |
| H57+ | 4 | 6.93 | 6.78 | 149.608 | 148.759104 | 151.10435 | 2.345 (89) | 134.14498 | 154.469736 | 20.325(694) |
| H57+ | 6 | 6.93 | 9.46 | 23.458 | 23.217313 | 23.815913 | 0.599 (14) | 8.662195 | 37.539042 | 28.877(355) |
| TransB | 3 | 7.59 | 7.84 | 129.76 | 128.983837 | 130.017759 | 1.031 (30) | 107.488451 | 146.110756 | 38.622(604) |
Figure 5.
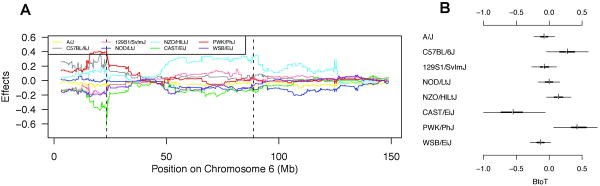
Founder strain effects at QTL for B/T ratio (log scale). Plot (A) shows the estimated dosage effect of each founder strain's haplotype at each locus along chr 6, with identified significant or suggestive QTL indicated by dashed vertical lines. Plot (B) gives Bayesian confidence intervals for effects within the QTL identified between 23.397-24.369Mb.
Low levels of CD23 are detected among the founders
Three of the CC founder strains, 129S1, CAST, and PWK had a consistently low-CD23 phenotype. Interestingly, in contrast to previous observations in the NZB and 129X1, we found that only two of the three CC founders with low-CD23 had relatively high serum-IgE (Figure 6c). To characterize further the overall genetic architecture of CD23 surface expression, we analyzed our available F1 data jointly with the founder data as an incomplete diallel.21. This showed that inheritance of CD23 MFI could be largely explained by an additive model of strain effects, wherein 129S1, CAST, and PWK contribute strong negative effects and WSB contributes a strong positive effect on the phenotype (Supplementary Figures 9.8 and 9.9). To confirm the observations made in our analysis of the diallel, we specifically analyzed CD23 MFI of F1 crosses of 129S1, CAST, and PWK with the B6. In agreement with the additive effects observed in the incomplete diallel, we found that F1 hybrids from crosses of the low-CD23 strains with B6 have a CD23 MFI which is significantly higher than the low-CD23 strains (Bonferroni's Post-Test p < 0.05) and significantly lower than the B6, suggesting that the alleles responsible for low-CD23 in 129S1, CAST, and PWK are semidominant in their effect.
Figure 6.
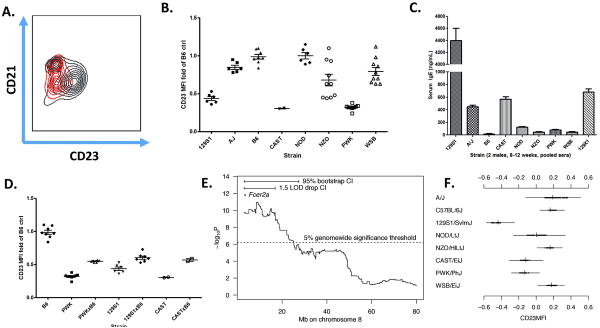
CD23 Mean Fluorescence Intensity in the Founder Strains and F1 crosses. A) B-cell subsets as defined by CD21 and CD23 in the B6 (gray) and PWK (red). Note that there is very little difference in CD21 expression between the two strains, while CD23 in the PWK is extremely reduced if not absent. B) CD23 surface expression in the founder strains. All MFI measurements are normalized to the B6 control in each experiment. Males and females are pooled. Note low CD23 levels in 129S1, CAST, and PWK. C) ELISA for serum IgE. Pooled sera were obtained from 8-12 week old males from each of the founder strains. Pooled serum from the 129X1 was also included as a positive control for high IgE. Error bars reflect assay precision. D) Inheritance of CD23 MFI in F1 crosses of the B6 with 129S1, CAST, and PWK. Reciprocal crosses, males and females are all pooled. E) Zoomed in region of genome scan for CD23 MFI indicating location of Fcer2a, confidence intervals, and genomewide significance threshold. F) Estimated strain effects for CD23MFI (log base 10 scale) at the Fcer2a locus.
Low levels of CD23 in pre-CC map to a QTL on chromosome 8
The ‘low-CD23’ phenotype was also observed in the pre-CC. This may be due to an allele common to the 129S1, CAST, and PWK founders: during analysis of the founders and the pre-CC, we found a surprising phenotype in B cells whereby CD23 surface expression appeared extremely reduced if not completely absent as measured by MFI (Figure 6a). CD23 is classically used together with CD21 to discriminate subsets of CD19 positive B-cells: Follicular B-cells (CD23hi, CD21mid), Marginal Zone B-cells (CD21hi, CD23lo) and Transitional B-cells (CD23lo, CD21lo). CD23 (Fcer2a) is the low affinity receptor for IgE. A low-CD23 phenotype has been described in the 129X1/SvJ and the NZB/BlN inbred strains which was associated with Hyper-IgE and enhanced parasite clearance17, 22. We determined that three of the CC founder strains, 129, CAST, and PWK have a low-CD23 phenotype (Figure 6b). Interestingly, in contrast to previous observations in the NZB and 129X1, we found that only 2 of the 3 CC founders with low-CD23 had relatively high serum-IgE (Figure 6c).
Previous studies of the low-CD23 phenotype in the NZB22 and 129X1 17 suggested that the phenotype was produced by a dominant negative mutation in CD23 (Fcer2a) in those strains. To understand better the genetic architecture of CD23 surface expression, we analyzed our available F1 data (Figure 6 d) jointly with the founder data as an incomplete diallel 21. We found that inheritance of CD23 MFI conformed well to an additive model of strain effects, wherein 129S1, CAST, and PWK had strong negative effects in the diallel. We found no evidence of heterosis, gender effects, or parent of origin effects (not shown) on the CD23 phenotype. Mapping indicated that the alleles responsible for low-CD23 in 129S1, CAST, and PWK are antimorphic and incompletely dominant.
Given that a low surface CD23 trait had been described in two other strains and associated with the coding region of CD23 itself (Fcer2a), we hypothesized the chromosomal region surrounding Fcer2a be significant in a linkage map17, 22. Indeed, mapping of CD23 MFI yielded a highly significant peak on the centromeric end of chromosome 8, the 1.5 LOD drop and bootstrap confidence intervals of which included Fcer2a (Figure 6e). Although there are many other genes (271-435, Table 2) in the major CD23 QTL, the afformentioned studies and the fact that Fcer2a codes for CD23 both made Fcer2a a strong candidate for further analysis. We estimated haplotype effects at all intervals across the chromosome, showing that 129S1, CAST, and PWK alleles in the region of Fcer2a appear to reduce CD23 MFI, consistent with observations made in the incomplete diallel (Figure 6f). Lastly, we visualized the inferred pattern of haplotypes at Fcer2a, which demonstrated an obvious pattern wherein 129S1, CAST and PWK alleles are present in mice with lower CD23 MFI and completely absent in mice with the highest CD23 MFI.
Coding mutations in Fcer2a potentially associated with low surface CD23: given the haplotype effects at the QTL and the strain effects in the incomplete diallel, we searched for mutations within the region Fcer2a in the 129S1, CAST, and PWK strains that might be associated with the low-CD23 phenotype using genomic sequence data for the founder strains of the CC from (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl) 23. We searched for single nucleotide polymorphisms (SNPs) and insertions/deletions present in the region Fcer2a in the 129S1, CAST, and/or PWK strains. This initial screen yielded five plausible SNPs inducing non-synonymous coding mutations in Fcer2a (Table 3). Four of the polymorphisms identified had been identified in the 129X1/SvJ15 and were all present in the closely related 129/SvImJ. We were able to exclude one polymorphism found in the 129X1/SvJ (K131E in the stalk region of CD23)15 since that mutation was also found to be present in the WSB/EiJ and the NZO/HlLtJ. We also found polymorphisms in the 3′ un-translated region of Fcer2a which were common to all three low-CD23 strains as well as the 129X1, but it has already been shown that Fcer2a mRNA expression in the 129X1 is not altered.17.
Table 3.
Coding polymorphisms in the gene Fcer2a. Conservation score ranges from 1 (highly conserved amino acid substitutions) to 0 (nonconserved). Merge scores rate the ability of the polymorphism to explain the association between CD23 MFI and the haplotypes at the Fcer2a locus, with positive scores indicating better support. The last column measures the strength of association (LogP) at the chr 8 QTL peak (originally 9.58) after controlling for potential effects of the Fcer2a polymorphism (see Methods and Results for details).
| A.A.# | Human | A/J | B6 | 129S1 | NOD | NZO | CAST | PWK | WSB | Protein Region | Conservation among founders (inc. human) | Merge Score | Conditional LogP at QTL peak |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 82 | A | A | A | T | A | A | A | A | A | Stalk | 0.809 (0.847) | -1.83 | 2.55 |
| 87 | S | V | V | A | V | V | A | A | V | Stalk | 0.648 (0.540) | 5.49 | 1.91 |
| 117 | G | G | G | G | G | G | E | E | G | Stalk | 0.648 (0.731) | -27.25 | 7.08 |
| 131 | - | K | K | E | K | E | E | E | E | Stalk | 0.673 (0.439) | -22.22 | 6.37 |
| 258 | V | S | S | L | S | S | L | L | S | Lectin | 0.531 (0.475) | 4.66 | 1.91 |
| 301 | G | D | D | N | D | D | N | D | D | Lectin | 0.710 (0.599) | -3.73 | 3.20 |
To help narrow further the list of plausible candidates within Fcer2a, we used three approaches: merge analysis 24, conditional association, and residue conservation25, 26. Our merge score (Table 3; see Methods for calculation) compares the ability of each Fcer2a coding polymorphism to explain CD23 MFI variation to the ability of the 8-haplotype mosaic in the region of Fcer2a to do the same. It gives a positive value when the coding polymorphism explains the association more succinctly than the haplotype mosaic, and a negative value when it explains less. It shows that, for example, whereas G117E and K131E explain much less than the haplotype mosaic, V87A and S258L explain more.
Conditional association tests to what extent the Fcer2a coding polymorphism can explain the association signal at the QTL peak at 15Mb, and in doing so helps to clarify the relationship between the CD23 MFI associations at Fcer2a and the QTL peak by testing whether they are independent or correlated. Specifically, the conditional LogP measures the strength of association at the QTL peak after controlling for a potential effect of the Fcer2a polymorphism. In Table 3 this shows that controlling for any of A82T, V87A, S258L and D301N drastically reduces the significance of the observed QTL. Although this is consistent with there being two separate QTL whose association signals are correlated in the tested set of 66 pre-CC lines, the small sample size of this experiment precludes a more thorough exploration of this hypothesis.
Discussion
The CC has been proposed as a powerful new tool for system genetics 5. As such, it is intended to provide a basis for experiments that aim to study traits under complex genetic control in ways that will generalize across, or highlight difference among, multiple genetic backgrounds. The CC is also the basis for the Diversity Outbred (DO) population27, a randomly mated population similar to heterogeneous stocks (e.g., 28), that shares its initial ancestry with the CC but then through extended outbreeding provides a non-replicable high resolution mapping resource that complements the replicable medium-resolution CC panel. Thus, phenotypes showing useful variation in the pre-CC are likely also to show useful variation in the DO.
The present study shows that the genetic diversity of the CC mice leads to phenotypic diversity in the immune system that is not only of potential value for future experiments, e.g., for the identification of reliably dysregulated mice, but also in itself for identifying genetic variants modulating immune function. Notably, the phenotypic diversity we have observed in the pre-CC includes not only that driven by genetic polymorphisms of known (or expected) effect, but also that driven by novel or as yet detected genetic variants.
We successfully mapped several immunophenotypic traits in the pre-CC using very few mice, a testament not only to the genetic and phenotypic diversity of the pre-CC panel, but also the randomized and balanced way in which alternative genetic combinations appear in that population. Nonetheless, these pre-CC experiments were subject to several limitations which will be remedied in future experiments using the completed CC strains. Principally, we were not able to take advantage of the reproducibility of Recombinant Inbred Lines (RIL), one of the key strengths of the CC. Residual heterozygosity in the pre-CC strains meant that it was not possible to obtain genetically identical mice from any one incipient strain 13. More importantly, the number of mice available for the pre-CC experiments was relatively small 13. Each strain was available for immunophenotyping only once, and significant coordination was required between disparate research groups to distribute tissues appropriately. In future experiments with the completed CC, analysis of multiple genetically identical mice would increase detection power by increasing apparent heritability of measured traits 29. Indeed, successful mapping of susceptibility to Aspergillus fumigatus infection has been done using replicates from 66 incipient CC lines from the Wellcome Trust Cohort 13, 30. Increased availability of mice will also permit selecting an appropriate number of strains for analysis and reanalyzing strains when sample processing errors occur. Thus, experiments using the completed CC will have greater detection power and will undoubtedly uncover more significant loci.
The QTL mapping of the MFI of CD23 is of interest. Previously it was suggested that CD23 expression levels regulated circulating IgE, and so was a major regulator of the IgE responses.17 The previous data suggested that a coding difference between 129 and BALB/c was the major cause of the difference, although a formal genetic analysis was not performed. Here we find a major QTL located over the structural gene Fcer2. Further work will be required to determine if the QTL we observed is mediated by the Fcer2 gene, another locus or an interaction.
We were unable to map some known loci for immunophenotypic traits we analyzed, particularly CD4/CD8 ratios. For example, several linkage studies of CD4/CD8 ratios in the mouse have pointed to significant regions on chromosome 17 28, 31. Several of those studies were done on lymphocyte populations in peripheral blood, and the correlation of CD4/CD8 ratios between spleen and blood is poor (secondary analysis of primary data available from phenome.jax.org, plots not shown here). However, work with reciprocal congenic strains with the B6 and NOD derived MHC (B6-H2g7 and NOD-H2b) has shown that high CD4 numbers in the lymph nodes were caused by a locus on Chr 17 outside of the MHC 32. Future mapping studies in the completed CC and in its complementary resources such as the DO are highly likely to map many additional loci not observed in the present study.
Methods
Mice
We collected phenotypes on three sets of mice: those belonging to founder lines of the CC (CC founders), F1 crosses of the CC founders, and the incipient Collaborative Cross (pre-CC).
The CC founder inbred lines are (short names in parentheses): 129S1/SvImJ (129S1), A/J (AJ), C57BL/6J (B6), NOD/ShiLtJ (NOD), NZO/HlLtJ (NZO), CAST/EiJ (CAST), PWK/PhJ (PWK), and WSB/EiJ (WSB). We obtained mice belonging to these lines from the Jackson Laboratory. In total, 112 founder mice were used for phenotyping, with these distributed among the strains as (number of mice in parentheses): AJ (6), B6 (70), 129 (6), NOD (5), NZO (5), CAST (2), PWK (9), and WSB (9). Of the 70 B6 mice, 61 were designated as controls for batch. A further 133 mice were generated from 31 distinct F1 crosses of (additional and unphenotyped) CC founders, and these F1s were also phenotyped. Founders and F1s used for analysis were bred and maintained in house at UNC-Chapel Hill.
The Collaborative Cross is a recombinant inbred (RI) panel derived from the CC founders. Each RI line arises from a distinct breeding funnel originating from a distinct 8-way cross of the original founder lines. Initiation of the CC breeding funnels, description of the CC strains, and animal housing are described elsewhere 10, 11, 13 The CC lines used in this study were initiated at The Oak Ridge National Laboratory (ORNL) and transferred to UNC-Chapel Hill at 9-13 weeks of age. The present work uses mice drawn from the incipient CC lines (pre-CC) at UNC, that is, mice from the CC breeding funnels before those funnels have reached the 20 generations required for standard inbreeding. We obtained spleens for one mouse from each of 66 pre-CC independent lines. As described in 13, the maturity of those lines ranged from generation G2:F5 (i.e., 4 generations of inbreeding after initial mixing) to G2:F12 (11 generations of inbreeding). All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at UNC-Chapel Hill.
Cell Preparation and Flow Cytometry
Founder strains, F1 offspring and selected pre-CC strains were euthanized and spleens were taken for analysis. One B6 mouse, obtained from The Jackson Laboratory, was incorporated into every group of samples as a control. Briefly, spleens were ground into suspension in cold R10 (RPMI supplemented with 10% v/v fetal bovine serum). Splenocytes were then pelleted and resuspended by flicking. One to two milliliters of cold ACK buffer (buffer) were added to the cell suspension for 45-60 seconds to lyse erythrocytes. Following lysis, ACK buffer was diluted with R10 to approximately 10 mL. Cells were repelleted, resuspended in R10 and counted with a hemocytometer. Finally, cells were pelleted once more and resuspended in FC blocking solution (2.4G2 hybridoma supernatant with 0.1% azide) prior to aliquoting into appropriate staining solutions.
Splenocytes were stained with a 9-color panel consisting of CD23 (clone B3B4), CD25 (clone PC-61.5), CD11c (clone N418), CD8 (clone 63-6.7) PerCP, CD19 (clone 6D5), CD21 (clone 7E9), CD4 (clone GK1.5), CD11b (clone M1/70), and TCR-Beta (clone H57-597). The standard panel with the fluorophores used is shown in Supplemental Table 1. Antibodies were used at appropriate dilutions as determined by titration and all appropriate single color and fluorophore-minus-one (FMO) controls were included with the samples. Cells were stained in FACS wash (PBS supplemented with 2% v/v fetal bovine serum and 0.1% sodium azide) and cells were fixed with 2% formaldehyde in PBS following the staining procedure. Samples were run on a Dako CyAn using the standard 9-color instrument configuration and data were collected using Summit 4.3 (Dako). Data were analyzed using FlowJo 7.6 (TreeStar) and numerical data were exported to spreadsheets for analysis.
Quantitation of Serum IgE. We measured serum IgE of the eight founder strains using a pretitrated kit (Biolegend). Pooled serum was obtained from Jackson Laboratories (Bar Harbor, Maine). Sera were pooled from two 8-12 week old male mice from each of the eight founder strains and 129X1/SvJ as a control. Samples were frozen and shipped on dry ice prior to analysis.
Statistical analysis of trait variation in the CC founders and F1 hybrids
To assess the extent to which phenotypes varied among mice from the CC founders we used a linear mixed model, testing for the effect of strain on the phenotype while controlling for the effect of sex and batch. Prior to analysis, phenotype values were transformed to approximate residual normality in order to match the assumptions of the statistical models used (Table 1). To maintain interpretability of effects, we achieved this by transforming proportions (eg, CD4+) to their logit, ie, to log[y/(1-y)]; applying base 10 logarithms (log10) to ratio quantities, which would otherwise be highly left-skewed; and using the Bliss-corrected inverse normal transformation (INT) for quantities whose distributions were otherwise problematic (eg, long-tailed but unsuitable for logit). Strain and sex were modeled as fixed effects, while batch was modeled as a random effect. Sex was uncertain for 88 mice, including the 61 B6 used as controls (although incomplete records strongly suggested these were male); for such mice we set the sex covariate to an intermediate value. The pre-CC mice were all male. Significance testing for the effect of strain was performed using a partial F-test. To estimate the significance of differences between specific strains, we used the Tukey-Kramer test 33. Mixed modeling of strain effects was performed in 34 using the packages nlme 35 and multcomp 36. Recognizing that the F1 hybrid data when considered jointly with the CC founder data corresponds to an incomplete diallel 37, we estimated effects of genetic additivity, heterosis and parent of origin on each phenotype using the R package BayesDiallel 21, where batch effects were included as above.
Genotyping
Genotyping of the pre-CC mice and the SNP arrays used are described thoroughly elsewhere. 13, 38 The mice used in this study were described in the preivous reports. 13, 38 In short, pre-CC mice were genotyped using test arrays developed as an intermediate step toward the development of the Mouse Diversity Array.38 A total of roughly 180,000 SNP assays in each array performed well and targeted loci which were polymorphic among the founder strains. The number of available SNP markers greatly exceeded the number of recombination events 13 allowing accurate inference of founder haplotypes. Genotyping data for many pre-CC strains are available for public access at the CC status website (http://csbio.unc.edu/CCstatus/index.py).
Haplotype reconstruction
Haplotype descent was inferred using HAPPY 39,, which implements a hidden Markov model (HMM) that calculates for each individual at each locus along the genome the relative proportion of genetic material in that region descended from any given pair of founder strains. Specifically, define locus m as the genomic interval between marker m and m + 1, and define pi,m(s,t) as the proportion of that interval in mouse i containing the s and t haplotypes, such that if pi,m(s,t)=1 and s=t the region is entirely homozygous for strain s. HAPPY computes at locus m for each mouse i, the 36 proportions: pi,m(AJ,AJ), pi,m(AJ,B6), …, pi,m(WSB,WSB). Because these proportions are calculated as averaged probabilities over the interval, we hereafter refer to them as “HAPPY probabilities”. Similar to ref 13 we mitigate the effects of undetected genotyping error on the reliability of the haplotype reconstruction, we used modified haplotype priors that allowed a conservative genotyping error rate of 0.01.
QTL mapping
We performed genome wide linkage disequilibrium (LD) mapping of QTL in the pre-CC by testing association at each location along the genome and calculated the statistical association between the inferred assignment of founder haplotypes and the phenotype of interest. We did this using BAGPIPE (http://valdarlab.unc.edu/software/bagpipe)40, as has been used in a number of other pre-CC studies.13, 15, 16 BAGPIPE performs an ANOVA-like test for how well the haplotypes at each locus explain the variation in phenotype. Moreover, to moderate the potentially disruptive effect of residual heterozygosity on additive association testing in such a small sample, we used observation weighting based on homozygote probabilities. These are described below.
The HAPPY probabilities described earlier (see Haplotype Reconstruction) are used to construct two derived quantities for mouse i at locus m. First, the “haplotype dosage” is the estimated number of haplotypes ai,m(s) for each haplotype s, calculated as . Second, the “homozygote probability” is the probability hi,m that the locus is fully inbred, calculated as . We then model the effect of a putative QTL at locus m on the transformed phenotype yi collected in batch b for mouse i in a linear model:
| Eq 1 |
where is the QTL effect, with βAJ,…, βWSB being the eight haplotype effects (or “additive strain effects”) that contribute to the effect of that QTL, c is a constant intercept term. Bb is the batch effect modeled as a standard random effect for non-MFI phenotypes (as in, eg, ref 41 but as a “plug-in” random effect for MFI phenotypes (see below), and εi is a mouse-specific noise term. The mouse specific noise is modeled as εi ∼ N(0,σ2/hi,m), that is, as having come from a normal distribution with variance scaled according to homozygosity. This has a filtering effect, acting to down-weight observations from mice to the extent that are considered heterozygous at the locus, and improves our power to detect additive-only genetic effects in the pre-CC. The regression in Eq 1 was fitted to each locus in the genome in turn, and in each case its goodness of fit was compared using a likelihood ratio test with a fit to a null model, namely Eq 1 with the QTL effect removed (ie, setting Qi,m = 0).
Batch effects were modeled differently for MFI and non-MFI phenotypes. In non-MFI phenotypes, batch effects are minimized by construction: these phenotypes are based on relative assessments of intensity through a gating procedure. In MFI phenotypes, day-to-day calibration --- in our case, using B6 control mice --- is required, such that the batch effect defines the day-specific baseline for the phenotype. In order to obtain stable baseline estimates for MFI when often only one B6 control was measured per batch, we used a mixed model procedure. Specifically, we first fit a linear mixed model to the log MFI of all mice phenotyped (founders, F1s, pre-CCs and B6 controls) with fixed effects of sex and random effects for batch and strain. This allowed us to obtained very stable shrinkage estimates, known in agricultural statistics as BLUPs 42, for the effect Bb of each batch b using data from ∼400 animals. Those Bb estimates, now informed not only by pre-CC mice from the same batch but also the B6 controls, were then substituted as fixed “plug-in” values into Eq 1, and mapping proceeded as above.
Genome-wide significance thresholds were determined by parametric bootstrap for non-MFI phenotypes (as in REF 41, 43) and by permutation for MFI phenotypes (as in 13). Confidence intervals for QTL location were estimated using the 1.5 LOD-drop method 44 and the more conservative positional bootstrap procedure (e.g. 45, 46).
Estimation of strain effects at QTL
Our mapping procedure tests the association between the phenotype under study and the inheritance pattern of haplotypes at the QTL. The genetic architecture at a given QTL is therefore most appropriately described in terms of its haplotype effects, i.e., the relative substitution effect of each founder strain's genetic material at that locus. To estimate haplotype effects at identified QTL, we used a mixed model similar to the Bayesian multiple imputation procedure in 30 , but incorporating additional parameters that allowed for residual heterozygosity and batch effects as described above.
Merge analysis at coding polymorphisms
Although mapping using an 8-haplotype model of association is powerful for detecting QTL, it is most plausible that the number of functional alleles giving rise to a detected signal is fewer than eight, with several strain haplotypes possessing the same allele. We used merge analysis 24 to measure the statistical support for a coding polymorphism affecting a phenotype in a genomic region whose haplotypes already show a strong association. Merge analysis helps narrow down the list of potential causal alleles by asking how well the observed haplotype association can be explained by a more parsimonious grouping of those haplotypes, specifically a grouping that corresponds to a particular coding SNP.
Our merge analysis proceeds as follows. Consider a coding SNP w with alleles v and V occurring within the locus m (ie, between genotyped marker m and m+1), and suppose this SNP has been genotyped in the 8 CC founder strains but not in the 66 pre-CC mice. Define allele count G(s,w) to equal 2 if strain s is known to have only V's at SNP w, 0 if strain s is known to have only v's, and 1 if it is a heterozygote. The estimated “allele dosage” at SNP w for mouse i can be calculated from the HAPPY probabilities, as . If SNP w was entirely responsible for the QTL effect at locus m, then we should be able to restate Eq 1 in terms of only the alleles at that SNP. That is, we could replace the 8 haplotype dosages, ai,m(AJ), …, ai,m(WSB) with the single allele dosage gi,m(w), so that Qi,m = βwgi,m(w), and this latter “merged” model should provide as good an explanation of how the data arose but with fewer parameters. Using this principle, to evaluate the candidacy of a SNP w within its locus m we compare the goodness of fit of the merged model at w with that of the haplotype model at m. Goodness of fit in our case was judged using the Bayes Information Criterion (BIC) 47, which offsets the deviance of the model (ie, its lack of fit) by its parsimony (how few parameters were needed). For each SNP w, this leads to a “merge score” ΔBIC(w,m) = BIC(m)-BIC(v,m). Positive values of this merge score indicate statistical support for the coding polymorphism; specifically, that the SNP at w more concisely explains the QTL effect on the phenotype than does the haplotype-based model at that locus.
Sequence Analysis
Analyses for specific polymorphisms in selected regions were done using gene information in MGI and publicly available genome sequences of the CC founder strains available from the Sanger Mouse Genomes Project (http://www.sanger.ac.uk/resources/mouse/genomes). In particular we made extensive use of the SNP/Indel viewer available for the Sanger Mouse Genomes Project (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl).
Supplementary Material
{kind=link}
Figure S1. Genome scans of pre-CC mice with no QTL detected at the 0.05 genomewide significance level. The x-axis plots location in the genome, y-axis gives statistical significance of association with the phenotype. Dashed horizontal lines indicate genomewide significance at the 0.05, 0.1, and 0.2 level.
Acknowledgments
The authors thank Dr. Ellen Young, Cindy Hensley, Shaun Steele for their substantial assistance in data collection. The authors acknowledge NIH grants GM070683 (genotyping), P50 MH0903380 (partial support for AB, FPMV, WV), GM104125 (partial support for AB, WV), F32 GM090667 (DLA) and the Lineberger Cancer Research Fund (JAF).
References
- 1.Conley ME. Genetics of primary immunodeficiency diseases. Rev Immunogenet. 2000;2(2):231–42. [PubMed] [Google Scholar]
- 2.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242(1):10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32(4):457–67. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Flint J, Eskin E. Genome-wide association studies in mice. Nat Rev Genet. 2012;13(11):807–17. doi: 10.1038/nrg3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Consortium CC. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012;190(2):389–401. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36(11):1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 7.Threadgill DW, Hunter KW, Williams RW. Genetic dissection of complex and quantitative traits: from fantasy to reality via a community effort. Mamm Genome. 2002;13(4):175–8. doi: 10.1007/s00335-001-4001-Y. [DOI] [PubMed] [Google Scholar]
- 8.Broman KW. The genomes of recombinant inbred lines. Genetics. 2005;169(2):1133–46. doi: 10.1534/genetics.104.035212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome. 2007;18(6-7):473–81. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesler EJ, Miller DR, Branstetter LR, Galloway LD, Jackson BL, Philip VM, et al. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome. 2008;19(6):382–9. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iraqi FA, Churchill G, Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Wellcome Trust cohort. Mamm Genome. 2008;19(6):379–81. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 12.Morahan G, Balmer L, Monley D. Establishment of “The Gene Mine”: a resource for rapid identification of complex trait genes. Mamm Genome. 2008;19(6):390–3. doi: 10.1007/s00335-008-9134-9. [DOI] [PubMed] [Google Scholar]
- 13.Aylor DL, Valdar W, Foulds-Mathes W, Buus RJ, Verdugo RA, Baric RS, et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011;21(8):1213–22. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welsh CE, Miller DR, Manly KF, Wang J, McMillan L, Morahan G, et al. Status and access to the Collaborative Cross population. Mamm Genome. 2012;23(9-10):706–12. doi: 10.1007/s00335-012-9410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelada SN, Aylor DL, Peck BC, Ryan JF, Tavarez U, Buus RJ, et al. Genetic analysis of hematological parameters in incipient lines of the collaborative cross. G3 (Bethesda) 2012;2(2):157–65. doi: 10.1534/g3.111.001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013;9(2):e1003196. doi: 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ford JW, Sturgill JL, Conrad DH. 129/SvJ mice have mutated CD23 and hyper IgE. Cell Immunol. 2009;254(2):124–34. doi: 10.1016/j.cellimm.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med. 2011;208(10):2033–42. doi: 10.1084/jem.20110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich Syndrome: Immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology. 2009;214(9-10):778–90. doi: 10.1016/j.imbio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kliche S, Worbs T, Wang X, Degen J, Patzak I, Meineke B, et al. CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood. 2012;119(3):777–85. doi: 10.1182/blood-2011-06-362269. [DOI] [PubMed] [Google Scholar]
- 21.Lenarcic AB, Svenson KL, Churchill GA, Valdar W. A general Bayesian approach to analyzing diallel crosses of inbred strains. Genetics. 2012;190(2):413–35. doi: 10.1534/genetics.111.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis G, Rapsomaniki E, Bouriez T, Crockford T, Ferry H, Rigby R, et al. Hyper IgE in New Zealand black mice due to a dominant-negative CD23 mutation. Immunogenetics. 2004;56(8):564–71. doi: 10.1007/s00251-004-0728-4. [DOI] [PubMed] [Google Scholar]
- 23.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289–94. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yalcin B, Flint J, Mott R. Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics. 2005;171(2):673–81. doi: 10.1534/genetics.104.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valdar WS. Scoring residue conservation. Proteins. 2002;48(2):227–41. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 26.Binkley J, Karra K, Kirby A, Hosobuchi M, Stone EA, Sidow A. ProPhylER: a curated online resource for protein function and structure based on evolutionary constraint analyses. Genome Res. 2010;20(1):142–54. doi: 10.1101/gr.097121.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, et al. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics. 2012;190(2):437–47. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valdar W, Solberg LC, Gauguier D, Burnett S, Klenerman P, Cookson WO, et al. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet. 2006;38(8):879–87. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- 29.Belknap JK. Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav Genet. 1998;28(1):29–38. doi: 10.1023/a:1021404714631. [DOI] [PubMed] [Google Scholar]
- 30.Durrant C, Tayem H, Yalcin B, Cleak J, Goodstadt L, de Villena FP, et al. Collaborative Cross mice and their power to map host susceptibility to Aspergillus fumigatus infection. Genome Res. 2011;21(8):1239–48. doi: 10.1101/gr.118786.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yalcin B, Nicod J, Bhomra A, Davidson S, Cleak J, Farinelli L, et al. Commercially available outbred mice for genome-wide association studies. PLoS Genet. 2010;6(9) doi: 10.1371/journal.pgen.1001085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koarada S, Wu Y, Yim YS, Wakeland EW, Ridgway WM. Nonobese diabetic CD4 lymphocytosis maps outside the MHC locus on chromosome 17. Immunogenetics. 2004;56(5):333–7. doi: 10.1007/s00251-004-0702-1. [DOI] [PubMed] [Google Scholar]
- 33.Hsu J. Multiple comparisons: Theory and Methods. Chapman and Hall/CRC; 1996. [Google Scholar]
- 34.Team RD. A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation of Statistical Computing; 2012. [Google Scholar]
- 35.Pinheiro J, Bates D. Mixed-Effects Models in S and S-PLUS. Springer; 2009. [Google Scholar]
- 36.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50(3):346–63. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 37.Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sinauer; Sunderland, Mass: 1998. [Google Scholar]
- 38.Yang H, Ding Y, Hutchins LN, Szatkiewicz J, Bell TA, Paigen BJ, et al. A customized and versatile high-density genotyping array for the mouse. Nat Methods. 2009;6(9):663–6. doi: 10.1038/nmeth.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mott R, Talbot CJ, Turri MG, Collins AC, Flint J. A method for fine mapping quantitative trait loci in outbred animal stocks. Proc Natl Acad Sci U S A. 2000;97(23):12649–54. doi: 10.1073/pnas.230304397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valdar W, Holmes CC, Mott R, Flint J. Mapping in structured populations by resample model averaging. Genetics. 2009;182(4):1263–77. doi: 10.1534/genetics.109.100727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnsen AK, Valdar W, Golden L, Ortiz-Lopez A, Hitzemann R, Flint J, et al. Genome-wide and species-wide dissection of the genetics of arthritis severity in heterogeneous stock mice. Arthritis Rheum. 2011;63(9):2630–40. doi: 10.1002/art.30425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson G. That BLUP is a good thing: the estimation of random effects. Statistical Science. 1991;6(1):15–51. [Google Scholar]
- 43.Solberg Woods LC, Holl KL, Oreper D, Xie Y, Tsaih SW, Valdar W. Fine-mapping diabetes-related traits, including insulin resistance, in heterogeneous stock rats. Physiol Genomics. 2012;44(21):1013–26. doi: 10.1152/physiolgenomics.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupuis J, Siegmund D. Statistical methods for mapping quantitative trait loci from a dense set of markers. Genetics. 1999;151(1):373–86. doi: 10.1093/genetics/151.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solberg Woods LC, Holl K, Tschannen M, Valdar W. Fine-mapping a locus for glucose tolerance using heterogeneous stock rats. Physiol Genomics. 2010;41(1):102–8. doi: 10.1152/physiolgenomics.00178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Visscher PM, Thompson R, Haley CS. Confidence intervals in QTL mapping by bootstrapping. Genetics. 1996;143(2):1013–20. doi: 10.1093/genetics/143.2.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwarz G. Estimating Dimension of a Model. Annals of Statistics. 1978;6(2):461–464. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genome scans of pre-CC mice with no QTL detected at the 0.05 genomewide significance level. The x-axis plots location in the genome, y-axis gives statistical significance of association with the phenotype. Dashed horizontal lines indicate genomewide significance at the 0.05, 0.1, and 0.2 level.