

Abstract
c-Abl is activated in the brain of Parkinson's disease (PD) patients and in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated mice where it inhibits parkin through tyrosine phosphorylation leading to the accumulation of parkin substrates, and neuronal cell death. In the present study, we evaluated the in vivo efficacy of nilotinib, a brain penetrant c-Abl inhibitor, in the acute MPTP-induced model of PD. Our results show that administration of nilotinib reduces c-Abl activation and the levels of the parkin substrate, PARIS, resulting in prevention of dopamine (DA) neuron loss and behavioral deficits following MPTP intoxication. On the other hand, we observe no reduction in the tyrosine phosphorylation of parkin and the parkin substrate, AIMP2 suggesting that the protective effect of nilotinib may, in part, be parkin-independent or to the pharmacodynamics properties of nilotinib. This study provides a strong rationale for testing other brain permeable c-Abl inhibitors as potential therapeutic agents for the treatment of PD.
Parkinson's disease (PD) is a progressive neurodegenerative disorder due to a selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), which leads to a decrease in the synthesis of dopamine (DA). Reductions in the SNpc and striatal DA contributes to the cardinal symptoms observed in the PD1. Current treatments for PD are symptomatic therapies with many limitations2,3. Although the etiology of PD is not clear, emerging evidence suggest that increased oxidative stress in dopaminergic neurons of the SNpc significantly contributes to the pathogenesis of PD. Studies in animal models of PD, as well as in postmortem PD human brains indicate the involvement of oxidative stress in the disease pathology4,5. c-Abl tyrosine kinase activation is a key indicator of oxidative stress6,7. c-Abl activation is associated with many neurodegenerative disorders such as Alzheimer's disease and PD7,8,9,10,11. c-Abl phosphorylation is robustly increased in PD brain samples, animal models of α-synucleinopathies and also in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced preclinical animal model of PD9,10,12,13,14. Activated c-Abl can phosphorylate parkin at tyrosine 143 leading to inhibition of parkin's E3 ligase function and accumulation of its toxic substrates, such as PARIS (PARkin Interacting Substrate)15, aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2) and far upstream element-binding protein 1 (FBP1)9,15,16. PARIS and AIMP2 are potentially important pathogenic parkin substrates since they accumulate in familial PD with parkin mutations, sporadic PD, adult conditional parkin knockout mice and MPTP intoxicated mice9,15,16,17. Under pathogenic conditions, where parkin is inactivated, PARIS levels increase, which leads to mitochondrial dysfunction through down-regulation of PGC-1α and eventually results in the loss of dopamine neurons that is PARIS-dependent15. Recently we showed that overexpression of AIMP2 leads to an age-dependent, selective neurodegeneration of dopamine neurons through activation of poly (ADP-ribose) polymerase 1 (PARP1) initiating parthanatos suggesting that AIMP2 is an important contributor to the loss of DA neurons due to parkin inactivation16.
STI-571 (Imatinib), a c-Abl inhibitor, restores parkin's E3 ligase activity, reduces the accumulation of parkin substrates, and thereby protects against 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in vitro9,10. Conditional knockdown of c-Abl protects against MPTP-induced DA neuronal loss in mice. Accompanying the neuroprotection was an absence of tyrosine phosphorylation of parkin10. Consistent with maintenance of parkin activity, the upregulation of the parkin substrates, AIMP2 and FBP-1, was suppressed, suggesting that c-Abl inhibition was, in part, protective through maintaining parkin in a catalytically active state10. Taken together these results suggest that inhibition of c-Abl activation could be an effective disease modifying therapy for PD.
Nilotinib (AMN107) (Tasigna®) is a second-generation c-Abl tyrosine kinase inhibitor. Compared to the other c-Abl inhibitors, nilotinib is more selective and potent with moderate brain penetration18. Nilotinib is currently used clinically in the treatment of chronic myeloid leukemia (CML). In the present study, we evaluated the in vivo efficacy of nilotinib in the acute MPTP intoxication model of PD. Our results show that administration of nilotinib results in substantial protection against DA neuronal loss following MPTP intoxication. This study provides a rationale for use of potent and brain penetrant c-Abl inhibitors as potential therapeutic agents to slow the progression of PD.
Results
Experimental Protocol
In this study we examined whether the inhibition of activated c-Abl can protect DA neurons in a pre-clinical model of PD. The experimental strategy shown in Fig. 1 indicates the time schedule of intervention and analyses performed. Initially, a post-treatment strategy of nilotinib was carried out, followed by MPTP injections. However, no significant protective effects of nilotinib were observed (data not shown). To address the possibility that when the c-Abl inhibitor was administered, c-Abl may have already exerted its adverse effect on dopaminergic neurons, a pre- and post-treatment strategy was used as illustrated in the Fig. 1.
Figure 1. The schematic diagram depicts the time schedule of intervention and analyses performed.
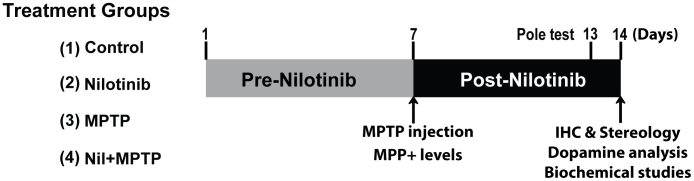
A schematic diagram depicts the experimental design of present study. Numerals represent the days experiments were conducted. Mice were pretreated with vehicle or nilotinib a week before MPTP injections and continued to be treated until experiments. On 7th day we injected saline or MPTP (2 h interval, 4 times, 20 mg/kg free base) in respective treatment groups. On 13th day the pole test was performed. On 14th day, mice were sacrificed for indicated studies. Following are animal numbers used for this studies, behavioural (n = 10), neurochemical (n = 5), immunohistochemistry (n = 5) and biochemical studies (n = 4) per each treatment group.
Effects of Nilotinib on the Blood Brain Barrier Penetration, DAT and MPP+ Levels, and c-Abl Activation
Since the majority of c-Abl inhibitors have poor brain penetration, we sought to determine whether nilotinib can penetrate the blood brain barrier to exert its effect in the brain. For in vivo studies, brain penetration is required for pharmacological inhibition of c-Abl. Accordingly, we analysed whether nilotinib can penetrate the blood brain barrier in mice. The concentration of nilotinib after oral gavage administration of the drug was estimated in whole brain by employing high-performance liquid chromatography (HPLC)-UV following administration of a single dose of nilotinib (25 mg/kg). Two hours after oral gavage administration of nilotinib, the mice were euthanized and brains were processed for HPLC-UV analysis. Administration of nilotinib resulted in approximately 7 pg/mg protein of nilotinib in the brain, thus indicating that nilotinib can cross the blood brain barrier (Fig. 2a).
Figure 2. In vivo assessments of nilotinib in mouse brain.

(a) In vivo measurement of nilotinib levels in the brain: Nilotinib was dissolved in vehicle (10% NMP and 90% PEG 300) and administrated by oral gavage to mice. At 2 h the concentration of nilotinib was measured by using HPLC-UV analysis. Nilotinib crosses the blood brain barrier and shows pharmacological effective levels in brain. Oral gavage of vehicle treated mice brain showed no peaks corresponding to reference to nilotinib standards. (b) Nilotinib has no effect on the levels of MPP+: Levels of MPP+ in the striatum of vehicle and nilotinib treated mice injected with saline and MPTP (2 h interval, 4 times, 20 mg/kg free base). 90 min after the final injection of MPTP, MPP+ was measured by using HPLC-UV analysis. (c) Effect of nilotinib on DAT levels following MPTP-intoxication in the striatum: Immunoblots of striatal lysates from C57BL/6 mice treated with saline, MPTP, MPTP with nilotinib (25 mg/kg body wt, p.o.), or nilotinib alone. The striatal lysates were immunoblotted with anti-DAT and anti-β-actin antibodies. β-actin serves as a loading control. (d) Relative DAT levels normalized to total β-actin are indicated. (e) Nilotinib inhibits c-Abl activation following MPTP-intoxication in the ventral midbrain: Immunoblots of ventral midbrain lysates from C57BL/6 mice treated with saline, MPTP, MPTP with nilotinib (25 mg/kg body wt, p.o.), or nilotinib alone. The ventral midbrain lysates were immunoblotted with anti-phospho-c-Abl, anti-c-Abl, and anti-β-actin antibodies. β-actin serves as a loading control. (f) Relative phospho-c-Abl levels normalized to total c-Abl are indicated. All experiments were repeated three times and representative images of the immunoblots are shown. The Error bars represent the mean ± SEM. (n = 3–4 mice per group). Statistical significance was determined by performing two-way ANOVA followed by Bonferroni post test. ** p < 0.01, ***p < 0.001 for MPTP compared with Saline group, ###p<0.001 for Nil + MPTP compared with MPTP group. Full length blots are presented in the supplementary Figure S1. (ns: not significant).
We further assessed whether nilotinib can alter the conversion of MPTP to its active metabolite MPP+ in the brain. Since the major determinant of MPTP toxicity is the amount of the active metabolite, MPP+, which is selectively up taken by DA neurons, MPP+ levels were assessed in MPTP intoxicated mice treated with vehicle versus nilotinib. Equivalent levels of MPP+ are observed in vehicle and nilotinib treated animals (Fig. 2b). To determine whether nilotinib administration itself affects dopamine transporter (DAT) levels, the levels of DAT after the administration of nilotinib were monitored via immunoblot analysis. There is no significant difference in the levels of DAT between nilotinib groups and vehicle groups, suggesting that nilotinib does not affect on DAT activity (Fig. 2c and d). Notably, nilotinib partially prevents the loss of DAT due to MPTP treatment (Fig. 2c and d). Since MPP+ binds to the DAT with high affinity and is taken up into DA neurons via the DAT19 and there is no reduction in the levels of DAT as well as no difference in the levels of MPP+, it indicates that the conversion to MPP+ from MPTP and the subsequent uptake of MPP+ into DA neurons is not affected by nilotinib treatment.
To examine whether nilotinib inhibits c-Abl activity in vivo, we analysed the effect of nilotinib treatment on c-Abl activity in the ventral midbrain of MPTP-intoxicated mice. MPTP treatment robustly activates c-Abl leading to an approximate 3-fold increase in phosphorylated c-Abl (Fig. 2e and f). Nilotinib pre- and post-treatment substantially prevents the MPTP-induced increase in c-Abl activity compared to MPTP treated mice and there is no effect on c-Abl activity in the mice with nilotinib only (Fig. 2e and f).
Nilotinib Restores the MPTP-Induced Depletion of Total Striatal Dopamine and Its Metabolites
The levels of DA and its metabolites in the striatum were measured by employing reverse-phase HPLC-electrochemical detection (ECD) to determine whether nilotinib can prevent the MPTP-induced reductions in DA and its metabolites. MPTP induces a significant reduction in DA levels in MPTP injected mice (Fig. 3a). Oral administration of nilotinib rescues the MPTP-induced DA loss in the striatum compared to MPTP treated mice (Fig. 3a). MPTP also causes a significant reduction in 3,4-dihydroxyphenylacetic acid (DOPAC) (Fig. 3b), homovanillic acid (HVA) (Fig. 3c) and 3-methoxytyramine (3MT) (Fig. 3d). Notably, administration of nilotinib significantly restores the reductions in DOPAC in MPTP injected mice (Fig. 3b). To analyse whether the catabolism of DA is altered by nilotinib treatment, we assessed the DA turnover ratio. In the MPTP group, the DA turnover ratio is increased significantly, whereas these effects are significantly attenuated by nilotinib (Fig. 3e and f). Nilotinib alone has no effect on the levels of dopamine and its metabolites and turnover.
Figure 3. c-Abl inhibitor, nilotinib protects against MPTP-induced dopamine depletion.

Nilotinib or vehicle administered mice were subjected to acute MPTP injections (20 mg/kg, MPTP free base X 4, every 2 h). Striatal dopamine (DA) and metabolites levels were analysed 7 days after the last MPTP injection by HPLC-ECD analysis. (a) Nilotinib rescues DA loss and (b) DOPAC in the striatum of MPTP mice. (c) Striatal levels of HVA and (d) 3MT were restored in mice treated with nilotinib. (e) There is a significant decrease in DA turnover [(DOPAC+HVA/DA) and (f) (DOPAC+3MT/DA)] in the striatum of nilotinib treated mice. Error bars represent the mean ± SEM, n = 5 mice per group. Two-way ANOVA was used to test significant and followed with post-hoc Bonferroni test to compare with the multiple group. **p<0.0002, *** p<0.0001 for MPTP with compare control group, #p<0.05, ### p<0.001 for Nil+ MPTP compared MPTP group. (ns: not significant).
Nilotinib Protects Against MPTP-Induced Dopaminergic Neurodegeneration
To determine whether nilotinib protects against the loss of DA neurons following MPTP administration, the number of tyrosine hydroxylase (TH)-positive neurons in the SNpc was assessed via an unbiased stereological counting analysis. Immunostaining of SNpc sections (Fig. 4a) and quantification of TH- and Nissl-positive stained DA neurons (Fig. 4b and c) show a significant loss of dopaminergic neurons in mice treated with MPTP compared to saline-treated controls. Administration of nilotinib significantly protects against MPTP-induced TH-neuronal loss (Fig. 4a, b and c). Administration of nilotinib alone does not have any effect on TH positive DA neurons number (Fig. 4a, b and c).
Figure 4. Nilotinib prevents MPTP-induced dopaminergic neurodegeneration.

Seven days following the last MPTP injection, the number of TH-positive neurons was analysed in the substantia nigra pars compacta (SNpc) of vehicle and nilotinib treated mice in the presence or absence of MPTP treatment by unbiased stereologic counting. (a) Representative photomicrographs from coronal mesencephalon sections containing tyrosine hydroxylase (TH)-positive neurons of (a and c) vehicle and (b and d) nilotinib mice treated with saline or MPTP. (b) Stereology counts of TH and (c) Nissl-positive neurons in the SNpc from mice injected with saline and MPTP with or without treatment of nilotinib. Error bars represent the mean ± SEM, n = 5 mice per group. Two-way ANOVA was used to test significance and followed with post-hoc Bonferroni test to compare with the multiple group. *** p<0.0001 for MPTP compared to control group, #p<0.01 for Nil+ MPTP compared to MPTP group.
Nilotinib Prevents Against MPTP-Induced Dopaminergic Terminal Loss
Striatal TH-immunopositive fiber density was examined via optical densitometry using Image J software (NIH). MPTP treatment significantly reduces dopaminergic nerve terminal integrity compared to saline-treated controls (Fig. 5a and b). Nilotinib significantly restores the MPTP-induced reduction in TH-fiber density (Fig. 5a and b). Administration of nilotinib alone does not have any effect on TH- fiber density (Fig. 5a and b).
Figure 5. Nilotinib prevents MPTP-induced dopaminergic terminal loss.

Seven days following the last MPTP injection, striatal TH-immunopositive fiber density was estimated. (a) Representative photomicrograph of striatal sections stained for TH immunoreactivity. From top to bottom panel, (a and c) vehicle and (b and d) nilotinib mice treated with saline or MPTP respectively. (b) Quantification of dopaminergic fiber densities in the striatum by using Image J software (NIH). Significant TH fiber density loss was observed in striatum after MPTP intoxication. Administration of nilotinib significantly decreased the loss of striatal TH-positive fibers after MPTP treatment. Error bars represent the mean +/−SEM, n = 5 mice per group for Striatal TH-fiber density. Two-way ANOVA was used to test significance and followed with Bonferrioni post hoc test to compare with the multiple groups. ***p < 0.0001 for MPTP compared with control group. ###p < 0.001 for Nil plus MPTP compared to the MPTP group.
Nilotinib Prevents Against MPTP-Induced Dopaminergic Behavioural Deficits
To examine the effect of nilotinib on the behavioral deficits induced by the MPTP administration, motor deficits were assessed by the pole test. MPTP injections results in a significant increase in the time to reach the base of the pole compared to saline treated controls (Fig. 6). In MPTP-treated mice, administration of nilotinib significantly reduces the time (Fig. 6). These results indicate that nilotinib has neuroprotective effects against MPTP-induced behavioral deficits.
Figure 6. Nilotinib protects against MPTP-induced behavioral deficits.
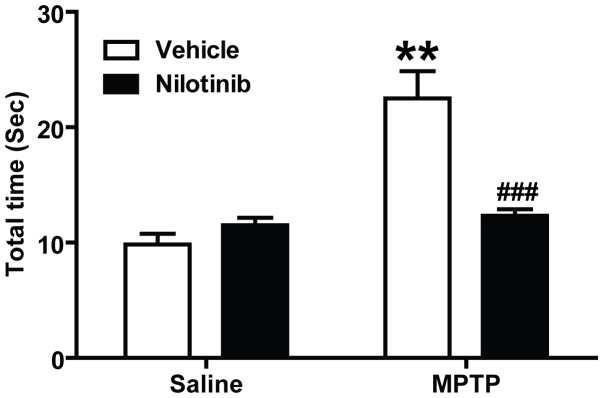
The sixth day after the last MPTP injection, the pole test was performed in vehicle or nilotinib treated mice. Behavioral abnormalities were improved in mice administered with nilotinib. Error bars represent the mean ± SEM, n = 10 mice per group for behavioural studies. Two-way ANOVA was used to test significance and followed with post-hoc Bonferroni test to compare with the multiple groups. ** p<0.001 for MPTP with compared to the control group, ###p<0.001 for Nil plus MPTP compared to the MPTP group.
The Neuroprotective Effects of Nilotinib in MPTP-Induced DA Neurodegeneration May Not Require Parkin
To determine whether nilotinib neuroprotection requires parkin, tyrosine phosphorylation of parkin and the levels of AIMP2 and PARIS were monitored. As previously reported9,10,15, the levels of tyrosine phosphorylated parkin and AIMP2 are increased in the midbrain after MPTP treatment compared with saline-treated controls (Fig. 7a and b). PARIS levels also increase after MPTP treatment (Fig. 7a and b). In MPTP injected mice, administration of nilotinib significantly reduces the increase in PARIS levels. Unexpectedly, nilotinib has no effect on the levels of tyrosine phosphorylated parkin and AIMP2 (Fig. 7a and b).
Figure 7. Nilotinib is unable to prevent MPTP-induced parkin phosphorylation and accumulation of AIMP2, but reduces PARIS in the ventral midbrain of MPTP-treated mice.

(a) Immunoblots of parkin immunoprecipitation samples from C57/BL6 mice treated with saline, MPTP only, MPTP with nilotinib (25 mg/kg body wt), or nilotinib alone (25 mg/kg body wt). Immunoblotting with an anti-phosphotyrosine antibody shows tyrosine-phosphorylated parkin and an anti-parkin antibody shows immunoprecipitated parkin. Brain lysates were immunoblotted with anti-AIMP2 and anti-PARIS antibodies to monitor their levels with β-actin serving as a loading control. (b) Normalized levels of tyrosine-phosphorylated parkin (p-parkin), AIMP2, and PARIS are indicated. The Error bars represent the mean ± SEM (n = 4 mice per group). Two-way ANOVA was used to test significance and followed with post-hoc Bonferroni test to compare with the multiple groups. ** p<0.001 for MPTP with compared to the control group, ##p<0.001 for Nil plus MPTP compared to the MPTP group. All experiments were repeated three times and representative images of the immunoblots are shown. Full length blots are presented in the supplementary Figure S2.
Discussion
This manuscript confirms and extends prior studies that demonstrated that c-Abl is activated following MPTP intoxication in mice9,10. Moreover inhibition of c-Abl with the brain-penetrant c-Abl inhibitor, nilotinib, protects against MPTP-induced loss of DA neurons, prevents the MPTP-induced reduction in DA levels and its metabolites, inhibits the MPTP-induced reduction in striatal DA terminal density and restores the behavioural deficits induced by MPTP. The degree of protection afforded by nilotinib against MPTP-induced deficits is greater than the c-Abl inhibitor STI-571, which is probably due to better brain penetration of nilotinib9. Nilotinib's protection against MPTP induced deficits seems to be equivalent to the protection afforded by conditional knockout of c-Abl10.
Prior studies indicated that protection against MPTP-induced deficits afforded by inhibiting or knocking out c-Abl was possibly due to preventing c-Abl induced parkin tyrosine phosphorylation, which leads to inactivation of parkin and accumulation of parkin substrates9,10. Consistent with this notion was the observation that in conditional c-Abl knockout mice there was no parkin tyrosine phosphorylation, and no demonstrable accumulation of parkin substrates following MPTP intoxication10. Moreover, knockdown of parkin prevented the protective effects of c-Abl inhibition against MPP+ toxicity in vitro10. In addition, a recent study examining the effect of INNO 406, another brain penetrant c-Abl inhibitor protected against MPTP intoxication via preventing parkin tyrosine phosphorylation, and accumulation of the parkin substrate AIMP213. In the current study, we show that c-Abl inhibition via nilotinib is able to protect against MPTP-induced dopaminergic deficits without reducing tyrosine phosphorylation of parkin and AIMP2 levels. On the other hand PARIS levels are significantly reduced. Since knockout of c-Abl and INNO-406 protect against MPTP toxicity by reducing tyrosine phosphorylation of parkin and accumulation of parkin substrates, it is conceivable that nilotinib's protection is occurring through similar mechanisms. The inability to see a reduction in tyrosine phosphorylation of parkin and AIMP2 levels despite a reduction in tyrosine-phosphorylation of c-Abl by nilotinib is probably due to different experimental paradigms between the nilotinib study reported here and the INNO 406 study13. Since the knockout of c-Abl protects against MPTP toxicity through reducing tyrosine phosphorylation of parkin and the levels of parkin substrates, the different readouts of parkin function between the two inhibitors may also be due to different pharmacodynamic properties between nilotinib and INNO 406, which account for INNO 406 prevention of tyrosine phosphorylation of parkin but not nilotinib. Although there is no significance decrease in tyrosine phosphorylation of parkin and accumulation of AIMP2 in nilotinib plus MPTP treated mice compared to MPTP plus vehicle treated mice, we observed an approximate 20% reduction in the tyrosine phosphorylation of parkin and AIMP2 levels suggesting that nilotinib's inhibition of c-Abl was incomplete.
Despite nilotinib failing to reduce tyrosine phosphorylation of parkin and AIMP2 levels, it protected against MPTP toxicity. Thus it is possible that PARIS is the predominant parkin substrate that mediates loss of DA neurons due to parkin inactivation since PARIS levels were reduced by c-Abl inhibition, but AIMP2 levels were not. Moreover, we cannot exclude the possibility that c-Abl toxicity involves both parkin dependent and parkin-independent mechanisms. In addition, nilotinib inhibits other kinases and it is possible that it provides protection against MPTP via inhibition of these other kinases20. Future studies will be required to clarify these issues.
c-Abl is active in sporadic PD leading to parkin tyrosine phosphorylation and accumulation of parkin substrates suggesting that c-Abl activation could contribute to the degenerative process of PD9,10. Thus, c-Abl inhibitors with brain penetrating properties are attractive disease modifying agents for PD treatment. Our findings that nilotinib protects against MPTP-induced dopaminergic deficits suggest that nilotinib and other brain penetrant c-Abl inhibitors could be used as disease modifying therapies in PD. Consistent with this notion is the observation that nilotinib reverses the loss of dopamine neurons and improves motor behavior in α-synuclein PD models12.
Based on the findings reported here and elsewhere, future studies are needed to identify the optimal dosing and administration to take advantage of the full neuroprotective potential of nilotinib. Alternatively other c-Abl inhibitors with better pharmacokinetic properties and safety profiles may need to be identified to take advantage of inhibition of c-Abl as a disease modifying therapy for PD. In addition, since post-treatment with nilotinib did not protect against MPTP toxicity, it will be important to determine whether c-Abl inhibition can protect against degeneration in α-synuclein PD models when there is ongoing degeneration. This is important because future treatment in PD patients will be done in the setting of active degeneration.
In summary, brain permeable c-Abl inhibitors are promising drug candidates for PD therapy to halt the progression of neuronal loss in this disorder. Since c-Abl activation has been implicated in other neurodegenerative diseases such as Alzheimer's disease and human tauopathies7, c-Abl inhibitors may have broad clinical implications. Our current finding coupled with other recent observations provides a strong rationale for identifying brain permeable c-Abl inhibitors with optimal pharmacodynamics properties to slow down the progression of PD and potentially other degenerative diseases.
Methods
Animals
All procedures involving animals were approved by and conformed to the guidelines of the Institutional Animal Care Committee of Johns Hopkins University. Male C57BL/6 mice (3-month old, 25–30 g) were obtained from Charles River. Animals were housed in a 12 h dark and light cycle with free access to water and food. All mice were acclimatised for 3d in the procedure room before starting any experiments. We have taken great measure to reduce the number of animal used in these studies and also taken effort to reduce animal suffering from pain and discomfort.
Treatment groups and nilotinib administration
Mice (n = 5 per group) were divided into 4 treatment group as follows, saline, nilotinib, MPTP and nilotinib along with MPTP (see Fig. 1 for more detail). Nilotinib (25 mg/kg) was solubilized in the vehicle containing (10% N-Methyl-2-pyrrolidone (NMP) and 90% polyethylene glycol 300 (PEG 300)). Saline and MPTP group were pre-treated with vehicle and nilotinib alone and nilotinib plus MPTP group mice received nilotinib for a week administered by oral gavage (once a day). On the seventh day, the saline and nilotinib groups received four intra-peritoneal (i.p.) injections of saline at 2 h intervals. The MPTP and nilotinib plus MPTP groups received four intra-peritoneal (i.p.) injections of MPTP·HCl (20 mg/kg free base) at 2 h intervals10,21. Following day 7, saline and MPTP groups received only vehicle for an additional one week. Nilotinib alone and nilotinib plus MPTP groups received an additional one week of nilotinib. Pole test were performed on day 13. All the mice were sacrificed on day 14 and brain samples were processed for neurochemical, biochemical and immunohistochemistry studies.
Pharmacokinetic analysis
The nilotinib concentration was estimated by employing HPLC-UV detection at 260 nm22. Mice were subjected to oral gavage with a single dose of nilotinib (25 mg/kg). Whole brain was extracted and processed for estimation of nilotinib at 2 h after oral gavage. Tissues were sonicated in 0.1 M ice cold percholric acid followed by centrifugation at 14,000 rpm for 30 min. A 20 µl supernatant was injected in to a cation-exchange Ultracyl-CS fitted with a guard column. The isocratic mobile phase consisted of methanol: 50 mM ammonium acetate (pH 8) (65:35, v/v) at room temperature under a constant flow rate of 1 ml/min employing HPLC-UV (Antec Leyden). Chromatograms were analysed using Clarity ® software. Results are presented as pg/mg protein.
MPP+ analysis
MPP+ was estimated following the method described previously10 using HPLC-UV detection at 295 nm. Vehicle and nilotinib groups were injected i.p. with four doses of MPTP (20 mg/kg, i.p. every 2 h) or saline and euthanized at 2 h after the final saline or MPTP injection. Striatum were dissected out and stored at −80°C until analysis. Striatal tissues were sonicated in 10 volumes of 5% trichloroacetic acid containing 5 μg/mL of 4-phenylpyridine (Sigma) as an internal standard. The samples were centrifuged at 14,000 rpm for 10 min and the supernatant was injected onto a cation-exchange Ultracyl- CS column (Beckman). The mobile phase consisted of 90% of a solution of 0.1M acetic acid and 75 mM triethylamine·HCl (pH 2.35 adjusted with formic acid), and 10% acetonitrile. The flow rate was 1.5 mL. Results are presented as μg/mg protein.
Monoamine analysis
Biogenic amine concentrations were measured by high-performance liquid chromatography with electrochemical detection (HPLC-ECD). Briefly, mice were sacrificed by decapitation and the striatum was quickly removed. Striatal tissue was weighed and sonicated in 0.2 ml ice cold 0.01 mM perchloric acid containing 0.01% EDTA and 60 ng 3,4-dihydroxybenzylamine (DHBA) as an internal standard. After centrifugation (15,000 × g, 30 min, 4°C), the supernatant was passed through a 0.2 µm filter. Twenty microliters of the supernatant were analysed in the HPLC column (4.6 mm × 150 mm C-18 reverse phase column, MC Medical, Tokyo, Japan) by a dual channel coulchem III electrochemical detector (Model 5300, ESA, Inc Chelmsford, MA, USA). The protein concentrations of tissue homogenates were measured using the BCA protein assay kit (Pierce, Rockford, IL, USA). Data were normalized to protein concentrations and expressed in ng/mg protein10.
Pole test
Animals were acclimatized in the behavioural procedure room for 30 min. The pole is made up 2.5 ft metal rod with 9 mm diameter and wrapped with bandage gauze. Briefly, the mice were placed on the top of the pole (3 inch from the top of the pole) facing the head-up. Total time taken to reach the base of the pole was recorded. Before the actual test the mice were trained for two consecutive days and each training session consists of three test trials. On the day of the test mice were evaluated in three sessions and total times were recorded. The maximum cutoff of time to stop the test and recording was 120 sec. Results were expressed in total time (in sec)23.
Immunohistochemistry and quantitative analysis
Mice were perfused with ice-cold phosphate buffered saline (PBS) and followed by 4% paraformaldehyde/PBS (pH 7.4). Brains were removed and post fixed for 4 h in the same fixative. After cryoprotection in 30% sucrose/PBS (pH 7.4), brains were frozen and serial coronal sections (60 μm sections) were cut with a microtome. Free-floating 60 μm sections were blocked with 4% goat serum/PBS plus 0.2% Triton X-100 and incubated with an antibody against TH (rabbit polyclonal; Novus Biologicals) followed by incubation with biotin-conjugated anti-rabbit antibody (anti-rabbit polyclonal; Vector Labs), ABC reagents (Vector Labs), and SigmaFast DAB Peroxidase Substrate (Sigma-Aldrich). Sections were counterstained with Nissl (0.09% thionin).
TH-positive and nissl positive DA neurons from the SNpc region were counted through optical fractionators, the unbiased method for cell counting. This method was carried out by using a computer- assisted image analysis system consisting of an Axiophot photomicroscope (Carl Zeiss Vision) equipped with a computer controlled motorized stage (Ludl Electronics), a Hitachi HV C20 camera, and Stereo Investigator software (MicroBright-Field). The total number of TH- stained neurons and nissl counts was calculated as described24,25,26. Serial striatal sections were processed for TH staining following the same procedure as above. Fiber density in the striatum was quantified by optical density (OD). ImagJ software (NIH) was used to analyse the OD as previously described27.
Immunoprecipitation and immunoblot analysis
The mouse brain tissues were homogenized in lysis buffer (10 mM Tris-HCL, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 10 mM Na-β-glycerophosphate, Phosphate inhibitor mixture I and II (Sigma), and complete protease inhibitor mixture (Roche)], using a Diax 900 homogenizer (Sigma). After homogenization, samples were rotated at 4°C for 30 min for complete lysis, the homogenate was centrifuged at 52,000 rpm for 20 min, and the supernatants were collected. Protein levels were quantified using the BCA Kit (Pierce) with BSA standards and analysed by immunoblot. The supernatant was used for immunoprecipitation with anti-parkin. The immunocomplexes were then washed with immunopreciptation buffer six times and separated by SDS/PAGE and subjected to immunoblot analysis with indicated antibodies. Immunoblot signals were visualized with chemiluminescence.
Statistical analysis
All quantitative data are expressed as the mean ± SEM. Statistical significance was determined by two-way ANOVA followed by Bonferroni post-hoc analysis for comparison among multiple treatment groups. p values lower than 0.05 were considered to be significant. Respective p values are indicated in figure legends. *Asterisks sign indicate statistical significance between control versus MPTP groups and # pound sign indicate statistical significance between MPTP versus nilotinib plus MPTP groups.
Author Contributions
V.L.D., H.S.K. and T.M.D. supervised the project. S.S.K., V.L.D., H.S.K. and T.M.D. formulated the hypothesis. S.S.K., V.L.D., H.S.K. and T.M.D. designed in vivo experiments. S.S.K. performed the MPTP injections, HPLC analysis, and behaviour tests. S.S.K. and Y.L. performed blinded stereological counting of tyrosine hydroxylase–positive neurons and nissl counts. S.B. performed the immunoblots and analysed the results. S.S.K., V.L.D., H.S.K. and T.M.D. initiated and organized the study and wrote the manuscript. All authors contributed to writing the final manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grants from the NIH NS38377 and the JPB Foundation. Y.L. was supported by the Samsung Scholarship Foundation. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Programs.
References
- Savitt J. M., Dawson V. L. & Dawson T. M. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 116, 1744–1754 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W., Mahlknecht P. & Jankovic J. Emerging therapies for Parkinson's disease. Curr Opin Neurol 25, 448–459 (2012). [DOI] [PubMed] [Google Scholar]
- Jankovic J. & Poewe W. Therapies in Parkinson's disease. Curr Opin Neurol 25, 433–447 (2012). [DOI] [PubMed] [Google Scholar]
- Dawson T. M. & Dawson V. L. Molecular pathways of neurodegeneration in Parkinson's disease. Science 302, 819–822 (2003). [DOI] [PubMed] [Google Scholar]
- Zhang Y., Dawson V. L. & Dawson T. M. Oxidative stress and genetics in the pathogenesis of Parkinson's disease. Neurobiol Dis 7, 240–250 (2000). [DOI] [PubMed] [Google Scholar]
- Li B. c-Abl in oxidative stress, aging and cancer. Cell Cycle 4, 246–248 (2005). [PubMed] [Google Scholar]
- Schlatterer S. D., Acker C. M. & Davies P. c-Abl in neurodegenerative disease. J Mol Neurosci 45, 445–452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez A. R., Sandoval P. C., Leal N. R., Castro P. U. & Kosik K. S. Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species. Neurobiol Dis 17, 326–336 (2004). [DOI] [PubMed] [Google Scholar]
- Imam S. Z. et al. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci 31, 157–163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H. S. et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A 107, 16691–16696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatterer S. D., Tremblay M. A., Acker C. M. & Davies P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J Alzheimers Dis 25, 119–133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron M. L., Lonskaya I. & Moussa C. E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum Mol Genet 22, 3315–3328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam S. Z. et al. Neuroprotective efficacy of a new brain-penetrating C-Abl inhibitor in a murine Parkinson's disease model. PLoS One 8, e65129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahul-Mellier A. L. et al. c-Abl phosphorylates alpha-syn and regulates its degradation, implication for alpha-syn clearance and contribution to the pathogenesis of Parkinson's Disease. Hum Mol Genet, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. H. et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144, 689–702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci 16, 1392–1400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson T. M. & Dawson V. L. Parkin Plays a Role in Sporadic Parkinson's Disease. Neurodegener Dis, (2013). [Google Scholar]
- Soverini S., Martinelli G., Rosti G., Iacobucci I. & Baccarani M. Advances in treatment of chronic myeloid leukemia with tyrosine kinase inhibitors: the evolving role of Bcr-Abl mutations and mutational analysis. Pharmacogenomics 13, 1271–1284 (2012). [DOI] [PubMed] [Google Scholar]
- Javitch J. A. & Snyder S. H. Uptake of MPP(+) by dopamine neurons explains selectivity of parkinsonism-inducing neurotoxin, MPTP. Eur J Pharmacol 106, 455–456 (1984). [DOI] [PubMed] [Google Scholar]
- Steegmann J. L., Cervantes F., le Coutre P., Porkka K. & Saglio G. Off-target effects of BCR-ABL1 inhibitors and their potential long-term implications in patients with chronic myeloid leukemia. Leuk Lymphoma 53, 2351–2361 (2012). [DOI] [PubMed] [Google Scholar]
- Thomas B. et al. Resistance to MPTP-neurotoxicity in alpha-synuclein knockout mice is complemented by human alpha-synuclein and associated with increased beta-synuclein and Akt activation. PLoS One 6, e16706 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A. et al. Simultaneous determination of nilotinib, imatinib and its main metabolite (CGP-74588) in human plasma by ultra-violet high performance liquid chromatography. Leuk Res 34, 702–707 (2010). [DOI] [PubMed] [Google Scholar]
- Karl T., Pabst R. & von Horsten S. Behavioral phenotyping of mice in pharmacological and toxicological research. Exp Toxicol Pathol 55, 69–83 (2003). [DOI] [PubMed] [Google Scholar]
- Dauer W. et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A 99, 14524–14529 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S. et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A 101, 665–670 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk K. C. et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane E. L., Winkler C., Brundin P. & Cenci M. A. The impact of graft size on the development of dyskinesia following intrastriatal grafting of embryonic dopamine neurons in the rat. Neurobiol Dis 22, 334–345 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information