

Abstract
Background
Placebo controlled trials of nonsteroidal antinflammatory drugs (NSAIDs) selective for inhibition of COX-2 reveal an emergent cardiovascular hazard in patients selected for low risk of heart disease. Postnatal global deletion of COX-2 accelerates atherogenesis in hyperlipidemic mice, a process delayed by selective enzyme deletion in macrophages.
Methods and Results
Here, selective depletion of COX-2 in vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) depressed biosynthesis of prostaglandin (PG)I2 and PGE2, elevated blood pressure and accelerated atherogenesis in Ldlr knockout (KO) mice. Deletion of COX-2 in VSMCs and ECs coincided with an increase in COX-2 expression in lesional macrophages and increased biosynthesis of thromboxane. Increased accumulation of less organized intimal collagen, laminin, α-smooth muscle actin and matrix-rich fibrosis was also apparent in lesions of the mutants.
Conclusions
Although atherogenesis is accelerated in global COX-2 KOs, consistent with evidence of risk transformation during chronic NSAID administration, this masks the contrasting effects of enzyme depletion in macrophages versus VSMCs and ECs. Targeting delivery of COX-2 inhibitors to macrophages may conserve their efficacy while limiting cardiovascular risk.
Keywords: cyclooxygenase, prostaglandin, atherogenesis, endothelial cell, vascular smooth muscle
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) designed specifically to inhibit cyclooxygenase (COX)-2 relieve pain and inflammation but expose patients to a cardiovascular hazard comprising myocardial infarction and stroke, hypertension, heart failure, arrhythmogenesis and sudden cardiac death.1 These effects are attributable to suppression of COX-2 derived cardioprotective prostaglandins (PGs), particularly prostacyclin (PGI2) in the vasculature and in cardiomyocytes.2 More controversial has been the potential impact of COX-2 inhibition on atherosclerosis. Extended dosing with COX-2 inhibitors in three placebo controlled trials was associated with emergence of a detectable increase in cardiovascular events in patients initially selected to be at low risk of heart disease.3-5 Consistent with this observation, deletion of the PGI2 receptor (the IP) fosters initiation and early development of atherogenesis in hyperlipidemic mice,6, 7 offering a potential mechanism for risk transformation during chronic drug exposure. However, experiments with inhibitors of COX-2 and conventional COX-2 knockout (KO) mice accelerated, delayed or had no effect on atherosclerosis.8 As this confusion may have reflected the failure to characterize the pharmacological specificity of enzyme inhibition and the diverse consequences of missing COX-2 during development, we induced global deletion of COX-2 postnatally. In these mice, atherogenesis is accelerated,9 consistent with the result in IPKOs. Cells tend to make one or two dominant COX products, often with contrasting biological effects - such as the divergent effects of platelet COX-1 derived thromboxane (Tx)A2 and COX-2 derived endothelial PGI2 on platelet activation.2, 10 Activated macrophages make predominantly TxA2 and PGE2 and ligation of the TxA2 receptor (TP) or the E prostanoid (EP) receptor 3 fosters atherogenesis.11, 12 Consistent with these observations, deletion of myeloid cell COX-2 restrains atherogenesis – an effect most likely attributable to macrophage gene deficiency as COX-2 is not expressed in mature platelets and minimally in dendritic cells and neutrophils.13 Given the contrasting effects of global and macrophage COX-2 deletion on disease evolution and the causative implication of enzyme inhibition in vascular cells in other aspects of the NSAID related cardiovascular hazard, we sought to elucidate the impact of COX-2 in ECs and VSMCs on atherosclerosis.
Material and Methods
Materials
All reagents used were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Generation of Vascular COX-2 KO Hyperlipidemic Mice
Endothelial cell COX-2 knockout (Tie2Cre/ COX-2F/F, EC KO), vascular smooth muscle cell COX-2 knockout (SM22Cre/ COX-2F/F, VSMC KO), and endothelial/ vascular smooth muscle cell double COX-2 knockout (Tie2Cre/ SM22Cre/ COX-2F/F, E/V DKO) mouse lines were generated as described.10, 14-16 These mouse lines were crossed with Ldlr−/− mice fully backcrossed onto a C57BL/6 background (The Jackson Laboratory, Bar Harbor, ME). All KOs were genotyped with tail DNA to confirm the presence of promoter specific Cre, COX-2F/F recombination, and an intact COX-2 allele to exclude Cre germline leakage. COX-2F/F/ Ldlr−/− mice were used as wild type (WT) littermate controls.
Animals
In all experiments, COX-2 deficient transgenic mice were compared with appropriate strain-, age-, and sex-matched control animals. Single nucleotide polymorphism (SNP) analyses showed EC KO, VSMC KO, and E/V DKO mouse lines for the atherosclerosis study achieved at least 90.5-92.5% purity on the C57BL/6 background. Mice of both genders were fed a high fat diet (HFD, 21.2% fat, 0.2% cholesterol, TD.88137, Harlan Teklad, Madison, WI) from 8 weeks of age for 3 and 6 months. Mice were weighed before and after the HFD feeding. All animals in this study were housed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. All experimental protocols were approved by IACUC.
Preparation of Mouse Aortas and En Face Quantification of Atherosclerosis
Mice were transferred after HFD feeding to new cages without food from 8am to 12pm. Water was provided ad libitum. All mice were sacrificed between 12pm to 4pm by CO2 over-exposure. The extent of atherosclerosis (Phase 3 Imaging Systems, Glen Mills, PA) was determined by the en face method and by assessment of aortic root lesion burden as previously described.17
Blood Pressure Measurement
Systolic blood pressure was measured in conscious mice using a computerized non-invasive tail-cuff system (Visitech Systems, Apex, NC) as described.18 Blood pressure was recorded once each day from 8am to 11am for 5-7 consecutive days after 3 days of training. Average systolic blood pressure was reported.
Mass Spectrometric Analysis of Prostanoids
Urinary prostanoid metabolites were measured by liquid chromatography / mass spectrometry as described.19 Such measurements provide a noninvasive, time integrated measurement of systemic prostanoid biosynthesis,20 reflective of short term minor alterations in product formation21 and of vascular stimulation.22 Briefly, mouse urine samples were collected using metabolic cages over a 15 hour period (6pm to 9am). Systemic production of PGI2, PGE2, PGD2, and TxA2 was determined by quantifying their major urinary metabolites - 2, 3-dinor 6-keto PGF1α (PGI-M), 7-hydroxy-5, 11-diketotetranorprostane-1, 16-dioic acid (PGE-M), 11, 15-dioxo-9α-hydroxy-2, 3, 4, 5-tetranorprostan-1, 20-dioic acid (tetranor PGD-M) and 2, 3-dinor TxB2 (Tx-M), respectively. Results were normalized with creatinine.
Immunohistochemical Examination of Lesion Morphology
Mouse hearts were embedded in OCT, and 10 μm serial sections of the aortic root were cut and mounted on Superfrost Plus slides (Fisher Scientific) for analysis of lesion morphology. Samples were fixed in acetone for 15 min at −20°C. Prior to treatment with the first antibody, sections were consecutively treated to block endogenous peroxidase (3% H2O2 for 15 min), with 10% normal serum blocking solution (dependent on host of secondary antibody, in 1%BSA/PBS for 15 min) and for endogenous biotin (streptavidin-biotin blocking kit, #SP-2002, Vector Laboratories). Sections were then incubated with the desired primary antibody in blocking solution overnight at 4°C. Samples were individually stained for collagen type-I (1 μg/ml, #1310-01, Southern Biotech), laminin (2 μg/ml, #L9393, Sigma), α-SMA (12.3 μg/ml, #F3777, Sigma), VCAM-1 (10 μg/ml, #553331, BD Bioscience), COX-1 (1 μg/ml, #160109, Cayman Chemicals), COX-2 (1 μg/ml, #160106, Cayman Chemicals), CD11b (5 μg/ml, #557395, BD Bioscience) and CD11c (5 μg/ml, #553800, BD Bioscience), all with isotype-matched controls. Where required, sections were then incubated with biotinylated-IgG secondary antibody (specific to host of primary antibody, all 1 μg/ml, Vector Laboratories) diluted in 1% BSA/PBS for 1 hr at RT. Sections were then incubated with Streptavidin-Horseradish Peroxidase (1 μg/ml, #016-030-084, Jackson Immunoresearch) diluted in 1% BSA/PBS for 30 min at RT. Slides were equilibrated in sterile H2O for 5 min at RT, then developed using the DAB substrate kit (#K3468, Dako) as per manufacturers’ protocol. Samples were counterstained with hematoxylin, dehydrated and mounted in Cytoseal-60 (#12-547, Fisher Scientific). Isotype-match controls were performed in parallel and showed negligible staining in all cases.
COX-2- CD11b Stepwise Double Staining
Ten μm frozen sections were left to air-dry for 5 min at room temperature (RT). Samples were then fixed in acetone for 15 min at −20°C. Prior to treatment with the first antibody, samples were consecutively treated to block endogenous peroxidase (3% H2O2 for 15 min), with 10% normal goat serum blocking solution (in 1% BSA/PBS for 15 min) and for endogenous biotin (streptavidin-biotin blocking kit, #SP-2002, Vector Laboratories). The COX-2 primary antibody (1 μg/ml, #160106, Cayman Chemicals) or COX-2 IgG control (1 μg/ml, #011-000-003, Jackson Immunoresearch) diluted in blocking solution, was then added and samples were incubated overnight at 4°C. Following three 5 min washes with 0.05% Tween-20/PBS, samples were incubated with biotinylated goat-anti-rabbit IgG secondary antibody (1 μg/ml, #BA-1000, Vector Laboratories) diluted in 1% BSA/PBS for 1 hr at RT. Three more washes were carried out before samples were next incubated with Streptavidin-Alkaline Phosphatase (1 μg/ml, #016-050-084, Jackson Immunoresearch) diluted in 1% BSA/PBS for 30 min at RT. Samples were again washed 3 times before being equilibrated in 0.1 M Tris-HCl (pH 8.2) for 5 min at RT, then developed using the Vector Red substrate kit (#SK-5100, Vector Laboratories) as per the manufacturer’s protocol. Prior to double-staining with CD11b, samples were once again treated with the streptavidin-biotin blocking kit. Biotinylated-CD11b primary antibody (5 μg/ml, #557395, BD Pharmingen) or biotinylated-IgG control (5 μg/ml, #13-4031, eBioscience) diluted in 1% BSA/PBS were then added and samples were incubated overnight at 4°C. After 3 washes, samples were incubated with Streptavidin-Horseradish Peroxidase (1 μg/ml, #016-030-084, Jackson Immunoresearch) diluted in 1% BSA/PBS for 1 h at RT. Following 3 more washes, samples were equilibrated in sterile H2O for 5 min at RT, and developed using the DAB substrate kit (#SK-4100, Vector Laboratories) as per manufacturers’ protocol. Samples were then counterstained with hematoxylin, dehydrated and mounted in Cytoseal-60 (#12-547, Fisher Scientific).
Second Harmonic Generation Analysis of Fibrillar Collagen Structure
Ten μm aortic root frozen sections were left to air-dry for 5 min at RT prior to second harmonic generation (SHG) microscopy analysis of fibrillar collagen structure. Samples were then fixed in acetone for 15 min at −20°C and submerged in PBS in a tissue culture dish. SHG images of the fibrillar collagen and background tissue autofluorescence (AF) images were captured at 10× magnification using a Prairie Technologies Ultima 2-Photon Microscope system (Middleton, WI). Images were taken with an excitation wavelength of 910 nm, and captured through emission filters of 457–487 nm (SHG signal) and 525-570 nm (AF). The SHG and AF signals were pseudo-colored in green and red, respectively for observation of structure and morphology. Quantification of fibrillar collagen content, intensity and organization was calculated using Fiji Image Analysis Software. Firstly, the fibrillar collagen signal was isolated by subtracting the background AF signal from the original SHG image. Collagen content (area and intensity) was then quantified as percentage of total lesion area and total integrated density of the collagen signal, respectively. To measure fibrillar collagen organization, the Directionality macro (within Fiji) was first used to generate an FFT (Fast Fourier transform) powerplot of the fibrillar collagen signal. An ellipse was then superimposed over the positive signal generated within each powerplot, and the aspect ratio (AR) of the ellipse for each lesion was calculated. An AR with a value closer to 0 indicated random orientation; closer to 1 indicating orientated collagen fibers. For each lesion, 6 sections (equally spaced over the entire aortic root, ~350 μm) were analyzed for fibrillar collagen content, intensity and organization.
Statistical Analysis
For data analyzed by ANOVA, Holm Sidak’s or Dunnett’s post-test was used to compare the differences between the means only if the ANOVA returned a p < 0.05. Sample sizes were based on variability of the test measurement and the desire to detect a minimal 10% difference in the variables assessed with α = 0.05 and the power (1-β) = 0.8.
Results
Aortic COX-2 expression is modulated by diet and gene deletion
Expression of COX-2 in the aortic arch (AA) and thoracic aorta (TA) of WT mice increased with the high fat diet (HFD) as compared to normal chow fed animals (online-only Data Supplement Figure IA). Vascular tissue specific COX-2 KOs on chow diet revealed significant reductions in aortic COX-2 expression compared to WT mice on both a chow diet (online-only Data Supplement Figure IB and IC) and after HFD feeding for both 3 or 6 months (online-only Data Supplement Figure IF and IG). Although data from female mice at 6 months of HFD are illustrated, regardless of gender and/or study duration COX-1 expression in AA or TA was not significantly altered in the mutants on chow (online-only Data Supplement Figure ID and IE) or HFD (online-only Data Supplement Figure IH and II).
Deletion of Cox-2 in Vascular Cells Modulates Prostaglandin Biosynthesis in Mice on a High Fat Diet
There were no significant effects of genotype or gender on plasma cholesterol, triglycerides or glucose or on weight gain at different times on a HFD. Thereafter, we focused our analysis on males at 3 months and on females at 6 months of a HFD as the extent of their atherosclerotic lesions were similar and their lesional morphology not advanced to the point where genotype dependent effects are often undetectable. Deletion of COX-2 in vascular cells generally depressed biosynthesis of PGI2 (Figures 1A and 1E) and PGE2 (Figures 1B and 1F) as reflected by their urinary metabolites. However, this was not observed with EC deletion alone in female mice. This may reflect the imperfect matching of lesional development between males at 3 months and females at 6 months on a HFD. Biosynthesis of PGD2 was depressed by COX-2 deletion only in males (Figures 1C). By contrast thromboxane biosynthesis was increased in the single vascular mutants in both genders and in the female compound mutants (Figures 1D and 1H). However, it was depressed in the compound male mutants.
Figure 1.

Impact of COX-2 deletion on prostaglandin biosynthesis in mice on high fat diet. Overnight (5pm to 10am) fasting urine from WT and vascular COX-2 mutants were collected at the end of HFD feeding, and prostanoid metabolites were analyzed by liquid chromatography-mass spectrometry as described in the Methods. Urinary PGI-M (A and E) and PGE-M (B and F) were depressed in male and female COX-2 KOs after 3 and 6 months on HFD, respectively. Tx-M was increased in female KOs fed 6 months HFD. One-way ANOVA showed a significant effect of genotype on urinary prostanoid levels in both genders fed a HFD. Holm Sidak’s multiple comparison tests were used to test significant differences between WT and COX-2 mutants. Data are means ± SEMs. *p< 0.05, ***p<0.001, ****p<0.0001; n=18-22 per genotype.
Vascular COX-2 Depletion Elevates Systolic Blood Pressure and Restrains Atherogenesis
While systolic blood pressure (SBP) was not altered in single EC or VSMC KOs male mice after 3 months on HFD, a significant elevation was observed in the compound E/V mutants compared to WT mice (WT vs E/V DKO, 114± 1 mmHg vs 124± 2 mmHg, P= 0.0002, Figure 2A). For female mice fed HFD for 6 months, SBP was also significantly elevated in both the compound mutants and in those lacking COX-2 in VSMCs (WT vs VSMC KO, E/V DKO, 111± 2 mmHg vs 121± 2 mmHg, 120± 3 mmHg, P= 0.0001, Figure 2B). Atherosclerotic lesion burden was increased by deletion of COX-2 in vascular cells in both males at 3 months (WT vs EC, VSMC, E/V DKOs, 3.72± 0.4% vs 7.17± 0.7%, 5.19± 0.3%, 4.73± 0.6%, P= 0.0001, Figure 3A) and in females at 6 months (10.15± 0.7% vs 13.36± 0.6%, 11.76± 0.8%, 13.19± 0.6%, P= 0.003, Figure 3B) on a HFD. As lesions became advanced in males at 6 and 11 months on a HFD (online-only Supplement Figure II), these genotype dependent changes were lost.
Figure 2.

Vascular specific COX-2 deletion elevates systolic blood pressure. Systolic blood pressure (SBP) was measured in conscious mice using the computerized non-invasive tail-cuff system as described in the Methods. SBP of male mice was elevated after 3 months on a HFD in E/V DKO compared to WT mice (A). This hypertensive phenotype was further augmented in all mutants after 6 months on a HFD, with the exception of female EC KOs (B). One-way ANOVA showed a significant effect of genotype on SBP in both genders. Holm Sidak’s multiple comparison tests were used to test significant differences between WT and COX-2 mutants. Data are means ± SEMs. **p< 0.01, ***p< 0.001; n=18-22 per genotype.
Figure 3.

Vascular COX-2 restrains atherogenesis. Aortic atherosclerotic lesion burden, represented by the percentage of lesion area to total aortic area, was quantified by en face analysis of aortas from male mice fed a HFD for 3 and of female mice at 6 months. Representative en face preparations are shown (Lower panels). Lesion area tended to increase in male or female COX-2 mutants fed a HFD for 3 or 6 months respectively (A and B). One-way ANOVA revealed a significant effect of genotype (male, P= 0.0001 and female, P= 0.003) on lesion progression. Holm Sidak’s multiple comparison tests were used to test significant differences between WT and COX-2 KOs. Data are means ± SEMs. **p< 0.01, ****p<0.0001; n=18-22 per genotype.
Lesional Morphology Consequent to COX-2 Deletion in Vascular KOs
Consistent with the lesion burden analyzed by the en face method, cross-sectional analysis of aortic root samples in female mice on a HFD for 6 months revealed an increase in the total lesion area in both single and double COX-2 KOs compared to WTs (Figure 4A). Morphological analysis revealed fibrosis with increased accumulation of collagen and matrix-rich fibrotic areas detected by laminin staining in the COX-2 mutants compared to WT mice (Figure 5A and 5B). However, second harmonic generation (SHG) two-photon microscopy showed minimal changes to collagen content and collagen signal intensity between WT and E/V DKO lesions (online-only Data Supplement Figure IIIA and B) and a more random orientation/ less organized structure of lesional collagen fibrils in the DKO, as revealed by the Fast Fourier transform (FFT)-based analyses (Figure 4B). Representative SHG-collagen images of WT and E/V DKO were shown (Figure 4C and 4D).
Figure 4.
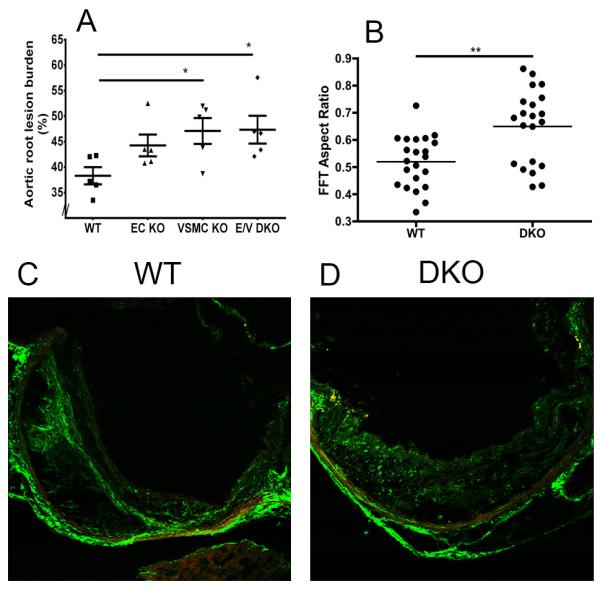
Vascular COX-2 restrains aortic root lesion burden and orientation of structural collagen in lesions. A. Quantification of cross-sectional analysis of aortic root samples from female mice fed 6 months HFD, was performed by measuring total lesion area across the aortic root as detailed in Methods. One-way ANOVA (Kruskal-Wallis test) revealed a significant effect of genotype (P=0.048) on lesion progression. Dunnett’s multiple comparison tests were used to test significant differences between WT and COX-2 KOs. *p< 0.05. Data are means ± SEMs. n=5 per group and 7-9 sections from each animal were averaged. B. Second harmonic generation (SHG) detection of structural collagen in WT (n=22) and E/V DKO (n= 21) lesions from female mice on a HFD for 6 months. Fast Fourier transform (FFT)-based analyses revealed a more random orientation of collagen fibers in E/V DKO lesions. Data are means ± SEMs. **p < 0.01 (Mann-Whitney test, two-tails). C and D. Representative WT and E/V DKO SHG images are shown.
Figure 5.

Morphometric consequences of vascular COX-2 deletion on lesion development. Lesion morphology in aortic roots from female mice fed HFD for 6 months were analyzed. Quantification of immunohistochemical staining of collagen (A), laminin (B), α-SMA (C) and VCAM-1 (D) from each genotype are shown in parallel with their representative aortic root sections. One-way ANOVA (Kruskal-Wallis test) revealed a significant effect of genotype (P<0.05) on lesion morphology. Dunnett’s multiple comparison tests were used to test significant differences between WT and COX-2 KOs. Data are means ± SEMs. n=4-5 per genotype. L- lumen, m- media, nc- necrotic core, fc- foam cell.
Alpha-smooth muscle actin (α-SMA) staining, a marker of differentiated VSMCs, was increased in both single and compound COX-2 KO lesions (Figure 5C). Necrotic cores in COX-2 KOs did not differ significantly in size as compared to those in WTs (Figure 5A-5C). However, only single COX-2 KOs showed an up-regulation of vascular cell adhesion molecule (VCAM-1) for activated VSMCs (Figure 5D). COX-2 positive lesional cells in the vascular mutants (online-only Data Supplement Figure IVD) co-stained with the inflammatory macrophage marker CD11b (Figure 6) in compound vascular mutants, while COX-1 expression was unaltered (online-only Data Supplement Figure IVC).
Figure 6.

Vascular COX-2 deletion increases COX-2 expression in lesional macrophages. Lesion morphology in aortic roots from female mice fed a HFD for 6 months were analyzed. Quantification of immunohistochemistry staining of COX-2 (A) and CD11b (B) from WT and E/V DKO are shown. A Mann-Whitney test (two-tailed) revealed a significant increase in COX-2 positive cells in DKO compared to WT, **p< 0.01; n=8-12 per genotype. Data are means ± SEMs. C. Representative aortic root sections from COX-2 staining (left panel) and CD11b (right panel) from E/V DKO are shown. Arrows indicate lesional cells that are stained positive for both COX-2 and CD11b. L- lumen, m- media.
Discussion
Nine placebo-controlled trials of COX-2 selective NSAIDs (rofecoxib, celecoxib and valdecoxib) have revealed a cardiovascular risk of myocardial infarction, hypertension, stroke and heart failure.23, 24 These risks are explicable in terms of the suppression of cardioprotective products of COX-2, particularly PGI2. In mice, inhibition or deletion of COX-2 dependent PGI2 formation augments the response to thrombogenic stimuli, elevates systemic and pulmonary blood pressure, disrupts vascular remodeling, and predisposes the animals to cardiac failure and arrhythmogenesis.12, 14, 18, 25
Three of these placebo controlled trials were performed in patients selected to be at low demographic risk of cardiovascular disease.3-5 Despite this, an increase in cardiovascular events became detectable with extended dosing with either celecoxib or rofecoxib, for more than one year. The time course of emergent risk would be consistent with a drug effect on atherogenesis, such as was observed in prostacyclin receptor (IP) deficient mice. However, the results of studies of COX-2 deletion or inhibition in hyperlipidemic mice have been conflicting. This may reflect the systemic consequences of COX-2 deficiency in utero, in the case of the conventional knockouts and of a failure to characterize the actual biochemical selectivity for COX-2 inhibition of the pharmacological regimens employed. This is an important point, as COX-1 inhibition or deletion attenuates atherogenesis.26, 27 More recently, we generated mice in which global COX-2 deletion was accomplished postnatally and here atherogenesis was accelerated in both genders when they were crossed into ApoE deficient mice.9
We have previously reported that both EC and VSMC COX-2 contribute substantially to systemic PGI2 formation under physiological conditions in normolipidemic mice as reflected by urinary PGI-M.10 Here, we extended these observations, showing that on a hyperlipidemic background, biosynthesis of both PGI2 and PGE2 derived in both genders substantially from COX-2 in vascular cells and that the enzyme contributed to PGD2 biosynthesis in males. Expression of COX-2 was up-regulated both in the aortic arch and in thoracic aortas of littermate control mice fed a HFD, consistent with the increase in urinary PGI-M in ApoE-deficient and LdlR-deficient mice.28
Platelet and neutrophil-vessel wall interactions during the development of atherosclerosis are reflected by the urinary thromboxane metabolite 2,3-dinor TxB2 (Tx-M).28, 29 TxA2 is also a dominant product of activated macrophages. Theoretically, cells outside the vascular compartment – glial cells and glomeruli for example – have the capacity to generate Tx and may contribute to urinary TxM. Mice lacking the IP fed a HFD excrete higher levels of Tx-M in both genders.6 Moreover, previous studies have shown that COX-2-dependent formation of PGI2 in the vasculature restrains TxA2 biosynthesis.13, 28 Here, urinary Tx-M was increased in COX-2 mutants fed a HFD, which perhaps reflects removal of a regulatory constraint on myeloid cell activation by suppression of the biosynthesis of PGI2.30 This phenomenon was not observed in male E/V DKOs, in which urinary TxM was depressed. Whether this reflects a feedback response to the differential impact of male sex hormones on thromboxane receptor expression31 that would be more exaggerated in the compound mutant or some other mechanism is unknown. Previous studies in which COX-2 is deleted in a cell specific fashion have highlighted the importance of prostaglandins in regulation of expression of their biosynthetic enzymes. Thus, deletion of COX-2 in cardiomyocytes results in COX-2 upregulation in cardiac fibroblasts, with a shift in the dominant enzyme product from PGI2 in the former to PGF2α in the latter.14 Similarly, COX-2 deletion in plaque macrophages was associated with enzyme upregulation in vascular smooth muscle cells, implying a shift from TxA2 and PGE2 as the dominant product in the former to PGI2 in the latter.13 In both cases, the shift in product formation may have contributed to the resultant phenotype – cardiac fibrosis and arrhythmogenesis in the heart14 and attenuated atherogenesis in the vasculature.13 Here, we observed increased expression of COX-2 in lesional macrophages of the mice in which COX-2 had been deleted in vascular cells – consistent with both the reduction in PGIM and increase TxM excretion and the accelerated atherogenesis observed in these mutants.
We compared lesions of roughly similar size – at 3 months on a HFD in males and 6 months in females - and at a maturity where the impact of genotype was likely to be apparent. The COX-2 mutants developed more fibrotic lesions than did WT controls, as reflected by staining for collagen and laminin. This may be attributable to suppression of PGI2 formation. COX-2-derived PGI2 is an important negative regulator of the collagen synthesis that contributes to arterial stiffness.32 The accumulation of collagen and laminin in the atherosclerotic lesions can presumably be formed by the migration of VSMCs from the media, although the origin of the VSMCs is debatable.33 Interestingly, unlike previously reported proliferative/ activated VSMCs (decrease in α-SMA and increase in VCAM-1 staining) in atherosclerotic lesions,9, 13, 34, 35 subsets of lesional cells in the single mutants differed with respect to elevated expression of α-SMA (a marker of differentiated VSMCs) or of VCAM-1 (a marker of proliferative VSMCs).33,36 However, a proportion of these cells at discrete locations were stained positive for both α-SMA and VCAM-1. In contrast, VSMCs in the double E/V DKOs resembled the differentiated phenotype that displays a uniform α-SMA immunoreactivity and minimal VCAM-1. These findings suggest that VCAM-1 expression is dependent on COX-2 in the vascular cells and that the impact of COX-2 can be compensated in the single mutants, but not when the enzyme is completely depleted in vascular cells. Mature differentiated VSMCs retain the capacity for phenotypic plasticity.33 For example, in a rat model of balloon injury of carotid artery, medial VSMCs dedifferentiate after endothelial injury and migrate to the intima where they proliferate and secrete extracellular matrix components, such as collagen fibrils and elastin.37 Here, the proliferative VSMCs in mature neointima (after 14 days of endothelial injury) gradually redifferentiate and attain a contractile-like phenotype as observed in the media. Whereas immunohistochemical analysis clearly revealed an increase in lesional collagen content, SHG two-photon microscopy analyses indicate that lesion size, fibrillar collagen content and intensity were apparently unaltered between WT and E/V DKO. This disparity could be explained by the limitation of the SHG microscope in detecting the finer fibrillar collagen of lower order structure. In addition to higher order collagen structures, the antibody recognized the epitopes of fine collagen I structures, thus giving the higher signal. Despite this, we observed changes in the orientation of fibrillar collagens that accumulated in the atherosclerotic lesions. Specifically, fibrillar collagens in COX-2 E/V DKO lesions were significantly more random or less organized compared to WT lesions.
COX-2-derived prostanoids contribute to blood pressure control by regulating vascular tone and renal sodium transport,38 consistent with the risk of COX inhibitors in causing hypertension.39, 40 Indeed, we have previously shown that vascular deletion of COX-2 renders mice susceptible to dietary salt induced hypertension. Here, vascular COX-2 mutants on a hyperlipidemic background show a time-dependent increase in SBP, when placed on HFD. This hypertensive phenotype is consistent with suppressed biosynthesis of PGI2 and/ or PGE2 41-44 : deletion of either IP or EP2 results in susceptibility to dietary salt induced hypertension.44, 45
A picture thus emerges that products of COX-2 may restrain or accelerate atherogenesis depending on the cell type in which they are formed. Global postnatal deletion of COX-2 may most closely resemble the consequences of systemic administration of COX-2 inhibitors and indeed, the acceleration of atherogenesis in these mice is consistent with the apparent gradual cardiovascular risk transformation observed in placebo controlled, randomized trials of NSAIDs specific for inhibition of COX-2.23 However, this masks divergent effects of macrophage COX-2 derived TxA2 fostering disease and PGI2 derived from COX-2 in ECs and VSMCs acting as restraint. These observations and others 46-51 suggest that targeting inhibitors of COX-2 selectively to the macrophage13 may shift fundamentally the balance of cardiovascular efficacy and risk for NSAIDs.
Supplementary Material
Acknowledgements
We gratefully acknowledge the technical support and advice of Helen Zou, Wenxuan Li-Feng, Weili Yan and Gregory Grant. We thank Irene Crichton, Jennifer Bruce and Zhou Yu for assistance in the early stage of this work.
Funding Sources: This work was supported by a grant (HL062250) from the National Institutes of Health.
Footnotes
Conflict of Interest Disclosures: None.
Journal Subject Codes: [96] Mechanism of atherosclerosis/growth factors, [134] Pathophysiology
References
- 1.FitzGerald GA. Cox-2 in play at the aha and the fda. Trends Pharmacol Sci. 2007;28:303–307. doi: 10.1016/j.tips.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET, Investigators APCS Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 4.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA, Adenomatous Polyp Prevention on Vioxx Trial I Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 5.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M, Adenoma Prevention with Celecoxib Study I Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 6.Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, Fitzgerald GA. Cox-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane a(2) and prostacyclin in the development of atherosclerosis in apoe-deficient mice. J Clin Invest. 2004;114:784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilley SL, Coffman TM, Koller BH. Mixed messages: Modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Z, Crichton I, Tang SY, Hui Y, Ricciotti E, Levin MD, Lawson JA, Pure E, Fitzgerald GA. Disruption of the 5-lipoxygenase pathway attenuates atherogenesis consequent to cox-2 deletion in mice. Proc Natl Acad Sci U S A. 2012;109:6727–6732. doi: 10.1073/pnas.1115313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular cox-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3003787. 132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross S, Tilly P, Hentsch D, Vonesch JL, Fabre JE. Vascular wall-produced prostaglandin e2 exacerbates arterial thrombosis and atherothrombosis through platelet ep3 receptors. J Exp Med. 2007;204:311–320. doi: 10.1084/jem.20061617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudic RD, Brinster D, Cheng Y, Fries S, Song WL, Austin S, Coffman TM, FitzGerald GA. Cox-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96:1240–1247. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 13.Hui Y, Ricciotti E, Crichton I, Yu Z, Wang D, Stubbe J, Wang M, Pure E, FitzGerald GA. Targeted deletions of cyclooxygenase-2 and atherogenesis in mice. Circulation. 2010;121:2654–2660. doi: 10.1161/CIRCULATIONAHA.109.910687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–7552. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lepore JJ, Cheng L, Min Lu M, Mericko PA, Morrisey EE, Parmacek MS. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in sm22alpha-cre transgenic mice. Genesis. 2005;41:179–184. doi: 10.1002/gene.20112. [DOI] [PubMed] [Google Scholar]
- 16.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 17.Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murine models: Correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in ldl receptor-deficient and apolipoprotein e-deficient mice. J Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 18.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin e synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song WL, Lawson JA, Wang M, Zou H, FitzGerald GA. Noninvasive assessment of the role of cyclooxygenases in cardiovascular health: A detailed hplc/ms/ms method. Methods Enzymol. 2007;433:51–72. doi: 10.1016/S0076-6879(07)33003-6. [DOI] [PubMed] [Google Scholar]
- 20.FitzGerald GA, Pedersen AK, Patrono C. Analysis of prostacyclin and thromboxane biosynthesis in cardiovascular disease. Circulation. 1983;67:1174–1177. doi: 10.1161/01.cir.67.6.1174. [DOI] [PubMed] [Google Scholar]
- 21.Ciabattoni G, Maclouf J, Catella F, FitzGerald GA, Patrono C. Radioimmunoassay of 11-dehydrothromboxane b2 in human plasma and urine. Biochim Biophys Acta. 1987;918:293–297. doi: 10.1016/0005-2760(87)90233-5. [DOI] [PubMed] [Google Scholar]
- 22.Roy L, Knapp HR, Robertson RM, FitzGerald GA. Endogenous biosynthesis of prostacyclin during cardiac catheterization and angiography in man. Circulation. 1985;71:434–440. doi: 10.1161/01.cir.71.3.434. [DOI] [PubMed] [Google Scholar]
- 23.Grosser T, Yu Y, Fitzgerald GA. Emotion recollected in tranquility: Lessons learned from the cox-2 saga. Annu Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 24.Kang HJ, Oh IY, Chung JW, Yang HM, Suh JW, Park KW, Kwon TK, Lee HY, Cho YS, Youn TJ, Koo BK, Kang WY, Kim W, Rha SW, Bae JH, Chae IH, Choi DJ, Kim HS. Effects of celecoxib on restenosis after coronary intervention and evolution of atherosclerosis (mini-corea) trial: Celecoxib, a double-edged sword for patients with angina. Eur Heart J. 2012;33:2653–2661. doi: 10.1093/eurheartj/ehs001. [DOI] [PubMed] [Google Scholar]
- 25.Cathcart MC, Tamosiuniene R, Chen G, Neilan TG, Bradford A, O’Byrne KJ, Fitzgerald DJ, Pidgeon GP. Cyclooxygenase-2-linked attenuation of hypoxia-induced pulmonary hypertension and intravascular thrombosis. J Pharmacol Exp Ther. 2008;326:51–58. doi: 10.1124/jpet.107.134221. [DOI] [PubMed] [Google Scholar]
- 26.Pratico D, Tillmann C, Zhang ZB, Li H, FitzGerald GA. Acceleration of atherogenesis by cox-1-dependent prostanoid formation in low density lipoprotein receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:3358–3363. doi: 10.1073/pnas.061607398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McClelland S, Gawaz M, Kennerknecht E, Konrad CS, Sauer S, Schuerzinger K, Massberg S, Fitzgerald DJ, Belton O. Contribution of cyclooxygenase-1 to thromboxane formation, platelet-vessel wall interactions and atherosclerosis in the apoe null mouse. Atherosclerosis. 2009;202:84–91. doi: 10.1016/j.atherosclerosis.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 28.Pratico D, Cyrus T, Li H, FitzGerald GA. Endogenous biosynthesis of thromboxane and prostacyclin in 2 distinct murine models of atherosclerosis. Blood. 2000;96:3823–3826. [PubMed] [Google Scholar]
- 29.FitzGerald GA, Smith B, Pedersen AK, Brash AR. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N Engl J Med. 1984;310:1065–1068. doi: 10.1056/NEJM198404263101701. [DOI] [PubMed] [Google Scholar]
- 30.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane a2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 31.Pratico D, FitzGerald GA. Testosterone and thromboxane. Of muscles, mice, and men. Circulation. 1995;91:2694–2698. doi: 10.1161/01.cir.91.11.2694. [DOI] [PubMed] [Google Scholar]
- 32.Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, Pure E, Phillips MC, Lund-Katz S, Janmey PA, Assoian RK. Cardiovascular protection by apoe and apoe-hdl linked to suppression of ecm gene expression and arterial stiffening. Cell Rep. 2012;2:1259–1271. doi: 10.1016/j.celrep.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuff CA, Kothapalli D, Azonobi I, Chun S, Zhang Y, Belkin R, Yeh C, Secreto A, Assoian RK, Rader DJ, Pure E. The adhesion receptor cd44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J Clin Invest. 2001;108:1031–1040. doi: 10.1172/JCI12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babaev VR, Bobryshev YV, Stenina OV, Tararak EM, Gabbiani G. Heterogeneity of smooth muscle cells in atheromatous plaque of human aorta. Am J Pathol. 1990;136:1031–1042. [PMC free article] [PubMed] [Google Scholar]
- 36.Braun M, Pietsch P, Schror K, Baumann G, Felix SB. Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc Res. 1999;41:395–401. doi: 10.1016/s0008-6363(98)00302-2. [DOI] [PubMed] [Google Scholar]
- 37.Thyberg J, Blomgren K, Hedin U, Dryjski M. Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: An electron-microscopic and stereological study. Cell Tissue Res. 1995;281:421–433. doi: 10.1007/BF00417860. [DOI] [PubMed] [Google Scholar]
- 38.Smith WL. Prostanoid biosynthesis and mechanisms of action. Am J Physiol. 1992;263:F181–191. doi: 10.1152/ajprenal.1992.263.2.F181. [DOI] [PubMed] [Google Scholar]
- 39.FitzGerald GA. Cox-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov. 2003;2:879–890. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- 40.Aw TJ, Haas SJ, Liew D, Krum H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch Intern Med. 2005;165:490–496. doi: 10.1001/archinte.165.5.IOI50013. [DOI] [PubMed] [Google Scholar]
- 41.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin ii. J Clin Invest. 2002;110:61–69. doi: 10.1172/JCI14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Audoly LP, Ruan X, Wagner VA, Goulet JL, Tilley SL, Koller BH, Coffman TM, Arendshorst WJ. Role of ep(2) and ep(3) pge(2) receptors in control of murine renal hemodynamics. Am J Physiol Heart Circ Physiol. 2001;280:H327–333. doi: 10.1152/ajpheart.2001.280.1.H327. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin ep2 receptor. Nat Med. 1999;5:217–220. doi: 10.1038/5583. [DOI] [PubMed] [Google Scholar]
- 44.Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, Rockman HA, Fitzgerald GA, Koller BH, Coffman TM. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2005;2:201–207. doi: 10.1016/j.cmet.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Tilley SL, Audoly LP, Hicks EH, Kim HS, Flannery PJ, Coffman TM, Koller BH. Reproductive failure and reduced blood pressure in mice lacking the ep2 prostaglandin e2 receptor. J Clin Invest. 1999;103:1539–1545. doi: 10.1172/JCI6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Yang G, Xu X, Grant G, Lawson JA, Bohlooly YM, FitzGerald GA. Cell selective cardiovascular biology of microsomal prostaglandin e synthase-1. Circulation. 2013;127:233–243. doi: 10.1161/CIRCULATIONAHA.112.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers C, Edelman ER, Simon DI. A mab to the beta2-leukocyte integrin mac-1 (cd11b/cd18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc Natl Acad Sci U S A. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simon DI, Dhen Z, Seifert P, Edelman ER, Ballantyne CM, Rogers C. Decreased neointimal formation in mac-1(-/-) mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barron MK, Lake RS, Buda AJ, Tenaglia AN. Intimal hyperplasia after balloon injury is attenuated by blocking selectins. Circulation. 1997;96:3587–3592. doi: 10.1161/01.cir.96.10.3587. [DOI] [PubMed] [Google Scholar]
- 50.Danenberg HD, Fishbein I, Gao J, Monkkonen J, Reich R, Gati I, Moerman E, Golomb G. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106:599–605. doi: 10.1161/01.cir.0000023532.98469.48. [DOI] [PubMed] [Google Scholar]
- 51.Hoch JR, Stark VK, van Rooijen N, Kim JL, Nutt MP, Warner TF. Macrophage depletion alters vein graft intimal hyperplasia. Surgery. 1999;126:428–437. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.