

Abstract
Vitamin K-dependent proteases generated in response to vascular injury and infection enable fibrin clot formation, but also trigger distinct immuno-regulatory signaling pathways on myeloid cells. Factor Xa, a protease crucial for blood coagulation, also induces protease-activated, receptor-dependent cell signaling. Factor Xa can bind both monocytes and macrophages, but whether factor Xa-dependent signaling stimulates or suppresses myeloid cell cytokine production in response to Toll-like receptor activation is not known. In this study, exposure to factor Xa significantly impaired pro-inflammatory cytokine production from lipopolysaccharide-treated peripheral blood mononuclear cells, THP-1 monocytic cells and murine macrophages. Furthermore, factor Xa inhibited nuclear factor-kappa B activation in THP-1 reporter cells, requiring phosphatidylinositide 3-kinase activity for its anti-inflammatory effect. Active-site blockade, γ-carboxyglutamic acid domain truncation and a peptide mimic of the factor Xa inter-epidermal growth factor-like region prevented factor Xa inhibition of lipopolysaccharide-induced tumor necrosis factor-α release. In addition, factor Xa anti-inflammatory activity was markedly attenuated by the presence of an antagonist of protease-activated receptor 2, but not protease-activated receptor 1. The key role of protease-activated receptor 2 in eliciting factor Xa-dependent anti-inflammatory signaling on macrophages was further underscored by the inability of factor Xa to mediate inhibition of tumor necrosis factor-α and interleukin-6 release from murine bone marrow-derived protease-activated receptor 2-deficient macrophages. We also show for the first time that, in addition to protease-activated receptor 2, factor Xa requires a receptor-associated protein-sensitive low-density lipoprotein receptor to inhibit lipopolysaccharide-induced cytokine production. Collectively, the findings of this study support a novel function for factor Xa as an endogenous, receptor-associated protein-sensitive, protease-activated receptor 2-dependent regulator of myeloid cell pro-inflammatory cytokine production.
Introduction
During sepsis, invading pathogens activate pattern recognition receptors expressed on a variety of cell types using specific pathogen-association molecular patterns present in bacteria, viruses, fungi and parasites.1 Toll-like receptors (TLR) are the most studied family of pattern recognition receptors, and their activation triggers signal transduction pathways that up-regulate pro-inflammatory cytokine expression vital for the resolution of infection.2 Lipopolysaccharide (LPS) from Gram-negative bacteria activates TLR4 to induce pro-inflammatory cytokine generation and leads to rapid induction of tissue factor (TF) expression on leukocytes,3 triggering blood coagulation in the absence of blood vessel damage.4 In sepsis, LPS-induced aberrant TF expression, depletion of anticoagulant plasma proteins5 and down-regulation of vascular cell surface receptors6 leads to unregulated coagulation protease activation and disseminated intravascular coagulopathy, often causing multiorgan failure and death.7
Coagulation proteases generated as a consequence of infection can interact with vascular and leukocyte surface receptors to either promote or inhibit pro-inflammatory signaling pathways. Inhibition of TF8 and thrombin9 is protective in murine endotoxemia. In contrast, the anticoagulant protease activated protein C (APC) suppresses LPS or cytokine-induced inflammation on monocytes,10 macrophages11,12 and vascular endothelial cells.13 Deficiency14 or impaired generation15,16 of APC increases sensitivity to LPS challenge in mice and recombinant APC has been used in the treatment of individuals with severe sepsis.17
Activated factor X (FXa) is a vitamin K-dependent protease generated rapidly upon exposure to TF. FXa, as part of the prothrombinase complex, catalyzes thrombin generation, leading to fibrin deposition. FXa is critical for effective blood coagulation, as evidenced by the severe bleeding phenotype of FX-deficient individuals18 and the embryonic or perinatal lethality exhibited by FX−/− mice.19
Like other coagulation proteases, FXa cell signaling is transduced by protease-activated receptors (PAR). Although structurally homologous to APC, FXa has been described both as a driver20,21 and an inhibitor22,23 of TLR- and cytokine-induced inflammation depending on the cell type and signaling receptors activated. FXa can activate both PAR1, PAR2 and to a lesser extent, PAR4.24 Co-receptors for FXa activation of PAR appear crucial in dictating FXa signaling specificity and multiple non-PAR cell receptors for FXa have been identified. Effector protease receptor 1 (EPR-1) was originally characterized as a high-affinity FXa receptor on platelets, endothelial cells and various leukocyte subsets.25–27 However, the molecular mechanism through which EPR-1-bound FXa exerts these cellular effects has not been described, and the identity of EPR-1 is itself controversial.28 FXa also has affinity for the endothelial cell protein C receptor (EPCR).29 Blockade of the EPCR-FXa interaction with an anti-EPCR monoclonal antibody prevents PAR1 activation by FXa and inhibits FXa cytoprotective signaling on endothelial cells.29 Furthermore, annexin-2 has been shown to bind specifically to an FXa isoform (FXa-β) and to facilitate PAR1 activation on endothelial cells, but its role in response to inflammatory stimuli is unknown.30 The receptor signaling requirements and downstream cellular consequences of FXa signal transduction are, therefore, complex, cell-type dependent and often divergent, and the determinants of this signaling diversity are not fully understood.
FX(a) binds both monocytes and macrophages with high affinity31 and is rapidly activated upon LPS activation and TF decryption, playing a crucial role in thrombin generation and subsequently fibrin deposition. However, whether FXa generated on the surface of myeloid cells contributes to the innate immune response beyond catalyzing coagulation is not known, as PAR-dependent signaling by FXa on myeloid cells and its role in response to TLR activation have not been characterized. In this study, we show that FXa inhibits a pro-inflammatory cytokine response to TLR stimulation in monocytes and macrophages via PAR2 activation and downstream activation and suppression of phosphatidylinositide 3-kinase (PI3K) and nuclear factor-kappa B (NF-κB) signaling, respectively. Furthermore, FXa structural determinants and cell surface receptor(s) required to facilitate FXa-mediated hyposensitivity to TLR stimulation are identified, thus delineating a novel role for FXa in regulating the inflammatory response to TLR activation on myeloid cells.
Methods
Materials
Detailed information relating to plasma-derived purified proteins, synthetic peptides and antibodies used in this study is given in the Online Supplementary Methods.
Isolation of human peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coat whole blood component obtained from healthy donor pools provided by the Irish Blood Transfusion Service. PBMC were isolated by centrifugation at 2000 rpm in Ficoll-Hypaque density gradient using the Boyum method32 then cultured in RPMI containing 10% fetal bovine serum. More details on PBMC isolation and culture are given in the Online Supplementary Methods.
Quantification of tumor necrosis factor-α secretion from THP-1 monocytic cells
THP-1 cells were re-suspended in serum-free RPMI 1640 medium (Life Technologies, Paisley, UK) supplemented with 3 mM CaCl2 and 0.6 mM MgCl2 at a density of 3×106 cells/mL and seeded in 96-well microtiter plates. Cells were incubated with vitamin K-dependent proteases (FVIIa, FIXa, FXa, FXaDERG, FXaDESGLA or APC; 0.313–20 nM) for 1–3 h as described for each assay, then stimulated with TLR agonists for 4 h. Cell viability was not compromised by FXa incubation (Online Supplementary Figure S2). Supernatants were collected thereafter and tumor necrosis factor alpha (TNFα) generation was determined using a human TNFα DuoSet enzyme-linked immunosorbent assay (R&D Systems, MN, USA) or a HEK Blue TNFα/IL-1β cell line (Invivogen, Toulouse, France). Exposure of the HEK Blue TNFα/IL-1β cells to TNFα resulted in dose-dependent activation of the NF-κB/AP-1 pathways and expression of the secreted alkaline phosphatase (ALP) reporter gene (Online Supplementary Figure S1). ALP activity in the supernatant was detected using QUANTI-Blue medium (Invivogen, Toulouse, France) containing a colorimetric ALP substrate. Colorimetric measurements were taken at 650 nm. ALP activity relative to that of cells treated only this LPS was determined using the following equation:
Where X is the test sample, N is the untreated phosphate-buffered sample and P is the LPS-treated positive control sample.
Quantification of nuclear factor-κB activation by THP1-XBlueCD14 cells
NF-κB activation was measured using THP1-XBlueCD14 cells (Invivogen, Toulouse, France). These cells stably co-express CD14 and an NF-κB/AP-1-inducible secreted ALP reporter. The THP1-XBlueCD14 cells were re-suspended in serum-free RPMI 1640 medium supplemented with 3 mM CaCl2 and 0.6 mM MgCl2 at a density of 3×106 cells/mL and seeded in 96-well plates. The cells were incubated with FXa (20 nM) for 1 h and subsequently stimulated with LPS (31–500 ng/mL) for 6 h. ALP activity in the supernatant was detected with QUANTI-Blue, as described above.
Mice
PAR2−/− mice, originally from Jackson Laboratories, were generated on a BALB/c background and maintained in-house. Additional details are given in the Online Supplementary Methods section.
Isolation and culture of murine bone marrow-derived macrophages
Bone marrow-derived macrophages were prepared from mice by standard techniques.33 Additional details are given in the Online Supplementary Methods section.
Quantification of cytokine secretion from human peripheral blood mononuclear cells and murine bone marrow-derived macrophages
Human PBMC or murine bone marrow-derived macrophages were washed with phosphate-buffered saline and incubated with FXa/APC/FXaDEGR (20 nM) in serum-free RPMI 1640 medium supplemented with 1 mM CaCl2 and 0.2 mM MgCl2 for 3 h prior to stimulation with LPS (PBMC; 50 ng/mL and macrophages; 20 ng/mL) for 18 h. Supernatants were collected and TNFα and IL-6 detected using DuoSet enzyme-linked immunosorbent assays (R&D Systems, MN, USA) for human and murine cytokines.
Results
Factor Xa attenuates pro-inflammatory cytokine production from myeloid cells in response to lipopolysaccharide
To examine whether FXa exposure promotes or suppresses the pro-inflammatory cytokine response of myeloid cells to LPS, the effect of FXa on TNFα production from LPS-treated primary monocytes (PBMC) was assessed. FXa inhibited LPS-induced TNFα secretion from PBMC in a significant and concentration-dependent manner (IC50~3 nM FXa; Figure 1A). FXa alone, however, did not induce TNFα production, or negatively affect cell viability (Online Supplementary Figure S2). The extent of FXa-mediated inhibition of TNFα release was similar to that observed in the presence of the homologous anti-inflammatory enzyme APC (Figure 1A). When monocytic THP-1 cells were used in place of PBMC in the same assay, prior exposure to FXa resulted in a significant (60±9% at 20 nM FXa; P<0.001) and dose-dependent (IC50~3 nM) reduction in LPS-induced TNFα secretion, which was again comparable to that observed with APC in the same assay (Figure 1B). A comparison of FXa anti-inflammatory activity with that of other homologous vitamin K-dependent proteases showed that, of the procoagulant vitamin K-dependent proteases tested, only FXa could replicate APC anti-inflammatory activity on monocytes (Figure 1C).
Figure 1.
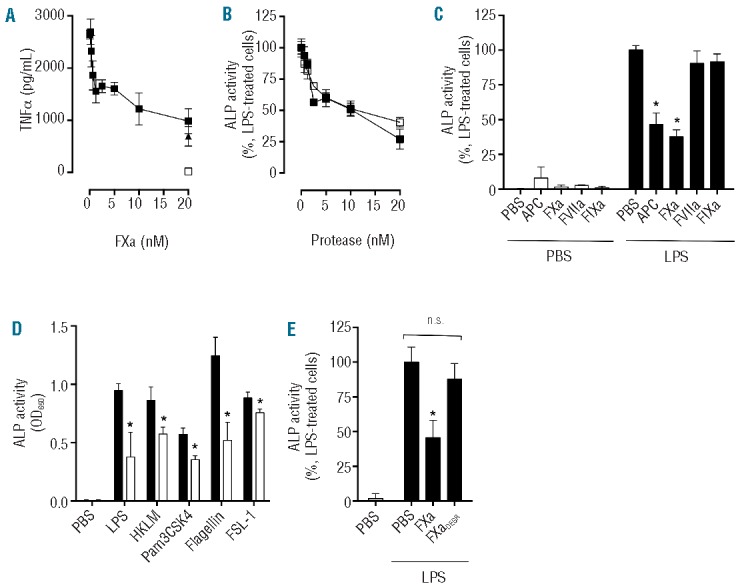
FXa inhibits LPS-induced monocyte TNFα secretion. (A) PBMC were exposed to FXa (0.313–20 nM;■)/ APC (20 nM;▲) for 3 h prior to stimulation with LPS (50 ng/mL; 18 h). TNFα production was determined by ELISA. No TNFα was detected in the absence of LPS (□). (B) The relative anti-inflammatory activity of APC (0.625–20 nM;□) and FXa (0.313–20 nM;■) was determined by incubation with THP-1 cells for 3 h, prior to stimulation with LPS (500 ng/mL; 4 h). TNFα secretion was detected using HEK Blue TNFα reporter cells (ALP activity) as described in the Methods section. (C) THP-1 cells were exposed to vitamin K-dependent proteases (APC, FXa, FVIIa, FIXa, all 20 nM) and then treated with PBS or LPS (500 ng/mL; 4 h) before TNFα release was determined using HEK Blue TNFα reporter cells (D) THP-1 cells were exposed to PBS (black bars) or FXa (white bars, 20 nM) for 1 h prior to stimulation with TLR agonists LPS (500 ng/mL), HKLM (5×106 cells), Pam3CSK4 (500 ng/mL), flagellin (50 ng/mL) or FSL-1 (50 ng/mL) for 4 h and TNFα was then measured. (E) THP-1 cells were exposed to FXa or FXaDEGR (20 nM) for 1 h prior to stimulation with LPS (500 ng/mL) for 4 h and then released TNFα was detected using HEK Blue TNFα reporter cells. All results represent the mean of at least three independent experiments ± SD.
Pro-inflammatory gene activation and cytokine expression can be initiated upon detection of TLR ligands other than LPS, by activation of TLR family members with different ligand specificity. Coagulation protease modulation of cytokine production induced by activation of TLR other than TLR4 has not, however, been determined. To investigate this, FXa was added to THP-1 cells prior to stimulation with activating ligands for the TLR2/2 homodimer (HKLM), TLR1/2 heterodimer (Pam3CSK4), TLR5/5 homodimer (flagellin) and TLR2/6 heterodimer (FLS-1). TNFα generation in response to each TLR agonist was significantly inhibited by FXa (P<0.05, Figure 1D). Importantly, co-incubation of FXa with a chloromethyl ketone inhibitor (DEGR), essentially eliminated FXa inhibition of LPS-induced cytokine release from THP-1 cells (Figure 1E). This demonstrated that FXa proteolytic activity was required for attenuation of cytokine release in response to LPS and, furthermore, that LPS contamination of plasma-derived FXa was not responsible for the reduced cytokine response induced by FXa.
Factor Xa attenuation of lipopolysaccharide-induced pro-inflammatory cytokine secretion on monocytes and macrophages requires PAR2 and is sensitive to receptor-associated protein
FXa can activate either PAR1 or PAR2 to mediate FXa-dependent anti-inflammatory or cytoprotective activity on endothelial cells. Active-site inhibition diminished the ability of FXa to inhibit cytokine release upon TLR4 activation by LPS from THP-1 cells (Figure 1E), suggesting of a key role for PAR proteolysis in mediating FXa anti-inflammatory signaling on myeloid cells. To determine which PAR was required to mediate this phenomenon, PBMC were treated with either a PAR1 or PAR2 antagonist (FR131117 and GB83, respectively) in conjunction with FXa and then challenged with LPS. PAR1 antagonism did not affect FXa impairment of LPS-induced TNFα production at any concentration tested; however, PAR2 antagonism dose-dependently inhibited FXa anti-inflammatory function until complete inhibition was observed at approximately 1 μM GB83 (Figure 2A). As expected, GB83 had no effect on APC-mediated TNFα inhibition. Replacement of PBMC with THP-1 cells in the same assay illustrated a similar dependence on PAR2, rather than PAR1, to enable FXa anti-inflammatory signaling on this type of cell (Figure 2B). To further characterize the role of PAR2 in FXa anti-inflammatory signaling on myeloid cells, macrophages isolated from wild-type and PAR2−/− BALB/c mice were exposed to FXa, FXaDEGR or APC prior to LPS stimulation. The inhibitory activity of FXa upon TNFα and IL-6 production from both LPS-treated wild-type macrophages was approximately double that observed when the same macrophages were incubated with FXaDEGR (Figure 2C,D). Accordingly, when the same assay was performed in the presence of PAR2−/− macrophages, both FXa and FXaDEGR failed to inhibit TNFα and IL-6 production significantly upon LPS challenge (Figure 2E,F). In contrast, APC significantly restricted TNFα and IL-6 release from LPS-stimulated wild-type and PAR2−/− macrophages (Figure 2C–F), in agreement with previous studies highlighting the importance of PAR1, rather than PAR2, in mediating APC anti-inflammatory activity on this cell type.12
Figure 2.

FXa inhibits LPS-induced pro-inflammatory cytokine production from myeloid cells via PAR2 activation. (A) PBMC were exposed to PAR1 (FR131117;○) or PAR2 (GB83;■) antagonists for 30 min prior to FXa (20nM) co-incubation for 3 h, followed by LPS treatment (50 ng/mL; 18 h). TNFα was then measured by ELISA. LPS-induced TNFα in the absence of FXa (▲) and LPS-induced TNFα in the presence of APC/GB83 is also shown (♦). (B) THP-1 cells were exposed to GB83 (0.125–1 μM) for 30 min prior to incubation with FXa (20nM;■) or APC (20nM;♦) for 3 h. Similarly, THP-1 cells were exposed to FR131117 (1.25 μM) for 30 min prior to incubation with FXa (20nM;○) for 3 h. Cells were treated with LPS (500 ng/mL; 4 h) and TNFα secretion was detected using HEK Blue TNFα reporter cells. LPS-induced TNFα in the absence of FXa or PAR antagonists is shown (▲). GB83 did not induce TNFα production in the absence of LPS (□). Murine bone marrow-derived macrophages were isolated from wild-type (C and D) and PAR2−/− (E and F) BALB/c mice and exposed to APC/FXa/FXaDEGR (all 20 nM) for 3 h prior to addition of PBS or LPS (20 ng/mL) for 18 h. Murine TNFα (C and E) and IL-6 (D and F) were determined by ELISA and the mean ± SD from three independent experiments is shown.
FXa can utilize a number of co-receptors to elicit PAR-dependent signaling, but those required for suppression of the pro-inflammatory cytokine response to TLR activation are not known. Low density lipoprotein (LDL) receptor family members have been identified as potential mediators of vitamin K-dependent protease signaling on monocytes.34 In order to determine whether an LDL family receptor interaction might also modulate PAR2-dependent FXa anti-inflammatory activity on myeloid cells, receptor-associated protein (RAP; which binds to the ligand-binding region of all LDL family receptors and prevents ligand interaction), was added in combination with FXa. RAP sensitivity of FXa-mediated attenuation of cytokine production from LPS-treated THP-1 monocytes and murine macrophages was then determined. RAP alone had no effect upon LPS-induced pro-inflammatory cytokine production in the absence of FXa, but completely ablated FXa anti-inflammatory activity on THP-1 monocytes (Figure 3A). Similarly, incubation solely with RAP did not alter LPS-induced cytokine production from LPS-treated macrophages, but completely inhibited FXa inhibition of TNF and IL-6 release (Figure 3B,C), highlighting a novel role of RAP-sensitive LDL receptors in mediating FXa anti-inflammatory cell signaling. Given the important role of ApoER2 in mediating APC anti-inflammatory signaling on monocytes, we sought to determine whether ApoER2 was the target for RAP inhibition of FXa anti-inflammatory activity on THP-1 cells. To achieve this, FXa was incubated with THP-1 cells in the presence of a mouse anti-ApoER2 monoclonal antibody directed against the extracellular region of human ApoER2 (anti-ApoER2 mAb 1). Anti-ApoER2 mAb 1 dose-dependently attenuated FXa impairment of LPS-induced TNFα production (Figure 5D and inset). In contrast, anti-ApoER2 mAb 2 (directed against the ApoER2 intracellular region) and a mouse IgG1 antibody isotype control had no effect upon FXa anti-inflammatory activity, suggesting a novel role for the extracellular region of ApoER2 in enabling FXa anti-inflammatory activity on THP-1 monocytes. These studies indicate that PAR2, in conjunction with a RAP-sensitive membrane receptor, is necessary for optimal FXa-mediated suppression of LPS-induced pro-inflammatory cytokine production from myeloid cells.
Figure 3.
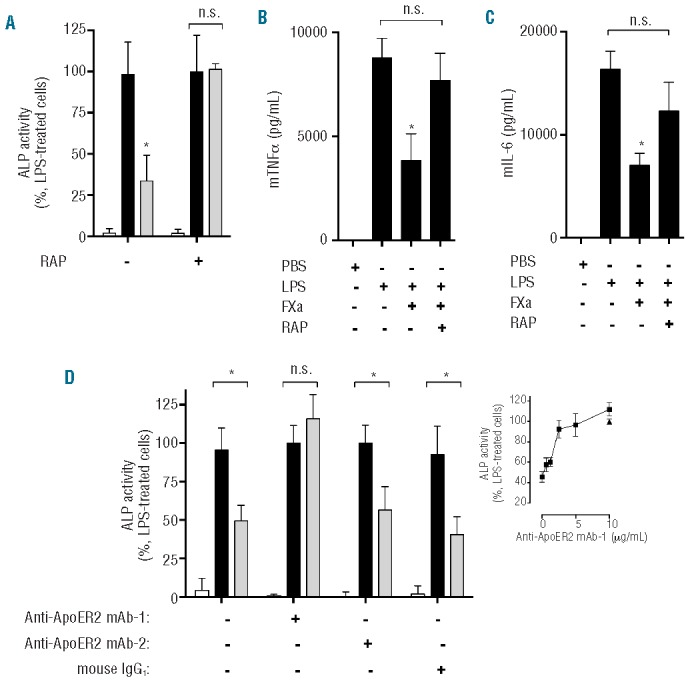
PAR2-dependent FXa suppression of LPS-induced cytokine production from myeloid cells is sensitive to RAP. (A) THP-1 cells were incubated with RAP (80 mM) for 30 min, prior to incubation with PBS (black bars) or FXa (gray bars, 20 nM) for 3 h. Cells were then treated with LPS (500 ng/mL; 4 h) and TNFα secretion was detected using HEK Blue TNFα reporter cells. Treatment with PBS alone (white bars) was used as a negative control. (B and C) Murine bone marrow-derived macrophages were treated with RAP (80 mM) for 30 min prior to FXa incubation (20 nM) for 3 h and stimulated with LPS (20 ng/mL; 18 h). Murine (B) TNFα and (C) IL-6 production was determined by ELISA. (D) THP-1 cells were incubated with two anti-ApoER2 (10 μg/mL; inset 0–10 μg/mL) or mouse IgG (10 μg/mL) antibodies for 30 min, prior to incubation with PBS (black bars; inset -▲) or FXa (gray bars; inset -■, 20 nM) for 3 h. Cells were then treated with LPS (500 ng/mL; 4 h) and TNFα secretion was detected using HEK Blue TNFα reporter cells. Treatment with PBS alone (white bars) was used as a negative control.
Figure 5.
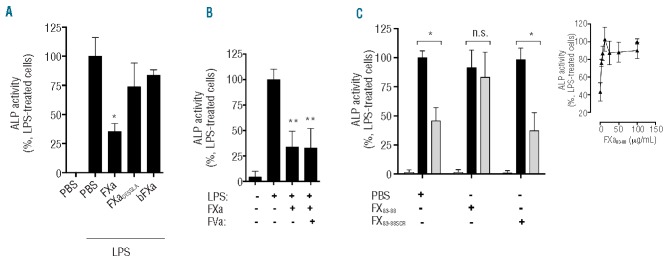
Structural and domain requirements for FXa anti-inflammatory activity on myeloid cells. (A) THP-1 cells were exposed to FXa, FXaDESGLA or bovine FXa (bFXa; all 20 nM) for 3 h prior to stimulation with LPS (500 ng/mL) for 4 h then TNFα in the THP-1 supernatant was measured. (B) THP-1 cells were exposed to FXa (20 nM) in the presence or absence of FVa (20 nM) for 3 h prior to stimulation with LPS (500 ng/mL) for 4 h. (C) THP-1 cells were treated with FXa83–88 or FXa83–88SCR (100 μg/mL) for 30 min prior to incubation with either PBS (black bars) or FXa (gray bars, 20 nM) for 3 h and then with LPS (500 ng/mL) for 4 h. PBS in the absence of FXa/LPS (white bars) had no effect on cytokine production when assessed using HEK Blue TNFα reporter cells. The dose-dependent inhibition of FXa83–88 on FXa anti-inflammatory activity on THP-1 cells is also shown (inset).
Factor Xa anti-inflammatory signaling causes inhibition of lipopolysaccharide-induced nuclear factor-κB activation and is sensitive to wortmannin
Activation of the transcription factor NF-κB controls the expression of an array of pro-inflammatory cytokine genes and is a shared downstream effector of TLR-activated signaling pathways. To examine whether NF-κB activation by LPS was impaired by prior exposure to FXa, THP-1 cells stably transfected with an NF-κB-dependent secreted ALP reporter construct were incubated with FXa and then stimulated with LPS. FXa was found to inhibit NF-κB activation significantly at all LPS concentrations tested (P<0.05; Figure 4A). Numerous negative regulatory mechanisms exist in order to control the magnitude of the pro-inflammatory response upon TLR-mediated NF-κB activation. Pertinently, FXa homolog APC induces PI3K activation and Akt phosphorylation to negatively regulate LPS-TLR4 signaling on U937 monocytes.34 To investigate whether PI3K/Akt pathway activation is similarly required for FXa regulation of LPS-dependent cytokine production, THP-1 cells were treated with FXa and LPS in the presence of the PI3K inhibitor wortmannin and the production of TNFα was measured. Wortmannin completely inhibited FXa-mediated suppression of TNFα production in response to LPS, with half-maximal inhibition of FXa activity observed at 0.5 μM (Figure 4B,C). Similarly, PI3K inhibition by wortmannin ablated the negative regulation of TNFα production by FXa following stimulation with Pam3CSK4 as a ligand for TLR1/2 (data not shown). To further characterize the receptor requirements for FXa-mediated inhibition of NF-κB activation in THP-1 cells, the assay was repeated in the presence of GB83 and RAP. Similar to the effect of wortmannin, both GB83 and RAP completely prevented inhibition of NF-κB activation by FXa in THP-1 cells (Figure 4D). FXaDEGR and FXa truncated at the N-terminal Gla domain (FXaDESGLA) were unable to inhibit NF-κB activation in a similar manner to FXa (Figure 4E), highlighting the importance of FXa enzymatic activity and the Gla domain in mediating FXa anti-inflammatory activity.
Figure 4.
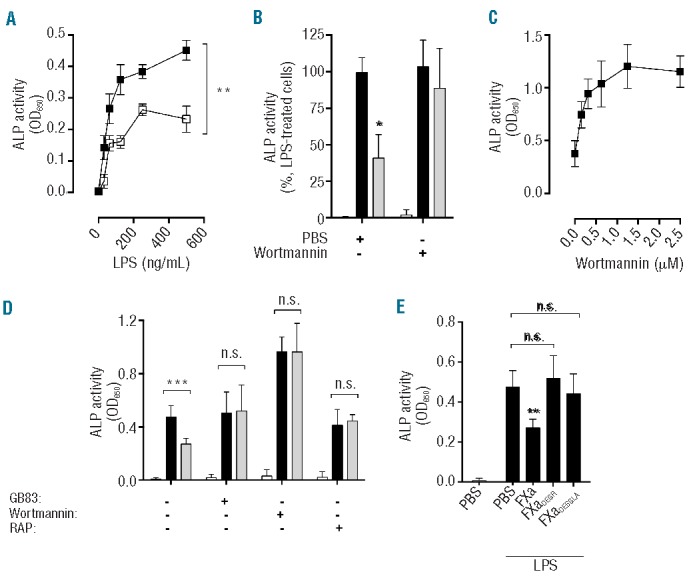
FXa anti-inflammatory PAR2-dependent signaling results in inhibition of LPS-induced NF-κB activation and is sensitive to wortmannin. (A) THP1-Blue CD14 NF-κB reporter cells were incubated with FXa (□; 20 nM) or PBS (■) for 1 h prior to stimulation with LPS (31.3–500 ng/mL; 6 h) and measurement of ALP activity. (B, C) THP-1 cells were treated with wortmannin (B – 1.25 μM, C – 0.313–2.5 μM) for 30 min prior to co-incubation with PBS (black bars) or FXa (gray bars, 20 nM; 1 h), and then stimulated with LPS (500 ng/mL) for 4 h. PBS treatment (white bars, B) did not elicit TNFα secretion. (D) THP1-Blue CD14 NF-κB reporter cells were incubated with GB83 (1 μM), wortmannin (1.25 μM) or RAP (80 μM) for 30 min prior to exposure to co-incubation with PBS (black bars) or FXa (gray bars, 20 nM; 1 h), and then stimulated with LPS (500ng/mL) for 4 h. PBS treatment (white bars) did not elicit NF-κB activation. (E) THP1-Blue CD14 NF-κB reporter cells were incubated with FXa, FXaDEGR or FXaDESGLA for 3 h prior to stimulation with LPS (500 ng/mL) for 4 h then ALP activity was detected. Results represent the mean ± SD of at least three independent experiments.
Structural determinants of factor Xa anti-inflammatory activity on myeloid cells
A range of FXa isoforms and truncations were used to characterize the molecular requirements for FXa inhibition of pro-inflammatory cytokine release upon TLR activation. The ability of FXaDESGLA to prevent LPS-induced TNFα production from THP-1 cells was significantly impaired compared to that of full-length FXa (Figure 5A). A similar response was observed when FXaDESGLA was used to prime cells prior to Pam3CSK4 treatment (data not shown). Bovine FXa, a close structural homolog of human FXa possessing approximately 70% amino acid sequence similarity to its human counterpart, was surprisingly ineffective at reducing LPS and Pam3CSK4-induced TNFα expression, suggesting that the molecular requirements for FXa anti-inflammatory activity on THP-1 monocytes are not conserved between these species (Figure 5A).
Once activated, FXa associates with activated factor V (FVa) on plasma membrane negatively charged phospholipids to activate prothrombin. FVa could theoretically inhibit the FXa interaction with cell surface receptors necessary for anti-inflammatory signaling. We found, however, that FVa had no effect on the ability of FXa to suppress LPS-induced cytokine production from THP-1 cells (Figure 5B), indicating that, in the presence of its procoagulant cofactor, the ability of FXa to negatively regulate LPS-stimulated cytokine production is maintained.
An intermediary amino acid sequence connecting the two FXa EGF-like domains (amino acid residues 83–88) has previously been shown to be important for FXa cell signaling.35 To investigate the contribution of the inter-EGF region to FXa inhibition of LPS-induced cytokine production on THP-1 monocytes, a short synthetic peptide mimicking this region (FX83–88) was co-incubated with FXa and LPS-induced TNFα production was measured (Figure 5C). The peptide alone had no effect on TNFα production, but its presence alongside FXa dose-dependently attenuated FXa-mediated inhibition of TNFα secretion in response to LPS (IC50 = 1.3 mg/mL FX83–88 peptide; Figure 5C, inset). To ensure specificity, a scrambled version of this peptide was tested but, unlike FX83–88 peptide, it was unable to inhibit FXa anti-inflammatory activity (Figure 5C).
Discussion
FXa signaling can induce disparate downstream outcomes depending upon the PAR activated, the cell type assessed and the cellular output measured. Consequently, precise determination of the role of FXa cell signaling in vivo has been difficult to ascertain. Moreover, despite the observation of robust FXa signaling in vitro, rapid inhibition of FXa by plasma serpins may limit the role of FXa signaling in its free form under normal physiological conditions. However, there are a number of pathological states, particularly disseminated intravascular coagulopathy, in which procoagulant proteases such as FXa are excessively generated and not subject to the same strict regulatory mechanisms that exist in the absence of infection. In such instances, FXa generated as a consequence of persistent coagulation activation has increased potential to bind leukocytes and trigger cell signaling.
Recent studies have highlighted the role of proteases associated with hemostasis in modulating innate immunity, either by simultaneously promoting both coagulation and inflammation,9 or by regulating inflammation and thus limiting mortality in preclinical animal models of endotoxemia or in patients with severe sepsis.15 The role of FXa in this context is currently not well understood, and has been described to induce cell signaling conducive to both the promotion and attenuation of inflammation. Therefore, in this study, we sought to determine how FXa signals on innate immune cells whose primary physiological role is to initiate a pro-inflammatory response upon pathogen detection. In doing so, we demonstrated that FXa induces a refractory state to TLR stimulation via PAR2 activation, and thereby impairs pro-inflammatory cytokine production from TLR-stimulated myeloid cells.
FXa did not induce the generation of pro-inflammatory cytokines in any of the cell types tested when administered alone, or when co-administered simultaneously with TLR ligands. In contrast, prior exposure to FXa produced significant and dose-dependent inhibition of LPS-induced cytokine production in THP-1 cells, PBMC and primary murine macrophages. A half-maximal reduction of TNFα production was elicited at ~3 nM FXa in all myeloid cell types tested.
The ability of FXa to regulate cytokine expression was not limited to that initiated by TLR4 activation and experiments using synthetic agonists directed against specific TLR demonstrated that FXa regulates the pro-inflammatory cytokine response upon activation of multiple TLR (including TLR 1, TLR2, TLR5 and TLR6), with varying degrees of efficacy. Given the enzymatic activity and broad range of identified substrates for FXa, we considered whether reduced sensitivity to LPS on FXa-treated myeloid cells was a consequence of FXa shedding of TLR from the cell surface. However, conserved FXa cleavage sites were not identified in the extracellular portions of any of the TLR under investigation, indicating that receptor proteolysis from the cell surface is unlikely to contribute to FXa anti-inflammatory activity.
Proteolytic activation of either PAR1 or PAR2 by FXa confers cytoprotective effects on endothelial cells, but it is not known whether they mediate FXa signaling on myeloid cells and, if so, which PAR is required to mediate anti-inflammatory FXa activity. A likely role for PAR in enabling FXa suppression of pro-inflammatory production was demonstrated by the diminished capacity of FXaDEGR to attenuate LPS-induced TNFα expression on both monocytes and macrophages. Furthermore, we found that the ability of FXa to inhibit LPS-induced cytokine production from THP-1 monocytes was significantly inhibited in the presence of PAR2, but not PAR1, antagonists. Moreover, LPS-induced TNFα/IL-6 production from PAR2−/− macrophages, in contrast to that from murine wild-type macrophages, was largely impervious to the anti-inflammatory activity of FXa. This is consistent with the proposed molecular mechanism of action of FXa cytoprotection on endothelial cells, given that previous studies have shown that anti-PAR2 antibodies block FXa-dependent maintenance of endothelial cell barrier integrity, down-regulate cell surface adhesion protein expression upon TNFα treatment and diminish NF-κB activation.23 Interestingly, PAR2 activation by non-physiological synthetic peptides also results in diminished LPS-induced TNFα and IL-6 expression from primary murine macrophages.36 Our study thus identifies a novel role for FXa as an endogenous PAR2 activator with similar anti-inflammatory activity on myeloid cells.
The downstream signaling induced upon PAR2 activation by FXa to limit inflammatory cytokine generation is not known and the intracellular pathways that mediate decreased sensitivity to LPS have not yet been fully delineated. However, we observed that FXa significantly inhibited NF-κB activation in LPS-activated THP-1 reporter cells. Furthermore, FXa anti-inflammatory signaling requires functional PI3K and is inhibited by the presence of wortmannin. The PI3K-Akt pathway has a well-characterized role in mediating cell survival, and has been implicated in both negative and positive regulation of TLR-induced pro-inflammatory cell signaling.37,38 Activation of the PI3K-Akt pathway in human monocytes and THP-1 cells has been demonstrated to limit LPS-induced up-regulated NF-κB, AP-1 and TNFα expression.37 Furthermore, pharmacological inhibition of PI3K/Akt increases LPS-induced coagulation and inflammation in murine models of endotoxemia.39 Our data imply that FXa activation of PI3K/Akt-dependent pathways in THP-1 cells is an important step in enabling FXa anti-inflammatory signaling on myeloid cells. Furthermore, this suggests that FXa uses a similar downstream signaling mechanism to its homolog APC on monocytes, which also induces PI3K and Akt phosphorylation to attenuate LPS-TLR4 pro-inflammatory signaling.34
The molecular determinants of specific PAR cleavage by FXa are incompletely understood, but appear to be regulated by FXa cell surface co-receptor interactions. For example, the interaction between FXaβ and annexin 2 facilitates activation of PAR1, but not PAR2, on endothelial cells, whereas EPCR binding is proposed to support FXa activation of both PAR1 and PAR2 on the same cell type.29,30,40 Given the ability of RAP to inhibit APC anti-inflammatory signaling on U937 monocytic cells, we sought to identify whether FXa anti-inflammatory signaling on THP-1 monocytes was similarly RAP-sensitive. FXa anti-inflammatory signaling was strongly inhibited by the presence of RAP on both monocytes and macrophages, indicating a crucial role for an LDL receptor family member in enabling FXa signaling activity on these cells. In addition, a mouse monoclonal antibody directed against the extracellular region of ApoER2 blocked FXa inhibition of TNFα release from THP-1 monocytes. Extensive studies to characterize the role of ApoER2 and other potential LDL receptor family members that may contribute to FXa anti-inflammatory activity on myeloid cells are required. This is particularly true given that FXa anti-inflammatory activity on bone marrow-derived murine macrophages derived from BALB/c mice was also found to be sensitive to RAP inhibition despite previous studies indicating that ApoER2 is not expressed on bone marrow-derived murine macrophages.12 This suggests that an as-yet-unidentified LDL receptor family member expressed on bone-marrow-derived macrophages, distinct from ApoER2, may represent an alternative target for the potent RAP-mediated inhibition of FXa anti-inflammatory activity observed on LPS-treated bone marrow-derived macrophages.
In this study, a peptide mimicking this FXa amino acid sequence (FX83–88), previously shown to be crucial for PAR2-dependent barrier protective and anti-inflammatory activity of FXa on endothelial cells,22,23 produced a dose-dependent decrease in FXa regulation of LPS-stimulated cytokine production from THP-1 cells, such that FXa anti-inflammatory activity could be completely blocked by the presence of this peptide. Interestingly, bovine FXa, whose inter-EGF region amino acid sequence is not similar to its human counterpart, was unable to mount an anti-inflammatory response similar to that of human FXa, providing a possible explanation for the observed species-specific loss of function.
The anti-inflammatory activity of FXa on myeloid cells was inhibited by N-terminal Gla domain truncation. In contrast, the FXa Gla domain may not be required for PAR2-dependent FXa signaling on endothelial cells.22 It is not clear at this stage whether FXa Gla domain truncation confers long-range structural changes that disrupt FXa myeloid receptor binding sites on the Gla domainless protease, or itself represents a crucial binding site for FXa myeloid cell surface receptors.
Collectively, this study suggests that FXa acts in a similar manner to APC in limiting pro-inflammatory cytokine production on monocytes and macrophages. Unlike APC, FXa uses PAR2, rather than PAR1, to initiate downstream anti-inflammatory signaling on macrophages but, similarly, attenuates NF-κB activation and activates PI3K signaling pathways to inhibit inflammation in LPS-treated monocytes. Myeloid cell-specific APC anti-inflammatory signaling is crucial in protecting mice from LPS-induced lethality.12,41 Assessment of the efficacy of FXa in this setting is, however, complicated by multiple confounding factors, including the potent procoagulant activity of FXa, its rapid inhibition by serpins in plasma42 and its pleiotropic cell-dependent signaling properties. However, the success of prior studies utilizing modified recombinant APC variants15 with signaling-selective activity to regulate murine endotoxemic response provides a useful insight as to how the anti-inflammatory activity of exogenous FXa could be investigated in vivo. Recombinant FXa variants with impaired ability to assemble into the prothrombinase complex and/or associate with inhibitors, but retained ability to interact with myeloid cell surface signaling receptors, may yield important insights into the role of FXa anti-inflammatory signaling when applied to pre-clinical animal models of acute inflammatory disease.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was funded by a Science Foundation Ireland Starting Investigator Research Grant (EMG and RJSP), The National Children’s Research Centre Paediatric Research in Translational Immunology (PRiTI) Programme (RJSP and PGF) and SFI Principal Investigator Programme Grants (JSO′D and PGF).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010; 11(5):373–84 [DOI] [PubMed] [Google Scholar]
- 2.Hargreaves DC, Medzhitov R. Innate sensors of microbial infection. J Clin Immunol. 2005;25(6):503–10 [DOI] [PubMed] [Google Scholar]
- 3.Schwartz BS, Levy GA, Edgington TS. Immune complex-induced human monocyte procoagulant activity. II. Cellular kinetics and metabolic requirements. J Immunol. 1982;128(3):1037–42 [PubMed] [Google Scholar]
- 4.Steinemann S, Ulevitch RJ, Mackman N. Role of the lipopolysaccharide (LPS)-binding protein/CD14 pathway in LPS induction of tissue factor expression in monocytic cells. Arterioscler Thromb. 1994;14(7):1202–9 [DOI] [PubMed] [Google Scholar]
- 5.Fourrier F, Chopin C, Goudemand J, Hendrycx S, Caron C, Rime A, et al. Septic shock, multiple organ failure, and disseminated intravascular coagulation. Compared patterns of antithrombin III, protein C, and protein S deficiencies. Chest. 1992;101(3):816–23 [DOI] [PubMed] [Google Scholar]
- 6.Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondaveeti S, et al. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med. 2001;345(6):408–16 [DOI] [PubMed] [Google Scholar]
- 7.Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341(8):586–92 [DOI] [PubMed] [Google Scholar]
- 8.Taylor FB, Jr, Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R, et al. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock. 1991;33(3):127–34 [PubMed] [Google Scholar]
- 9.Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, et al. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;452(7187):654–8 [DOI] [PubMed] [Google Scholar]
- 10.Stephenson DA, Toltl LJ, Beaudin S, Liaw PC. Modulation of monocyte function by activated protein C, a natural anticoagulant. J Immunol. 2006;177(4):2115–22 [DOI] [PubMed] [Google Scholar]
- 11.Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW. Selective inhibitory effects of the anticoagulant activated protein C on the responses of human mononuclear phagocytes to LPS, IFN-gamma, or phorbol ester. J Immunol. 1994;153(8):3664–72 [PubMed] [Google Scholar]
- 12.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010;120(6):1971–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riewald M, Ruf W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J Biol Chem. 2005;280(20):19808–14 [DOI] [PubMed] [Google Scholar]
- 14.Lay AJ, Donahue D, Tsai MJ, Castellino FJ. Acute inflammation is exacerbated in mice genetically predisposed to a severe protein C deficiency. Blood. 2007;109(5):1984–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204(10):2439–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng X, Li W, Song Y, Hu Y, Ferrell GL, Esmon NL, et al. Non-hematopoietic EPCR regulates the coagulation and inflammatory responses during endotoxemia. J Thromb Haemost. 2007;5(7):1394–400 [DOI] [PubMed] [Google Scholar]
- 17.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709 [DOI] [PubMed] [Google Scholar]
- 18.Menegatti M, Peyvandi F. Factor X deficiency. Semin Thromb Hemost. 2009;35(4):407–15 [DOI] [PubMed] [Google Scholar]
- 19.Tai SJ, Herzog RW, Margaritis P, Arruda VR, Chu K, Golden JA, et al. A viable mouse model of factor X deficiency provides evidence for maternal transfer of factor X. J Thromb Haemost. 2008;6(2):339–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papapetropoulos A, Piccardoni P, Cirino G, Bucci M, Sorrentino R, Cicala C, et al. Hypotension and inflammatory cytokine gene expression triggered by factor Xa-nitric oxide signaling. Proc Natl Acad Sci USA. 1998;95(8):4738–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones A, Geczy CL. Thrombin and factor Xa enhance the production of interleukin-1. Immunology. 1990;71(2):236–41 [PMC free article] [PubMed] [Google Scholar]
- 22.Rana S, Yang L, Hassanian SM, Rezaie AR. Determinants of the specificity of protease-activated receptors 1 and 2 signaling by factor Xa and thrombin. J Cell Biochem. 2012;113(3):977–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bae JS, Yang L, Rezaie AR. Factor X/Xa elicits protective signaling responses in endothelial cells directly via PAR-2 and indirectly via endothelial protein C receptor-dependent recruitment of PAR-1. J Biol Chem. 2010; 285(45):34803–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camerer E, Kataoka H, Kahn M, Lease K, Coughlin SR. Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. J Biol Chem. 2002;277(18):16081–7 [DOI] [PubMed] [Google Scholar]
- 25.Altieri DC, Edgington TS. Identification of effector cell protease receptor-1. A leukocyte-distributed receptor for the serine protease factor Xa. J Immunol. 1990;145(1):246–53 [PubMed] [Google Scholar]
- 26.Bouchard BA, Catcher CS, Thrash BR, Adida C, Tracy PB. Effector cell protease receptor-1, a platelet activation-dependent membrane protein, regulates prothrombinase-catalyzed thrombin generation. J Biol Chem. 1997;272(14):9244–51 [DOI] [PubMed] [Google Scholar]
- 27.Nicholson AC, Nachman RL, Altieri DC, Summers BD, Ruf W, Edgington TS, et al. Effector cell protease receptor-1 is a vascular receptor for coagulation factor Xa. J Biol Chem. 1996;271(45):28407–13 [DOI] [PubMed] [Google Scholar]
- 28.Zaman GJ, Conway EM. The elusive factor Xa receptor: failure to detect transcripts that correspond to the published sequence of EPR-1. Blood. 2000;96(1):145–8 [PubMed] [Google Scholar]
- 29.Schuepbach RA, Riewald M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C receptor. J Thromb Haemost. 2010;8(2):379–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharjee G, Ahamed J, Pawlinski R, Liu C, Mackman N, Ruf W, et al. Factor Xa binding to annexin 2 mediates signal transduction via protease-activated receptor 1. Circ Res. 2008;102(4):457–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altieri DC, Edgington TS. The saturable high affinity association of factor X to ADP-stimulated monocytes defines a novel function of the Mac-1 receptor. J Biol Chem. 1988;263(15):7007–15 [PubMed] [Google Scholar]
- 32.Boyum A. Isolation of lymphocytes, granulocytes and macrophages. Scand J Immunol. 1976;Suppl 5:9–15 [PubMed] [Google Scholar]
- 33.Hams E, Saunders SP, Cummins EP, O’Connor A, Tambuwala MT, Gallagher WM, et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock. 2011;36(3):295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang XV, Banerjee Y, Fernandez JA, Deguchi H, Xu X, Mosnier LO, et al. Activated protein C ligation of ApoER2 (LRP8) causes Dab1-dependent signaling in U937 cells. Proc Natl Acad Sci USA. 2009;106(1):274–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ambrosini G, Plescia J, Chu KC, High KA, Altieri DC. Activation-dependent exposure of the inter-EGF sequence Leu83-Leu88 in factor Xa mediates ligand binding to effector cell protease receptor-1. J Biol Chem. 1997;272(13):8340–5 [DOI] [PubMed] [Google Scholar]
- 36.Nhu QM, Shirey KA, Pennini ME, Stiltz J, Vogel SN. Proteinase-activated receptor 2 activation promotes an anti-inflammatory and alternatively activated phenotype in LPS-stimulated murine macrophages. Innate Immun. 2012;18(2):193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277(35):32124–32 [DOI] [PubMed] [Google Scholar]
- 38.Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-kappa B activation. J Immunol. 2000;164(3):1355–63 [DOI] [PubMed] [Google Scholar]
- 39.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24(10):1963–9 [DOI] [PubMed] [Google Scholar]
- 40.Feistritzer C, Lenta R, Riewald M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost. 2005;3(12):2798–805 [DOI] [PubMed] [Google Scholar]
- 41.Kerschen E, Hernandez I, Zogg M, Jia S, Hessner MJ, Fernandez JA, et al. Activated protein C targets CD8+ dendritic cells to reduce the mortality of endotoxemia in mice. J Clin Invest. 2010;120(9):3167–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivanciu L, Toso R, Margaritis P, Pavani G, Kim H, Schlachterman A, et al. A zymogen-like factor Xa variant corrects the coagulation defect in hemophilia. Nat Biotechnol. 2011;29(11):1028–33 [DOI] [PMC free article] [PubMed] [Google Scholar]