

Abstract
Galectin-3 (Gal-3), a member of the β-galactoside binding protein family containing the NWGR antideath motif of the Bcl-2 protein family, is involved in various aspects of cancer progression. Previously, it has been shown that the antiapoptotic activity of Gal-3 is regulated by the phosphorylation at Ser6 by casein kinase 1 (CK1). Here we questioned how phosphorylation at Ser6 regulates Gal-3 function. We have generated serine-to-alanine (S6A) and serine-to-glutamic acid (S6E) Gal-3 mutants and transfected them into the BT-549 human breast carcinoma cell line, which does not express Gal-3. BT-549 cell clones expressing wild-type (wt) and mutant Gal-3 were exposed to chemotherapeutic anticancer drugs. In response to the apoptotic insults, phosphorylated wt Gal-3 was exported from the nucleus to the cytoplasm and protected the BT-549 cells from drug-induced apoptosis while nonphosphorylated mutant Gal-3 neither was exported from the nucleus nor protected BT-549 cells from drug-induced apoptosis. Furthermore, leptomycin B, a nuclear export inhibitor, increased the cisplatin-induced apoptosis of Gal-3 expressing BT-549 cells. These results suggest that Ser6 phosphoryaltion acts as a molecular switch for its cellular translocation from the nucleus to the cytoplasm and, as a result, regulates the antiapoptotic activity of Gal-3.
Galectins are a family of proteins characterized by their affinity for β-galactosides and by the conserved sequence of a carbohydrate recognition domain (5). Gal-3 is a chimeric protein consisting of three structural domains, a globular COOH terminal encompassing the carbohydrate binding motif; a repetitive collagen-like sequence rich in proline, glycine, and tyrosine, and 12 amino acids at the NH2 terminus. Gal-3 was shown to be involved in the cellular adhesion process, cell proliferation, and apoptosis (17, 20, 58).
Several members of the galectin protein family were implicated in the promotion of apoptosis. Exogenous Gal-1 and Gal-9 induce the apoptosis of immune cells and melanoma cells, respectively (28, 43). Intracellular Gal-7 induces the apoptosis of colon cancer cells (32). In contrast, Gal-3 was reported to exhibit an antiapoptotic function (1, 29, 31, 57, 61). It was previously reported that Gal-3 inhibits T-cell apoptosis induced by anti-Fas antibody (57) and epithelial cell apoptosis induced by staurosporine, cisplatin, genistein, and anoikis (1, 29, 31, 61). The antiapoptotic activity of Gal-3 was also demonstrated in Gal-3-deficient mice. Peritoneal macrophages from Gal-3-deficient mice were more sensitive to apoptotic stimuli than were those from control mice (23). Moreover, introducing Gal-3 into epithelial cells endowed them with resistance to apoptotic insult (1, 29, 31, 57, 61). The ability of Gal-3 to protect cells against apoptosis induced by agents working through different mechanisms suggests that Gal-3 regulates a common apoptosis commitment step. Recent reports have demonstrated that two sites in the Gal-3 sequence are important for this function. Gal-3 contains the antideath Asp-Trp-Gly-Arg (NWGR) motif that is conserved in the Bcl-2 homology domain (BH1) of the Bcl-2 family (1, 52). The other is Ser6, a CK1 phosphorylation site (25, 61). Experiments employing site-directed mutagenesis revealed that mutations of either of these sites abrogate the antiapoptotic function of Gal-3, an effect similar to that of Bcl-2 (1, 29, 61). However, the regulation of the posttranslational modification of Gal-3 N-terminal-domain phosphorylation in relation to its antiapoptotic function is still unclear. Recent studies have shown that Bcl-2 is phosphorylated at Ser70 by protein kinase C (47) and that the phosphorylation may activate or inactivate its antiapoptotic function, depending on cell type and death-signaling molecules (19, 26, 38, 44, 56, 60). Bcl-2 may be phosphorylated at a specific site, and the phosphorylated Bcl-2 loses the ability to heterodimerize with Bax (48). The apparent similarity between Bcl-2 and Gal-3 in their antiapoptotic functions and the posttranslational modification of serine phosphorylation prompted the investigation of the role of Gal-3 phosphorylation and its subcellular localization in its antiapoptotic activity.
Gal-3 undergoes posttranslational modification by addition of a phosphate group catalyzed by CK1, and its phosphorylation serves as a molecular switch for the sugar binding ability (10, 24, 34, 39). It was previously reported that among the cytoplasmic, nuclear, and nuclear export fractions, only the nuclear export fraction contained exclusively phosphorylated Gal-3, while the nuclear fraction had both phosphorylated and nonphosphorylated isoforms of Gal-3 (51). These results imply that phosphorylation is important for the cellular translocation of the Gal-3.
Previous reports have indicated the relevance of cell cycle regulation by Gal-3 and antiapoptotic activity of Gal-3. Overexpression of Gal-3 in breast cancer cells induced G1 or G2 arrest in response to apoptotic stimuli like CDDP (cisplatin), genistein, and suspension culture and, at the same time, protected the cells from apoptosis (29, 31, 61). Previous report studies have demonstrated that Gal-3 changes cyclins and cyclin inhibitor expression and that phosphorylation at Ser6 of Gal-3 induces cell cycle arrest, resulting in inhibition of apoptosis (61). Because cell cycle regulators, such as cyclins and cyclin inhibitors, work in the nucleus, subcellular localization of Gal-3 may be important for its cell cycle regulation and its antiapoptotic activity.
The nuclear localization of Gal-3 is presumably associated with normal in vitro cell proliferation since it is a required splicing factor interacting with Gemin4 (10, 24, 39, 40). In vivo, the expression of Gal-3 is associated with tumor invasion and metastatic potential in various human cancers (11). Loss of nuclear Gal-3 expression is associated with tumor progression of colon carcinoma (33); in prostate carcinomas, Gal-3 is excluded from the cell nuclei (53), and in squamous carcinoma cells of the tongue, the levels of nuclear Gal-3 markedly decreased during the progression from normal to cancerous states while the cytoplasmic expression level increased (22).
Here we question the relationship among human Gal-3 posttranslational modification, subcellular localization, antiapoptotic activity, and signaling pathway, wt human Gal-3 and Ser6 mutant Gal-3 cDNAs were transfected into the breast cancer cell line BT-549; this was followed by cloning and establishment of the levels of Gal-3 expression. All cell clones expressed Gal-3 both in the nucleus and in the cytoplasm, but only wt Gal-3 translocated from the nucleus to the cytoplasm and protected the BT-549 cells from drug-induced apoptosis; mutant Gal-3 neither translocated nor protected the BT-549 cells from apoptosis. In addition, inhibition of nuclear export of Gal-3 abrogated its antiapoptotic activity.
MATERIALS AND METHODS
Abbreviations.
The following abbreviations are used in this work: Ac, acetyl, Ala, alanine; Arg, Arginine; Asp, aspartic acid; CK1, casein kinase 1; CDDP, cis-diamminedichloroplatinum (cisplatin); DEVD, Asp-Glu-Val-Asp; DTT, dithiothreitol; FITC, fluorescein isothiocyanate; 5-FU, fluorouracil; DMEM, Dulbecco modified Eagle medium; ERK, extracellular signal-regulated kinase; FBS, fetal bovine serum; Gal-3, galectin-3; Gly, glycine; Glu, glutamic acid; His, histidine; HRP, horseradish peroxidase; JNK, Jun NH 2-terminal kinase; LEHD, Leu-Glu-His-Asp; Leu, leucine; LMB, leptomycin B; NES, nuclear export signal; NWGR, asparagine-tryptophan-glycine-arginine; MAPK, mitogen-activated protein kinase; MTT, 3-(4,5-dimethyl-thiazole-2-yl)-2,5-diphenyl tetrozolium bromide; NES, nuclear export signal; PAGE, polyacrylamide gel electrophoresis; PBS, phosphate-buffered saline; PNA, p-nitroaniline; SD, standard deviation; Ser, serine; SDS, sodium dodecyl sulfate; SMN complex, survival of motor neuron complex; Trp, typtophan; Val, valine; and wt, wild type.
Cells and culture conditions.
The human breast carcinoma cell line BT-549 was obtained from E. W. Thompson. Human cervix carcinoma cell line HeLa (ATCC CCL-2) and murine fibroblast cell line NIH 3T3 (ATCC CRL-1658) were purchased from the American Type Culture Collection (Manassas, Va.). All cell lines were grown as monolayers on plastic tissue culture dishes containing DMEM supplemented with 10% heat-inactivated FBS and antibiotics and maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Establishment of stable BT-549 Gal-3 transfectants.
The wt and Ser6 mutant Gal-3 transfectants were generated previously (61). Briefly, Ser6 mutant Gal-3 was generated using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, Calif.) as described previously (17). wt and mutant Gal-3 cDNA were excised from pGEM (7+) and inserted into pBK-CMV (Stratagene). Each plasmid was transfected into BT-549 cells with Lipofectamine (Invitrogen, Carlsbad, Calif.) and incubated with 800 μg of G418 per ml. Two weeks later, several clones were subcloned for each transfection. For further experiments, BT-549 parental, BT-549/V (control vector-transfected clone), BT-549/WT1 and WT2 (wild-type Gal-3-transfected clones), and BT-549/S6A and S6E (mutant Gal-3-transfected clones) were used. These transfected cells were maintained in complete DMEM containing 400 μg of G418 per ml.
Immunofluorescence.
Immunofluorescent staining of cells was performed as described previously (51). Briefly, the cells were fixed with 4% paraformaldehyde for 5 min, permeabilized with 0.5% Triton X-100 for 5 min, and blocked with 1% bovine serum albumin in PBS for 30 min. After the blocking step, rabbit polyclonal anti Gal-3 antibody was added at 1:50 dilution and the mixture was incubated for 1 h. Secondary antibody (FITC-conjugated goat anti-rabbit immunoglobulin G Zymed) was added at a 1:200 dilution and incubated for 1 h.
Western blot analysis of cytoplasmic and nuclear protein.
Cytoplasmic and nuclear proteins were extracted as described previously (13). Cells were grown to subconfluency on 150-mm dishes and exposed to anticancer drugs; adriamycin, CDDP, 5-FU, and VP-16 (Sigma, St. Louis, Mo.). At 48 h later, all cells were collected, washed with PBS, and resuspended in hypotonic buffer (20 mM HEPES, 10 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 0.1% Triton X-100, 20% glycerol, 5 μg of leupeptin per ml, 10 μg of aprotinin per ml, 500 μM phenylmethylsulfonyl fluoride σ). The cells were centrifuged at 850 × g for 5 min. The supernatant was used as the cytoplasmic fraction. The pellet was suspended in extraction buffer (20 mM HEPES, 10 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 0.1% Triton X-100, 20% glycerol, 420 mM NaCl, 5 μg of leupeptin per ml, 10 μg of aprotinin per ml, 500 μM phenylmethylsulfonyl fluoride). This suspension was centrifuged at 15,000 × g for 30 min. The supernatant contained the nuclear fraction. Protein concentrations were measured using a protein assay reagent (Bio-Rad). Cytoplasmic protein (10 μg) and nuclear protein (20 μg) were separated by SDS-PAGE (12.5% polyacrylamide) by the method of Laemmli and transferred to a polyvinylidene difluoride membrane. This membrane was subjected to immunoblot analysis using rat anti-Gal-3 antibody TIB166 (American Type Culture Collection), anti-phospho-MAPK antibody, anti-phospho-p38 antibody, anti-active JNK antibody (Promega, Madison, Wis.) anti-histone H1 antibody (Calbiochem, San Diego, Calif.), anti-cytochrome c antibody (Zymed, San Francisco, Calif.), antiphosphoserine antibody (Sigma), anti-β-actin antibody (Sigma), HRP-conjugated anti-rat antibody, HRP-conjugated anti-mouse antibody, and HRP-conjugated anti-rabbit antibody (Zymed). Expression levels were detected using the enhanced chemiluminescence system. For densitometric analysis of Western blots, Scion Image (Scion, Frederick, Md.) was used.
Metabolic labeling and immunoprecipitation.
Labeling in vivo with 32P was performed as described previously (61). Briefly, 8 × 106 cells were grown in 100-mm dishes. The cells were washed with phosphate-free DMEM (Invitrogen), incubated for 2 h at 37°C in phosphate-free DMEM supplemented with 5% dialyzed FBS per ml and labeled for 5 h at 37°C with 0.2 mCi of 32P (ICN Biomedicals, Costa Mesa, Calif.) in phosphate-free DMEM supplemented with 10% dialyzed FBS. Cytoplasmic and nuclear fractions were extracted as described above. The protein from the cytoplasmic and nuclear fractions was incubated with protein A-Sepharose (Amersham, Piscataway, N.J.) for 1 h at 4°C. Then 2 μg of rabbit polyclonal anti-Gal-3 antibody (17) was added, and the mixture was further incubated for 1 h. The mixture was washed three times. The washed precipitates were boiled for 5 min in SDS-PAGE sample buffer and then separated by SDS-PAGE (12.5% polyacrylamide). Gal-3 phosphorylation state was analyzed with a PhosphorImager (Molecular Dynamics, Sunnyvale, Calif.), and immunoprecipitated Gal-3 was detected by immunoblotting using TIB166. CKI-7 (Seikagaku Corp., Falmouth, Mass.) was used to inhibit CK1 activity.
Nuclear export assay.
Nuclear export assays using digitonin-permeabilized cells were performed as described previously (51). Cells were grown in 60-mm dishes. The cells were washed with transport buffer (20 mM HEPES [pH 7.3], 2 mM EGTA, 110 mM potassium acetate, 2 mM magnesium acetate) and then permeabilized for 5 min at 4°C in transport buffer containing 50 ng of digitionin (Sigma) per ml. After the cells were washed with transport buffer, transport buffer containing 1 mM DTT and proteinase inhibitors was added and the mixture was incubated for 30 min at room temperature. The supernatant was collected as nuclear export fractions. Then the cells were incubated with extraction buffer for 30 min at 4°C. The supernatant was collected as the nuclear residue fraction.
Inhibition of nuclear export.
Cells were seeded into 96-well plates, and CDDP and the nuclear export inhibitor LMB (Sigma) were added. At 48 h later, cell viability was assessed by the MTT assay. 10 μl of 5 mg/ml MTT solution (Sigma) was added to each well. After 3 h of incubation, 200 μl of dimethyl sulfoxide was added to the each well. The absorbance of each well was measured at 495 and 650 nm using a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, Calif.).
Apoptosis assay.
Apoptosis was assessed by the accumulation of cells in the sub-G 1 fraction. After a 48-h exposure to anticancer drugs, cells were fixed with 70% ethanol for 15 min at 4°C, incubated with RNase A for 30 min at 37°C, and stained with 50 μg of propidium iodide per ml for 20 min. The samples were analyzed by using FACScalibur (Beckton Dickinson, San Jose, Calif.) and Cell Quest (Beckton Dickinson).
Measurement of caspase activity.
Caspase-3 activity was measured as described previously (2). Briefly, cells were fixed with 1% formaldehyde for 15 min at 4°C, washed twice with PBS, permeabilized with 0.1% Triton X-100 in PBS for 15 min at 4°C, and incubated with goat serum. Then cells were stained with FITC-conjugated anti-active caspase-3 antibody (PharMingen, San Diego, Calif.). After being washed with PBS, the cells were fixed with 1% formaldehyde again and analyzed with FACScalibur.
To further investigate caspase activity, we measured DEVDase and LEHDase activity in CDDP-treated cells as described previously (62). The cells were lysed with cell extract buffer (20 mM HEPES [pH 7.5], 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT) containing 0.03% Nonidet P-40. Lysates were centrifuged at 15,000 × g for 10 min, and 50 μl of the cytosolic fraction was incubated for 60 min at 37°C in a total volume of 200 μl of caspase buffer (10 mM HEPES [pH 7.5], 50 mM NaCl, 2.5 mM DTT) containing 25 μM Ac-DEVD-pNA or Ac-LEHD-pNA (Biosource, Camarillo, Calif.) p-Nitroaniline, released by caspase activity, was measured at 405 nm using a Vmax kinetic microplate reader (Molecular Devices).
Inhibition of ERK and/or JNK.
Cells were seeded in 60-mm plates. The next day, the medium was changed to 1% BSA-DMEM containing 30 μM ERK inhibitor, U0126 (Calbiochem) and/or 25 μM JNK inhibitor, SP600125 (Calbiochem). After a 24-h treatment with inhibitors, CDDP was added to a final concentration of 500 μM. At 3 days later, the cells were collected, stained with propidium iodide, and analyzed by flow cytometry as described above.
RESULTS
Gal-3 translocates from the nucleus to the cytoplasm.
Since Gal-3 shuttles between the nucleus and cytoplasm and translocates to the perinuclear membrane following a variety of apoptotic stimuli, such as CDDP, serum withdrawal, and staurosporine (12, 62), we investigated the regulation of subcellular expression and localization of Gal-3 in response to apoptotic insult. Initially, we performed immunofluorescent staining for Gal-3 localization in commonly used mouse and human cell lines, NIH 3T3 and HeLa, respectively, following exposure to the antimetabolite drug, 5-FU. Although Gal-3 was randomly localized in both nucleus and cytoplasm compartments (Fig. 1, panels IA and C), it was observed to be expressed predominantly in the cytoplasm after a 48-h exposure to 100 μM 5-FU at 37°C (Fig. 1, panels IB and D). The observed change in Gal-3 localization was confirmed by Western blot analysis. In both NIH 3T3 and HeLa cells, the level of nuclear Gal-3 decreased in a dose-dependent manner (Fig. 1, panel IIB). In HeLa cells, the level of cytoplasmic Gal-3 increased in a dose-dependent manner, while in NIH 3T3 cells, the increase was not obvious (Fig. 1, panel IIA). Densitometric tracing analysis determined that the level of cytoplasmic Gal-3 in NIH 3T3 and HeLa cells increased by 1.1- and 1.3-fold, respectively (Fig. 1, panel IIA) while the level of nuclear Gal-3 in NIH 3T3 and HeLa cells decreased by 9.4- and 3.4-fold, respectively (P < 0.05) (Fig. 1, panel IIB). Western blot analysis of whole-cell lysates revealed that exposure to 5-FU did not change the total levels of Gal-3 (data not shown), indicating that down-regulation of nuclear Gal-3 expression may not be due to degradation of Gal-3 but, rather, may be due to its nuclear export. To examine whether only 5-FU, the antimetabolite drug, induces Gal-3 translocation or whether it is a general phenomenon, we have tested other anticancer drugs with different mode of actions, including the antibiotics anthracyclin and adriamycin the DNA-cross-linking agent CDDP, and the topoisomerase II inhibitor VP-16. Adriamycin, CDDP, and VP-16 increased the level of cytoplasmic Gal-3 in HeLa cells by 1.5-, 1.5-, and 1.3-fold, respectively (Fig. 1, panel IIIA) and decreased the level of nuclear Gal-3 by 2.4-, 3.2-, and 2.7-fold, respectively (Fig. 1, panel IIIB). To conclude, all drugs tested here induced Gal-3 translocation from the nucleus to the cytoplasm.
FIG. 1.

Gal-3 translocates from the nucleus to the cytoplasm. (I) intracellular localization of Gal-3. NIH 3T3 (A and B) and HeLa (C and D) cells were stained with polyclonal anti-Gal-3 antibody without (A and C) or with (B and D) exposure to 100 μM 5-FU for 48 h. (II) Western blot analysis of cytoplasmic (A) and nuclear (B) Gal-3 expression in NIH 3T3 and HeLa cells with exposure to 5-FU for 48 h. β-Actin and histone H1 were used for the internal control of the cytoplasmic and nuclear fractions as well as to determine possible cross-contamination between the fractions. The amount of Gal-3 was determined by densitometric analyses. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD (III) Western blot analysis of cytoplasmic (A) and nuclear (B) Gal-3 in HeLa cells exposed to anticancer drugs. A, C, and V represent adriamycin, CDDP, and VP-16, respectively; 1 μg of adriamycin per ml, 20 μM CDDP, and 20 μM of VP-16 were used. The amount of Gal-3 was determined by densitometric analyses. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD.
Gal-3 expression and phosphorylation in stable transfectants of BT-549 cells.
To investigate the role of Gal-3 phosphorylation at Ser6 in translocation and apoptosis, we established stable wt Gal-3 and Ser6 mutant Gal-3 transfectants. It is well known that mutation of Ser to Glu mimics Ser phosphorylation in certain proteins. In contrast, the Ser6 → Glu mutation in Gal-3 as well as the Ser6 → Ala mutation mimics dephosphorylated Ser. We have shown that mutation at Ser6 → Glu in Gal-3 results in retention of the sugar binding ability in vitro although phosphorylated Gla-3 loses the sugar binding ability, and we have shown that cells expressing Gal-3 with the Ser6 → Glu mutation are not resistant to CDDP, suggesting that Gal-3 with the Ser6 → Glu mutation mimics dephosphorylated Gal-3 in vitro and in vivo (34, 61). We transfected wt and Ser6 mutant Gal-3 into the breast cancer cell line BT-549, which expresses no Gal-3. After 2 weeks of selection with G418, several clones were obtained. A control clone (BT-549/V), two wt Gal-3 clones (BT-549/WT1 and BT-549/WT2), and two Ser6 mutant Gal-3 clones (BT-549/S6A and BT-549/S6E) were used for further analysis. Expression of Gal-3 in these transfectants was determined by Western blot analysis (Fig. 2 panel I). All Gal-3 transfectants (BT-549/WT1, BT-549/WT2, BT-549/S6A, and BT-549/S6E) expressed similar levels of Gal-3, while the control BT-549/V cells did not express any detectable Gal-3. Although the level of cytoplasmic Gal-3 was similar in all Gal-3 transfectants (Fig. 2, panel IA), the nuclear Gal-3 levels in wt cell clones were significantly lower than those in mutant clones (P < 0.01) (Fig. 2, panel IB). Next, we examined whether Gal-3 underwent phosphorylation in vivo. Following metabolic labeling with 32P, Gal-3 was immunoprecipitated with polyclonal anti-Gal-3 antibody from total-cell lysates, subjected to SDS-PAGE, and either analyzed by PhosphorImager analysis or transferred to a polyvinylidene difluoride membrane and immunoblotted with anti-Gal-3 monoclonal antibodies (Fig. 2, panel II). Although immunoprecipitates of both BT-549/WT1 and BT-549/S6A cells contained Gal-3, only BT-549/WT1 cells expressed phosphorylated Gal-3. Phosphorylation at Ser6 was confirmed by immunoblotting with antiphosphoserine antibody (data not shown). These results are consistent with the previous report, which describes phosphorylation of Gal-3 at Ser6 in vitro (34). Since CK1 is reported to phosphorylate Gal-3 in vitro, we examined the effect of the CK1 inhibitor CKI-7 on in vivo phosphorylation. As shown in Fig. 2, panel II, in vivo Gal-3 phosphorylation was inhibited by CKI-7. These results indicate that CK1 phosphorylates Gal-3 at Ser6 in vivo.
FIG. 2.

Gal-3 expression and phosphorylation in stable transfectants of BT-549 cells. (I) Western blot analysis of cytoplasmic (A) and nuclear (B) Gal-3 in a control clone (BT-549/V), wt clones (BT-549/WT1 and BT-549/WT2), and Ser6 mutant clones (BT-549/S6A and BT-549/S6E). The amount of Gal-3 was determined by densitometric analysis. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD. β-Actin and histone H1 were used for the internal control of the cytoplasmic and nuclear fractions as well as to determine possible cross-contamination between the fractions. (II) Detection of phosphorylated Gal-3 in BT-549/V, BT-549/WT1, and BT-549/S6A cells. The cells were labeled with 32P. Gal-3 was immunoprecipitated with polyclonal anti-Gal-3 antibody. Phosphorylated Gal-3 was detected by PhosphorImager, analysis. CKI-7 (50 μM) was used to inhibit CK1 activity.
Phosphorylation of Gal-3 is required for its nuclear export.
Previously it was shown that phosphorylation of Gal-3 is important for its nuclear export in normal mouse fibroblasts (51). Thus, we examined the effect of Ser6 phosphorylation on the translocation of human Gal-3. Cell clones were treated with 25 μM CDDP for 48 h at 37°C and subsequently immunostained with polyclonal anti-Gal-3 antibody. As shown in Fig. 3, panel IA and C, Gal-3 was localized in both the cytoplasm and the nucleus of BT-549/WT1 and BT-549/S6A control cells. After exposure to CDDP, Gal-3 in BT-549/WT1 cells was found predominantly in the cytoplasm while the mutant Gal-3 in BT-549/S6A cells was present in both the cytoplasm and the nucleus (Fig. 3, panels IB and D). This phenomenon was confirmed by Western blot analysis (Fig. 3, panel II). Exposure to 12.5 and 25 μM CDDP decreased the level of nuclear Gal-3 in BT-549/WT1 cells by 1.3- and 2.1-fold, respectively, although no change in the level of nuclear Gal-3 was observed in BT-549/S6A cells (Fig. 3, panel II). Similar results were obtained when BT-549/WT2 and BT-549/S6E cells were used (data not shown). To exclude the possibility of Gal-3 degradation in the nucleus, we have examined the Gal-3 expression in total-cell lysates. There was no change in Gal-3 expression level before and after CDDP treatment (data not shown). These results suggest that Gal-3 is excluded from the nucleus presumably by nuclear export.
FIG. 3.

Phosphorylation of Gal-3 is required for its translocation. (I) immunofluorescent staining of Gal-3 transfectants. BT-549/WT1 (A and B) and BT-549/S6A (C and D) cells were stained with polyclonal anti-Gal-3 antibody without (A and C) or with (B and D) exposure to 25 μM CDDP for 48 h. (II) Western blot analysis of cytoplasmic (A) and nuclear (B) Gal-3 in BT-549 transfectants with exposure to CDDP for 48 h. The amount of Gal-3 was determined by densitometric analyses. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD. β-Actin and histone H1 were used for the internal control of cytoplasmic and nuclear fractions as well as to determine possible cross-contamination between the fractions.
Next, we examined the phosphorylation status of Gal-3 in cytoplasmic and nuclear fraction after exposure to 25 μM CDDP for 24 h. CDDP treatment decreased the amount of nuclear Gal-3 in BT-549/WT1 cells (Fig. 4, panel IB). However, the amount of P-Gal-3 remained the same. Densitometric analysis revealed that CDDP treatment increased the rate of phosphorylation (P-Gal-3/Gal-3) in nuclear and cytoplasmic fraction of BT-549/WT1 cells by 1.7- and 1.1-fold, respectively. Similar results were obtained when BT-549/WT2 and BT-549/S6E were used (data not shown). These results suggest that phosphorylation ships Gal-3 to the cytoplasm, where it could be detected in the phosphorylated form.
FIG. 4.
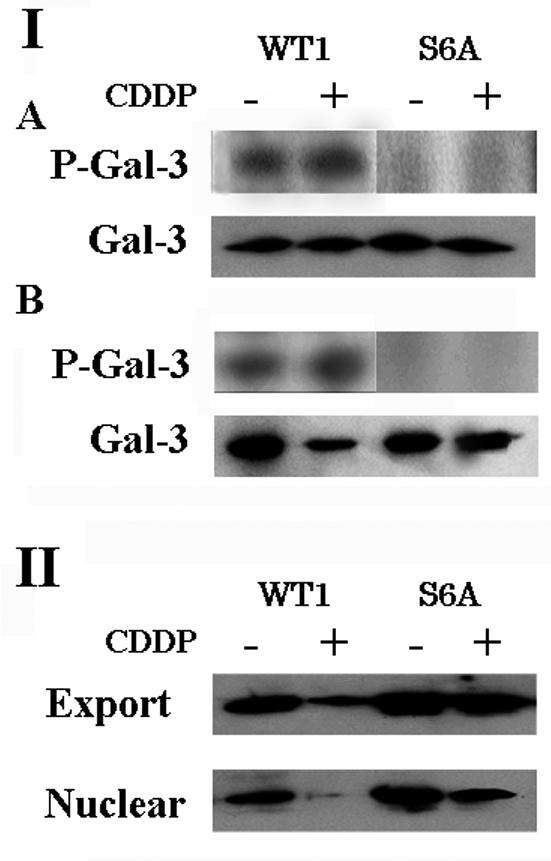
Phosphorylation of Gal-3 enhances nuclear export. (I) Phosphorylation status of Gal-3 in the cytoplasmic (A) and nuclear (B) fractions of CDDP- treated BT-549 clones. BT-549/WT1 and BT-549/S6A were exposed to 25 μM CDDP for 24 h and labeled with 32P. Cytoplasmic and nuclear Gal-3 was immunoprecipitated. (II) Nuclear export of Gal-3. BT-549/WT1 and BT-549/S6A were exposed to 25 μM CDDP for 24 h and permeabilized with 50 ng of digitonin per ml. The export fraction and nuclear residue (5 μg each) were subjected to SDS-PAGE and immunoblotted with anti-Gal-3 antibody.
To examine whether Gal-3 translocation was caused by increased nuclear export, a nuclear export assay using digitonin-permeabilized cells was performed. Gal-3 in the nuclear export fraction and the nuclear residue was detected by Western blotting (Fig. 4, panel II). Although the levels of Gal-3 in the export fractions of the cell clones were comparable, the level of Gal-3 in the nuclear residue of CDDP-treated BT-549/WT1 was remarkably reduced. Similar results were obtained when BT-549/WT2 and BT-549/S6E cells were used (data not shown). Densitometric analysis revealed that the amount of exported Gal-3 (Gal-3 in the export fraction) in CDDP-treated BT-549/WT1 cells was 85% of the amount of total nuclear Gal-3 (Gal-3 in nuclear residue + Gal-3 in export fraction), while the amounts in nontreated BT-549/WT1 and nontreated and CDDP-treated BT-549/S6A cells were 60 to 70% of the amount of total nuclear Gal-3.
These results indicate that phosphorylation of Gal-3 promotes its nuclear export after apoptotic stimuli through enhanced nuclear export.
The Ser6 mutation abrogates the antiapoptotic function of Gal-3.
It was previously reported that Gal-3 inhibits CDDP-induced apoptosis (1, 61) and that the antiapoptotic function requires the phosphorylation of Ser6 (61). Here, we investigated whether the antiapoptotic function of phosphorylation-dependent Gal-3 is restricted only to DNA-cross-linking agents or is effective for other targets of anticancer drugs, similarly to Bcl-2. To address this, we tested the antibiotics anthracyclin and adriamycin, the DNA-cross-linking agent CDDP, the antimetabolite 5-FU, and the topoisomerase II inhibitor VP-16. BT-549 control and Gal-3-harboring cells were exposed to different concentrations of the anticancer drugs for 48 h at 37°C, stained with propidium iodide and analyzed by flow cytometry. Apoptosis was assessed by the accumulation of cells at the sub-G1 fraction. As shown in Fig. 5, BT-549/WT1 and BT-549/WT2 cells were more resistant to all drugs tested than were BT-549/V, BT-549/S6A, and BT-549/S6E cells. When CDDP or adriamycin was used, the percentages of BT-549/WT1 and BT-549/WT2 cells in the sub-G1 fraction (1.7 and 1% for CDDP and 1.7 and 2.7% for adriamycin, respectively) were significantly lower than those of BT-549/V, BT-549/S6A, and BT-549/S6E cells (28.9, 42.7, and 58.4% for CDDP and 20.1, 23.1, and 25.1% for adriamycin, respectively) (P < 0.01). In addition, BT-549/S6A and BT-549/S6E cells were more sensitive to anticancer drugs than were BT-549/V cells. These results indicate that wt Gal-3 confers drug resistance to cells and that the Gal-3-mediated drug resistance involves Ser6 phosphorylation.
FIG. 5.

Ser6 mutation abrogates the antiapoptotic function of Gal-3. The sub-G1 fraction of BT-549 transfectants after exposure to anticancer drugs was determined by flow cytometry. V, WT1, WT2, S6A, and S6E represent BT-549/V, BT-549/WT1, BT-549/WT2, BT-549/S6A, and BT-549/S6E, respectively. ADR, CDDP, 5-FU, and VP-16 represent adriamycin, cisplatin, fluorouracil, and VP-16, respectively; 1 μg of adriamycin per ml, 25 μM CDDP, 800 μM, fluorouracil, and 10 μM VP-16 were used. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD.
Gal-3 translocation is required for its antiapoptotic activity.
Recently, CRM1 protein was identified as the export receptor for the leucine-rich NES. Gal-3 has a putative leucine-rich NES in its sequence (51). Therefore, we tested the effect of LMB, which inhibits CRM1-NES-dependent nuclear export, on Gal-3 translocation. As shown in Fig. 6, panels IA to C, LMB inhibited CDDP-induced Gal-3 translocation in BT-549/WT1 cells but had no significant effect on the distribution of Gal-3 in BT-549/S6A cells (Fig. 6, panels ID to F). Next we examined the effect of LMB on the antiapoptotic activity of Gal-3. BT-549/V, BT-549/WT1, and BT-549/S6A cells were exposed to 25 μM CDDP in the presence of LMB (0 to 32 ng/ml) for 48 h and than the cell viability was assessed by the MTT assay. LMB decreased the cell viability of CDDP-treated BT-549/WT1 cells in a dose-dependent manner (Fig. 6, panel II). This effect of LMB does not result only from the inhibition of Gal-3 export, since LMB is known to inhibit the export of other proteins such as ERK as well as that of Gal-3. On the other hand, it did not affect the cell viability of CDDP-treated BT-549/V and BT-549/S6A cells. Similar results were obtained when BT-549/WT2 and BT-549/S6E cells were used (data not shown). These results indicate that Gal-3 translocation by Ser6 phosphorylation is a prerequisite for its antiapoptotic activity.
FIG. 6.

Effect of LMB on Gal-3 translocation and antiapoptotic activity. (I) Immunofluorescent staining of Gal-3 transfectants. BT-549/WT1 (A to C) and BT-549/S6A (D to F) were exposed to 25 μM CDDP for 48 h (B, C, E, and F) in the absence (B and E) or presence (C and F) of 8 ng of LMB per ml. (A and D) No treatment. (II) Cell viability of CDDP- and LMB-treated cells. BT-549/V (squares), BT-549/WT1 (circles), and BT-549/S6A (triangles) were treated with 25 μM CDDP in the presence of LMB (0, 2, 8, or 32 ng/ml) for 24 h. Cell viability was assessed by the MTT assay. The relative values for CDDP- and LMB-treated cells with respect to untreated cells are plotted in the graph.
Cytochrome c release and subsequent caspase activation are inhibited by wt Gal-3 but not Ser6 mutant Gal-3.
Death signals initiated by anticancer drugs induce cytochrome c release from mitochondria and subsequent caspase 3 activation, resulting in apoptosis (36). Recently, it was reported that Gal-3 inhibits cytochrome c release and caspase 3 activation of breast cancer cells (62). Therefore, we investigated the molecular regulation of the Ser6 mutation in these processes. BT-549 cells expressing wt and mutant Gal-3 were exposed to CDDP for 48 h at 37°C, and cytochrome c released from the mitochondria to the cytoplasm was determined by Western blot analysis. As shown in Fig. 7, panels IA and B, CDDP increased cytochrome c release in BT-549/V and BT-549/S6A cells by 1.8- and 2.5-fold, respectively. However, no detectable amount of cytochrome c was observed in drug-treated BT-549/WT1 cells, suggesting that only wt Gal-3 inhibits cytochrome c release. Similar results were obtained when BT-549/parental, BT-549/WT2, and BT-549/S6E cells were used (data not shown). Next, caspase-3 activation was examined using flow cytometry (Fig. 7, panel II). Treatment with 25 μM CDDP for 48 h increased the number of caspase 3-positive cells in BT-549/V and BT-549/S6A cells from 1.2 to 20.2% and from 0.8 to 20.2%, respectively, while the number of active caspase 3-positive cells was about 1 log unit lower in BT-549/WT1 cells (0.2 to 3.1%).
FIG. 7.

Cytochrome c release and subsequent caspase activation were inhibited by wt Gal-3 but not Ser6 mutant Gal-3. (I) (A) Western blot analysis of cytochrome c release in BT-549/V, BT-549/WT1, and BT-549/S6A, cells. BT-549/V, BT-549/WT1, and BT-549/S6A cells were exposed to CDDP for 48 h. Cytoplasmic fractions of these cells were subjected to Western blot analysis. The amount of Cytochrome c was determined by densitometric analysis. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD. (II) Caspase 3 activity in BT-549/V, BT-549/WT1, and BT-549/S6A cells. Cells without (shaded histogram) or with (open histogram) exposure to 25 μM CDDP for 48 h were fixed, incubated with FITC-conjugated anti-active caspase 3 antibody, and analyzed by flow cytometry. M1 indicates active caspase 3-positive cells. (III) DEVDase and LEHDase activity in BT-549/V, BT-549/WT1, and BT-549/S6A, cells. The cells were exposed to 25 μM CDDP for 0, 24, and 48 h. DEVDase and LEHDase activity was measured with colorimetric DEVD and LEHD substrates. Squares, circles, and triangles represent BT-549/V, BT-549/WT1, and BT-549/S6A, respectively. Data are means and SD from three separate experiments performed in duplicate.
To further study the effect of Gal-3 on caspase activation, we measured DEVDase and LEHDase activities, which represent caspase 3 and caspase 9 activity, respectively. After a 48-h treatment with 25 μ M CDDP, there was a 13- and 25-fold increase in DEVDase activity in BT-549/V and BT/549/S6A cells, respectively, and an 8- and 10-fold increase in LEHDase activity in BT-549/V and BT/549/S6A cells, respectively, whereas, both DEVDase and LEHDase activities were unaltered in BT-549/WT1 cells in response to the drug insult. Similar results were obtained when BT-549/parental, BT-549/WT2, and BT-549/S6E were used (data not shown).
These results suggest that phosphorylation of Gal-3 is essential for the inhibition of cytochrome c release and subsequent caspase 9 and caspase 3 activation in response to cytotoxic drugs.
Up-regulation of MAPK pathways by Gal-3.
MAPK signaling pathways regulate cell survival, differentiation, and death and have been implicated in the response of tumor cells to chemotherapeutic drug (15). Recently, it was shown that intracellular galectin-7 up-regulates JNK pathways (30) and intracellular galectin-1 binds H-ras, resulting in activation of the ERK pathway (42). Therefore, we investigated the activation of three major MAPK pathways, ERK, p38, and JNK, using antibodies that recognize only activated forms of ERK, p38, and JNK, respectively (Fig. 8). The activation levels of ERK, which promotes cell survival, in BT-549/WT1 were 2.4- and 5-fold higher than those in BT-549/V or BT-549/S6A (Fig. 8A and B). In all clones (BT-549/V, BT-549/WT1, and BT-549/S6A), p38 was activated after exposure to CDDP and there was no difference in activation levels among the cell clones (data not shown). Activation of JNK was observed only in BT-549/WT1 cells. In BT-549/WT1 cells, JNK1 was inactivated after exposure to CDDP (1.5-fold decrease) while JNK2 was activated (3.1-fold increase). These results indicate that similarly to Gal-1 and Gal-7, Gal-3 up-regulates the ERK and JNK pathways.
FIG. 8.

Up-regulation of MAPK pathways by Gal-3. (A) Activation of MAPK pathways in BT-549/V, BT-549/WT1, and BT-549/S6A cells with or without treatment with 25 μM CDDP was evaluated by Western blot analysis using anti-active ERK and anti-active JNK antibody. (B and C) Densitometric analyses of the results in panel A. The experiments were repeated three times, and the data are means of the experiments. Bars indicate SD.
Effect of ERK and JNK inhibition on CDDP-induced apoptosis.
To further investigate the role of ERK and JNK pathways in the ability of Gal-3 to protect cells against apoptosis induction, we used the ERK inhibitor U0126 (46) and the JNK inhibitor SP600125 (16) and then evaluated CDDP-induced apoptosis of BT-549/WT1 cells. The inhibition of ERK and JNK activation was confirmed by Western blotting (Data not shown). As shown in Fig. 9, both U0126 and SP600125 had a tendency to increase apoptosis, by 1.34- and 1.17-fold, respectively. U0126 and SP600125 had no effect on BT-549/V and BT-549/S6A cells (data not shown). These results indicate that Gal-3 protects cells from apoptosis, at least in part by the activation of ERK and JNK pathways.
FIG. 9.
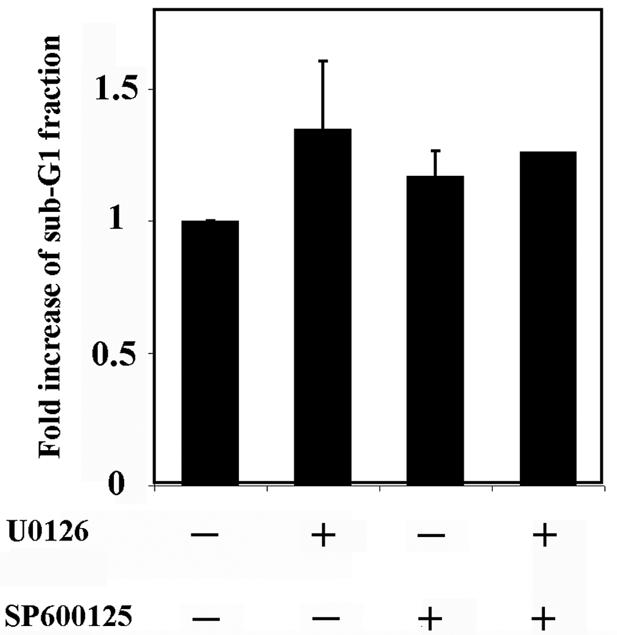
Inhibition of ERK and JNK increase the apoptosis of BT-549/WT1 cells. BT-549/WT1 cells were treated with U0126 and/or SP600125, incubated with 500 μM CDDP for 3 days, and analyzed by flow cytometry. Data are means of three independent experiments. Bars indicate SD. P < 0.005.
DISCUSSION
Galectins are a 14-member family of evolutionarily conserved carbohydrate binding proteins, some of which were reported to be novel regulators of apoptosis. Extracellular Gal-1 and Gal-9 induce the apoptosis of thymocytes and activated T cells (28, 43). Gal-7, an early transcriptional target of p53, enhances the apoptosis of keratinocytes and colon cancer cells (6, 30), while intracellular Gal-1 promotes transformation in cooperation with Ras, resulting in cell survival (42). In contrast, our studies (1, 29, 31, 61) and others have shown that intracellular, but not extracellular Gal-3 inhibits apoptosis. Recent studies have revealed that Gal-3 protects cells from apoptotic stimuli by CDDP (1, 61), anti-Fas antibody, staurosporine (57), suspension culture (29, 61) and nitric oxide (35). In this study, we demonstrated that Gal-3 translocated from the nucleus to the cytoplasm after apoptotic stimuli and that the enhanced nuclear export by phosphorylation at Ser6 is required for antiapoptotic activity. In addition, cells with the Ser6 mutant Gal-3, which have more Gal-3 in their nucleus than do cells with wt Gal-3, are more sensitive to apoptotic stimuli than are Gal-3-null cells. This is consistent with previous findings from immunohistochemical clinical studies, showing that the increase in the level of cytoplasmic Gal-3 and the loss of nuclear Gal-3 correlate with tumor progression (22, 33, 53). From these findings and the findings in this study, we propose the following model: Gal-3 phosphorylated by CK1 translocates from the nucleus to the cytoplasm and activates ERK and JNK; these activations of MAPK pathways, at least in part, inhibit cytochrome c release and subsequent caspase activation, resulting in suppression of apoptosis (Fig. 10).
FIG. 10.

Model of the role of intracellular Gal-3 in apoptosis. Gal-3 phosphorylated by CK1 translocates from the nucleus to the cytoplasm and activates ERK and JNK. These activations of the MAPK pathways inhibit cytochrome c release and subsequent caspase activation, resulting in suppression of apoptosis.
In response to apoptotic stimuli, Gal-3 is translocated to intracellular membranes including the mitochondria, a key target site for apoptotic stimuli (52). Although Gal-3 does not belong to the Bcl-2 family, it effectively protects mitochondrial integrity and down-regulates the caspase cascade in response to apoptotic signals. Gal-3 contains sequence and functional similarities to Bcl-2; it has the NWGR motif, an antideath motif which is highly conserved within the BH1 domain of the Bc1-2 family (52, 59), and both Gal-3 and Bcl-2 have antiapoptotic functions regulated, in part, by serine phosphorylation. The functional role of Bcl-2 phosphorylation at Ser70 in apoptosis is debatable due to conflicting reports (19, 26, 38, 44, 56, 60). These reports have shown that Bcl-2 phosphorylation at Ser70 activates or inactivates its antiapoptotic activity. It is possible that the apparent contradiction results from the differences among cell types and apoptotic stimuli used and from the effect of other kinases implicated in Bcl-2 phosphorylation including protein kinase C (47), JNK (56), Raf-1 (8), and protein kinase A (50). Gal-3 has acidic residues on both sides of Ser6, making it a likely substrate for either CK1 and/or CK2, but only CK1 was found to phosphorylate Gal-3 in vitro (25). CK1 is a member of a subgroup of mammalian protein kinase family homologues of budding yeast Hrr25, a nuclear Ser/Thr protein kinase that phosphorylates the Swi6 transcription factor (21). A large number of CK1 isoforms with different functions have been identified and are found in all cellular components. Although the majority of CK1 is found in the cytoplasm, CK1 translocates to the nucleus after irradiation (21). The phosphorylation of Gal-3 by CK1 is not a unique event. It has already been been described that CK1 phosphorylates a large number of proteins including tumor necrosis factor alpha receptor, cytoskeletal components such as spectrin, neurofilaments, and enzymes such as the regulatory component of protein phosphatase I (18). Moreover, there are a number of nuclear substrates for CK1 such as RNA polymerase II, simian virus 40 large T antigen, and p53 (18). In addition, it was shown that Snail phosphorylation translocated it out of the nucleus into the cytoplasm (14) and that a cluster of serine phosphorylation sites in FKHR appear to accelerate its nuclear export (7). Since it was concluded that only the phosphorylated form of Gal-3 could be found in the nuclear export fraction of digitionin-permeabilized cells (51), we may speculate that the nuclear localization of Gal-3 is required for its phosphorylation by CK1 with subsequent export to the cytoplasm.
Here, we show that phosphorylation of Gal-3 at Ser6 regulates its subcellular localization and its antiapoptotic function. Phosphorylation is a common signaling mechanism regulating subcellular compartmentalization of a large number of proteins. Serine phosphorylation of several apoptosis-related proteins changes their subcellular localization. Bad, one of the proapoptotic Bcl-2 family proteins, translocates from the mitochondria to the cytosol after phosphorylation at Ser136 by phosphatidylinositide-3-OH kinase (41). Phosphorylation of β-catenin by CK1 promotes its proteasomal degradation, and only nonphosphorylated β-catenin translocates from the cytosol to the nucleus, resulting in its accumulation in the nucleus (4). P38-regulated/activated protein kinase can shuttle between the cytoplasm and the nucleus, and its nuclear export requires p38-mediated phosphorylation (37). Raf-1 phosphorylated at Ser338 and Ser339 by p21-activated protein kinase 1 translocates to the mitochondria and protects endothelial cells from apoptosis (3). Thus, it appears that serine phosphorylation plays a key role in regulation of Gal-3 function as well as that of other apoptosis-related proteins.
Although it is unclear how Gal-3 exerts this antiapoptotic function, previous studies have demonstrated that Gal-3 influences mitochondrial homeostasis, resulting in antiapoptosis (62). In this study, we showed that Gal-3 activates MAPK pathways. Moreover, we demonstrated that independent inhibition of these pathways results in an increase of apoptosis. These results suggest that activation of ERK and JNK pathways may explain, in part, the antiapoptotic activity of Gal-3, although other mechanisms of Gal-3 protection of cells from apoptosis are not known yet. MAPKs, originally found as kinases activated by a mitogen such as insulin, regulate cell growth as well as differentiation, survival, and apoptosis (9). Previous reports implicated galectins in MAPK pathways. It was reported that Gal-7, a p53-inducible protein, up-regulates the JNK pathway and induces apoptosis (30). Intracellular Gal-1 mediates Ras membrane anchorage, resulting in activation of the Ras-ERK pathway (42). Galectin family members have homology of the carbohydrate-binding motif, and all intracellular galectins may recognize a common ligand(s) (11). Thus, Gal-3 also may affect MAPK pathways. We show here that Gal-3 up-regulated the ERK pathway while Ser 6 mutant Gal-3 did not. Since activation of the ERK pathway induces Bad phosphorylation, stabilizes mitochondria, and protects cells from apoptosis (54), activation of the ERK pathway by Gal-3 may explain its antiapoptotic activity. We also demonstrated that Gal-3 activated another member of the MAPK family protein, JNK, shown by others to promote apoptosis (15). However, c-jun-deficient mouse embryo fibroblasts are more sensitive to apoptosis induced by UV irradiation than are normal controls, suggesting that JNK pathway activation promotes cell survival under certain conditions (55). On the other hand, involvement of JNK pathway in carcinogenesis has been evidenced by genetic analyses of mice. c-jun-deficient MEFs exhibit a profound senescence-like growth arrest in culture (49) and are resistant to transformation by the Ras oncogene (27). A recent report showed that both ERK and JNK activation are required for transformation by Ras (45). Therefore, activation of JNK and ERK by Gal-3 may promote transformation.
Although the cytoplasmic and the nuclear localization of Gal-3 is well known (10, 24, 39, 51), the mechanism by which Gal-3 is shuttled to, retained within, or exported from the nucleus is unclear. It was suggested that Gal-3 might be associated with Gemin4, a component of the macromolecular complex designated the survival of motor neuron SMN complex, and Gal-3 may be taken along when SMN is imported into the nucleus. Once in the nucleus, Gal-3 may participate with SMN in the assembly of the spliceosome complex (40). It is also possible that Gal-3 translocation to the nucleus is passive, since it does not contain a canonical basic nuclear localization sequence and its small size allows passive diffusion through the nuclear pore complex. Phosphorylation of serine is the only known posttranslational modification of Gal-3 and thus may play a key role in its function. Since phosphorylation abrogates its sugar-binding ability (34), it is possible that it leads to its release from the nuclear complex, followed by export into the cytoplasm. In the cytoplasm, Gal-3 may interact with either Bcl-2 (57) or synexin (62), allowing it to be translocated to the perinuclear mitochondrial membrane, where it regulates mitochondrial integrity critical to apoptosis regulation. In summary, we show here that Gal-3 phosphorylation regulates its subcellular localization and antiapoptotic function.
Acknowledgments
We thank Victor Hogan and Vivian Powell for editing the manuscript.
This work was supported in part by grant CA46120 from the NIH/NCI (to A.R.), by DOD grant DAMD17-99-1-9442 (to H.-R.C.K.), and by grant NIH/NCI CA 69480 (to R.S.B.).
REFERENCES
- 1.Akahani, S., P. N. Makker, H. Inohara, H. R. Kim, and A. Raz. 1997. Galectin-3: a novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res. 57:5272-5276. [PubMed] [Google Scholar]
- 2.Akari, H., S. Bour, S. Kao, A. Adachi, and K. Strebel. 2001. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J. Exp. Med. 194:1299-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alavi, A., J. D. Hood, R. Frausto, D. G. Stupack, and D. A. Cheresh. 2003. Role of Raf in vascular protection from distinct apoptotic stimuli. Science 301:94-96. [DOI] [PubMed] [Google Scholar]
- 4.Amit, S., A. Hatzubai, Y. Birman., J. S. Andersen, E. Ben-Shushan, M. Mann, Y. Ben-Neriah, and I. Alkalay. 2002. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 16:1066-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barondes, S. H., D. N. Cooper, M. A. Gitt, and H. Leffler. 1994. Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem. 269:20807-20810. [PubMed] [Google Scholar]
- 6.Bernerd, F., A. Sarasin, and T. Magnaldo. 1999. Galectin-7 overexpression is associated with the apoptotic process in UVB-induced sunburn keratinocytes. Proc. Natl. Acad. Sci. USA 96:11329-11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biggs W. H., III, J. Meisenhelder, T. Hunter, W. K. Cavenee, and K. C. Arden. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 96:7421-7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blagosklonny, M. V., P. Giannakakou, W. S. el- Deiry, D. G. I. Kingston, P. I. Higgs, L. Neckers, and T. Fojo. 1997. Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death. Cancer Res. 57:130-135. [PubMed] [Google Scholar]
- 9.Blenis, J. 2001. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 12:397-408 [PubMed] [Google Scholar]
- 10.Cowles, E. A., N. Agrwal, R. L. Anderson, and J. L. Wang. 1990. Carbohydrate-binding protein 35. Isoelectric points of the polypeptide and a phosphorylated derivative. J. Biol. Chem. 265:17706-17712. [PubMed] [Google Scholar]
- 11.Danguy, A., I. Camby, and R. Kiss. 2002. Galectins and cancer. Biochim. Biophys. Acta 1572:285-293. [DOI] [PubMed] [Google Scholar]
- 12.Davidson, P. J., M. J. Davis, R. J. Patterson, M. A. Ripoche, F. Poirier, and J. L. Wang. 2002. Shuttling of galectin-3 between the nucleus and cytoplasm. Glycobiology 12:329-337. [DOI] [PubMed] [Google Scholar]
- 13.Dignam, J. D., P. L. Martin, B. S. Shastry, and R. G. Roeder. 1983. Eukaryotic gene transcription with purified components. Methods Enzymol. 101:582-598. [DOI] [PubMed] [Google Scholar]
- 14.Dominguez, D., B. Montserrat-Sentis, A. Virgos-Soler, S. Guaita, J. Grueso, M. Porta, I. Puig, J. Baulida, C. Franci, and A. Garcia de Herreros. 2003. Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol. Cell. Biol. 23:5078-5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan, M., and T. C. Chambers. 2001. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist. Update 4:253-267. [DOI] [PubMed] [Google Scholar]
- 16.Ghose, A., J. Fleming, K. El-Bayoumy, and P. R. Harrison. 2001. Enhanced sensitivity of human oral carcinomas to induction of apoptosis by selenium compounds: involvement of mitogen-activated protein kinase and Fas pathways. Cancer Res. 61:7479-7487. [PubMed] [Google Scholar]
- 17.Gong, H. C., Y. Honjo, P. N. Makker, V. Hogan, N. Mazurak, R. S. Bresalier, and A. Raz. 1999. The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res. 59:6239-6245. [PubMed] [Google Scholar]
- 18.Gross, S. D., and R. A. Anderson. 1998. Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signaling 10:699-711. [DOI] [PubMed] [Google Scholar]
- 19.Haldar, S., A. Basu, and C. M. Croce. 1998. Serine-70 is one of the critical sites for drug-induced Bcl2 phosphorylation in cancer cells. Cancer Res. 58:1609-1615. [PubMed] [Google Scholar]
- 20.Herrmann, J., C. W. Turck, R. E. Atchison, M. E. Huflejt, L. Poulter, M. A. Gitt, A. L. Burlingame, S. H. Barondes, and H. Leffler. 1993. Primary structure of the soluble lactose binding lectin L-29 from rat and dog and interaction of its non-collagenous proline-, glycine-, tyrosine-rich sequence with bacterial and tissue collagenase. J. Biol. Chem. 268:26704-26711. [PubMed] [Google Scholar]
- 21.Ho, Y., S. Mason, R. Kobayashi, M. Hoekstra, and B. Andrews. 1997. Role of the casein kinase I isoform, Hrr25, and the cell cycle-regulatory transcription factor, SBF, in the transcriptional response to DNA damage in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 94:581-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honjo, Y., H. Inohara, S. Akahani T. Yoshii, Y. Takenaka, J. Yoshida, K. Hattori, Y. Tomiyama, A. Raz, and T. Kubo. 2000. Expression of cytoplasmic galectin-3 as a prognostic marker in tongue carcinoma. Clin. Cancer Res. 6:4635-4640. [PubMed] [Google Scholar]
- 23.Hsu, D. K., R. Y. Yang, Z. Pan, L. Yu, D. R. Salomon, W. P. Fung Leung, and F. T. Liu. 2000. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am. J. Pathol. 156:1073-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubert, M., S. Y. Wang, J. L. Wang, A. P. Seve, and J. Hubert. 1995. Intranuclear distribution of galectin-3 in mouse 3T3 fibroblasts: comparative analyses by immunofluorescence and immunoelectron. Exp. Cell Res. 220:397-406. [DOI] [PubMed] [Google Scholar]
- 25.Huflejt, M. E., C. W. Turck, R. Lindstedt, S. H. Barondes, and H. Leffler. 1993. L-29, a soluble lactose-binding lectin, is phosphorylated on serine 6 and serine 12 in vivo and by casein kinase I. J. Biol. Chem. 268:26712-26718. [PubMed] [Google Scholar]
- 26.Ito, T., X. Deng, B. Carr, and W. S. May. 1997. Bcl-2 phosphorylation required for anti-apoptosis function. J. Biol. Chem. 272:11671-11673. [DOI] [PubMed] [Google Scholar]
- 27.Johnson, R., B. Spiegelman, D. Hanahan, and R. Wisdom. 1996. Cellular transformation and malignancy induced by ras require c-jun. Mol. Cell. Biol. 16:4504-4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kageshita, T., Y. Kashio, A. Yamauchi, M. Seki, M. J. Abedin, N. Nishi, H. Shoji, T. Nakamura, T. Ono, and M. Hirashima. 2002. Possible role of galectin-9 in cell aggregation and apoptosis of human melanoma cell lines and its clinical significance. Int. J. Cancer 99:809-816. [DOI] [PubMed] [Google Scholar]
- 29.Kim, H. R., H. M. Lin, H. Biliran, and A. Raz. 1999. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 59:4148-4154. [PubMed] [Google Scholar]
- 30.Kuwabara, I., Y. Kuwabara, R. Y. Yang, M. Schuler, D. R. Green, B. L. Zuraw, D. K. Hsu, and F. T. Liu. 2001. Galectin-7 (PIG1:p53-induced gene 1) exhibits pro-apoptotic function through JNK activation and mitochondrial cytochrome c release J. Biol. Chem. 277:3487-3497. [DOI] [PubMed] [Google Scholar]
- 31.Lin, H. M., B. K. Moon, F. Yu, and H. R. Kim. 2000. Galectin-3 mediates genistein-induced G(2)/M arrest and inhibits apoptosis. Carcinogenesis 21:1941-1945. [DOI] [PubMed] [Google Scholar]
- 32.Liu, F. T., R. J. Patterson, and J. L. Wang. 2002. Intracellular functions of galectins. Biochim. Biophys. Acta 1572:263-273. [DOI] [PubMed] [Google Scholar]
- 33.Lotz, M. M., C. W. Andrews, C. A. Korzelius, E. C. Lee, G. D. Steele, A. Clarke, and A. M. Mercurio. 1993. Decreased expression of Mac-2 (carbohydrate binding protein 35) and loss of its nuclear localization are associated with the neoplastic. Proc. Natl. Acad. Sci. USA 90:3466-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazurek, N., J. Conklin, J. C. Byrd, A. Raz, and R. S. Bresalier. 2000. Phosphorylation of the beta-galactoside-binding protein galectin-3 modulates binding to its ligands. J. Biol. Chem. 275:36311-36315. [DOI] [PubMed] [Google Scholar]
- 35.Moon, B. K., Y. J. Lee, P. Battle, J. M. Jessup, A. Raz, and H. R. C. Kim. 2001. Galectin-3 protects human breast carcinoma cells against nitric oxide-induced apoptosis: implication of galectin-3 function during metastasis. Am. J. Pathol. 159:1055-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mow, B. M., A. L. Blajeski, J. Chandra, and S. H. Kaufmann. 2001. Apoptosis and the response to anticancer therapy. Curr. Opin. Oncol. 13:453-462. [DOI] [PubMed] [Google Scholar]
- 37.New, L., Y. Jiang, and J. Han. 2003. Regulation of PRAK subcellular location by p38 MAP kinases. Mol. Biol. Cell 14:2603-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ojala, P. M., K. Yamamoto, E. Castanos-Velez, P. Biberfeld, S. J. Korsmeyer, and T. P. Makela. 2000. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat. Cell Biol. 11:819-825. [DOI] [PubMed] [Google Scholar]
- 39.Openo, K. P., M. M. Kadrofske, R. J. Patterson, and J. L. Wang. 2000. Galectin-3 expression and subcellular localization in senescent human fibroblasts. Exp. Cell Res. 255:278-290. [DOI] [PubMed] [Google Scholar]
- 40.Park, J. W., P. G. Voss, S. Grabski, J. L. Wang, and R. J. Patterson. 2001. Association of galectin-1 and galectin-3 with Gemin4 in complexes containing the SMN protein. Nucleic Acids Res. 29:3595-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pastorino, J. G., M. Tafani, and J. L. Farber. 1999. Tumor necrosis factor induces phosphorylation and translocation of BAD through a phosphatidylinositide-3-OH kinase-dependent pathway. J. Biol. Chem. 274:19411-19416. [DOI] [PubMed] [Google Scholar]
- 42.Paz, A., R. Haklai, G. Elad-Sfadia, E. Ballan, and Y. Kloog. 2001. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 20:7486-7493. [DOI] [PubMed] [Google Scholar]
- 43.Perillo, N. L., K. E. Pace, J. J. Seilhamer, and L. G. Baum. 1995. Apoptosis of T cells mediated by galectin-1. Nature 378:736-739. [DOI] [PubMed] [Google Scholar]
- 44.Pratesi, G., P. Perego, and F. Zunino. 2001. Role of Bcl-2 and its post-transcriptional modification in response to antitumor therapy. Biochem. Pharmacol. 61:381-386. [DOI] [PubMed] [Google Scholar]
- 45.Pruitt, K., W. M. Pruitt, G. K. Bilter, J. K. Westwick, and C. J. Der. 2002. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J. Biol. Chem. 277:31808-31817. [DOI] [PubMed] [Google Scholar]
- 46.Rice, P. L., R. J. Goldberg, E. C. Ray, L. J. Driggers, and D. J. Ahnen. 2001. Inhibition of extracellular signal-regulated kinase 1/2 phosphorylation and induction of apoptosis by sulindac metabolites. Cancer Res. 61:1541-1547. [PubMed] [Google Scholar]
- 47.Ruvolo, P. P., X. Deng, B. K. Carr, and W. S. May. 1998. A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J. Biol. Chem. 273:25436-25442. [DOI] [PubMed] [Google Scholar]
- 48.Salah-Eldin, A. E., S. Inoue, S. Tsukamoto, H. Aoi, and M. Tsuda. 2003. An association of Bcl-2 phosphorylation and Bax localization with their functions after hyperthermia and paclitaxel treatment. Int. J. Cancer 103:53-60. [DOI] [PubMed] [Google Scholar]
- 49.Schreiher, M., A. Kolbus, F. Piu, A. Szabowski, U. Mohle-Steinlein, J. Tian, M. Karin, P. Angel, and E. F. Wagner. 1999. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 13:607-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Srivastava, R. K., A. R. Srivastava, S. J. Korsmeyer, M. Nesterova, Y. S. Cho-Chung, and D. L. Longo. 1998. Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol. Cell. Biol. 18:3509-3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsay, Y. G., N. Y. Lin, P. G. Voss, R. J. Patterson, and J. L. Wang. 1999. Export of galectin-3 from nuclei of digitonin-permeabilized mouse 3T3 fibroblasts. Exp. Cell Res. 252:250-261. [DOI] [PubMed] [Google Scholar]
- 52.Tsujimoto, Y., and S. Shimizu. 2000. Bcl-2 family: life-or-death switch. FEBS Lett. 466:6-10. [DOI] [PubMed] [Google Scholar]
- 53.Van den Brule, F. A., D. Waltregny, F. T. Liu, and V. Castronovo. 2000. Alteration of the cytoplasmic/nuclear expression pattern of galectin-3 correlates with prostate carcinoma progression. Int. J. Cancer 89:361-367. [DOI] [PubMed] [Google Scholar]
- 54.Wang, H. G., U. R. Rapp, and J. C. Reed. 1996. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell 87:629-638. [DOI] [PubMed] [Google Scholar]
- 55.Wisdom, R., R. S. Johnson, and C. Moore. 1999. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 18:188-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto, K., H. Ichijo, and S. Korsmeyer. 1999. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 19:8469-8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang, R. Y., D. K. Hsu, and F. T. Liu. 1996. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. USA 93:6737-6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang, R. Y., and F. T. Liu. 2003. Galectins in cell growth and apoptosis. Cell Mol. Life Sci. 60:267-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin, X. M., Z. N. Oltvai, and S. J. Korsmeyer. 1994. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369:321-323. [DOI] [PubMed] [Google Scholar]
- 60.Yokote, H., T. Terada, H. Matsumoto, K. Kakishita, Y. Kinoshita, N. Nakao, K. Nakai, and T. Itakura. 2000. Dephosphorylation-induced decrease of anti-apoptotic function of Bcl-2 in neuronally differentiated P19 cells following ischemic insults. Brain Res. 857:78-86. [DOI] [PubMed] [Google Scholar]
- 61.Yoshii, T., T. Fukumori, Y. Honjo, H. Inohara, H. R. C. Kim, and A. Raz. 2002. Galectin-3 phosphorylation is required for its anti-apoptotic function and cell cycle arrest. J. Biol. Chem. 277:6852-6857. [DOI] [PubMed] [Google Scholar]
- 62.Yu, F., R. L. Finley, A. Raz, and H. R. C. Kim. 2002. Galectin-3 translocates to the perinuclear membranes and inhibits cytochrome c release from the mitochondria. A role for synexin in galectin-3 translocation. J. Biol. Chem. 277:15819-15827. [DOI] [PubMed] [Google Scholar]