

Abstract
Although farnesyltransferase inhibitors have shown promising activity in relapsed lymphoma and sporadic activity in acute myelogenous leukemia, their mechanism of cytotoxicity is incompletely understood, making development of predictive biomarkers difficult. In the present study, we examined the action of tipifarnib in human acute myelogenous leukemia cell lines and clinical samples. In contrast to the Ras/MEK/ERK pathway-mediated Bim upregulation that is responsible for tipifarnib-induced killing of malignant lymphoid cells, inhibition of Rheb-induced mTOR signaling followed by dose-dependent upregulation of Bax and Puma occurred in acute myelogenous leukemia cell lines undergoing tipifarnib-induced apoptosis. Similar Bax and Puma upregulation occurred in serial bone marrow samples harvested from a subset of acute myelogenous leukemia patients during tipifarnib treatment. Expression of FTI-resistant Rheb M184L, like knockdown of Bax or Puma, diminished tipifarnib-induced killing. Further analysis demonstrated that increased Bax and Puma levels reflect protein stabilization rather than increased gene expression. In U937 cells selected for tipifarnib resistance, neither inhibition of signaling downstream of Rheb nor Bax and Puma stabilization occurred. Collectively, these results not only identify a pathway downstream from Rheb that contributes to tipifarnib cytotoxicity in human acute myelogenous leukemia cells, but also demonstrate that FTI-induced killing of lymphoid versus myeloid cells reflects distinct biochemical mechanisms downstream of different farnesylated substrates. (ClinicalTrials.gov identifier NCT00602771)
Introduction
Farnesyltransferase inhibitors (FTIs) have undergone extensive clinical testing in various hematologic malignancies.1–3 A recent multi-institution phase II study demonstrated that the orally bioavailable non-peptidomimetic FTI tipifarnib4 induces partial and complete remissions, including highly durable remissions, in up to 50% of patients with relapsed Hodgkin’s disease and peripheral T-cell lymphomas.5 Phase II and phase III studies have also demonstrated responses of 11–23% in elderly patients with previously untreated poor risk AML.6,7 Moreover, a recent multicenter phase II trial has shown that tipifarnib improves early survival when administered as maintenance therapy in remission.8 Accordingly, there is considerable interest in achieving a better understanding of the mechanism of cytotoxicity of FTIs.
Previous studies have suggested that many chemotherapeutic agents induce apoptosis through the mitochondrial or intrinsic pathway.9,10 This pathway is regulated by interactions between pro- and anti-apoptotic members of the Bcl-2 family which modulate mitochondrial outer membrane integrity.11–13 In particular, Bax and Bak are responsible for mitochondrial outer membrane permeabilization, leading to cytochrome c release, caspase 9 activation and subsequent apoptotic events.12–14 Bax and Bak are held in check by binding to antiapoptotic proteins such as Bcl-2, Bcl-xL and Mcl-1. These interactions are in turn regulated by a variety of posttranslational modifications as well as the expression and activation of BH3-only family members such as Bim, Bid, Puma, and Noxa, which neutralize antiapoptotic Bcl-2 family members and/or directly activate Bax and Bak.11–13,15
In a recent study, we demonstrated that tipifarnib-induced killing of malignant lymphoid cells results from diminished signaling through the Raf/MEK/ERK pathway leading to upregulation of Bim, its trafficking to mitochondria, and subsequent cytochrome c release.16 The observation that downregulation RasGRP1, a Ras guanine nucleotide exchange factor,17 protects malignant lymphoid lines from tipifarnib-induced killing implicated H-Ras in the cytotoxic effects of FTIs in these cells.16 Although only a small number of samples were available for analysis, clinical responses also appeared to occur preferentially in lymphomas with high RasGRP1 and low Bcl-2.5
Whether inhibition of the Ras/Raf/MEK/ERK pathway leading to Bim upregulation is also responsible for the sporadic activity of tipifarnib in AML has been unclear. Earlier studies failed to demonstrate a correlation between inhibition of ERK phosphorylation during tipifarnib treatment and response of AMLs to therapy,6 suggesting that AML response might differ from that in lymphoid cells. Although gene expression studies initially identified a correlation between RasGRP1 mRNA and response of AML to tipifarnib,18 subsequent selection of AML patients based on RasGRP1 expression failed to enrich for tipifarnib responders,19 again arguing that human AML might be responding differently from lymphoma.
Investigations have shown that Rheb, a small GTPase involved in activating the serine/threonine kinase mammalian target of rapamycin (mTOR), also requires farnesylation for proper membrane association.20,21 As a result, lymphomas induced by Rheb overexpression are sensitive to FTI treatment.22 In accord with these studies, our prior studies have shown that tipifarnib inhibits phosphorylation of p70 S6 kinase and its substrate ribosomal protein S6 in clinical AML samples without affecting phosphorylation of Akt substrates,23 pointing to Rheb as a potentially important FTI target in AML as well. How Rheb inhibition leads to antineoplastic effects, however, has remained uncertain.
Building on these earlier observations, the present studies were designed to explore the mechanism by which tipifarnib induces apoptosis in AML cells, focusing on Rheb and subsequent changes in Bcl-2 family members. Results of these studies identified a pathway involving Puma and Bax stabilization in tipifarnib-induced killing of human myeloid cells, thereby demonstrating that the mechanism of cytotoxicity in AML differs markedly from that in malignant human lymphoid cells.
Methods
Materials
Tipifarnib was provided by David End (Johnson & Johnson, New Brunswick, NJ, USA). The broad spectrum caspase inhibitor Q-VD-OPh24 was from SM Biochemicals (Anaheim, CA, USA). Additional reagents and antibodies are described in the Online Supplementary Appendix.
Cell culture
AML cell lines were authenticated and passaged as described in the Online Supplementary Appendix. To generate resistant U937 cells, parental cells were exposed continuously to increasing tipifarnib concentrations over a 6-month period. Individual clones were generated by limiting dilution at 1 cell/10 wells in the continuous presence of 800 nM tipifarnib. U937 cells stably expressing Puma or Bax shRNA25 were generated by lentiviral transduction26 followed 48 h later by addition of 2 μg/mL puromycin to select stable transductants, which were then cloned by limiting dilution.
Clinical samples
After Institutional Review Board approval and patient informed consent, bone marrow aspirates were harvested prior to therapy and, if patients consented, again prior to drug administration on Day 8 of treatment with twice daily oral tipifarnib, which was administrated as part of a phase II trial of single-agent tipifarnib27 or a phase II trial of the tipifarnib/etoposide combination28 in AML. Marrow mononuclear cells were prepared on Ficoll-Hypaque gradients, washed with serum-free RPMI 1640 medium, and prepared for electrophoresis.29
Immunoprecipitation and immunoblotting
Immunoprecipitation of active Bax was performed using methods previously described for the detection of active Bak16 but substituting the 6A7 anti-active Bax antibody. Blotting of immunoprecipitates was performed as described.16
Whole cell lysates were prepared from cell lines or clinical samples as described previously.29 Aliquots were resuspended in SDS sample buffer at 5 mg protein/mL as assayed by the bicinchoninic acid method, separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with various antibodies.30
MTS assay, detection of cell cycle distribution and apoptosis
MTS assays,16 as well as staining with propidium iodide followed by flow cytometry to determine cell cycle distribution and DNA fragmentation, annexin V staining and Hoechst staining for apoptotic nuclear changes, were performed as described.23,31,32
Microarray analyses and connectivity map assessment
Using methods described in the Online Supplementary Appendix, RNA was isolated from parental cells treated for 48 h with 0.1% DMSO or tipifarnib (800 nM, U937 and ML-1; 3200 nM, HL-60) or resistant cells growing in the same tipifarnib concentrations and hybridized to Affymetrix U133A 2.0 microarrays. After probes were corrected for GC content33 followed by non-linear normalization34 applied to the perfect match data from all chips together, probes were then summarized to the probeset level via RMA.35 Empirical Bayes linear models36 were then used to test the hypothesis of differential expression between samples treated with diluent versus tipifarnib. Probes with a 2-fold or more change in the tipifarnib treated cells and P<0.05 were analyzed using the Broad Institute’s Connectivity Map website (www.broadinstitute.org/cmap/newQuery). Enrichment of the induced and repressed genes of a signature within each Connectivity Map treatment profile was estimated with a metric based on the Kolmogorov-Smimov statistical analysis and combined to produce a “connectivity score” as described.37
Results
Antiproliferative effect of tipifarnib in AML cell lines
To identify a model system for studying tipifarnib action in AML, the human AML cell lines U937, ML-1, and HL-60 were exposed to increasing but therapeutically achievable tipifarnib concentrations.38 Compared to untreated cells, the tipifarnib treated cells continued proliferating for several days and then slowed (Figure 1A–C, left panels). Analysis of cell number on Day 6 revealed that 25–100 nM tipifarnib inhibited proliferation by 50% (Figure 1A–C, right panels). The Jurkat T-cell acute lymphocytic leukemia line, run at the same time, was slightly more sensitive, with over 50% inhibition of proliferation at 25 nM (Online Supplementary Figure S1). Flow cytometry performed on Day 6 (Figure 1D–F and Online Supplementary Figure S2) indicated a G1 arrest in two of the three AML cell lines. A subdiploid fraction, suggesting apoptosis, was also evident in U937 and ML-1 cells but not HL-60 (Figure 1G–I and Online Supplementary Figure S2A and B). U937 cells also bound annexin V (Online Supplementary Figure S2C and D) and displayed apoptotic morphological changes, including chromatin condensation and nuclear fragmentation (Online Supplementary Figure S2E and F), suggesting that tipifarnib was inducing apoptosis. Compared to lymphoid cells, which express RasGRP1 and respond to tipifarnib by suppressing Ras/Raf/MEK/ERK-mediated signaling and upregulating Bim,16 the AML cells expressed much less RasGRP1. RasGRP1 protein was undetectable in HL-60, U937, ML-1, ML-2, MV-4-11, Molm-13, HEL, THP.1, and SET2 AML cells as well as K562 and KBM5 CML-blast crisis cell lines even upon prolonged blot exposure (Figure 2A and data not shown). Moreover, quantitative RT-PCR indicated that RasGRP1 mRNA in HL-60, U937, and ML-1 cells (the only lines examined) was at least 150-fold lower than in Jurkat cells. Further analysis demonstrated that RasGRP1 protein levels were also very low (Figure 2B and C) in pre-treatment bone marrow samples from AML patients enrolled on a recent tipifarnib-etoposide trial.28 This low RasGRP1 expression at the protein level, coupled with our previous demonstration that tipifarnib-induced apoptosis in lymphoid lines is inhibited by RasGRP1 downregulation of this magnitude,16 suggested that the mechanism of tipifarnib-induced killing in the AML lines might differ from that in lymphoid cells.
Figure 1.
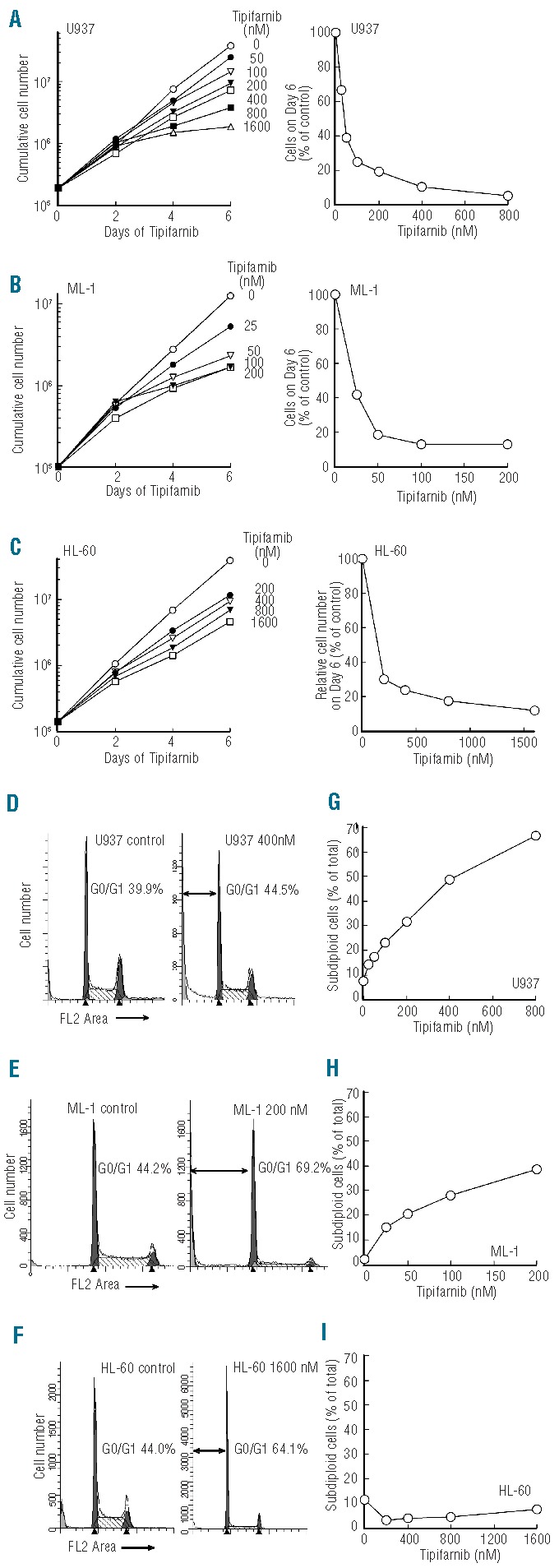
Tipifarnib inhibits proliferation and induces apoptosis in AML cell lines. (A–C) Aliquots containing 1−2×105 HL-60, U937, and ML-1 cells/mL were treated with the indicated concentrations of tipifarnib or diluent continuously, with cells being counted and diluted into fresh medium with drug every 2 days. Graphs on the left indicate cumulative cell number/mL after taking into account dilution. Graphs on the right show cell number on Day 6 relative to diluent-treated controls. (D–F) DNA histograms of cells treated with diluent or the indicated tipifarnib concentration for 6 days. Double-headed arrows indicate events with <2n DNA content. G-I, percentage of subdiploid events observed on Day 6 after analysis of FL2 height as illustrated in Online Supplementary Figure S2A. Results in all panels are representative of at least 3 independent experiments.
Figure 2.
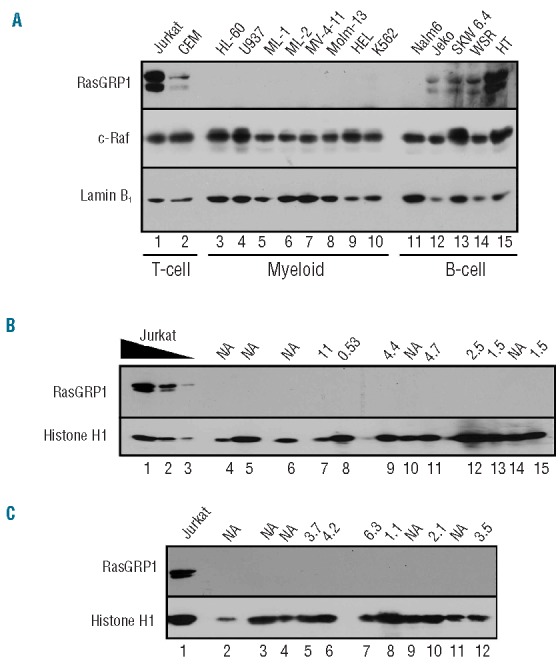
Low expression of RasGRP1 in myeloid cell lines and AML specimens. (A) Whole cell lysates (50μg protein) from the indicated lymphoid and AML lines were subjected to immunoblotting with a previously characterized anti-RasGRP1 antibody.16 c-Raf served as a loading control. (B and C) Aliquots containing 3×105 bone marrow mononuclear cells from pre-treatment bone marrow aspirates of AML patients enrolled on a recent tipifarnib/etoposide phase II trial28 were subjected to SDS-PAGE followed by immunoblotting for the indicated antigens. Jurkat cells, which served as a positive control for RasGRP1 expression, were loaded at 3×105/lane (lane 1, panels B and C) or 1×105 and 0.3×105/lane (B, lanes 2 and 3, respectively). Number above each lane indicates RasGRP1:aprataxin ratio previously reported for the corresponding mRNA sample.28 NA, not assayed due to lack of RNA sample.
Tipifarnib inhibits Rheb farnesylation and diminishes mTOR signaling
In further experiments, we examined alterations in signaling after 24 h of tipifarnib treatment in U937 cells. This analysis demonstrated that tipifarnib decreased the mobility of Rheb (Figure 2A), consistent with the inhibition of farnesylation and downstream farnesylation-dependent processing.21 Decreased phosphorylation of mTORSer2448 (a marker of active mTOR in mTORC1),39 p70S6 kinase at Thr389 and ribosomal S6 protein at Ser235,236 was also observed over the same tipifarnib concentration range (Figure 3A). In contrast, there was no decrease in phosphorylation of the Akt substrates TSC2, PRAS40 or GSK3β (Online Supplementary Figure S3). This is consistent with inhibition of signaling at the level of Rheb.
Figure 3.
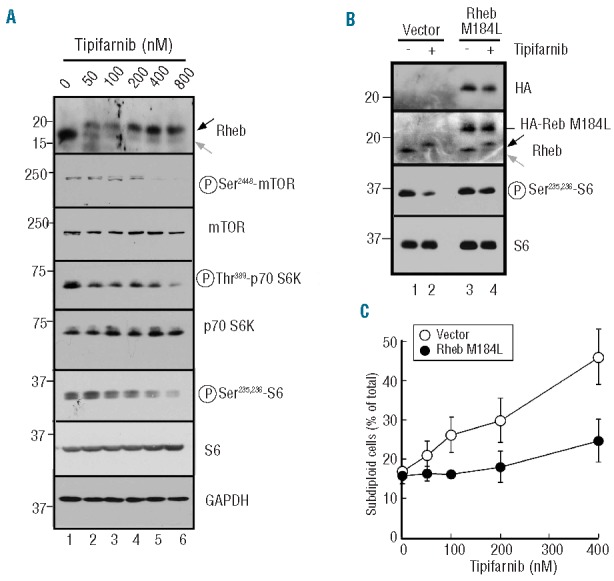
Role of Rheb in tipifarnib-induced apoptosis. (A) After U937 cells were treated for 24 h with the indicated tipifarnib concentration, whole cell lysates were subjected to immunoblotting with antibodies that recognize the indicated antigens. GAPDH served as a loading control. In this and subsequent figures, gray arrow indicates farnesylated antigen and black arrow indicates unfarnesylated antigen. (B) U937 cells stably transduced with empty vector or HA-tagged Rheb M184L were treated for 24 h with diluent (0.1% DMSO) or 800 nM tipifarnib. Whole cell lysates were then probed with antibodies to the indicated antigen. Note that S6 phosphorylation diminishes less after tipifarnib treatment in cells transduced with HA-tagged Rheb M184L than in cells transduced with empty vector. (C) U937 cells stably transduced with empty vector or HA-tagged Rheb M184L were treated for 6 days with the indicated tipifarnib concentration, stained with propidium iodide and subjected to flow microfluorimetry as depicted in Online Supplementary Figure S2A.
To further assess the potential importance of these changes, cells were transfected with a mutant Rheb construct that becomes membrane associated after geranylgeranylation (Rheb M184L) rather than farnesylation.21 This construct diminished tipifarnib-induced changes in ribosomal S6 phosphorylation (Figure 3B), tipifarnib-induced effects on proliferation (data not shown) and tipifarnib-induced killing (Figure 3C), suggesting a critical role for Rheb inhibition in all of these processes.
In an initial attempt to tie these effects to tipifarnib-induced cell cycle arrest and/or apoptosis, the three AML cell lines were treated with tipifarnib for 48 h and harvested for preparation of RNA, which was hybridized to Affymetrix U133 2.0 arrays. Comparison of diluent- and tipifarnib-treated cells revealed statistically significant (P<0.05) 2-fold or greater changes in 34, 27, and 759 transcripts in U937, ML-1, and HL-60 cells, respectively (Online Supplementary Figure S4A).
In view of our results indicating that tipifarnib diminishes mTOR-mediated phosphorylation of p70S6 kinase and its substrate S6 (Figure 3A), we asked whether the gene expression changes resembled the pattern observed after treatment with sirolimus (rapamycin) or any other agent. Comparison to publicly available Connectivity Maps (see Methods) revealed that the transcriptional changes in all three cell lines resembled those induced in other cell lines by sirolimus or the PI3 kinase inhibitors LY294002 and wortmanin (Online Supplementary Figure S4B). Interestingly, however, there were very few changes in common among the AML cell lines. For example, in U937 and ML-1 cells, which both underwent tipifarnib-induced apoptosis, only three transcripts were altered in parallel: CLN8 (which encodes a transmembrane protein involved in lipid transport or sensing), ZC3HAV1 (which encodes a zinc finger protein that is thought to inhibit viral gene expression and induce innate immunity to retroviruses), and TNFRSF10D (which encodes a decoy receptor for the death ligand TRAIL). Because the resulting decoy receptor would be expected to selectively inhibit TRAIL-induced apoptosis, it was unclear how the observed 4-fold upregulation in the two cell lines would contribute to tipifarnib-induced apoptosis. At the same time, the transcriptional profiling failed to identify changes in messages encoding Bcl-2 family members in any of the three cell lines, suggesting that the explanation for apoptosis must lie elsewhere.
Upregulation of Bax and Puma in U937 cells undergoing apoptosis
Further experiments focused on U937 cells because of their ease of lentiviral transduction relative to the other two lines. Immunoblotting for various Bcl-2 family members demonstrated increased Puma and Bax after six days of tipifarnib treatment (Figure 4A). This was accompanied by a marked increase in Bax activation (Figure 4B). Time course experiments demonstrated that inhibition of S6 phosphorylation occurred gradually, with accumulation of Bax and Puma beginning on Day 4 or 5 of treatment (Figure 4C). Knockdown of Bax or Puma by stable transduction with shRNA almost completely suppressed tipifarnib-induced apoptosis (Figure 4D), confirming the involvement of these proteins in tipifarnib-induced apoptosis. In contrast, the antiapoptotic proteins Mcl-1, Bcl-2, and Bcl-xLdid not change (Figure 4A).
Figure 4.

Tipifarnib-induced apoptosis in U937 cells is Bax- and Puma-dependent. (A) After U937 cells were treated for 6 days with the indicated tipifarnib concentration, whole cell lysates were subjected to immunoblotting with antibodies that recognize the indicated polypeptides. (B) After U937 cells were treated with the indicated tipifarnib concentration for 6 days, cell lysates were subjected to immunoprecipitation (IP) with 6A7 antibody, which recognizes Bax only in its active conformation. Blots were then probed with an antibody that recognizes total Bax. GAPDH served as a loading control for the input. (C) After U937 cells were treated for the indicated length of time with 800 nM, whole cell lysates were subjected to immunoblotting. (D) Independent clones of U937 cells stably transduced with empty vector (C2, C3), Puma shRNA (H4, H5) or Bax shRNA (A2, A7) were treated for 6 days with the indicated tipifarnib concentration, lysed, and stained with propidium iodide as illustrated in Online Supplementary Figure S2A. Inset in (D), immunoblot showing whole cell lysates probed for Puma (left panels) or Bax (right panels). β-actin served as a loading control.
In further experiments, Bax and Puma mRNA levels were examined by semiquantitative and quantitive RT-PCR. In agreement with the transcriptional profiling results (Online Supplementary Figure S4), Bax and Puma message levels did not appreciably change (Figure 5A and B). Instead, tipifarnib altered turnover of Bax and Puma. Upon treatment with cycloheximide, Bax and Puma levels in diluent-treated cells decreased with a half-life of approximately 12 h (Figure 5C, lanes 1–4). In contrast, after three days of tipifarnib, this half-life was extended to beyond 48 h (Figure 5C, lanes 5–8), indicating that tipifarnib enhances the stability of these proteins.
Figure 5.
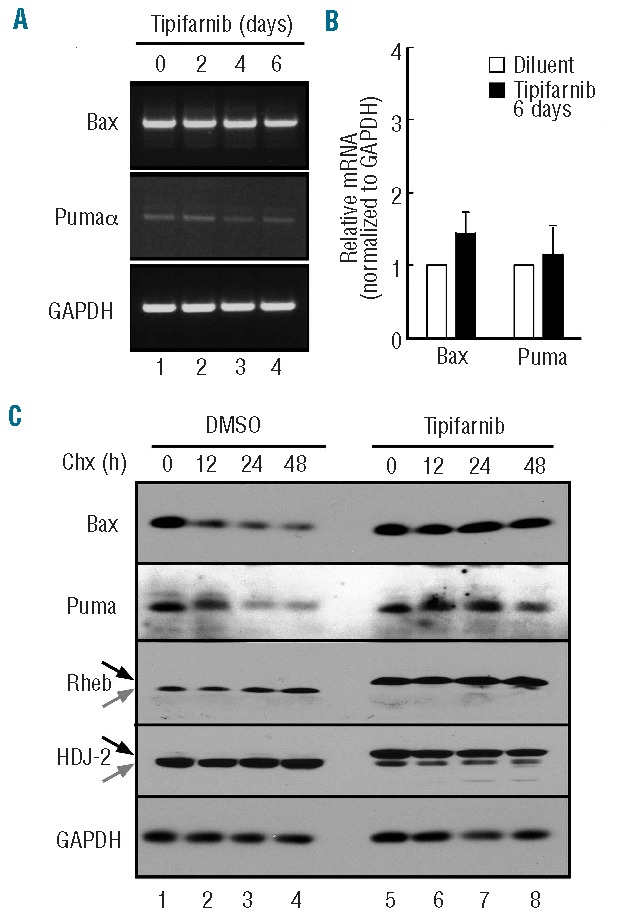
Posttranslational stabilization of Bax and Puma. (A and B) after U937 cells were treated with 800 nM tipifarnib for the indicated time in the presence of 5 μM Q-VD-OPh, a broad spectrum caspase inhibitor, RNA was isolated and subjected to semiquantitative (A) or quantitative RT-PCR (B) using GAPDH as a loading control. (C) After treatment for 3 days with diluent (lanes 1–4) or 800 nM tipifarnib (lanes 5–8), cells were supplemented with 30 μM cycloheximide for 0–48 h in the continued presence of diluent or tipifarnib, then subjected to SDS-PAGE followed by immunoblotting with antibodies that recognize the indicated antigens. GAPDH served as a loading control. The similar levels of Bax and Puma in lanes 1 and 5 are consistent with time course experiments in which a 6-day tipifarnib treatment up-regulated Bax or Puma (Figure 4A) but a 3-day treatment (Figure 4C) did not.
Characterization of tipifarnib-resistant U937 cells
To further evaluate the importance of the Rheb/mTOR/p70S6K pathway in tipifarnib-induced apoptosis, U937 cells were selected for the ability to grow during continuous tipifarnib exposure. The resulting cells were approximately 50-fold resistant to tipifarnib (Figure 6A) and cross-resistant to the farnesyltransferase inhibitor lonafarnib (Figure 6B). Immunoblotting demonstrated that FT was still inhibited in the resistant cells, as indicated by the effect of tipifarnib on posttranslational processing of the chaperone HDJ-2 and Rheb (Figure 6C). Nonetheless, the resistant cells had restored phosphorylation of p70S6K and ribosomal protein S6 despite the presence of tipifarnib (compare Figure 6C, lanes 1 and 3). RT-PCR followed by sequencing failed to identify any mutations in mTOR or p70S6 kinase.
Figure 6.
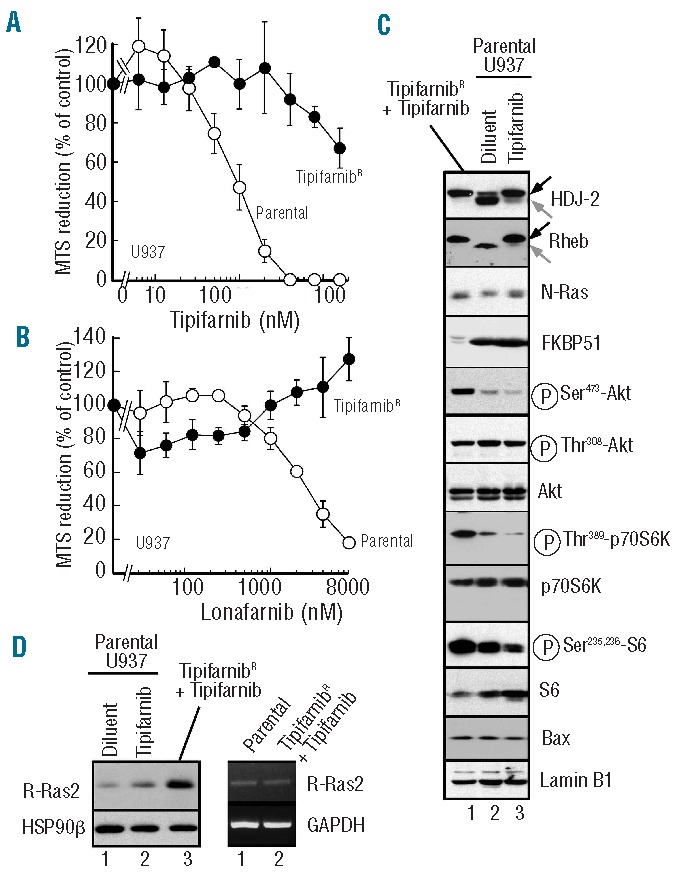
Characterization of tipifarnib-resistant U937 cells. (A and B) parental U937 cells or cells growing continuously in 800 nM tipifarnib were washed free of drug, diluted to 20,000 cells/mL and incubated for 6 days in the presence of the indicated concentration of tipifarnib (A) or lonafarnib (B) before incubation with MTS. (C) Whole cell lysates prepared from parental U937 cells treated for 48 h with diluent or 800 nM tipifarnib (lanes 2 and 3, respectively) or from tipifarnib-resistant cells growing in 800 nM tipifarnib (lane 1) were subjected to SDS-PAGE followed by immunoblotting with antibodies that recognize the indicated antigen. (D) Immunoblots (left) or semiquantitative PCR (right) using protein or RNA, respectively, from parental or tipifarnib-resistant U937 cells. For the immunoblot, parental cells were treated with diluent or 800 nM tipifarnib for 48 h. HSP90β and GAPDH served as loading controls, respectively.
Further analysis identified multiple changes in the PI3K/Akt/mTOR pathway in the resistant cells. These included diminished expression of FKBP51 (Figure 6C), which has been reported to act as a chaperone for the Akt phosphatase PHLPP, accompanied by increased activating phosphorylation of Akt on Ser473 (Figure 6C) and an increase in Akt-mediated inhibitory phosphorylation of the Rheb inhibitor TSC2 (data not shown). In addition, increased expression of R-Ras2, a small G-protein previously implicated in FTI resistance in fibroblasts,40 was also observed at the protein level without any alteration in R-Ras2 message (Figure 6D), suggesting a change in R-Ras2 translation or stability. Similar results were observed in multiple clones (Online Supplementary Figure S5A), suggesting that the resistance might be multifactorial.
In contrast to parental cells, where prolonged tipifarnib treatment up-regulated Bax (Figure 4A), Bax was not up-regulated in the resistant cells relative to untreated parental cells (compare Online Supplementary Figure S5B, lanes 1 and 5), and Bax turnover was restored in the resistant cells despite the continued presence of tipifarnib (Online Supplementary Figure S5B). Collectively, results shown in Figures 3, 4, 6 and Online Supplementary Figure S5B suggest that tipifarnib inhibits signaling through Rheb, that this inhibition leads to Bax and Puma stabilization, and that changes resulting in restoration of signaling through the Rheb/mTOR pathway lead to Bax downregulation and tipifarnib resistance.
Bax upregulation in other AML lines and clinical AML samples
To extend these results to additional AML lines, ML-1 and HL-60 were examined in greater detail. Tipifarnib-induced apoptosis in ML-1 cells (Figure 7A and B) also accompanied Bax and Puma upregulation (Figure 7C). Once again, this reflected inhibition at the level of mTOR, as indicated by diminished mTOR phosphorylation at Ser2448, as well as decreased phosphorylation of p70S6 kinase and ribosomal protein S6 without any change in phosphorylation of Akt (Figure 7D) or its substrates (data not shown). In contrast, HL-60 cells, which up-regulated Puma but not Bax (Figure 7E), failed to undergo tipifarnib-induced apoptosis (Figures 1F and 7B).
Figure 7.
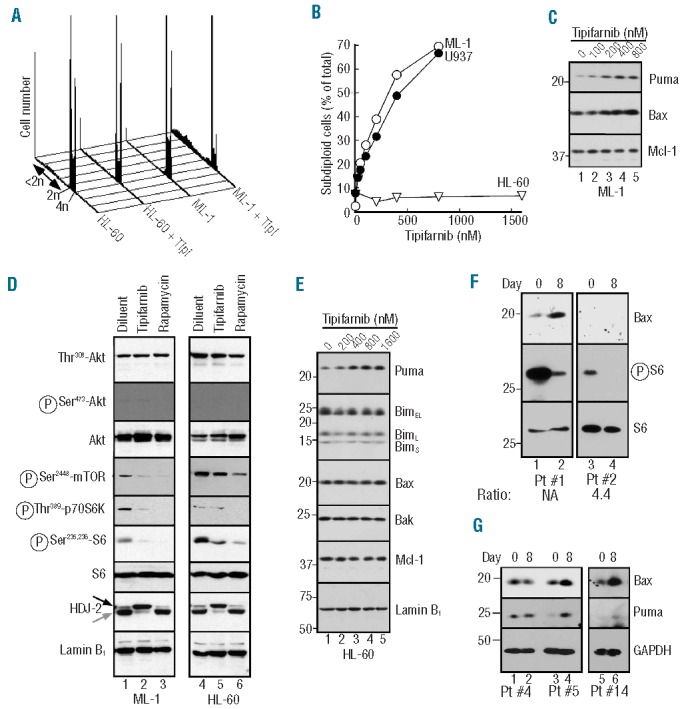
Correlation between tipifarnib-induced Bax upregulation and apoptosis. A. After ML-1 cells were treated for 6 days with the indicated tipifarnib concentration, samples were stained with propidium iodide and subjected to flow microfluorimetry. B. Summary of subdiploid events observed after U937, ML-1 or HL-60 cells were analyzed as illustrated in panel A. C. After ML-1 cells were treated with diluent (lane 1) or tipifarnib at 100, 200, 400 or 800 nM (lanes 2–5, respectively) for 6 days, whole cell lysates were subjected to SDS-PAGE followed by immunoblotting with antibodies to the indicated antigen. D. After ML-1 (lanes 1–3) or HL-60 (lanes 4–6) were treated for 48 h with 0.1% DMSO (lanes 1, 4), 800 or 1600 nM tipifarnib (lanes 2 and 5, respectively) or 100 nM rapamycin (lanes 3, 6), whole cell lysates were subjected to SDS-PAGE followed by immunoblotting with antibodies to the indicated antigen. For each antigen, all lanes were derived from corresponding signals on a single piece of x-ray film but were rearranged for clarity. E. After HL-60 cells were treated for 6 days with diluent (lane 1) or tipifarnib at 200, 400, 800 or 1600 nM (lanes 2–5, respectively), whole cell lysates were subjected to SDS-PAGE followed by immunoblotting with antibodies to the indicated antigens. The nucleoskeletal protein lamin B1 served as a loading controls in panels (D and E). (F and G). Aliquots containing 5 × 105 marrow mononuclear cells harvested prior to treatment (Day 0) or after 14 doses of tipifarnib (on Day 8 before further drug administration) on a phase II trial of tipifarnib (Days 1–14) + etoposide (Days 1–3 and 8–10)28 (F) or a phase II trial of single-agent tipifarnib27 (G) were subjected to SDS-PAGE followed by immunoblotting for the indicated antigens. Number below panel in F indicates RasGRP1:aprataxin ratio previously reported for the corresponding mRNA sample.28 NA: not assayed due to lack of RNA sample.
In further studies, serial bone marrow samples from a recently completed multi-center phase II trial of tipifarnib and etoposide28 were examined. Although paired samples were only available from a small number of patients, Bax levels were higher on Day 8 than on Day 1 in 4 of 10 clinical samples (Figure 7F, lane 2) and unchanged in the others. Likewise, examination of samples remaining from a previous single-agent tipifarnib trial27 revealed Bax and Puma upregulation on Day 8 (Figure 7G, lanes 4 and 6) relative to Day 1 in 2 of 16 paired specimens and no change in the others (Figure 7G, lane 2), indicating that the changes observed in vitro also occur in some AMLs in the clinical setting.
Discussion
Results of the present study demonstrate that tipifarnib, likely acting on Rheb, causes accumulation of Puma and Bax to induce apoptosis in AML cell lines. A number of observations, including the effects of Puma and Bax shRNAs, as well as the ability of Rheb M184L to protect cells, point to the importance of these changes in tipifarnib-induced killing of myeloid cells. Similar upregulation was also detectable in a subset of AML specimens in the clinical setting. Collectively, these results provide new insight into the cytotoxic action of tipifarnib in human AML.
Previous studies have identified a number of potential targets of FTIs, including members of the Ras, Rho and Rheb families.41,42 Which of these are important for FTI-mediated killing in various cell types has not been fully resolved. In fibroblasts, for example, effects on H-Ras appear to play a critical role in FTI-induced killing.43 On the other hand, the importance of these findings for AML has never been established.38 In the AML cell lines treated in this study, we observed inhibition of Rheb prenylation and mTOR phosphorylation on Ser2448 as well as diminished phosphorylation of p70S6 kinase and ribosomal S6 without any change in phosphorylation of the Rheb regulator TSC2 or other Akt substrates at early time points (Figures 3A, 6C and 7D and Online Supplementary Figure S3), consistent with Rheb inhibition. To our knowledge, this represents the first demonstration that prenylation of endogenous Rheb (as opposed to tagged over-expressed Rheb) is inhibited by FTI treatment. The ability of Rheb M184L to protect the cells from tipifarnib (Figure 3C) emphasizes the potential importance of Rheb as a farnesylation target in this cell type.
Our further studies demonstrate that tipifarnib up-regulates Bax in multiple AML cell lines (Figures 4A and 7), a finding consistent with an earlier report that tipifarnib induces Bax upregulation and killing in myeloma cell lines.44 Several additional observations point to the critical importance of this event. First, Bax downregulation protects U937 cells from the cytotoxic effects of tipifarnib (Figure 4D). Second, the HL-60 cell line, which fails to up-regulate Bax, fails to undergo tipifarnib-induced apoptosis (Figure 7B and E). Importantly, signaling downstream of mTOR and proliferation are inhibited in this cell line (Figures 1 and 7) despite the absence of apoptosis, indicating that the block to tipifarnib-induced apoptosis likely occurs somewhere between mTOR inhibition and Bax stabilization. Third, signaling changes that result from selection for tipifarnib resistance have reversed the tipifarnib-induced Bax upregulation (Figure 6). Collectively, these results provide the first evidence that Bax upregulation plays a critical role in FTI-induced killing in AML cell lines and highlight the potential importance of studying this process further.
Bim, tBid, and Puma are currently thought to be direct activators of Bax.45 Our studies demonstrated that tipifarnib also up-regulates Puma in AML cell lines (Figures 4A and 7C). Because U937 and HL-60 cells lack p53, this Puma upregulation must be p53-independent. Importantly, Puma shRNA inhibits tipifarnib-induced apoptosis in U937 cells (Figure 4D), demonstrating the importance of this Puma upregulation. On the other hand, Puma also increases in HL-60 cells, which are resistant to tipifarnib-induced apoptosis (Figure 7E). Thus, Puma upregulation might be necessary but not sufficient for tipifarnib-induced apoptosis in AML cell lines, which appear to require Bax upregulation as well.
Similar Bax and Puma upregulation was observed in sequential marrow samples from a subset of AML patients receiving tipifarnib (Figure 7G), demonstrating that these changes can occur in the clinical setting. Because simultaneous Bax and Puma upregulation in AML cell lines was accompanied by apoptosis, which would result in loss of cells during gradient purification, we surmised that any AML samples showing Bax and Puma upregulation on Day 8 would likely have a block in apoptosis46 that would protect the cells from loss during purification. Consistent with this prediction, the AML samples displaying increased Bax and Puma on Day 8 (like the vast majority of samples) came from patients who did not respond to therapy. Accordingly, the present study shows that Bax and Puma can be up-regulated by tipifarnib-based treatment in the clinical setting but is not meant to establish this change as a predictive marker of response.
Interestingly, the Bax and Puma upregulation observed after tipifarnib treatment did not result from changes in Bax or Puma mRNA (Figure 5A and B). Instead, both of these changes reflected increased protein half-life (Figure 5C). Although it is fashionable to examine drug-induced changes in mRNA profiles, the present demonstration that critical changes in Bcl-2 family members occurred without any change in message (compare Figures 3 and 5) serves as a reminder that signal transduction modulators can have lethal effects on target cells independent of any change in mRNA expression.
As this work was being initiated, Raponi and co-workers reported that high levels of mRNA encoding the Ras guanine nucleotide exchange factor RasGRP1 were associated with a higher probability of AML response to tipifarnib.18 While the authors speculated that high RasGRP1 might reflect activation of (and dependence on) the Ras pathway, mechanistic studies linking RasGRP1 to the response of AML cells were not reported. Earlier studies had shown that RasGRP1 is expressed at particularly high levels in lymphocytes17,47–49 but is absent from myeloid cells.50 Consistent with these results, we have observed that RasGRP1 protein (Figure 2) is below the limit of detection in multiple AML lines. Instead, the present study demonstrates that tipifarnib can inhibit proliferation and induce apoptosis in AML cells with markedly diminished or absent RasGRP1, consistent with recent observations that RasGRP1 might not always enrich for AML that is tipifarnib responsive.19 In the present study, tipifarnib sensitivity in AML cells depends more strongly on the ability to posttranslationally up-regulate Bax.
Finally, when combined with our simultaneously performed studies of tipifarnib action in malignant lymphoid cells,16 the present results also suggest that the mechanism of tipifarnib-induced killing depends upon the cellular context. In particular, the critical farnesylation target seems to vary from cell type to cell type. In malignant lymphoid cells, our studies point to the importance of FTI-induced inhibition of mitogen-activated protein kinase signaling downstream of Ras, leading to Bim upregulation and Bim-dependent killing.16 In contrast, we have not observed inhibition of Akt phosphorylation or ERK phosphorylation at 24–48 h in the AML cell lines examined here (e.g. Figures 6C and 7D), nor have we observed Bim upregulation (Figures 4A and 7E), arguing that AML cell killing might occur by a different mechanism. Instead, the present results suggest that tipifarnib inhibits Rheb-mediated signaling in AML cells (Figures 3A and 7D), leading to stabilization of Bax and Puma (Figures 4A and C and 7C) followed by induction of apoptosis.
Acknowledgments
The authors gratefully acknowledge discussions with H. Dai, gifts of reagents from M. Kastan, R. Abraham and D. Toft, the assistance of personnel in the Mayo Clinic Flow Cytometry and Gene Expression Analysis Core Facilities, the diligence of numerous clinical colleagues in providing the AML specimens used in this study, and the editorial assistance of Deb Strauss.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported in part by grants from the Leukemia and Lymphoma Society (6047–08 and R6233–13) and NIH (U01 CA70095).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Karp JE, Lancet JE. Development of farnesyltransferase inhibitors for clinical cancer therapy: focus on hematologic malignancies. Cancer Invest. 2007;25(6):484–94 [DOI] [PubMed] [Google Scholar]
- 2.Harousseau JL. Farnesyltransferase inihibitors in hematologic malignancies. Blood Rev. 2007;21(4):173–82 [DOI] [PubMed] [Google Scholar]
- 3.Stone RM. Targeted agents in AML: much more to do. Best Pract Res Clin Haematol. 2007;20(1):39–48 [DOI] [PubMed] [Google Scholar]
- 4.Doll RJ, Kirschmeier P, Bishop WR. Farnesyltransferase inhibitors as anticancer agents: critical crossroads. Curr Opin Drug Discov Devel. 2004;7(4):478–86 [PubMed] [Google Scholar]
- 5.Witzig TE, Tang H, Micallef IM, Ansell SM, Ding H, Link BK, et al. A Multi-Institution Phase 2 Study of the Farnesyltransferase Inhibitor Tipifarnib (R115777) in Patients with Relapsed and Refractory Lymphomas. Blood. 2011;118:4882–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancet JE, Gojo I, Gotlib J, Feldman EJ, Greer J, Liesveld JL, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;109(4):1387–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harousseau JL, Martinelli G, Jedrzejczak WW, Brandwein JM, Bordessoule D, Masszi T, et al. A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood. 2009;114(6):1166–73 [DOI] [PubMed] [Google Scholar]
- 8.Luger S, Zicki L, Paietta E, Ketterling RP, Litzow MR, Rowe JM, et al. R115777(tipifarnib) Improves Early Survival when Used As Maintenance Therapy for Elderly or Relapsed/Refractory Patients with Acute Myelogenous Leukemia in Remission. Blood (ASH Annual Meeting Abstracts). 2012;120((ASH Annual Meeting Abstracts)):676 [Google Scholar]
- 9.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–9 [DOI] [PubMed] [Google Scholar]
- 10.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis. A Link Between Cancer Genetics and Chemotherapy. Cell. 2002;108(2):153–64 [DOI] [PubMed] [Google Scholar]
- 11.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1):92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–41 [DOI] [PubMed] [Google Scholar]
- 15.Dai H, Smith A, Meng XW, Schneider PA, Pang Y-P, Kaufmann SH. Transient Binding of an Activator BH3 Domain to the Bak BH3-Binding Groove Initiates Bak Oligomerization. J Cell Biol. 2011;194:39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding H, Hackbarth J, Schneider PA, Lee S-H, Meng XW, Dai H, et al. Cytotoxicity of Farnesyltransferase Inhibitors in Lymphoid Cells Mediated by MAPK Pathway Inhibition and Bim Upregulation. Blood. 2011;(118):4872–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium-and diacylglycerol-binding motifs. Science. 1998;280(5366):1082–6 [DOI] [PubMed] [Google Scholar]
- 18.Raponi M, Lancet JE, Fan H, Dossey L, Lee G, Gojo I, et al. A 2-gene classifier for predicting response to the farnesyltransferase inhibitor tipifarnib in acute myeloid leukemia. Blood. 2008;111(5):2589–96 [DOI] [PubMed] [Google Scholar]
- 19.Lancet JE, Feldman E, Goster MC, Gojo I, Odenike O, Komrokji RS, et al. Phase 2 Trial of the Farnesyltransferase Inhibitor Tipifarnib in Previously Untreated Older Adults with AML and Baseline Presence of a Specific 2-Gene Expression Signature Ratio. Blood (ASH Annual Meeting Abstracts). 2012;120(ASH Annual Meeting Abstracts):1508 [Google Scholar]
- 20.Clark GJ, Kinch MS, Rogers-Graham K, Sebti SM, Hamilton AD, Der CJ. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J Biol Chem. 1997;272(16):10608–15 [DOI] [PubMed] [Google Scholar]
- 21.Basso AD, Mirza A, Liu G, Long BJ, Bishop WR, Kirschmeier P. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J Biol Chem. 2005;280(35):31101–8 [DOI] [PubMed] [Google Scholar]
- 22.Mavrakis KJ, Zhu H, Silva RL, Mills JR, Teruya-Feldstein J, Lowe SW, et al. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008; 22(16):2178–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karp JE, Flatten K, Feldman EJ, Greer JM, Loegering DA, Ricklis RM, et al. Active oral regimen for elderly adults with newly diagnosed acute myelogenous leukemia: a preclinical and phase 1 trial of the farnesyltransferase inhibitor tipifarnib (R115777, Zarnestra) combined with etoposide. Blood. 2009;113(20):4841–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent anti-apoptotic properties. Apoptosis. 2003;8(4):345–52 [DOI] [PubMed] [Google Scholar]
- 25.Huang S, Okumura K, Sinicrope FA. BH3 mimetic obatoclax enhances TRAIL-mediated apoptosis in human pancreatic cancer cells. Clin Cancer Res. 2009;15(1):150–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng XW, Peterson KL, Dai H, Schneider P, Lee SH, Zhang JS, et al. High cell surface death receptor expression determines type I versus type II signaling. J Biol Chem. 2011;286:35823–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lancet JE, Gojo I, Gotlib J, Feldman EJ, Greer J, Liesveld JL, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myeloid leukemia. Blood. 2006;109:1387–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karp JE, Vener TI, Raponi M, Flatten K, Greer JM, Smith BD, et al. Multi-Institution Phase II Clinical and Pharmacogenomic Trial of Tipifarnib Plus Etoposide for Elderly Adults with Newly Diagnosed Acute Myelogenous Leukemia. Blood. 2012;119:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufmann SH, Svingen PA, Gore SD, Armstrong DK, Cheng Y-C, Rowinsky EK. Altered Formation of Topotecan-Stabilized Topoisomerase I-DNA Adducts in Human Leukemia Cells. Blood. 1997;89:2098–104 [PubMed] [Google Scholar]
- 30.Kaufmann SH. Reutilization of Immunoblots After Chemiluminescent Detection. Analytical Biochemistry. 2001; 296:283–6 [DOI] [PubMed] [Google Scholar]
- 31.Mesa RA, Loegering D, Powell HL, Flatten K, Arlander SAH, Dai NT, et al. Heat Shock Protein 90 Inhibition Sensitizes Acute Myelogenous Leukemia Cells to Cytarabine. Blood. 2005;106:318–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K, et al. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43–9006)-induced TRAIL sensitization. J Biol Chem. 2007;282(41):29831–46 [DOI] [PubMed] [Google Scholar]
- 33.Wu Z. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;99:909–17 [Google Scholar]
- 34.Ballman KV, Grill DE, Oberg AL, Therneau TM. Faster cyclic loess: normalizing RNA arrays via linear models. Bioinformatics. 2004;20(16):2778–86 [DOI] [PubMed] [Google Scholar]
- 35.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64 [DOI] [PubMed] [Google Scholar]
- 36.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3(1):Article 1 [DOI] [PubMed] [Google Scholar]
- 37.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35 [DOI] [PubMed] [Google Scholar]
- 38.Karp JE, Lancet JE, Kaufmann SH, End DW, Wright JJ, Bol K, et al. Clinical and Biological Activity of the Farnesyltransferase Inhibitor R115777 in Adults with Refractory and Relapsed Acute Leukemias: A Phase I Clinical-Laboratory Correlative Trial. Blood. 2001;97(11):3361–9 [DOI] [PubMed] [Google Scholar]
- 39.Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69(5):1821–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carboni JM, Yan N, Cox AD, Bustelo X, Graham SM, Lynch MJ, et al. Farnesyltransferase inhibitors are inhibitors of Ras but not R-Ras2/TC21, transformation. Oncogene. 1995;10(10):1905–13 [PubMed] [Google Scholar]
- 41.Prendergast GC, Rane N. Farnesyltransferase inhibitors: mechanism and applications. Expert Opin Investig Drugs. 2001;10(12):2105–16 [DOI] [PubMed] [Google Scholar]
- 42.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11(11):775–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sebti SM, Der CJ. Opinion: Searching for the elusive targets of farnesyltransferase inhibitors. Nat Rev Cancer. 2003;3(12):945–51 [DOI] [PubMed] [Google Scholar]
- 44.Beaupre DM, Cepero E, Obeng EA, Boise LH, Lichtenheld MG. R115777 induces Ras-independent apoptosis of myeloma cells via multiple intrinsic pathways. Mol Ca Therap. 2004;3(2):179–86 [PubMed] [Google Scholar]
- 45.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330(6009):1390–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schimmer AD, Pedersen IM, Kitada S, Eksioglu-Demiralp E, Minden MD, Pinto R, et al. Functional blocks in caspase activation pathways are common in leukemia and predict patient response to induction chemotherapy. Cancer Res. 2003;63(6):1242–8 [PubMed] [Google Scholar]
- 47.Dower NA, Stang SL, Bottoroff DA, Ebinu JO, Dickie P, Ostergaaard HL, et al. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nature Immunol. 2000;1(4):317–21 [DOI] [PubMed] [Google Scholar]
- 48.Coughlin JJ, Stang SL, Dower NA, Stone JC. RasGRP1 and RasGRP3 regulate B cell proliferation by facilitating B cell receptor-Ras signaling. J Immunol. 2005;175(11):7179–84 [DOI] [PubMed] [Google Scholar]
- 49.Lee SH, Yun S, Lee J, Kim MJ, Piao ZH, Jeong M, et al. RasGRP1 is required for human NK cell function. Journal of Immunology. 2009;183(12):7931–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tognon CE, Kirk HE, Passmore LA, Whitehead IP, Der CJ, Kay RJ. Regulation of RasGRP via a phorbol ester-responsive C1 domain. Mol Cell Biol. 1998;18(12):6995–7008 [DOI] [PMC free article] [PubMed] [Google Scholar]