

Abstract
A series of air-stable nickel complexes of the form L2Ni(aryl) X (L = monodentate phosphine, X = Cl, Br) and LNi(aryl)X (L = bis-phosphine) have been synthesized and are presented as a library of precatalysts suitable for a wide variety of nickel-catalyzed transformations. These complexes are easily synthesized from low-cost NiCl2·6H2O or NiBr2·3H2O and the desired ligand followed by addition of 1 equiv of Grignard reagent. A selection of these complexes were characterized by single-crystal X-ray diffraction, and an analysis of their structural features is provided. A case study of their use as precatalysts for the nickel-catalyzed carbonyl-ene reaction is presented, showing superior reactivity in comparison to reactions using Ni(cod)2. Furthermore, as the precatalysts are all stable to air, no glovebox or inert-atmosphere techniques are required to make use of these complexes for nickel-catalyzed reactions.
Introduction
Homogeneous nickel catalysis has continued to develop in recent years as a powerful set of tools for the construction of a wide variety of carbon–carbon and carbon–heteroatom bonds.1 Nickel, a base metal, is a low-cost, versatile, and attractive metal for use in catalytic transformations. Arguably the greatest barrier to the wider adoption of homogeneous nickel catalysis for synthesis, however, is the difficulty and cost of synthesizing and handling nickel(0) sources and the phosphines often used in conjunction with such complexes. It is for this reason we aim to further develop nickel(II)-based precatalysts, as this would greatly increase the accessibility of homogeneous nickel catalysis in both academic as well as industrial settings.
Ideally, these precatalysts would (1) be indefinitely air stable, (2) have a low molecular weight, (3) be highly active, (4) be simple to synthesize from low-cost materials, (5) be readily activated under a variety of conditions without producing interfering byproducts of activation, and (6) be applicable to virtually any nickel-catalyzed transformation. Precatalysts possessing many or all of these qualities would greatly add to the value of new as well as previously established nickel-catalyzed transformations.
Although nickel metal itself is extremely low in cost (∼15 USD/kg at commodity prices), the cost of nickel(0) sources such as Ni(cod)2 (cod = 1,5-cyclooctadiene) easily exceeds 20000 USD/kg, which often weakens the economic argument for using nickel in catalytic transformations. Indeed, Ni(cod)2 is only marginally less expensive than comparable palladium(0) sources such as Pd2(dba)3 (dba = dibenzylideneacetone) on a mole-for-mole basis, despite the fact that palladium metal is far more expensive than nickel metal.2 Taken in conjunction with the fact that a high catalyst loading of nickel is often required (5–20 mol %), there is little economic incentive to use nickel in place of precious metals such as palladium unless cheaper sources of nickel can be used. Regardless of cost and ease of use issues, however, any comparison between nickel and precious metals (particularly palladium) should recognize that nickel has demonstrated valuable and unique reactivity and behavior, which enables an entirely different set of chemical transformations.1
The use of precatalysts in transition-metal catalysis is not a new idea: indeed, Pd(OAc)2 has been in use as a precursor to Pd(0) species for close to 50 years,3 though the concept of the single-component, discrete precatalyst is a somewhat newer development.4 Several groups (Nolan,5 Buchwald,6 Organ,7 and others) have greatly advanced the ubiquity of precatalysts in organic synthesis, with much of the effort focused on palladium catalysts for cross couplings, amination, and related transformations.8 Likewise, though by far less established than those for palladium-catalyzed reactions, single-component nickel precatalysts are not new, with the most frequently employed being complexes such as (PPh3)2NiCl2.9 Though air stable, these precatalysts are usually limited to activation by nucleophilic organometallic reagents, and as such, they must often be preactivated by addition of an exogenous reductant.10 Such a strategy has been shown to be effective in many instances but is neither an ideal nor a universal solution.
Complexes such as trans-(PPh3)2Ni(1-naphthyl)Cl have been known to be air-stable since 1960, when they were first reported by Chatt and Shaw,11 but these complexes were not utilized in catalytic transformations until many years later. Relatively few phosphine ligands have been used to prepare such complexes for use in catalysis,12−14 although several new types of nickel precatalysts with varying degrees of air stability have also been developed in the past few years.15
During the development of trans-(PCy2Ph)2Ni(o-tolyl)Cl for use in the coupling reaction of benzyl chlorides with alkenes,16 it became apparent that such complexes could be used for numerous nickel-catalyzed transformations, since these precatalysts can be activated by nucleophilic reagents (RMgX, RZnX, R3B, R3SiH—by a transmetalation/reductive elimination sequence) as well as electrophilic reagents (R3SiOTf—by a Lewis acid induced Ni to Ni transmetalation followed by reductive elimination of a biaryl). Members of this class have been shown to possess significantly enhanced catalytic activity in comparison to the combination of Ni(cod)2 and the corresponding phosphine ligand due to the absence of cod, which is known to hinder catalysis in some instances.17
Results and Discussion
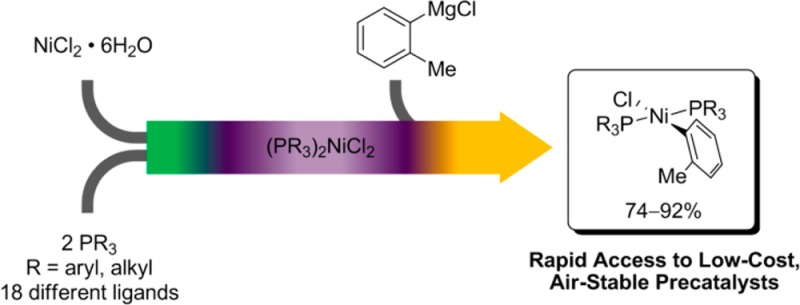
Synthesis of the precatalyst complexes is straightforward (see Scheme 1): NiCl2·6H2O and the desired mono- or bidentate phosphine are combined in ethanol and briefly refluxed, after which the L2NiCl2 complex is isolated by a simple vacuum filtration on a sintered-glass frit. After it is dried under vacuum to remove residual solvent, the complex is redissolved in THF or CH2Cl2 and 1 equiv of Grignard reagent (o-tolylmagnesium chloride, 2-mesitylmagnesium bromide, or similar) is added. Removal of the solvent by rotary evaporation and addition of methanol precipitates the complex and dissolves the magnesium chloride or bromide; isolation by vacuum filtration on a glass frit followed by washing with the appropriate solvent yields the complex. No further purification of the powder obtained this way is necessary, though recrystallization can be carried out if desired.
Scheme 1. Synthesis of Precatalysts.

X = Cl, Br. R = alkyl, aryl.
At present, we have prepared more than 20 such complexes, with the most significant examples shown in Table 1. The selection of ligands is intended to encompass a variety of mono- and bidentate phosphines commonly used in organic synthesis, as well as a number of less frequently employed ligands. Many of the ligands in the latter category, particularly the low-molecular-weight, liquid phosphines, find less frequent use in organic synthesis at least in part because they are difficult to synthesize and handle safely and because they are expensive to purchase due to the high cost of shipping pyrophoric and/or highly flammable goods. Triethylphosphine (10), dimethylphenylphosphine (8, 9), tricyclopentylphosphine (5), tri-n-butylphosphine (11), and tribenzylphosphine (6) all undergo reactions with air ranging from vigorous to violent, yet the precatalysts derived from each of these ligands are completely stable to oxygen in the solid phase and can be stored in air indefinitely.
Table 1. Nickel Phosphine Complexes Synthesizeda.
| isolated
yield (%) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | ligand | geometry | R | X | LnNiX2 | LnNi(R)X | overall |
| Monodentate Ligands | |||||||
| 1 | PPh3 | trans | o-tolyl | Cl | 91 | 89 | 81 |
| 2 | PCyPh2 | trans | o-tolyl | Cl | 92 | 81 | 75 |
| 3 | PCy2Ph | trans | o-tolyl | Cl | 95 | 88 | 84 |
| 4 | PCy3 | trans | o-tolyl | Cl | 97 | 87 | 84 |
| 5 | PCyp3 | trans | o-tolyl | Cl | 99 | 90 | 89 |
| 6 | PBn3 | trans | o-tolyl | Cl | 96 | 90 | 86 |
| 7 | PPh2Me | trans | o-tolyl | Cl | 99 | 81 | 80 |
| 8 | PMe2Ph | trans | 2,4,6-triisopropylphenyl | Cl | 95 | 83 | 79 |
| 9 | PMe2Ph | trans | 2,6-dimethoxyphenyl | Br | 95 | 87 | 83 |
| 10 | PEt3 | trans | 2-mesityl | Br | 95 | 88 | 84 |
| 11 | P(n-Bu)3 | trans | 2-mesityl | Br | 89 | 90 | 80 |
| Bidentate Ligands | |||||||
| 12 | dppe | cis | o-tolyl | Cl | 98 | 84 | 82 |
| 13 | dppp | cis | 2-mesityl | Br | 89 | 85 | 76 |
| 14 | dppb | trans | 2-mesityl | Br | 96 | 86 | 83 |
| 15 | (S)-BINAP | cis | o-tolyl | Cl | 94 | 97 | 91 |
| 16 | dppf | cis | o-tolyl | Cl | 97 | 95 | 92 |
| 17 | dcpf | trans | o-tolyl | Cl | 98 | 83 | 81 |
| 18 | Xantphos | trans | o-tolyl | Cl | 86 | 92 | 79 |
| 19 | pyphos | cis | o-tolyl | Cl | 90 | 82 | 74 |
Abbreviations: dppe, 1,2-bis(diphenylphosphino)ethane; dppp, 1,3-bis(diphenylphosphino)propane; dppb, 1,4-bis(diphenylphosphino)butane; BINAP, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; dppf, 1,1′-bis(diphenylphosphino)ferrocene; dcpf, 1,1′-bis(dicyclohexylphosphino)ferrocene; Xantphos, 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene; pyphos, 2-[2-(diphenylphosphino)ethyl]pyridine.
In some instances, the complexes containing the o-tolyl moiety were not adequately stable to allow isolation in good yield and/or did not form air-stable complexes. For example, trans-(PEt3)2Ni(o-tolyl)Cl can be isolated in good yield (>90%); however, upon standing in air for several days, it begins to show clear signs of decomposition. A solution to this problem was found by increasing the steric bulk of the aryl group on nickel, which is hypothesized to further shield nickel from associative substitution.
To synthesize these complexes with more substituted aryl groups, the phosphine was condensed with NiBr2·3H2O to yield the corresponding L2NiBr2 complex, which was then treated with commercially available 2-mesitylmagnesium bromide.18 In this way, several complexes which had proven elusive could be synthesized to form stable precatalysts. In the case of tri-n-butylphosphine, trans-(Pn-Bu3)2Ni(o-tolyl)Cl was found to be a liquid at room temperature that could not be stored for more than a few days, whereas trans-(Pn-Bu3)2Ni(2-mesityl)Br (11) is an indefinitely stable solid.
Additionally, [dppp]Ni(o-tolyl)Cl and [dppb]Ni(o-tolyl)Cl are difficult to synthesize in good yield and purity using this method. In both instances, the addition of another 1 equiv of o-tolylmagnesium chloride takes place very readily (which lowers the yield and purity of the isolated product) and neither complex is very stable to methanol, leading to a loss of yield during workup and purification. In both instances, however, changing the aryl group to a mesityl group solved this problem, allowing isolation of cis-[dppp]Ni(2-mesityl)Br (13) and trans-[dppb]Ni(2-mesityl)Br (14). It should be noted, however, that the complexes containing the o-tolyl ligand can be synthesized by metathesis starting from trans-(PPh3)2Ni(o-tolyl)Cl (1), indicating they are indeed stable complexes.13b
Unfortunately, the switch from o-tolyl to 2-mesityl did not enable isolation of a stable complex in one instance: PMe2Ph. Neither the o-tolyl nor the 2-mesityl complexes were stable under ambient conditions or in the presence of alcohols. Because PMe2Ph represents the least sterically demanding phosphine used in this study, it is perhaps unsurprising that its complex is in turn the most sensitive to nucleophilic attack by water or alcohols, since nickel is less shielded. As before, increasing the steric hindrance around nickel provided the solution. Reaction of trans-(PMe2Ph)2NiBr2 with 2,4,6-triisopropylphenylmagnesium bromide19 gave trans-(PMe2Ph)2Ni(2,4,6-triisopropylphenyl)Br (8) in 83% yield. This complex, in stark contrast to the corresponding o-tolyl and 2-mesityl complexes, demonstrates absolutely no air or water sensitivity.
However, due to the concern that activation of this precatalyst may be slow because of the extreme hindrance provided by the isopropyl groups at the 2- and 6-positions of the aryl ring, a precatalyst incorporating a 2,6-dimethoxyphenyl substituent (9) was also prepared and found to be air stable.
As the numerous entries in Table 1 demonstrate, complexes of this type can be made from a wide range of phosphines, including electron-rich and electron-poor as well as sterically demanding and undemanding phosphines. However, a number of phosphines were not able to be successfully incorporated into these types of complexes. Those ligands fall into two categories: electron-poor and sterically hindered (P(4-F-C6H4)3, P(o-tol)3, and P(o-anis)3) and extremely sterically hindered phosphines, regardless of their electronic nature (P(t-Bu)3,20 (9,9-dimethyl-9H-xanthene-4,5-diylbis(di-tert-butylphosphine)), and 1,2-bis((di-tert-butylphosphino)methyl)benzene). In all instances the L2NiX2 or LNiX2 complexes did not form, precluding attempts to synthesize the corresponding arylnickel complexes by this synthetic route.
Although our principal interest in these complexes is their catalytic activity, it is also important to understand their structural features, geometry, and bonding, as this may afford deeper insight that would enable further development of new complexes, types of precatalysts, or alternative modes of activation.
The complexes strongly favor a square-planar arrangement, and whether the two phosphorus atoms are in a cis or trans arrangement at nickel is readily discerned from inspection of each complex’s 31P NMR spectrum. Complexes derived from monodentate phosphines were universally found to adopt a trans geometry, as indicated by the presence of only one singlet in the 31P NMR spectrum. This arrangement presumably results from the minimization of steric interaction between the ligands on nickel, and the magnitude of the steric repulsion is evidently large enough to overwhelm any thermodynamic trans effects that might favor a cis arrangement.21
Conversely, complexes derived from bidentate phosphines were more often observed to adopt a cis arrangement, but several counterexamples were also seen. The preferred arrangement appears to depend on the bite angle of the ligand, its rigidity, and the identity of the substituents on phosphorus.
For example, the complex derived from dppf (16) exists only as the cis, square planar isomer in solution, whereas the closely related dcpf (17) adopts a distorted trans, square planar geometry, as illustrated in its single-crystal X-ray structure and in its 31P NMR spectrum. In this instance, the change from phenyl groups to cyclohexyl groups on phosphorus is enough to alter the preferred geometry, despite the fact that both complexes are built on the same ferrocene scaffold.
A selection of these precatalysts have been characterized by single-crystal X-ray diffraction (Figure 1). Crystal structures were determined following established procedures,22 and complete details are given in the Supporting Information. Complexes derived from PBn3 and PMe2Ph both adopt nearly ideal trans, square planar structures and are, for the most part, structurally unremarkable. Complex 15 (derived from (S)-BINAP) adopts a nearly ideal square planar structure with a cis arrangement, yielding a dihedral angle of 73.24(3)° between the two naphthalene rings of BINAP. The most interesting feature of this complex, though, is the fact that it forms diastereomers due to the two possible arrangements of the o-tolyl group. These diasteromers are both crystallographically and spectroscopically (1H and 31P NMR) observable, suggesting that interconversion is either slow or does not take place at any appreciable rate near room temperature.
Figure 1.

Complexes analyzed by single-crystal X-ray diffraction. Thermal ellipsoids are drawn at the 50% probability level, and hydrogen atoms are not included. Disorder of the o-tolyl ligand (6, 15, 18, 19) and solvent molecules of crystallization (6, 15, 17, 18) are not shown.
The complex derived from dcpf (17) is another interesting case: its 31P NMR spectrum exhibits one singlet, despite the fact that it is a bidentate phosphine. XRD analysis showed a geometry at nickel that is best described as square planar, but with significant distortion toward tetrahedral.23 For example, the P(1)–Ni–P(2) bond angle is ca. 145°, well shy of the ideal 180°. However, the P(1)–Ni–Cl and P(2)–Ni–Cl bond angles are 91.264(13) and 91.642(13)°, very close to the ideal 90° for a square plane. Because of this, it is appropriate to describe the two phosphorus atoms as trans to one another.
Complex 18 (derived from Xantphos) adopts a distorted-square-pyramidal geometry in the solid state. The oxygen atom of the ligand occupies the apical position and the two phosphorus atoms are in equatorial positions trans to each other. In solution, two isomers are observed by 1H and 31P NMR, the second perhaps being the true square-planar isomer, without oxygen coordinated at nickel.
Pyphos (19), being an unsymmetrical, bidentate ligand, can form at least two structural isomers—chloride could be trans to either phosphorus or to nitrogen. The 31P NMR spectrum shows only one, sharp singlet, which suggests that one isomer is dominant in solution. Single-crystal X-ray diffraction analysis showed 19 to adopt a square-planar structure with chlorine trans to phosphorus. This geometrical arrangement presumably indicates that the thermodynamic trans effect dominates the ground-state conformation, rather than any potential steric interaction between the diphenylphosphino moiety and the o-tolyl ligand.
To demonstrate the utility and advantages these precatalysts present over other means of entry into nickel(0), we have used the nickel-catalyzed carbonyl-ene reaction, which couples a terminal alkene (or ethylene), an aldehyde, and a silyl triflate to form allylic or homoallyic silyl ethers (Table 2).24 Preliminary experiments demonstrated that catalysts 3 and 1 were indeed catalytically competent and provided the desired allylic (20) and homoallylic (21) products, respectively. In both instances, the selectivity and yields were observed to be comparable to those of reactions using Ni(cod)2. However, the rate was observed to be higher than when cod is present—following studies demonstrated that the reaction reaches completion in ca. 18 h, rather than the 36–48 h required when using Ni(cod)2 as the nickel source.
Table 2. Screening of the Ni-Catalyzed Carbonyl-Ene Reactiona.
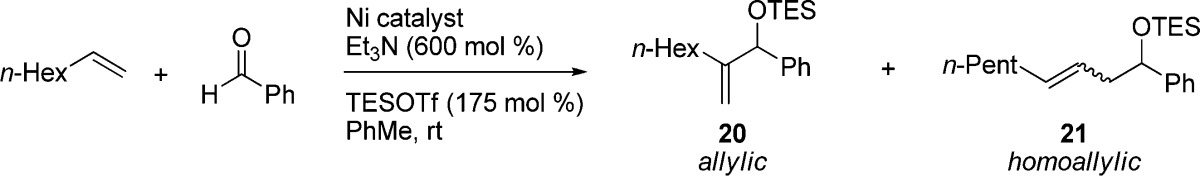
| yieldb (%) |
||||||
|---|---|---|---|---|---|---|
| entry | ligand | Ni source | 20 | 21 | combined | 20:21 |
| 1 | PPh3 | Ni(cod)2 | 6 | 78 | 84 | 7:93 |
| 2 | PPh3 | 1 | 7 | 81 | 88 | 8:92 |
| 3 | PCy2Ph | Ni(cod)2 | 52 | 21 | 73 | 71:29 |
| 4 | PCy2Ph | 3 | 54 | 20 | 74 | 73:27 |
| 5 | PCyPh2 | 2 | 17 | 56 | 73 | 23:77 |
| 6 | PCy3 | 4 | 18 | 2 | 20 | 90:10 |
See the Supporting Information for complete data for all complexes. Reactions were carried out on a 0.50 mmol scale with 20 mol % of catalyst and run for 48 h.
Yields determined by gas chromatography calibrated against an internal standard of n-dodecane.
A comprehensive screen of precatalysts 1–19 was carried out to demonstrate the ease with which screening of ligands can be accomplished (abbreviated results are shown in Table 2; see the Supporting Information for complete data). The use of these single-component, air-stable precatalysts reduces the effort involved to an exercise which can be carried out on the benchtop with no exclusion of air during setup of the reactions. This approach, while convenient, may however not be applicable in all instances, as it is necessary to have already synthesized the desired precatalysts. For researchers frequently involved in screening of nickel-catalyzed reactions, however, such an approach could be valuable.
Conclusion
In summary, we have synthesized and characterized 19 air-stable Ni(II) complexes derived from a range of mono- and bidentate phosphine ligands commonly used in synthesis. These complexes are accessed from low-cost NiCl2·6H2O rather than from an expensive and air-sensitive Ni(0) source such as Ni(cod)2. These complexes can function as precatalysts for a range of nickel-catalyzed reactions, as they are readily converted to Ni(0) phosphine complexes by treatment with reagents such as RMgX, RZnX, R3B, RLi, R3SiH, and R3SiOTf among others, allowing their convenient use in Ni(0)-catalyzed reactions. Many of these reactions, which previously employed Ni(cod)2 as the Ni(0) source and thus required the use of a glovebox or glovebag, can now be carried out with no exclusion of air or water during setup, which greatly facilitates the use of nickel catalysis as a tool for synthesis. These benefits have been demonstrated in the context of the nickel-catalyzed carbonyl-ene reaction, where the use of a precatalyst provided a significant rate enhancement for the target reaction while maintaining selectivity equivalent to that of reactions catalyzed by Ni(cod)2.
Experimental Section
General Considerations
Dichloromethane, THF, and acetonitrile were degassed by sparging with nitrogen and dried by passage through a column of activated alumina. Ethanol (200 proof, <0.1% water) and n-butanol (99.9%) were roughly degassed by sparging with nitrogen and were not further dried prior to use. Methanol (>99.8%, <0.1% water) was used as received. Manipulation of all air-sensitive reagents was carried out in a glovebox filled with dry nitrogen. Bis(1,5-cyclooctadiene)nickel(0) was purchased from Strem Chemicals (Newburyport, MA) and stored at −30 °C in a glovebox. All other chemicals were purchased from Sigma-Aldrich (Milwaukee, WI), Alfa Aesar (Ward Hill, MA), TCI America (Portland, OR), or Oakwood Products, Inc. (West Columbia, SC).
Melting points were determined on an electrothermal apparatus using glass capillaries open to air except where specified. The material used for the determinations was not recrystallized but was ground to a fine powder using a metal spatula before analysis.
NMR spectra were obtained in CDCl3 (99.8% atom D), C6D6 (99.5% atom D), or CD2Cl2 (99.9% atom D) purchased from Cambridge Isotope Laboratories (Andover, MA). 1H NMR spectra were obtained at either 300 or 500 MHz, 13C spectra were recorded at 126 MHz with 1H decoupling, and 31P spectra were recorded at 121 or 202 MHz with 1H decoupling. Chemical shifts (1H and 13C) are reported in parts per million relative to TMS (δ = 0.00 ppm) and were referenced to the residual solvent peak (1H, CDCl3 7.26 ppm, C6D6 7.16 ppm, CD2Cl2 5.32 ppm; 13C, CDCl3 77.16 ppm, C6D6 128.06 ppm, CD2Cl2 53.84 ppm); 31P NMR spectra are reported in parts per million relative to an external standard of 85% phosphoric acid (δ 0.00 ppm). The following designations are used to describe multiplicities: s (singlet), d (doublet), t (triplet), q (quartet), br (broad), app (apparent).
General Procedure
LnNiX2
NiCl2·6H2O or NiBr2·3H2O, EtOH, and a magnetic stir bar were placed in a round-bottom flask. The flask was sealed with a rubber septum, the solution was sparged with nitrogen for 15 min, the septum was removed, and then the phosphine was added in one portion. The flask was fitted with a reflux condenser, and the mixture was heated to 80 °C for 30 min and then cooled to room temperature. Once cool, the flask was chilled to 0 °C for 10 min, after which the solid was collected by vacuum filtration and washed twice with ethanol (and twice with ether in some instances). Drying under vacuum yielded the product.
LnNi(aryl)X
LnNiX2 was placed in an oven-dried round-bottom flask containing a magnetic stir bar. Solvent (THF or CH2Cl2) was added, the solution was cooled to 0 °C with an ice bath, and Grignard reagent was added dropwise with vigorous stirring. The solution was stirred for 15 min at 0 °C, after which the stir bar was removed and the solution was evaporated to dryness under reduced pressure. MeOH was added, and the mixture was sonicated until a uniform suspension was obtained (approximately 5 min). After the suspension was cooled to 0 °C, the precipitate was collected by vacuum filtration, washed with two portions of cold MeOH, and dried under high vacuum to yield the complex.
Representative Syntheses
(PMePh2)2NiCl2
NiCl2·6H2O (17.42 mmol, 4.141 g), EtOH (55 mL), and a magnetic stir bar were placed in a 100 mL round-bottom flask. The flask was sealed with a rubber septum, the solution was sparged with nitrogen for 15 min, and then PPh2Me (38.32 mmol, 7.672 g, 7.13 mL) was added portionwise over 5 min. The flask was equipped with a reflux condenser, and the mixture was heated to 80 °C for 30 min then cooled to room temperature. Once cool, the flask was chilled to 0 °C for 10 min, after which the solid was collected by vacuum filtration and washed twice with ethanol (5 mL). Drying under vacuum yielded S7 (9.111 g, 99%) as a deep maroon, crystalline solid.
trans-(PMePh2)2Ni(2-MePh)Cl (7)
(PMePh2)2NiCl2 (8.29 mmol, 4.394 g) was placed in an oven-dried, 100 mL round-bottom flask containing a magnetic stir bar. THF (55 mL) was added, the solution was cooled to 0 °C with an ice bath, and o-tolylmagnesium chloride (8.29 mmol, 0.856 M in THF, 9.68 mL) was added dropwise with vigorous stirring. Near the end of the addition, the solution began to turn orange. The solution was stirred for 15 min at 0 °C, after which the stir bar was removed and the solution was evaporated to dryness under reduced pressure. MeOH (25 mL) was added, and the mixture was sonicated until a uniform suspension was obtained (approximately 2 min). After the suspension was cooled to 0 °C, the bright yellow precipitate was collected by vacuum filtration, washed with two portions of cold MeOH (10 mL), and dried under high vacuum to yield 7 (3.940 g, 81%)25 as a fine, bright yellow powder. Mp: 139–140 °C dec. Anal. Calcd for C33H33ClNiP2: C, 67.67; H, 5.68. Found: C, 67.41; H, 5.78. 1H NMR (500 MHz, C6D6, δ): 7.84 (br s, 4H), 7.62 (br s, 4H), 7.12–6.94 (m, 13H), 6.72–6.64 (m, 1H), 6.64–6.53 (m, 2H), 2.75 (s, 3H), 1.08 (s, 6H). 31P{1H} NMR (121 MHz, C6D6, δ): 8.32. 13C{1H} NMR (126 MHz, C6D6, δ): 153.09 (t, J = 33.7 Hz), 143.27, 136.02 (t, J = 3.8 Hz), 134.99 (t, J = 20.3 Hz), 133.84 (t, J = 5.2 Hz), 133.46 (t, J = 20.8 Hz), 133.26 (t, J = 5.4 Hz), 129.79, 129.56, 124.03, 122.55, 26.56, 12.94 (t, J = 15.7 Hz). IR (ATR, cm–1): 3051 (w), 1570 (w), 1561 (w), 1484 (w), 1434 (m), 1372 (w), 1335 (w), 1309 (w), 1285 (w), 1186 (w), 1160 (w), 1098 (m), 1074 (w), 1027 (w), 1012 (w), 999 (w), 895 (m), 887 (s), 878 (s), 850 (w), 740 (s), 729 s), 692 (s).
[dppe]NiCl2
NiCl2·6H2O (9.6 mmol, 2.282 g), EtOH (30 mL), and a magnetic stir bar were placed in a 50 mL round-bottom flask. The flask was sealed with a rubber septum, the solution was sparged with nitrogen for 15 min, the septum was removed, and then dppe (9.66 mmol, 3.849 g) was added in one portion. The flask was fitted with a reflux condenser, and the mixture was heated to 80 °C for 30 min and then cooled to room temperature. Once cool, the flask was chilled to 0 °C for 10 min, after which the solid was collected by vacuum filtration and washed twice with ethanol (5 mL). Drying under vacuum yielded S12 (4.956 g, 98%) as a fine, orange powder.
cis-[dppe]Ni(2-MePh)Cl (12)
[dppe]NiCl2 (3.92 mmol, 2.07 g) was placed in an oven-dried, 250 mL round-bottom flask containing a magnetic stir bar. THF (200 mL) was added, the mixture was cooled to 0 °C with an ice bath, and o-tolylmagnesium chloride (3.92 mmol, 0.986 M in THF, 3.98 mL) was added dropwise with vigorous stirring. Partway through the addition, the solution became completely homogeneous and began to turn yellow. After complete addition of the Grignard reagent, the solution was stirred for 15 min at 0 °C, after which the stir bar was removed and the solution was evaporated to dryness under reduced pressure. MeOH (20 mL) was added, and the mixture was sonicated until a uniform suspension was obtained (approximately 5 min). After the suspension was cooled to 0 °C, the yellow precipitate was collected by vacuum filtration, washed with two portions of cold MeOH (5 mL), and dried under high vacuum to yield 12 (1.92 g, 84%) as a fine, bright yellow powder. Mp: 190–192 °C dec. Anal. Calcd for C33H31ClNiP2: C, 67.90; H, 5.35. Found: C, 68.28; H, 5.66. 1H NMR (500 MHz, CD2Cl2, δ): 8.16 (dd, J = 10.9, 7.3 Hz, 4H), 7.77 (ddd, J = 9.2, 7.1, 1.8 Hz, 2H), 7.62–7.41 (m, 9H), 7.31 (t, J = 7.1 Hz, 1H), 7.20 (dt, J = 8.4, 4.7 Hz, 1H), 7.07 (td, J = 7.7, 2.6 Hz, 2H), 6.71 (dd, J = 10.8, 7.6 Hz, 2H), 6.59 (dd, J = 6.0, 2.9 Hz, 2H), 6.45–6.39 (m, 1H), 2.58–2.34 (m, 2H), 2.34–2.09 (m, 4H), 1.60 (tdd, J = 14.4, 11.7, 6.7 Hz, 1H). 31P{1H} NMR (121 MHz, CD2Cl2, δ): 53.09 (d, J = 17.9 Hz), 35.78 (d, J = 17.8 Hz). 13C{1H} NMR (126 MHz, CD2Cl2, δ): 158.03 (dd, J = 86.1, 38.5 Hz), 143.71 (t, J = 2.0 Hz), 136.17 (dd, J = 3.1, 1.7 Hz), 134.97 (d, J = 11.2 Hz), 134.08 (d, J = 11.2 Hz), 133.14 (d, J = 10.3 Hz), 132.20 (d, J = 8.4 Hz), 131.92 (d, J = 2.6 Hz), 131.66 (d, J = 1.5 Hz), 131.28 (d, J = 2.1 Hz), 130.87 (dd, J = 47.8, 0.6 Hz), 130.73 (d, J = 2.3 Hz), 130.39 (dd, J = 31.9, 0.8 Hz), 130.35 (d, J = 2.7 Hz), 129.72 (dd, J = 56.0, 5.0 Hz), 129.38 (d, J = 9.2 Hz), 129.28 (d, J = 10.5 Hz), 129.02 (dd, J = 6.3, 2.4 Hz), 128.94 (d, J = 9.4 Hz), 127.94 (d, J = 10.1 Hz), 123.49 (dd, J = 6.3, 1.7 Hz), 122.71 (t, J = 1.2 Hz), 29.34 (dd, J = 27.7, 21.7 Hz), 25.50 (q, J = 1.7 Hz), 22.22 (dd, J = 26.0, 11.4 Hz). IR (ATR, cm–1): 3051 (w), 1561 (w), 1434 (m), 1421 (w), 1098 (m), 1026 (m), 1012 (w), 999 (w), 873 (w), 817 (m), 749 m), 742 (s), 708 (m), 692 (s), 679 (m), 652 (m).
Acknowledgments
Dr. Georgiy Teverovskiy and Prof. Stephen Buchwald are acknowledged for insightful discussion and advice. Financial support has been provided by the NIGMS (GM63755) and by an NSF Graduate Research Fellowship (E.A.S.). X-ray crystallography was carried out on instrumentation purchased with the help of NSF grant CHE-0946721. NMR spectroscopy was carried out on instruments purchased in part with funds provided by the NSF (CHE-9808061 and CHE-8915028).
Supporting Information Available
Text, figures, tables, and CIF files giving experimental procedures and spectral data for complexes 1–19, X-ray crystallographic data for complexes 6, 9, 15, 17, 18, 19, a description of single-crystal X-ray diffraction experiments, and full reaction screening information and procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry and Biochemistry, Brigham Young University, Provo, UT 84602.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Wilke G. Angew. Chem., Int. Ed. 1988, 27, 185–206. [Google Scholar]; b Modern Organonickel Chemistry; Tamaru Y., Ed.; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]; c Hu X. Chem. Sci. 2011, 2, 1867–1886. [Google Scholar]; d Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Chem. Rev. 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mesganaw T.; Garg N. K. Org. Process Res. Dev. 2013, 17, 29–39. [Google Scholar]; f Yamaguchi J.; Muto K.; Itami K. Eur. J. Org. Chem. 2013, 19–30. [Google Scholar]
- Palladium metal is more than 1500 times more expensive than nickel metal, yet the cost of Ni(cod)2 is only ca. 50% less than Pd2(dba)3 as a result of the reagents required for their synthesis, yield of the reaction, and cost of purification.
- Stephenson T. A.; Morehouse S. M.; Powell A. R.; Heffer J. P.; Wilkinson G. J. Chem. Soc. 1965, 3632–3640. [Google Scholar]
- For reviews of palladacycles in synthesis from 2001 and earlier, see:; a Herrmann W. A.; Böhm V. P. W.; Reisinger C.-P. J. Organomet. Chem. 1999, 576, 23–41. [Google Scholar]; b Dupont J.; Pfeffer M.; Spencer J. Eur. J. Inorg. Chem. 2001, 1917–1927. [Google Scholar]
- a Viciu M. S.; Kissling R. M.; Stevens E. D.; Nolan S. P. Org. Lett. 2002, 4, 2229–2231. [DOI] [PubMed] [Google Scholar]; b Viciu M. S.; Gemaneau R. F.; Navarro-Fernandez O.; Stevens E. D.; Nolan S. P. Organometallics 2002, 21, 5470–5472. [Google Scholar]
- a Zim D.; Buchwald S. L. Org. Lett. 2003, 5, 2413–2415. [DOI] [PubMed] [Google Scholar]; b Bruno N. C.; Tudge M. T.; Buchwald S. L. Chem. Sci. 2013, 4, 916–920and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Organ M. G.; Avola S.; Dubovyk I.; Hadei N.; Kantchev E. A. B.; O’Brien C. J.; Valente C. Chem. Eur. J. 2006, 12, 4749–4755. [DOI] [PubMed] [Google Scholar]; b O’Brien C. J.; Kantchev E. A. B.; Hadei N.; Valente C.; Chass G. A.; Nasielski J. C.; Lough A.; Hopkinson A. C.; Organ M. G. Chem. Eur. J. 2006, 12, 4743–4748. [DOI] [PubMed] [Google Scholar]; c Organ M. G.; Abdel-Hadi M.; Avola S.; Hadei N.; Nasielski J.; O’Brien C. J.; Valente C. Chem. Eur. J. 2007, 13, 150–157. [DOI] [PubMed] [Google Scholar]; d Kantchev E. A. B.; O’Brien C. J.; Organ M. G. Angew. Chem., Int. Ed. 2007, 46, 2768–2813. [DOI] [PubMed] [Google Scholar]; e Valente C.; Çalimsiz S.; Hoi K. H.; Mallik D.; Sayah M.; Organ M. G. Angew. Chem., Int. Ed. 2012, 51, 3314–3332. [DOI] [PubMed] [Google Scholar]
- An excellent overview of these precatalysts is provided in:; a Hill L. L.; Crowell J. L.; Tutwiler S. L.; Massie N. L.; Hines C. C.; Griffin S. T.; Rogers R. D.; Shaughnessy K. H.; Grasa G. A.; Johansson Seechurn C. C. C.; Li H.; Colacot T. J.; Chou J.; Woltermann C. J. J. Org. Chem. 2010, 75, 6477–6488. [DOI] [PubMed] [Google Scholar]; b Johansson Seechurn C. C. C.; Parisel S. L.; Colacot T. J. J. Org. Chem. 2011, 76, 7918–7932. [DOI] [PubMed] [Google Scholar]
- a For the first instance of zinc reduction of (PPh3)2NiCl2 to access Ni(0), see: Kende A. S.; Liebeskind L. S.; Braitsch D. M. Tetrahedron Lett. 1975, 39, 3375–3378. [Google Scholar]; b For an overview of the development of nickel precatalysts, see: Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Chem. Rev. 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an example of the use of a Grignard reagent to preactivate a Ni(II) catalyst, see:Wolfe J. P.; Buchwald S. L. J. Am. Chem. Soc. 1997, 119, 6054–6058. [Google Scholar]
- a Chatt J.; Shaw B. L. J. Chem. Soc. 1960, 1718–1729. [Google Scholar]; b Cross R. J.; Wardle R. J. Chem. Soc. A 1970, 840–845. [Google Scholar]
- For a selection of some recent examples, see:; a Chen C.; Yang L.-M. Tetrahedron Lett. 2007, 48, 2427–2430. [Google Scholar]; b Gao C.-Y.; Yang L.-M. J. Org. Chem. 2008, 73, 1624–1627. [DOI] [PubMed] [Google Scholar]; c Zhou L.; Feng X.; He R.; Bao M. J. Coord. Chem. 2009, 62, 2824–2831. [Google Scholar]; d Fan X.-H.; Yang L.-M. Eur. J. Org. Chem. 2010, 2457–2460. [Google Scholar]; e Fan X.-H.; Yang L.-M. Eur. J. Org. Chem. 2011, 1467–1471. [Google Scholar]; f Zhang N.; Hoffman D. J.; Gutsche N.; Gupta J.; Percec V. J. Org. Chem. 2012, 77, 5956–5964. [DOI] [PubMed] [Google Scholar]; g Leowanawat P.; Zhang N.; Safi M.; Hoffman D. J.; Fryberger M. C.; George A.; Percec V. J. Org. Chem. 2012, 77, 2885–2892. [DOI] [PubMed] [Google Scholar]; h Park N. H.; Teverovskiy G.; Buchwald S. L. Org. Lett. 2014, 16, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the use of these precatalysts for cross-coupling polymerization, see:; a Miyakoshi R.; Yokoyama A.; Yokozawa T. J. Am. Chem. Soc. 2005, 127, 17542–17547. [DOI] [PubMed] [Google Scholar]; b Bronstein H. A.; Luscombe C. K. J. Am. Chem. Soc. 2009, 131, 12894–12895. [DOI] [PubMed] [Google Scholar]
- McNeil and co-workers have carried out detailed studies into use of this type of precatalyst for the synthesis of poly(thiophene) and related polymers:; a Lanni E. L.; McNeil A. J. J. Am. Chem. Soc. 2009, 131, 16573–16579. [DOI] [PubMed] [Google Scholar]; b Lanni E. L.; McNeil A. J. Macromolecules 2010, 43, 8039–8044. [Google Scholar]; c Lanni E. L.; Locke J. R.; Gleave C. M.; McNeil A. J. Macromolecules 2011, 44, 5136–5145. [Google Scholar]; d Lee S. R.; Bryan Z. J.; Wagner A. M.; McNeil A. J. Chem. Sci. 2012, 3, 1562–1566. [Google Scholar]; e Lee S. R.; Bloom J. W. G.; Wheeler S. E.; McNeil A. J. Dalton Trans. 2013, 42, 4218–4222. [DOI] [PubMed] [Google Scholar]; f Bryan Z. J.; McNeil A. J. Chem. Sci. 2013, 4, 1620–1624. [Google Scholar]; g Bryan Z. J.; McNeil A. J. Macromolecules 2013, 46, 8395–8405. [Google Scholar]
- a Ge S.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ge S.; Hartwig J. F. J. Am. Chem. Soc. 2014, 136, 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also ref (17a).
- Standley E. A.; Jamison T. F. J. Am. Chem. Soc. 2013, 135, 1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ge S.; Hartwig J. F. J. Am. Chem. Soc. 2011, 133, 16330–16333. [DOI] [PMC free article] [PubMed] [Google Scholar]; b For a detailed report examining the role of dba in zerovalent palladium catalysis, see: Amatore C.; Jutand A.; Khalil F.; M’Barki M. A.; Mottier L. Organometallics 1993, 12, 3168–3178. [Google Scholar]
- The change from chloride to bromide is merely for convenience, since 2-mesitylmagnesium bromide is commercially available, while the chloride is not. The identity of the halogen does not appear to alter the behavior of the complexes as precatalysts.
- This particular Grignard reagent was selected for the isopropyl groups at the 2- and 6-positions, with the isopropyl group in the 4-position not expected to play a role in the complex’s stability. This reagent is commercially available as a 0.5 M solution in THF.
- It is known, for example, that P(t-Bu)3 reacts with NiBr2 in ethanol to form [P(t-Bu)3NiBr3]−[HP(t-Bu)3]+. See:Alyea E. C.; Costin A.; Ferguson G.; Fey G. T.; Goel R. G.; Restivo R. J. J. Chem. Soc., Dalton Trans. 1975, 1294–1298. [Google Scholar]
- Quagliano J. V.; Schubert L. Chem. Rev. 1952, 50, 201–260. [Google Scholar]
- Müller P. Crystallogr. Rev. 2009, 15, 57–83. [Google Scholar]
- This deviation in geometry is also likely to be the reason this complex is red, as opposed to the normal bright yellow.
- a Ng S.-S.; Jamison T. F. J. Am. Chem. Soc. 2005, 127, 14194–14195. [DOI] [PubMed] [Google Scholar]; b Ho C.-Y.; Ng S.-S.; Jamison T. F. J. Am. Chem. Soc. 2006, 128, 5362–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ng S.-S.; Ho C.-Y.; Jamison T. F. J. Am. Chem. Soc. 2006, 128, 11513–11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Complex 7 is somewhat soluble in methanol, causing the lower yield in comparison to other complexes. The yield can be further raised by collecting a second crop of solid from the filtrate.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.