

Abstract
Cerebral amyloid angiopathy (CAA) results from deposition of β-amyloid in the media and adventitia of small arteries and capillaries of the leptomeninges and cerebral cortex and is a major cause of lobar intracerebral hemorrhage and cognitive impairment in the elderly. CAA is associated with a high prevalence of magnetic resonance imaging markers of small vessel disease, including cerebral microbleeds and white matter hyperintensities. Although advanced CAA is present in approximately ¼ of brains with Alzheimer disease (AD), fewer than half of CAA cases meet pathologic criteria for AD. This review will discuss the pathophysiology of CAA and focus on new imaging modalities and laboratory biomarkers that may aid in the clinical diagnosis of individuals with the disease.
Cerebral amyloid angiopathy (CAA) is a major cause of lobar intracerebral hemorrhage (ICH) and cognitive impairment in the elderly and is associated with a high prevalence of markers of small vessel disease, including white matter hyperintensities and cerebral microbleeds.1
CAA is the most common cause of lobar intracerebral hemorrhage (ICH) in the elderly and results from cerebrovascular deposition of β-amyloid protein.2,3 CAA is present in nearly all brains with Alzheimer disease (AD),4 and advanced CAA is present in approximately 25% of AD brains.5 This may suggest a common β-amyloid– based pathogenesis for these diseases. However, despite the close molecular relationship between the 2 diseases, CAA remains a clinically distinct entity from AD. Fewer than 50% of CAA cases meet the pathologic criteria for AD.2,3 Furthermore, >75% of patients with AD have only mild or no CAA.5
This review will discuss the pathophysiology of CAA and focus on new imaging modalities and laboratory biomarkers that may aid in the clinical diagnosis of individuals with the disease. Future areas of research will also be discussed.
Vascular Pathophysiology and CAA-Related Brain Injury
Sporadic CAA is characterized by deposition of β-amyloid in the media and adventitia of small arteries and capillaries of the leptomeninges and cerebral cortex. The occipital regions are preferentially affected for unclear reasons. In contrast to Aβ deposition in AD, a substantial proportion of Aβ in vascular deposits is the shorter Aβ40 species.6 Accelerated vascular deposition of Aβ may occur through transcriptional regulation of the lipoprotein receptor LRP in vascular smooth muscle due to overexpression of the transcription factors serum response factor (SRF) and myocardin.7 Additionally, SRF and myocardin may also regulate contractile proteins in vascular smooth muscle cells, thus altering normal vessel physiology.8
Although the exact source of vascular amyloid has not been elucidated, it has been suggested to be predominantly generated by neurons and subsequently deposited in the vessel wall.9 Transgenic mouse models suggest that amyloid expressed in neurons can generate CAA,6,10 with possible additional contribution from ineffective transport of Aβ out of the central nervous system.7,11–13 An alternative or complementary mechanism is the possible role of peripheral Aβ in the development of CAA and Aβ-related brain pathology, as recently highlighted in a transgenic mouse model.14 There is some evidence to suggest that the liver may be a source of Aβ.15 Indeed, Aβ in circulating plasma may be an important precursor pool for brain Aβ, as it has been shown to cross the blood–brain barrier in a variety of animal models.16–22
Pathological examination of blood vessels in both sporadic and familial CAA show loss of smooth muscle cells, vessel wall thickening, lumenal narrowing, concentric splitting of the vessel wall, microaneurysm formation, and perivascular microhemorrhage.3,23–28 These results have been further extended to animal models of CAA.6,29–33 Even in mildly affected transgenic mice overexpressing mutant forms of amyloid precursor protein, Aβ deposition has been shown to affect resting vessel diameter27,34 and influence vessel dilatation in response to physiologic or pharmacologic stimuli.30,35–37 There appears to be decreased cortical vascular reactivity to carbon dioxide and whisker stimulation proportional to the severity of vascular amyloid.38
In line with these animal studies, there is evidence to suggest that Aβ vessel pathology may have similar effects in patients with CAA. In Dutch-type hereditary CAA, the presence of dementia is be predicted by the amount of severely stenotic amyloid-laden vessels (as opposed to the severity of AD pathology).39 More recent evidence has further suggested that amyloid deposition may also be associated with capillary vessel occlusion.40 Furthermore, decreased vascular reactivity in response to visual stimulation has recently been found in a small cohort of CAA subjects. This potentially reflects the occipital predilection of the disease.41
CAA-related impairments of perfusion may be responsible for the subcortical white matter lesions and tissue microstructural changes seen in the disease.42–45 Studies have suggested that advanced CAA is associated with a large burden of white matter lesions compared to healthy elders44 or patients with AD alone.45 Furthermore, damage to white matter in CAA is associated with cognitive impairment independent of the effects of the brain hemorrhage.42,43 Although CAA-related white matter hyperintensities preferentially affect the same periventricular regions affected by hypertensive small vessel disease,46 there is some suggestion that at least a subgroup of patients with CAA demonstrate predominantly posterior white matter involvement.47
Pathologic studies suggest that cortical microinfarctions are common in CAA, with reported frequencies ranging from 37% to nearly 100%.48–53 These microinfarctions are frequently multiple and are located in the cortical ribbon or underlying subcortical white matter. These microinfarctions may be related to impaired cerebral blood flow regulation in CAA38,41 due to smooth muscle degeneration and capillary occlusion.40 Recent studies have demonstrated that small diffusion-weighted imaging (DWI)-hyperintense lesions suggestive of subacute ischemic infarction are not infrequent in patients with CAA, occurring in approximately 15% of these patients.54,55 Their presence appears to be unrelated to conventional vascular risk factors, but instead is associated with the number of hemorrhagic lesions on gradient echo (GRE) magnetic resonance imaging (MRI), a marker of CAA severity.54 These lesions appear to be clinically silent events that occur as part of the ongoing pathogenesis of CAA. The signal characteristics, size, and location of these DWI-positive lesions suggest they may represent the neuroimaging correlates of the neuropathologic infarctions described in association with CAA.48–52 Given the transient appearance of DWI-positive signal after stroke, the finding of these DWI-positive lesions in 15% of subjects suggests these small infarctions may occur at a very high frequency. Based on this, and assuming a 10-day postinfarction period where diffusion changes remain visually detectable on DWI,56 their estimated annual prevalence would be approximately 8 new infarctions per person-year.54 This estimate is strikingly high relative to the estimated incidence of new microbleeds (~1.4 per year) or symptomatic ICH (~0.14 per year) calculated from other cohorts with advanced CAA.57 This suggests that the lifetime burden of ischemic infarction in advanced CAA could be substantial.
The genetic abnormalities underlying sporadic CAA have not been fully elucidated, although several inherited familial forms of CAA have been described.58 The only specific genetic risk factor consistently identified for the sporadic disease has been the apolipoprotein E (APOE) genotype as a risk for CAA-related ICH.59–61 In a population- based study, the presence of either APOE ε2 or ε4 alleles increased the risk of lobar ICH (odds ratio, 2.3; 95% confidence interval, 1.2–4.4).59 Furthermore, the presence of the ε2 or ε4 alleles of the apolipoprotein E gene is associated with an increased risk of recurrent lobar ICH (28% cumulative recurrence rate at 2 years compared to 10% in lobar ICH patients without either allele).62 The APOE ε2 and ε4 alleles have also been associated with an earlier age of onset of lobar hemorrhage. 63,64 Pathological studies have demonstrated that the ε2 allele may promote steps in CAA-related vessel breakdown including wall splitting, microhemorrhage, and fibrinoid necrosis,63,65 whereas APOE ε4 is associated with a dose-dependent increase in the amount of vascular amyloid.66–68 In contrast, in AD, APOE ε4, but not APOE ε2, is an established risk factor for late onset disease.69,70 Individuals with the APOE ε4 allele appear to have earlier onset and more rapid progression of AD-associated pathology.69,71 A current hypothesis suggests APOE ε4 may increase β-amyloid aggregation, impair β-amyloid clearance, or both.71
Cerebral Microbleeds and Significance in Sporadic CAA
Cerebral microbleeds were first described after the clinical use of GRE or T2*-weighted MRI.72–74 Old and recent cerebral hemorrhages can be detected with high sensitivity using this technique.73,74 The hypointense signal on GRE sequences is caused by hemosiderin, a blood breakdown product that causes magnetic susceptibility-induced dephasing, leading to T2* signal loss. The appearance of microbleeds on GRE sequences is larger than the actual tissue lesions because of the so-called blooming effect of the magnetic resonance signal at the border of these lesions.75,76 GRE MRI can detect millimeter-sized paramagnetic blood products (including hemosiderin) in brain parenchyma.77 As hemosiderin remains in macrophages for many years after hemorrhage, 78,79 GRE sequences allow for reliable assessment of an individual’s hemorrhagic burden over time. Furthermore, more recent technical advances in MRI software and hardware have yielded significant improvements in sensitivity, which have lead to increased detection of microbleeds in different populations.80–82 For example, novel techniques such as susceptibility-weighted imaging have considerably increased microbleed detection rates.57,74,80,81 Slice thickness and magnetic field strength may also influence detection rates.81 Older studies employing GRE, larger slice thickness, or lower magnetic field strength may have failed to detect microbleeds that would have been visualized with these higher resolution techniques. Microbleed detection does not appear to vary greatly based on choice of precise size parameters.83
Lobar microbleeds in CAA are likely caused by vessel fragility and rupture due to the deposition of amyloid within the media and adventitia of small- to medium-sized cerebral arteries3 and have been extensively studied in CAA.57,78 A set of validated criteria (termed the Boston criteria) have been established to diagnose CAA during life.84 The presence of multiple, strictly lobar hemorrhages (including microbleeds) detected by GRE MRI sequences has been shown to be highly specific for severe CAA in elderly patients with no other definite (Fig 1) cause of ICH, such as trauma, ischemic stroke, tumor, coagulopathy, or excessive anticoagulation (termed probable CAA-related ICH).84,85
FIGURE 1.
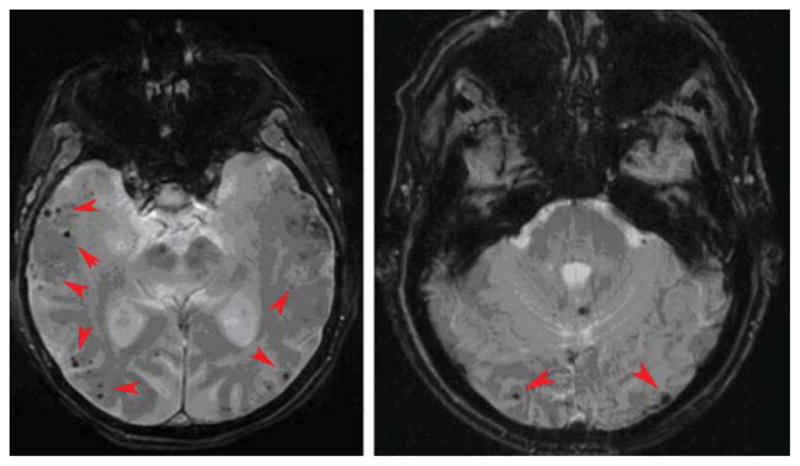
Two examples of patients with probable cerebral amyloid angiopathy. Magnetic resonance imaging demonstrates multiple strictly lobar microbleeds (red arrowheads).
Similar to the distribution of CAA pathology86 and CAA-related lobar macrohemorrhages,87,88 the distribution of microbleeds in CAA seems to show a posterior cortical predominance.89 In a study of patients with probable CAA, microbleeds occurred more frequently in the temporal and occipital lobes compared to other hemispheric regions. Additionally, lesions tended to cluster in the same lobe in subjects with multiple lesions.89
Microbleeds located in the lobar regions in CAA have been shown to be related to disease progression, recurrent ICH, and CAA-related impairment.57,85 In elderly patients (≥55 years old) presenting with lobar ICH, microbleeds appear >2× more frequently than macrohemorrhages.57 Among patients who underwent an MRI 16-months later, 50% experienced new, frequently multiple microbleeds. Large number of microbleeds at baseline and APOE ε2 or ε4 genotype were the only predictors of new microbleeds. Both the number of hemorrhages at baseline and the number of new microbleeds at follow-up increased the risk of recurrent hemorrhage (3-year cumulative risk, 14%, 17%, 38%, and 51% in subjects with 1, 2, 3–5, or ≥6 baseline hemorrhages, respectively). The distribution of new microbleeds at follow-up has been correlated with the distribution of baseline microbleeds.89 In those who experienced recurrent lobar ICH, the location of hematoma was positively associated with the distribution of baseline hemorrhages (including microbleeds). The number of baseline hemorrhages was also associated with increased incidence of cognitive impairment, functional dependence, or death at follow-up (mean follow-up, 27.9 months; hazard ratio, 1.9).57
The precise relationship between microbleeds and macrobleeds in CAA remains an active area of investigation. A recent study has suggested that CAA subjects with very high (>50) microbleed counts have increased vessel wall thickness compared to those with relatively few (<3) microbleeds.90 This suggests that increased wall thickness may predispose vessels to microbleeding when they rupture. In support of this hypothesis, severe wall thickening occurs in Iowa-type hereditary CAA, which is characterized by multiple microbleeds without symptomatic hemorrhage.23 Although increased vessel wall thickness and luminal narrowing in patients with CAA is a well-described phenomenon,24,91 little is known regarding the factors that determine degree of vessel wall thickness and how this relates to clinical impairment in the disease.27
CAA and AD
Neuropathological studies suggest that vascular and parenchymal Aβ deposits can occur either relatively independently of each other, or can overlap. The characteristics features of cerebrovascular and parenchymal Aβ deposition are summarized in Table 1. Of those individuals who die of CAA-related hemorrhage, approximately 50% meet AD criteria; approximately 25% of patients with AD also have severe CAA.4,5 Neuroimaging evidence further supports these neuropathological findings. Particularly striking is the recent observation of lobar microbleeds— a hallmark feature of CAA—in ⅕ or more of patients diagnosed with AD.92–96 Microbleed prevalence has been reported to range from 15 to 32% of AD patients presenting to memory disorders clinics (Table 2). Evidence suggests that both prevalence and number of microbleeds in subjects with AD93 are significantly higher than those reported in healthy populations.97,98 Furthermore, microbleeds are considerably more prevalent in AD compared to many other causes of dementia, such as frontotemporal dementia, corticobasal degeneration, dementia with Lewy bodies, and progressive supranuclear palsy.93
TABLE 1.
Comparison of Features of Cerebrovascular versus Parenchymal Senile Plaque Amyloid Deposition
| Feature | Cerebrovascular Amyloid Deposition | Senile Plaque Amyloid Deposition |
|---|---|---|
| Predominant Aβ type | Aβ40 | Aβ42 (particularly in diffuse plaques) |
| Location of Aβ deposition | Relative occipital lobe predominance | Frontal, parietal, temporal lobes |
| APOE allele risk factors | APOE ε4 (for amyloid deposition) and APOE ε2 (for vessel breakdown) | APOE ε4 |
| Inflammatory subtype with reversible white matter hyperintensities | Occurs spontaneously as CAA-related inflammation | May occur iatrogenically as a result of amyloid immunotherapy or other candidate treatments targeting amyloid |
| Cerebral microbleeds | Lobar predominant, particularly occipital | Not associated with senile plaques |
| Location of white matter disease | Equal distribution between anterior and posterior subcortical regions (subgroup may have posterior-dominant white matter disease) | Equal distribution between anterior and posterior subcortical regions, but less extensive than advanced CAA |
See text for more details discussion and details.
APOE = apolipoprotein E; CAA = cerebral amyloid angiopathy.
TABLE 2.
Microbleeds in Alzheimer Disease
The presence of CAA in patients with AD may also have important clinical relevance. This possibility is supported by clinical–pathological studies showing independent contributions of small vessel disease99,100 and CAA in particular101,102 to impairment in AD during life. This suggests that vascular brain injury acts additively or synergistically with concomitant AD pathology to produce more severe cognitive dysfunction than either process alone. This interpretation is supported by extensive clinical–pathologic data indicating that subjects with both vascular disease and AD pathology show either more severe cognitive impairment during life than those with pure AD100,103,104 or require less severe AD pathology to produce the same amount of cognitive impairment. 105–107 Most of the vascular lesions described in these studies were lacunar infarcts or microinfarcts rather than large territorial infarctions, supporting the importance of small vascular lesions also noted in population-based clinical–radiographic studies.108,109 The possibility of synergistic interaction between AD and microvascular pathology is particularly relevant to the potential cognitive effects of CAA, which occurs preferentially in conjunction with AD pathology.4,5,86
CAA may be the basis of the inflammatory response that halted a phase IIa trial of active Aβ42 immunization in AD.110 Both autopsied cases111,112 of subjects who died from this experimental treatment demonstrated advanced CAA and perivascular inflammation similar to spontaneous CAA-related inflammation.113 Further support for the possible role of CAA as trigger for the vaccine-associated reaction comes from the clinical, neuroimaging, and neuropathologic similarities between this syndrome and a spontaneous form of CAA-associated vascular inflammation.113,114 Most recently, MRI changes similar to those in CAA-associated vascular inflammation have been reported in early trials of passive anti-Aβ immunization in 3 subjects and associated cognitive changes in 1,115 suggesting that CAA remains an important obstacle even for passive immunotherapy and highlighting the potential importance of identifying AD subjects with accompanying CAA.
CAA in the General Population
Several recent studies in population-based cohorts have suggested that lobar cerebral microbleeds are common in healthy elderly individuals.82,116 For example, Vernooij et al found that microbleeds were present in >30% of individuals aged >70 years,82 the majority with a strictly lobar microbleed distribution. Individuals with the APOE ε4 allele were more likely to have lobar microbleeds. Although further studies are necessary, these data may suggest that there may be a large number of asymptomatic elders with CAA in the general population and raise the eventual possibility of identifying these individuals early in their clinical course, before they experience a devastating lobar ICH or cognitive impairment.
The presence of CAA in the general population may have an independent impact on the cognitive status of elders. In this issue of Annals of Neurology, Arvanitakis et al117 examine the relationship between CAA and cognitive impairment in community-dwelling elders from the clinicopathologic Religious Orders Study. They show that CAA is very common in this population, occurring in nearly 85% of subjects, and that CAA pathology is correlated with AD pathology. Multivariate analyses controlling for AD pathology and other potential confounding variables demonstrate that individuals with moderate-to-severe CAA have lower perceptual speed and episodic memory. These interesting results suggest that CAA pathology affects specific cognitive domains independently from AD pathology.
Other Methods of CAA Detection
Pittsburgh Compound B–Positron Emission Tomography Imaging
MRI detection of CAA-associated hemorrhages has proven extremely useful as a pathologically validated technique for diagnosing CAA during life. This approach is fundamentally limited, however, to detecting the pathologic effects of advanced CAA rather than the vascular amyloid itself.
An emerging technique is positron emission tomography (PET) imaging with Pittsburgh compound B (PiB) to measure the burden and location of fibrillar Aβ deposits. 118–122 In a recent study, a group of CAA subjects were compared with a group of probable AD subjects and a group of older normal control subjects. Global PiB retention in the nondemented CAA subjects was significantly increased in CAA relative to healthy control subjects (p = 0.0009), although lower in CAA than in AD subjects (p = 0.002) (example shown in Fig 2, left panel). Importantly, the occipital-to-global PiB ratio was found to be significantly greater in CAA than AD (p = 0.003) (see Fig 2, right panel).121 In addition to being able to detect the parenchymal Aβ deposits in AD, PiBPET appears to be able to specifically detect CAA pathology, as recently shown in Iowa-type hereditary CAA.123
FIGURE 2.
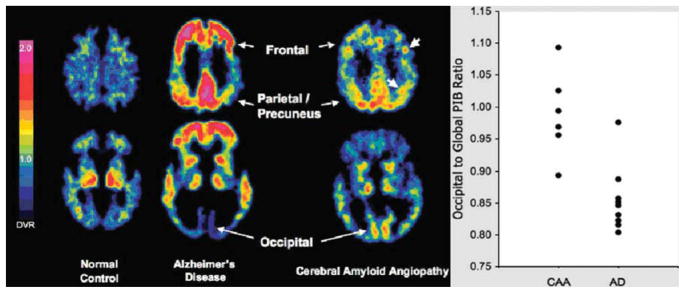
Cerebral amyloid angiopathy (CAA) subjects show intermediate level of global Pittsburgh compound B (PiB) retention with occipital predominance. Representative PiB positron emission tomographic images at 2 transaxial levels from normal control (NC) (PiB-negative), Alzheimer disease (AD), and CAA (left panel). Compared with AD and NC, CAA subjects had an intermediate level of global PiB retention, but had relatively increased occipital retention compared with AD (right panel). Modified from Johnson et al.121 DVR = distribution volume ratio.
CSF Biomarkers
There is convincing evidence to suggest that Aβ protein in cerebrospinal fluid (CSF) may serve as biomarkers in the diagnostic workup of dementia patients and may be able to reliably distinguish patients with AD pathology. 124–129 Patients with AD have consistently been shown to have decreased CSF concentrations of Aβ42 and increased tau protein.124,130,131 Therefore, measurement of CSF concentrations Aβ and tau protein potentially allows highly accurate differentiation between AD patients and controls (sensitivity and specificity generally >80%).
More recently, differences in Aβ40 and Aβ42 levels between healthy elderly subjects and patients with either AD or CAA have been investigated.132 Whereas concentrations of Aβ42 were reduced in both the AD and the CAA groups, Aβ40 levels were also lower in CAA compared to both the healthy controls and the AD subjects. These results are consistent with the hypothesis that the large component of Aβ40 deposited in vessels in advanced CAA would deplete this peptide from CSF in an analogous manner to AD-associated reductions in CSF Aβ42. Additionally, total and phosphorylated tau protein levels were not significantly different from controls in CAA subjects, consistent with a lower level of tau-containing pathology in CAA. The combination of Aβ42 and total tau strongly discriminated cerebral amyloid angiopathy from controls (area under the receiver operator curve, 0.98), although discrimination between CAA and AD was less distinct (area under the receiver operator curve, 0.82). These data suggest that Aβ40, Aβ42, and tau protein levels in the CSF may serve as sensitive biomarkers to identify patients with advanced CAA pathology.
Future Directions
There are many unanswered questions in CAA that represent potential important avenues of future research. We highlight some of the prominent areas below.
Mechanism of Small Vessel Injury
Microbleeds and white matter lesions appear to contribute to CAA-related brain injury as outlined above. There is also recent evidence to suggest that other MRI markers of cerebral small vessel disease may be important in CAA. These include ultrastructural damage detected by diffusion-tensor imaging42,133 and small ischemic infarcts.54,55 Untangling the independent contributions of these lesions to CAA-related neurologic dysfunction could be an important step toward prevention.
Effect of Blood Pressure on Clinical Course
A recent subanalysis from the randomized-controlled trial PROGRESS suggests that blood pressure lowering may have an important effect in reducing recurrent ICH in general and CAA-related ICH in particular.134 Although blood pressure appears not to be the primary causative agent in CAA, its impact on outcomes such as recurrent ICH, cognitive impairment, or progression of small vessel disease pathology remains to be determined.
Early Markers
Although the Boston criteria have been very useful in identifying patients with advanced CAA, these criteria are likely less specific in detecting individuals with early CAA. Furthermore, they remain indirect measures of the effects of Aβ-mediated pathology. Direct measures of vascular Aβ burden during life such as PiB-PET imaging or CSF Aβ may prove useful in identifying individuals in the early stages of the disease. Additionally, in vivo measures of abnormal vascular reactivity41 may represent other tools to help distinguish individuals with nascent CAA pathology.
Prescribing Antithrombotic Therapy
Anticoagulation appears generally unsafe following CAA-related ICH, even for strong indications such as nonvalvular atrial fibrilation.135 The relative risks and benefits are less clear for individuals with microbleeds only; a small case–control study suggested that these lesions may be an independent risk factor for warfarin-related ICH.136 For the (presumably safer) alternative of antiplatelet therapy, there is also evidence to suggest increased ICH risk in CAA, particularly for individuals with larger numbers of microbleeds.137,138 Larger, more definitive studies are required to settle these important questions, which could have considerable clinical implications.
Role of Antiamyloid Immunotherapy on Blood Vessels in CAA
The risks—and even possible benefits—of anti–β-amyloid therapy in individuals with CAA remain largely unknown. Animal studies have suggested that effective clearance of β-amyloid through immunotherapy may be possible.139–141 Whether similar results are achievable in humans, and how they might affect future clinical impairment, remain to be determined.
Role and Clinical Impact of Cerebrovascular Aβ Deposition in Patients with Both AD and Advanced CAA
It remains uncertain whether advanced CAA can be definitively recognized in AD patients. Additionally, apart from its potentially important role in reaction to immunotherapy, the possibility that advanced CAA represents severe enough small vessel disease to modify the clinical, cognitive, or neuroimaging profile of AD needs to be clarified.
Footnotes
Potential Conflicts of Interest
A.V. has served as a consultant for Athena Diagnostics and has grants/grants pending from the NIH. S.M.G. has served as a consultant for Hoffman-La Roche, Janssen Alzheimer Immunotherapy, and Bristol-Myers Squibb Company and has received honoraria from Medtronic and Pfizer.
References
- 1.Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009;40:2601–2606. doi: 10.1161/STROKEAHA.108.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viswanathan A, Greenberg SM. Chapter 38: Intracerebral hemorrhage. Handb Clin Neurol. 2008;93:767–790. doi: 10.1016/S0072-9752(08)93038-4. [DOI] [PubMed] [Google Scholar]
- 3.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 4.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 5.Ellis RJ, Olichney JM, Thal LJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 6.Herzig MC, Winkler DT, Burgermeister P, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 7.Bell RD, Deane R, Chow N, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow N, Bell RD, Deane R, et al. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc Natl Acad Sci U S A. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol. 2006;16:40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis J, Xu F, Deane R, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 11.Deane R, Wu Z, Sagare A, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Monro OR, Mackic JB, Yamada S, et al. Substitution at codon 22 reduces clearance of Alzheimer’s amyloid-beta peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol Aging. 2002;23:405–412. doi: 10.1016/s0197-4580(01)00317-7. [DOI] [PubMed] [Google Scholar]
- 13.Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 14.Eisele YS, Obermuller U, Heilbronner G, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of beta-amyloid is sufficient to reduce brain beta-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89:808–814. doi: 10.1002/jnr.22603. [DOI] [PubMed] [Google Scholar]
- 16.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10:463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 17.Mackic JB, Bading J, Ghiso J, et al. Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer’s disease lesions. Vascul Pharmacol. 2002;38:303–313. doi: 10.1016/s1537-1891(02)00198-2. [DOI] [PubMed] [Google Scholar]
- 18.Mackic JB, Weiss MH, Miao W, et al. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem. 1998;70:210–215. doi: 10.1046/j.1471-4159.1998.70010210.x. [DOI] [PubMed] [Google Scholar]
- 19.Poduslo JF, Curran GL, Haggard JJ, et al. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid beta-protein 1–40 at the blood-brain barrier. Neurobiol Dis. 1997;4:27–34. doi: 10.1006/nbdi.1997.0132. [DOI] [PubMed] [Google Scholar]
- 20.Martel CL, Mackic JB, McComb JG, et al. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer’s amyloid beta in guinea pigs. Neurosci Lett. 1996;206:157–160. doi: 10.1016/s0304-3940(96)12462-9. [DOI] [PubMed] [Google Scholar]
- 21.Ghilardi JR, Catton M, Stimson ER, et al. Intra-arterial infusion of [125I]A beta 1–40 labels amyloid deposits in the aged primate brain in vivo. Neuroreport. 1996;7:2607–2611. doi: 10.1097/00001756-199611040-00040. [DOI] [PubMed] [Google Scholar]
- 22.Zlokovic BV, Ghiso J, Mackic JB, et al. Blood-brain barrier transport of circulating Alzheimer’s amyloid beta. Biochem Biophys Res Commun. 1993;197:1034–1040. doi: 10.1006/bbrc.1993.2582. [DOI] [PubMed] [Google Scholar]
- 23.Grabowski TJ, Cho HS, Vonsattel JP, et al. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 24.Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol. 1986;45:79–90. [PubMed] [Google Scholar]
- 25.Vinters HV, Natte R, Maat-Schieman ML, et al. Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D) Acta Neuropathol. 1998;95:235–244. doi: 10.1007/s004010050793. [DOI] [PubMed] [Google Scholar]
- 26.Vonsattel JP, Myers RH, Hedley-Whyte ET, et al. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol. 1991;30:637–649. doi: 10.1002/ana.410300503. [DOI] [PubMed] [Google Scholar]
- 27.Zekry D, Duyckaerts C, Belmin J, et al. Cerebral amyloid angiopathy in the elderly: vessel walls changes and relationship with dementia. Acta Neuropathol. 2003;106:367–373. doi: 10.1007/s00401-003-0738-6. [DOI] [PubMed] [Google Scholar]
- 28.Vinters HV, Secor DL, Read SL, et al. Microvasculature in brain biopsy specimens from patients with Alzheimer’s disease: an immunohistochemical and ultrastructural study. Ultrastruct Pathol. 1994;18:333–348. doi: 10.3109/01913129409023202. [DOI] [PubMed] [Google Scholar]
- 29.Calhoun ME, Burgermeister P, Phinney AL, et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158:1065–1071. doi: 10.1016/S0002-9440(10)64053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fryer JD, Taylor JW, DeMattos RB, et al. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci. 2003;23:7889–7896. doi: 10.1523/JNEUROSCI.23-21-07889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Dorpe J, Smeijers L, Dewachter I, et al. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol. 2000;157:1283–1298. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winkler DT, Bondolfi L, Herzig MC, et al. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimchi EY, Kajdasz S, Bacskai BJ, Hyman BT. Analysis of cerebral amyloid angiopathy in a transgenic mouse model of Alzheimer disease using in vivo multiphoton microscopy. J Neuropathol Exp Neurol. 2001;60:274–279. doi: 10.1093/jnen/60.3.274. [DOI] [PubMed] [Google Scholar]
- 35.Niwa K, Carlson GA, Iadecola C. Exogenous A beta1–40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab. 2000;20:1659–1668. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Niwa K, Younkin L, Ebeling C, et al. Abeta 1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A. 2000;97:9735–9740. doi: 10.1073/pnas.97.17.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han BH, Zhou ML, Abousaleh F, et al. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J Neurosci. 2008;28:13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin HK, Jones PB, Garcia-Alloza M, et al. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain. 2007;130:2310–2319. doi: 10.1093/brain/awm156. [DOI] [PubMed] [Google Scholar]
- 39.Natte R, Maat-Schieman ML, Haan J, et al. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol. 2001;50:765–772. doi: 10.1002/ana.10040. [DOI] [PubMed] [Google Scholar]
- 40.Thal DR, Capetillo-Zarate E, Larionov S, et al. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 41.Smith EE, Vijayappa M, Lima F, et al. Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology. 2008;71:1424–1430. doi: 10.1212/01.wnl.0000327887.64299.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viswanathan A, Patel P, Rahman R, et al. Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke. 2008;39:1988–1992. doi: 10.1161/STROKEAHA.107.509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith EE, Gurol ME, Eng JA, et al. White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology. 2004;63:1606–1612. doi: 10.1212/01.wnl.0000142966.22886.20. [DOI] [PubMed] [Google Scholar]
- 44.Holland CM, Smith EE, Csapo I, et al. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke. 2008;39:1127–1133. doi: 10.1161/STROKEAHA.107.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurol ME, Irizarry MC, Smith EE, et al. Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology. 2006;66:23–29. doi: 10.1212/01.wnl.0000191403.95453.6a. [DOI] [PubMed] [Google Scholar]
- 46.Smith EE, Nandigam KR, Chen YW, et al. MRI markers of small vessel disease in lobar and deep hemispheric intracerebral hemorrhage. Stroke. 2010;41:1933–1938. doi: 10.1161/STROKEAHA.110.579078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, Chabriat H, Godin O, et al. Distribution of white matter hyperintensity in patients with intracerebral hemorrhage and healthy aging. Paper presented at: Second International CAA Conference: Cerebral Amyloid Angiopathy and Related Microangiopathies; May 13–15, 2010; Los Angeles, CA. [Google Scholar]
- 48.Olichney JM, Hansen LA, Hofstetter CR, et al. Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch Neurol. 2000;57:869–874. doi: 10.1001/archneur.57.6.869. [DOI] [PubMed] [Google Scholar]
- 49.Haglund M, Passant U, Sjobeck M, et al. Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry. 2006;21:681–687. doi: 10.1002/gps.1550. [DOI] [PubMed] [Google Scholar]
- 50.Suter OC, Sunthorn T, Kraftsik R, et al. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33:1986–1992. doi: 10.1161/01.str.0000024523.82311.77. [DOI] [PubMed] [Google Scholar]
- 51.Okamoto Y, Ihara M, Fujita Y, et al. Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport. 2009;20:990–996. doi: 10.1097/WNR.0b013e32832d2e6a. [DOI] [PubMed] [Google Scholar]
- 52.Okazaki H, Reagan TJ, Campbell RJ. Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc. 1979;54:22–31. [PubMed] [Google Scholar]
- 53.Soontornniyomkij V, Lynch MD, Mermash S, et al. Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol. 2010;20:459–467. doi: 10.1111/j.1750-3639.2009.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kimberly WT, Gilson A, Rost NS, et al. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology. 2009;72:1230–1235. doi: 10.1212/01.wnl.0000345666.83318.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Menon RS, Kidwell CS. Neuroimaging demonstration of evolving small vessel ischemic injury in cerebral amyloid angiopathy. Stroke. 2009;40:e675–e677. doi: 10.1161/STROKEAHA.109.552935. [DOI] [PubMed] [Google Scholar]
- 56.Lansberg MG, Thijs VN, O’Brien MW, et al. Evolution of apparent diffusion coefficient, diffusion-weighted, and T2-weighted signal intensity of acute stroke. AJNR Am J Neuroradiol. 2001;22:637–644. [PMC free article] [PubMed] [Google Scholar]
- 57.Greenberg SM, Eng JA, Ning M, et al. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke. 2004;35:1415–1420. doi: 10.1161/01.STR.0000126807.69758.0e. [DOI] [PubMed] [Google Scholar]
- 58.Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, et al. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woo D, Sauerbeck LR, Kissela BM, et al. Genetic and environmental risk factors for intracerebral hemorrhage: preliminary results of a population-based study. Stroke. 2002;33:1190–1195. doi: 10.1161/01.str.0000014774.88027.22. [DOI] [PubMed] [Google Scholar]
- 60.Biffi A, Ayres AM, Sonni A, et al. APOE Genotype Predicts Hematoma Volume and Expansion in Intracerebral Hemorrhage. Paper presented at: International Stroke Conference; February 9– 11, 2011; Los Angeles, CA. [Google Scholar]
- 61.Biffi A, Sonni A, Anderson CD, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. 2010;68:934–943. doi: 10.1002/ana.22134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342:240–245. doi: 10.1056/NEJM200001273420403. [DOI] [PubMed] [Google Scholar]
- 63.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology. 1998;50:961–965. doi: 10.1212/wnl.50.4.961. [DOI] [PubMed] [Google Scholar]
- 64.Greenberg SM, Briggs ME, Hyman BT, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke. 1996;27:1333–1337. doi: 10.1161/01.str.27.8.1333. [DOI] [PubMed] [Google Scholar]
- 65.McCarron MO, Nicoll JA, Stewart J, et al. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol. 1999;58:711–718. doi: 10.1097/00005072-199907000-00005. [DOI] [PubMed] [Google Scholar]
- 66.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Premkumar DR, Cohen DL, Hedera P, et al. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol. 1996;148:2083–2095. [PMC free article] [PubMed] [Google Scholar]
- 68.Greenberg SM, Rebeck GW, Vonsattel JP, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–259. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 69.Corder EH, Lannfelt L, Bogdanovic N, et al. The role of APOE polymorphisms in late-onset dementias. Cell Mol Life Sci. 1998;54:928–934. doi: 10.1007/s000180050223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saunders AM, Hulette O, Welsh-Bohmer KA, et al. Specificity, sensitivity, and predictive value of apolipoprotein-E genotyping for sporadic Alzheimer’s disease. Lancet. 1996;348:90–93. doi: 10.1016/s0140-6736(96)01251-2. [DOI] [PubMed] [Google Scholar]
- 71.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scharf J, Brauherr E, Forsting M, Sartor K. Significance of haemorrhagic lacunes on MRI in patients with hypertensive cerebrovascular disease and intracerebral haemorrhage. Neuroradiology. 1994;36:504–508. doi: 10.1007/BF00593508. [DOI] [PubMed] [Google Scholar]
- 73.Offenbacher H, Fazekas F, Schmidt R, et al. MR of cerebral abnormalities concomitant with primary intracerebral hematomas. AJNR Am J Neuroradiol. 1996;17:573–578. [PMC free article] [PubMed] [Google Scholar]
- 74.Fazekas F, Kleinert R, Roob G, et al. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: evidence of microangiopathy-related microbleeds. AJNR Am J Neuroradiol. 1999;20:637–642. [PMC free article] [PubMed] [Google Scholar]
- 75.Ripoll MA, Siosteen B, Hartman M, Raininko R. MR detectability and appearance of small experimental intracranial hematomas at 1.5 T and 0.5 T. A 6–7-month follow-up study. Acta Radiol. 2003;44:199–205. doi: 10.1080/j.1600-0455.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 76.Alemany Ripoll M, Stenborg A, Sonninen P, et al. Detection and appearance of intraparenchymal haematomas of the brain at 1. 5 T with spin-echo, FLAIR and GE sequences: poor relationship to the age of the haematoma. Neuroradiology. 2004;46:435–443. doi: 10.1007/s00234-004-1191-5. [DOI] [PubMed] [Google Scholar]
- 77.Atlas SW, Mark AS, Grossman RI, Gomori JM. Intracranial hemorrhage: gradient-echo MR imaging at 1.5 T. Comparison with spinecho imaging and clinical applications. Radiology. 1988;168:803–807. doi: 10.1148/radiology.168.3.3406410. [DOI] [PubMed] [Google Scholar]
- 78.Greenberg SM, Finklestein SP, Schaefer PW. Petechial hemorrhages accompanying lobar hemorrhage: detection by gradient-echo MRI. Neurology. 1996;46:1751–1754. doi: 10.1212/wnl.46.6.1751. [DOI] [PubMed] [Google Scholar]
- 79.Roob G, Fazekas F. Magnetic resonance imaging of cerebral microbleeds. Curr Opin Neurol. 2000;13:69–73. doi: 10.1097/00019052-200002000-00013. [DOI] [PubMed] [Google Scholar]
- 80.Haacke EM, DelProposto ZS, Chaturvedi S, et al. Imaging cerebral amyloid angiopathy with susceptibility-weighted imaging. AJNR Am J Neuroradiol. 2007;28:316–317. [PMC free article] [PubMed] [Google Scholar]
- 81.Nandigam K, Viswanathan A, Skehan ME, et al. Newer susceptibility-weighted MRI to detect cerebral microbleeds: examining the effect of field strength, sequence parameters, and slice thickness. A JNR Am J Neuroractology. 2009;30:338–343. doi: 10.3174/ajnr.A1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vernooij MW, van der Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70:1208–1214. doi: 10.1212/01.wnl.0000307750.41970.d9. [DOI] [PubMed] [Google Scholar]
- 83.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/wnl.56.4.537. [DOI] [PubMed] [Google Scholar]
- 85.Greenberg SM, O’Donnell HC, Schaefer PW, Kraft E. MRI detection of new hemorrhages: potential marker of progression in cerebral amyloid angiopathy. Neurology. 1999;53:1135–1138. doi: 10.1212/wnl.53.5.1135. [DOI] [PubMed] [Google Scholar]
- 86.Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke. 1983;14:924–928. doi: 10.1161/01.str.14.6.924. [DOI] [PubMed] [Google Scholar]
- 87.Ropper AH, Davis KR. Lobar cerebral hemorrhages: acute clinical syndromes in 26 cases. Ann Neurol. 1980;8:141–147. doi: 10.1002/ana.410080203. [DOI] [PubMed] [Google Scholar]
- 88.Kase CS. Lobar hemorrhage. In: Kase CS, Caplan LR, editors. Intracerebral hemorrhage. Boston, MA: Butterworth-Heinemann; 1994. pp. 363–382. [Google Scholar]
- 89.Rosand J, Muzikansky A, Kumar A, et al. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol. 2005;58:459–462. doi: 10.1002/ana.20596. [DOI] [PubMed] [Google Scholar]
- 90.Greenberg SM, Nandigam RN, Delgado P, et al. Microbleeds versus macrobleeds: evidence for distinct entities. Stroke. 2009;40:2382–2386. doi: 10.1161/STROKEAHA.109.548974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Okoye MI, Watanabe I. Ultrastructural features of cerebral amyloid angiopathy. Hum Pathol. 1982;13:1127–1132. doi: 10.1016/s0046-8177(82)80251-7. [DOI] [PubMed] [Google Scholar]
- 92.Atri A, Locascio JJ, Lin JM, et al. Prevalence and effects of lobar microhemorrhages in early-stage dementia. Neurodegener Dis. 2005;2:305–312. doi: 10.1159/000092317. [DOI] [PubMed] [Google Scholar]
- 93.Cordonnier C, van der Flier WM, Sluimer JD, et al. Prevalence and severity of microbleeds in a memory clinic setting. Neurology. 2006;66:1356–1360. doi: 10.1212/01.wnl.0000210535.20297.ae. [DOI] [PubMed] [Google Scholar]
- 94.Hanyu H, Tanaka Y, Shimizu S, et al. Cerebral microbleeds in Alzheimer’s disease. J Neurol. 2003;250:1496–1497. doi: 10.1007/s00415-003-0245-7. [DOI] [PubMed] [Google Scholar]
- 95.Nakata Y, Shiga K, Yoshikawa K, et al. Subclinical brain hemorrhages in Alzheimer’s disease: evaluation by magnetic resonance T2*-weighted images. Ann N Y Acad Sci. 2002;977:169–172. doi: 10.1111/j.1749-6632.2002.tb04813.x. [DOI] [PubMed] [Google Scholar]
- 96.Pettersen JA, Sathiyamoorthy G, Gao F-Q, et al. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008;65:790–795. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 97.Cordonnier C, Al-Shahi Salman R, Wardlaw J. Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain. 2007;130:1988–2003. doi: 10.1093/brain/awl387. [DOI] [PubMed] [Google Scholar]
- 98.Viswanathan A, Chabriat H. Cerebral microhemorrhage. Stroke. 2006;37:550–555. doi: 10.1161/01.STR.0000199847.96188.12. [DOI] [PubMed] [Google Scholar]
- 99.Petrovitch H, Ross GW, Steinhorn SC, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005;57:98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 100.Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- 101.Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). . Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 102.Pfeifer LA, White LR, Ross GW, et al. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–1634. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 103.Esiri MM, Nagy Z, Smith MZ, et al. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354:919–920. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- 104.Heyman A, Fillenbaum GG, Welsh-Bohmer KA, et al. Cerebral infarcts in patients with autopsy-proven Alzheimer’s disease: CERAD, part XVIII. Consortium to Establish a Registry for Alzheimer’s Disease. Neurology. 1998;51:159–162. doi: 10.1212/wnl.51.1.159. [DOI] [PubMed] [Google Scholar]
- 105.Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry. 1997;63:749–753. doi: 10.1136/jnnp.63.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee JH, Olichney JM, Hansen LA, et al. Small concomitant vascular lesions do not influence rates of cognitive decline in patients with Alzheimer disease. Arch Neurol. 2000;57:1474–1479. doi: 10.1001/archneur.57.10.1474. [DOI] [PubMed] [Google Scholar]
- 107.Zekry D, Duyckaerts C, Moulias R, et al. Degenerative and vascular lesions of the brain have synergistic effects in dementia of the elderly. Acta Neuropathol. 2002;103:481–487. doi: 10.1007/s00401-001-0493-5. [DOI] [PubMed] [Google Scholar]
- 108.Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–1222. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 109.Longstreth WT, Jr, Dulberg C, Manolio TA, et al. Incidence, manifestations, and predictors of brain infarcts defined by serial cranial magnetic resonance imaging in the elderly: the Cardiovascular Health Study. Stroke. 2002;33:2376–2382. doi: 10.1161/01.str.0000032241.58727.49. [DOI] [PubMed] [Google Scholar]
- 110.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 111.Ferrer I, Boada Rovira M, Sanchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nicoll JA, Wilkinson D, Holmes C, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 113.Eng JA, Frosch MP, Choi K, et al. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004;55:250–256. doi: 10.1002/ana.10810. [DOI] [PubMed] [Google Scholar]
- 114.Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68:1411–1416. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- 115.Black RS, Sperling R, Kirby L, et al. A single ascending dose study of bapinezumab, a humanized monoclonal antibody to A″. Paper presented at: Ninth Annual Geneva Spring-field Symposium on Advances in Alzheimer’s Therapy; April 19–22, 2006; Geneva, Switzerland. AD 006. [Google Scholar]
- 116.Sveinbjornsdottir S, Sigurdsson S, Aspelund T, et al. Cerebral microbleeds in the population based AGES-Reykjavik study: prevalence and location. J Neurol Neurosurg Psychiatry. 2008;79:1002–1006. doi: 10.1136/jnnp.2007.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arvanitakis Z, Leurgans S, Wang Z, et al. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011;69:320–327. doi: 10.1002/ana.22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 119.Bacskai BJ, Hickey GA, Skoch J, et al. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:12462–12467. doi: 10.1073/pnas.2034101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- 121.Johnson KA, Gregas M, Becker JA, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 122.Ly JV, Donnan GA, Villemagne VL, et al. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology. 2010;74:487–493. doi: 10.1212/WNL.0b013e3181cef7e3. [DOI] [PubMed] [Google Scholar]
- 123.Greenberg SM, Grabowski T, Gurol ME, et al. Detection of isolated cerebrovascular beta-amyloid with Pittsburgh compound B. Ann Neurol. 2008;64:587–591. doi: 10.1002/ana.21528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vemuri P, Wiste HJ, Weigand SD, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67:308–316. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vemuri P, Wiste HJ, Weigand SD, et al. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010;75:143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 128.de Jong D, Kremer BP, Olde Rikkert MG, Verbeek MM. Current state and future directions of neurochemical biomarkers for Alzheimer’s disease. Clin Chem Lab Med. 2007;45:1421–1434. doi: 10.1515/CCLM.2007.320. [DOI] [PubMed] [Google Scholar]
- 129.Hulstaert F, Blennow K, Ivanoiu A, et al. Improved discrimination of AD patients using beta-amyloid(1–42) and tau levels in CSF. Neurology. 1999;52:1555–1562. doi: 10.1212/wnl.52.8.1555. [DOI] [PubMed] [Google Scholar]
- 130.Verbeek MM, De Jong D, Kremer HP. Brain-specific proteins in cerebrospinal fluid for the diagnosis of neurodegenerative diseases. Ann Clin Biochem. 2003;40:25–40. doi: 10.1258/000456303321016141. [DOI] [PubMed] [Google Scholar]
- 131.Sunderland T, Linker G, Mirza N, et al. Decreased beta-amyloid1– 42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA. 2003;289:2094–2103. doi: 10.1001/jama.289.16.2094. [DOI] [PubMed] [Google Scholar]
- 132.Verbeek MM, Kremer BP, Rikkert MO, et al. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann Neurol. 2009;66:245–249. doi: 10.1002/ana.21694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Salat DH, Smith EE, Tuch DS, et al. White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. Stroke. 2006;37:1759–1764. doi: 10.1161/01.STR.0000227328.86353.a7. [DOI] [PubMed] [Google Scholar]
- 134.Arima H, Tzourio C, Anderson C, et al. Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the PROGRESS trial. Stroke. 2010;41:394–396. doi: 10.1161/STROKEAHA.109.563932. [DOI] [PubMed] [Google Scholar]
- 135.Eckman MH, Rosand J, Knudsen KA, et al. Can patients be anti-coagulated after intracerebral hemorrhage? A decision analysis. Stroke. 2003;34:1710–1716. doi: 10.1161/01.STR.0000078311.18928.16. [DOI] [PubMed] [Google Scholar]
- 136.Lee SH, Ryu WS, Roh JK. Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology. 2009;72:171–176. doi: 10.1212/01.wnl.0000339060.11702.dd. [DOI] [PubMed] [Google Scholar]
- 137.Soo YO, Yang SR, Lam WW, et al. Risk vs benefit of anti-thrombotic therapy in ischaemic stroke patients with cerebral microbleeds. J Neurol. 2008;255:1679–1686. doi: 10.1007/s00415-008-0967-7. [DOI] [PubMed] [Google Scholar]
- 138.Biffi A, Halpin A, Towfighi A, et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology. 2010;75:693–698. doi: 10.1212/WNL.0b013e3181eee40f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci. 2007;27:1973–1980. doi: 10.1523/JNEUROSCI.5426-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schroeter S, Khan K, Barbour R, et al. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci. 2008;28:6787–6793. doi: 10.1523/JNEUROSCI.2377-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thakker DR, Weatherspoon MR, Harrison J, et al. Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2009;106:4501–4506. doi: 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]