

Abstract
The adaptive immune system requires a diverse T-cell repertoire to be able to respond to a wide variety of pathogens. Worryingly, the repertoire diversity declines dramatically in old age. As thymic output generates novel T cells, the conventional view holds that a decrease in this output with age is responsible for the loss in the repertoire. However, many additional factors affect the repertoire such as homeostatic turnover and antigen-dependent expansion in response to infection. Mathematical models taking a population biology perspective are important tools for understanding how the interplay between these factors affects the immune repertoire. These models suggest that thymic decline is not a major factor but rather that some combination of virus-induced proliferation and T-cell-intrinsic genetic or epigenetic changes gives rise to the oligoclonal expansions that cause the decline in T-cell diversity. We also discuss consequences for strategies to rejuvenate the immune repertoire in old age.
Keywords: memory, repertoire evolution, T-cell receptors
Introduction
The adaptive immune system faces a challenging task: it must specifically recognize and destroy unknown, evolving pathogens. The immune system solves this problem by having a highly diverse population of very specific antigen receptors. Canonically, each cell has receptors of a single specificity, and different cells have different receptors. This diversity has a fundamental connection to health: the greater the diversity, the greater the chance of having a cell that can respond to a particular infection.1 A robust young adult human has an enormous population of ∼ 1012 T cells, which comprises at least 107 distinct clonal lineages as defined by their T-cell receptor (TCR).2–4
What causes the decline in the repertoire that is observed as people age? The most obvious change with age is a decrease in the rate at which T cells with novel specificities are generated in the thymus. Yet many additional factors affect the frequencies of T-cell clones within the repertoire, such as homeostatic turnover of cells and antigen-dependent expansion of a small number of clones in response to infections. The challenge lies in understanding the net effect on diversity of the complex non-linear interactions between these factors. In this situation, verbal models are insufficient and mathematical models that describe changes in interacting cell populations are needed to delineate the effects of multiple factors.
In this review, we link T-cell dynamics to directly analogous population processes in ecology and evolutionary biology. This linkage allows us to leverage the decades of research in population biology to bring new insight to this immunological puzzle of the generation, maintenance and loss of the T-cell repertoire with age. Using this quantitative population framework, we review different hypotheses for the mechanism underlying the decline in diversity with age. We outline why models using this framework force us to reject the conventional view that thymic influx plays the central role in maintaining the repertoire. Instead, these models support alternative hypotheses, which propose that the loss of the repertoire with age is due to the selective expansion of a small number of clones, which indirectly causes the extinction of many other clones. We explore two mechanisms that could give rise to such an oligoclonal expansion: either persistent stimulation by pathogens or somatic mutation(s)1 conferring a homeostatic advantage to particular clones. We also discuss why understanding the mechanism underlying the oligoclonal expansion is critical for the design of effective interventions to maintain or even rejuvenate an aging immune system.
The organization of the review is as follows. We begin by summarizing the population-scale processes responsible for the generation and maintenance of the diversity of the immune repertoire. Then, we present the currently available experimental data regarding aging T-cell repertoires. Third, we use mathematical models to discriminate between different hypotheses for how the processes described in the first section give rise to the observed changes in the repertoire. Finally, we consider the implications of these different mechanisms for treatment or avoidance of immunosenescence.
Processes affecting repertoire dynamics
Figure 1(a) provides a schematic of T-cell generation, migration between T-cell subpopulations, and the maintenance of subpopulations. Each of these processes affects different aspects of the repertoire with age as detailed below and indicated in Fig. 1(b). For our purposes, we focus on the dynamics of two subpopulations: naive and memory populations of both CD4 and CD8 T cells. As we are concerned with long-term trends, we ignore transient subpopulations such as effector cells.
Figure 1.
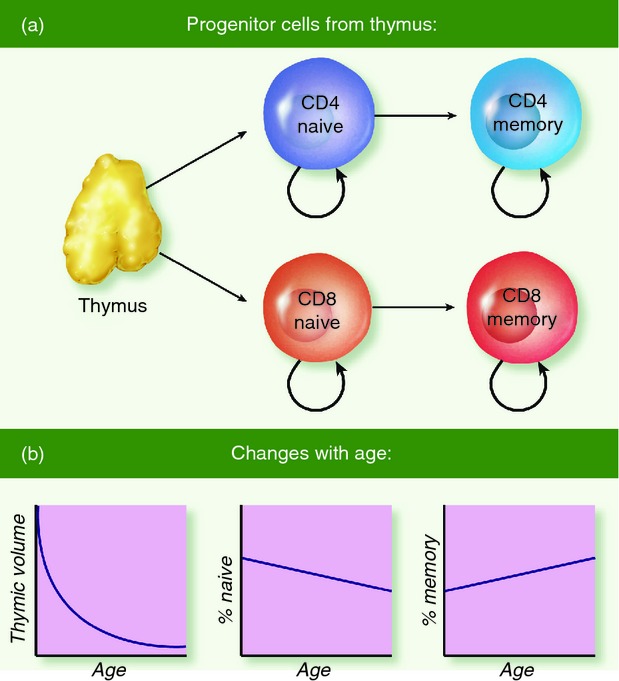
Population flows. In the thymus, progenitor cells express a unique functional T-cell receptor and either CD4 or CD8 co-receptors. These cells leave the thymus with a small clone size to enter their respective naive subpopulation (a). After exposure to cognate antigen, cells undergo clonal selection to proliferate and differentiate into their respective memory subpopulation. Homeostatic proliferation supplements these dynamic flows to maintain each subpopulation. Note that this figure does not show transient subpopulations such as effector cells. In (b), graphs illustrate changes with age: thymic output declines, the % naive declines, and the % memory increases correspondingly.
Before delving into the details of the repertoire dynamics, we briefly consider what, precisely, is meant by ‘diversity’. Ecologists divide diversity into two components: richness and evenness. The former corresponds to the number of different species (i.e. unique TCR lineages), while the latter accounts for the relative frequencies of different species (i.e. different TCR lineages). Both components of T-cell diversity will affect immune dynamics and hence potentially affect health.
Generation
The ultimate source of T-cell diversity is the thymus, where new T cells are generated with potentially novel TCRs produced by somatic recombination. Although the recombination process has a tremendous number of potential products,4,5 recent work suggests that biases may lead to repeated generation of the same TCR.3,6,7 After TCR creation, each new cell undergoes irreversible differentiation to become either a CD8 or CD4 T cell. Finally, these newly generated cells emigrate from the thymus to join their respective antigen-naive subpopulations in the periphery of the body (Fig. 1a).
Clearly, thymic emigration has a positive effect on the richness component of the diversity of the naive subpopulations. However, thymic involution causes the rate of emigration to decline, starting as young as 1 year old (left panel of Fig. 1b). The decline starts at a rate of roughly 4% per year and slows in mid-life.8,9
Inter-population dynamics: clonal selection, differentiation, expansion
Upon novel antigen exposure, clonal selection results in the differentiation and vigorous proliferation of those few T cells bearing TCRs complementary to the antigen (more precisely, specific for epitopes presented on the appropriate MHC).10,11 This process generates a large transient population of effector cells that control the pathogen and eventually give rise to memory cells (right side of Fig. 1a). The final clone size of the differentiated cells in the memory subpopulation is orders of magnitude greater than the original clone size in the naive subpopulation. For instance, with the acute lymphocytic choriomeningitis virus in mice, CD8 cells specific for a given epitope of lymphocytic choriomeningitis virus have a net 1000-fold increase from 102 naive cells to 105 memory cells.10 Analogous estimates in humans, and particularly for CD4 T cells, are generally lower and in the range of 10-fold to 100-fold.12,13 The expanded clone size enables a faster T-cell response to the same antigen in the future, whereas the differentiation from the naive to the memory phenotypes allows the maintenance of high levels of diversity (both richness and evenness) in both subpopulations. Finally, the proportion of memory versus naive T cells gradually increases with age (middle and right panels of Fig. 1b).
Intra-population dynamics: secondary expansions, homeostasis
The naive and memory subpopulations of CD4 and CD8 T cells have independent homeostatic mechanisms, which are crucial to the maintenance of diversity. This independence has been demonstrated experimentally,14,15 and its necessity is evident from ecological principles. If the only constraint were on the overall T-cell population size, then every time a naive cell expanded into the memory subpopulation, the memory subpopulation would grow bigger and the naive subpopulation smaller. This process would quickly reduce the diversity of the naive subpopulation as clone sizes would be squeezed to extinction. Fortunately, and probably as a result of natural selection, naive and memory populations avoid direct competition16 and so avoid such an adverse outcome.
However, within a subpopulation, T cells do compete with each other. For instance, a secondary infection will stimulate the expansion of cells already in the memory subpopulation. While the total memory subpopulation size may expand under some circumstances,17 over the course of life this subpopulation size is relatively constrained.18 This constraint implies that clonal expansions must often lead to a slight decrease in the average size of memory clones. In addition, naive and memory cells have half-lives (i.e. mean time between divisions) of about 500 days and 60 days, respectively,19 which means that competition during homeostatic turnover plays a significant role over a human lifespan. While the precise mechanisms underlying homeostasis in the different subpopulations remain unclear, they all involve competition for limited resources such as the cytokines interleukin-7 and interleukin-15.20–22
The net effect of homeostatic proliferation on T-cell diversity depends on the the role of antigen in the maintenance of homeostasis.
TCR-blind maintenance (drift)
If all T-cell lineages were equivalent when competing for survival/proliferation factors, then the number of cells bearing any particular TCR would drift up and down over time in a random manner during homeostasis. This process would slowly lead to the extinction of individual lineages as clone sizes reach 0, with the expected time to extinction increasing with cell half-life and initial clone size. The resulting decrease in diversity due to competition has been well-studied in the ecological literature and applied to T-cell dynamics,16 although, given the long half-lives of naive T cells and large clone sizes of memory cells, the decrease would be quite slow in both subpopulations. Experimental support for this potential mechanism includes work demonstrating that memory CD8 cells undergo stochastic homeostatic turnover23 and persist even in mice lacking the ability to present antigen to these cells (i.e. MHC class I knockouts24).
TCR-dependent maintenance (frequency-dependent selection)
If homeostatic proliferation were driven by stimulation from self antigens, then we would expect a form of frequency-dependent selection to confer stability to the repertoire. In this process, an increase in the frequency of lineages specific to a given self antigen would result in each cell in these lineages receiving less stimulation (i.e. a selective disadvantage), which would lead to a subsequent decline in their numbers. The converse would also hold, with a decrease in lineage frequency leading to each cell receiving more stimulation (i.e. a selective advantage) and a subsequent increase in their numbers.
Such a frequency-dependent selection process would be more effective at preserving diversity than a pure drift process, although the former would still include some stochastic drift because the number of T-cell clones exceeds the number of self antigens. As different self peptides will be present in different quantities and different T cells will have different cross-reactivities, this hypothesis taken by itself implies high variation in the frequencies of different TCRs. However, if this hypothesis were combined with a model of receptor tuning in which each new TCR calibrates its sensitivity to self peptides,25 then more uniform clone frequencies could be maintained.
The experiments of Hataye et al.26 suggest that the population of naive CD4 T cells is regulated by frequency-dependent selection for self antigen. They transferred different numbers of naive CD4 cells of a known specificity into congenic mice and closely monitored their frequencies over time.26 Transfers of low initial numbers of cells appeared, on average, to increase in frequency and transfers with high initial numbers of cells appear, on average, to decrease in frequency. Note that here the condition ‘on average’ is important, because proliferation and death are still acting stochastically.
Data from aging repertoires
Despite the homeostatic mechanisms outlined above, the diversity of the T-cell repertoire declines in aged mice,27–29 aged macaques,30 and aged humans.31 Recent work has started to quantify different aspects of this loss of diversity.
Naylor et al.31 examined human naive and memory CD4 cells in three age groups each containing three individuals: young (25–30 years), old (60–65 years), and very old (75–80 years) using two different hybridization assays. They found a peculiar pattern in which diversity changed relatively little between young and old individuals, but then diversity decreased sharply in the very old population. These results imply that the rate of diversity change is dramatically non-constant (Fig. 2). Perhaps most puzzling, this pattern held for both the naive and memory CD4 subpopulations, suggesting that the underlying reason for the sharp loss in diversity might be shared between the two subpopulations. Also surprising is that this pattern was visible even in the cross-sectional data used for this study, which implies that the variance in the age of diversity is relatively low. These results must be interpreted with caution because the sample size was small (frequencies of 50 random TCR-β chains) and, as a result, cannot be used to infer the richness of the repertoire. However, this sample does allow assessment of evenness, and, crucially, the same experimental limitations apply to all three age groups, so allowing for relative comparisons of diversity.
Figure 2.
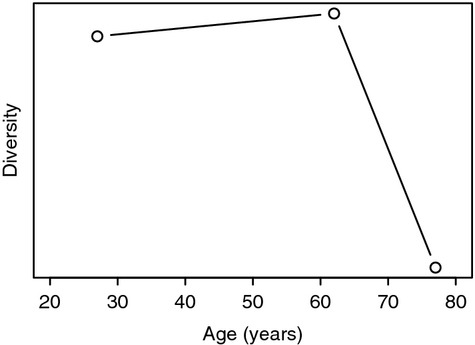
T-cell receptor diversity and age. Summary of naive CD4 diversity at different ages using data from Naylor et al.31 that analysed a cross-sectional sample of young (25–30 years), middle aged (60–65 years), and elderly (75–80 years) people. Values on y-axis are calculated using Simpson's formula50 applied to frequency spectrum data.31
The loss in diversity may arise from changes in many aspects of T-cell populations with age:
Thymic output decreases exponentially starting around age 1.8,9
The total T-cell population size declines slightly but significantly such that a 100-year-old has roughly half as many T cells per ml of peripheral blood as a child.18
The balance between naive and memory subpopulations shifts from an even 1 : 1 ratio in young adulthood to a skewed 1 : 2 ratio (CD4 cells) or 1 : 9 ratio (CD8 cells) in the elderly.32
Exposure to novel antigens shifts specific TCRs from the naive to memory subpopulations, which has the byproduct of potentially squeezing the existing memory lineages.17,33
Latent persistent infections such as Epstein–Barr virus and cytomegalovirus lead to oligoclonal expansions, particularly of CD8 T cells.34–37 Since many of these oligoclonal cells are phenotypically distinguished from the bulk of memory cells38 and appear to compete for different resources and space, their impact on the full repertoire of memory or naive cells is undetermined.
A quantitative approach is needed to discriminate between different hypotheses for how these factors interact to cause the loss of diversity.
Explaining the data
Four non-exclusive hypotheses could explain the observed loss in the repertoire with age (outlined in Fig. 3).
Figure 3.
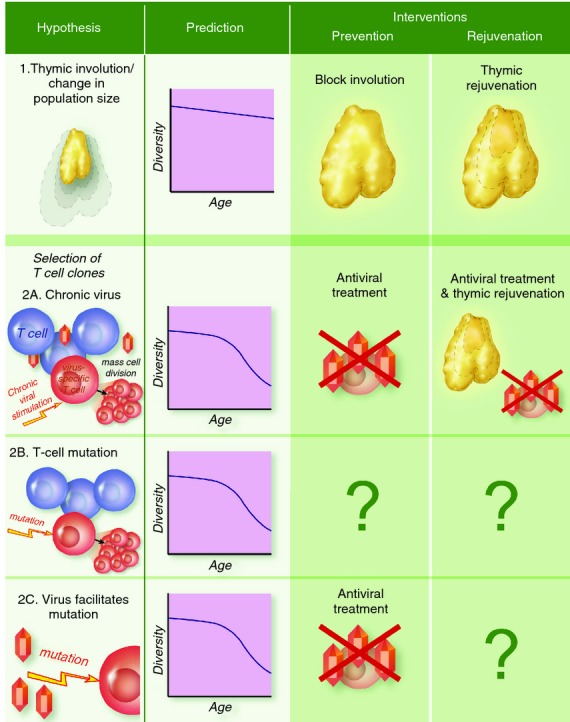
Mechanisms for diversity loss. Different mechanisms for the underlying cause of diversity loss with age make different predictions for the age-dependent pattern of loss (compare with data in Fig. 2). Correspondingly, they would require different interventions to reverse or avoid the loss.
Hypothesis 1: thymic involution
The most straightforward explanation for the decline in T-cell diversity involves a diminution in the generator of diversity – the thymus. As noted earlier, the thymus begins to shrink in volume starting as early as 1 year of age. Involution is accompanied by a corresponding reduction in the rate of emigration of novel T-cell lineages from the thymus such that, by middle age, the production of new lineages in the thymus is a fraction of its peak.8,9 Without this source of new TCRs, existing lineages cannot be replaced when they undergo stochastic extinction.
While intuitively appealing, thymic involution does not explain three significant observations. First, the naive CD4 and CD8 subpopulations shrink in size at different rates, with CD4 T cells much less affected by age.39 Second, thymic involution directly affects only the naive subpopulation yet the loss in diversity appears to occur roughly contemporaneously in the naive and memory CD4 subpopulations.31 Third, and most significantly, thymic involution primarily occurs early in life while the abrupt loss in diversity occurs later in life. This disconnect in timing has been confirmed with a mathematical model, which predicted that thymic involution combined with a TCR-blind homeostatic mechanism (see above) would lead to a constant and relatively low rate of decline in diversity rather than the observed abrupt decline in old age40 (Fig. 3 top row versus Fig. 2).
Hypothesis 2: selection
An alternative to the thymic involution hypothesis involves intra-individual selection acting on a fitness advantage held by a subset of T-cell lineages in the population. Note that we are ascribing an advantage to specific somatic cells and not to the organism as a whole. As long as the total T-cell population size remains constrained, any expansion by high-fitness lineages must lead to a decrease in the clone sizes of other lower-fitness lineages. As this process progresses, diversity would be lost as lower-fitness lineages are squeezed out of the population. In support of such a model, increased T-cell turnover rates at the age when repertoire contraction occurred were observed in humans31 as well as macaques.30 Lineages could acquire a fitness advantage in the following two possible ways.
Hypothesis 2a: virus-induced advantage (memory inflation)
Latent persistent infections such as those caused by the herpes viruses Epstein–Barr virus and cytomegalovirus induce oligoclonal expansions of CD8 memory cells in a process known as memory inflation.41 From a population biology perspective, repeated exposure to viral antigens confers a fitness advantage on the T-cell clones specific for these antigens (Fig. 3 second row). The precise mechanisms and timing underlying the pattern of viral reactivation and corresponding memory inflation remain unclear, as is whether such virus-induced inflation plays a role for CD4 memory cells. At least in principle, the frequency of virus reactivations could increase with age to yield the observed pattern of diversity loss later in life. As implied by the name ‘memory inflation’, this process should only affect memory diversity because antigen-experienced cells should not reside in the naive subpopulation unless they can phenotypically masquerade as naive cells. However, in a mouse model, persistent herpes virus infections have also been shown to affect the naive CD8 subpopulation.42
Hypothesis 2b: mutation-induced advantage
Instead of a virus-induced advantage, T cells could also gain an advantage through random somatic genetic or epigenetic mutations.40 Under this hypothesis, TCR lineages could, at very low rate, acquire heritable mutations that provide a selective advantage during homeostasis – perhaps the ability to better bind interleukin-7 or a lowered threshold for the signal to divide. This advantage would lead to clones bearing the mutation increasing in frequency and, as a consequence, clones lacking it decreasing in frequency with an increased risk of stochastic extinction. This model of mutation-induced advantage is similar to models of cancer,43,44 except that in our case the overall population size is still constrained. This mechanism could explain both the delay until older age and the sharp decline in diversity thereafter (Fig. 3, third row). While such a mutation-induced advantage could occur in either the naive or memory subpopulations, this model does not explain why the diversity loss would be synchronized between naive and memory subpopulations.
Hypothesis 2c: interaction between viruses and mutation
In the previous two hypotheses, we considered two ways in which oligoclonal populations of T cells can be generated. In the first, persistent virus infections cause expansion of virus-specific T-cell clones. In the second, mutations give a selective advantage to one or a few random T-cell clones. Rather then being distinct, these two mechanisms could act in synergy, with viruses facilitating the generation and accumulation of mutations that confer selective advantages that result in oligoclonal expansions.
Chronic infections might facilitate the generation of mutations by indirectly increasing the mutation rate in T cells in a number of ways. Since mutations occur predominantly during the process of cell division, the constant stimulation and rapid cycling of antigen-specific cells specific for chronic infections would result in a higher mutation rate in these cells. Quantifying such processes in models would require incorporating the details of T-cell differentiation and turnover23,45–47 and how these change during chronic infections. Further, if the tropism for a chronic virus included T cells, then it might directly increase the mutation rate.
Challenges and limitations
None of the above hypotheses explain the puzzling observation of the contemporaneous loss in naive and memory CD4 diversity. One explanation is phenotypic interconversion between the naive and memory subpopulations, as suggested by Naylor et al.,31 who found identical TCR sequences in both subpopulations in their oldest age group. Such interconversion could result in sloppy homeostasis, in which memory cells masquerading as naive cells might compete for the same homeostatic factors as naive cells; these lineages might have a further selective advantage over other naive cells if they were exposed to their cognate antigen.
Testing this explanation as well as the earlier hypotheses requires more accurate quantification of the repertoire. The advent of inexpensive high throughput sequencing of T-cell receptors greatly improves diversity measurements relative to earlier methods such as TCR spectratyping2 or hybridization.31 For the first time, these new techniques allow the quantification of diversity even in extremely diverse naive subpopulations.3,48,49 The resulting data also allow for improved methods for analysing diversity. Instead of simply summarizing the repertoire's richness or evenness in a single number such as the Simpson index,50 sequencing reveals the full frequency spectrum, which specifies the distribution of TCR frequencies in a population (i.e. how many distinct TCRs are present in only one cell, how many are present in exactly two cells, etc.). That said, even current bulk high-throughput sequencing is not perfect because it only captures one of the two genomic loci that jointly encode the specificity of the TCR. Potential solutions include methods for linking the two loci such as single cell sequencing or emulsion PCR.51 Further, in humans even high-throughput sequencing is limited by the proportion of the total T-cell population that can be sampled in a reasonable amount of peripheral blood, which means statistical methods must be developed to extrapolate from these samples to the entire repertoire (analogous to methods for estimating numbers of species in ecology52,53). To explore diversity change with age, these improved estimates for diversity in a single individual at a single time-point must be incorporated into population studies that encompass individuals of different ages. Practically, any population study will be cross-sectional in nature, so the number of individuals sampled must be large enough to detect the signal of repertoire decline despite inter-individual variation.
Another limitation is the lack of a good model system and the challenges associated with working with humans. The classic mouse model is not ideal for aging studies because the balance between thymic immigration and homeostatic proliferation appears to be fundamentally different in mice than in humans.54,55 Further, human studies tend to be restricted to sampling peripheral blood. Not only does peripheral blood include a relatively small fraction of total lymphocytes (roughly 2% in humans56), but the repertoire in blood samples may not be representative of all tissues.
Finally, uncertainty in our knowledge of basic immunology inhibits our understanding of aging. In particular, the effects of T-cell differentiation and population heterogeneity have yet to be incorporated into analyses of repertoire diversity. In the naive CD4 subpopulation, the CD31+ subset (associated with recent thymic emigrants) has greater diversity as assessed by TCR spectratyping than the CD31− subset57 but its functional significance is unknown. In the antigen-experienced subpopulations, recent studies have examined the differentiation of T cells during immune responses leading to the generation of memory;45–47 understanding how this process occurs in chronic infections such as Epstein–Barr virus may have implications for repertoire diversity.
Implications for prevention or rejuvenation
The design of effective interventions to halt or reverse the loss of the repertoire requires understanding the underlying cause of the loss. As we detail below, the different mechanistic hypotheses imply different intervention strategies.
Hypothesis 1
Immunologists have long dreamed of thymic rejuvenation as a universal means of improving the robustness of the adaptive immune system in the aged.41,58,59 Clearly, if thymic involution is responsible for the loss of diversity, then preventing or reversing the loss of the thymus would restore diversity. In principle, such rejuvenation could also be effective if the loss of diversity were caused by other factors such as radiation or chemotherapy treatment. The success of this approach depends on the rejuvenated thymic emigrants competing effectively with the remaining T-cell lineages in the body, and consequently this approach will not work in all situations, as we discuss below.
Hypothesis 2a
If a specific virus were responsible for the loss in diversity by causing expansion of a few virus-specific T-cell clones in the memory subpopulation, then the obvious intervention would be to find a way to remove the virus. Treatment of infection would halt but not reverse the decline in diversity. Since significant diversity loss only occurs relatively late in life (after the age of 60 years in Fig. 2) there is a potentially large timeframe in which treatment would preserve diversity. If naive diversity has been lost (e.g. by memory cells masquerading as naive cells as discussed earlier), then thymic rejuvenation after viral clearance could restore the diversity of the naive subpopulation. However, thymic rejuvenation will not directly impact the diversity of the memory subpopulation in which virus-dependent T-cell expansion occurs. The loss of the memory repertoire may be particularly problematic in elderly individuals to the extent that it compromises their ability to generate robust secondary responses to previously encountered pathogens.
Hypothesis 2b
If a somatic mutation conferring a fitness advantage underlies the loss in diversity, then no clear intervention strategy exists. In general, we have no means of lowering the rate of mutation. New lineages from a rejuvenated thymus would be at an inherent disadvantage relative to existing clones with the fitness advantage. Despite this disadvantage, a sufficiently large flux from a rejuvenated thymus might partially counter a loss of diversity in the naive subpopulation, although it would have little effect on the memory subpopulation.
Hypothesis 2c
Finally, if proliferation arising from a virus infection facilitates the generation of a novel mutation that confers a fitness advantage, then, as in hypothesis 2a, early clearance of the virus (before generation of a mutation) could halt the loss of diversity. However, once a mutation is generated, then viral clearance would have little effect on the loss of diversity, and we would have a situation similar to hypothesis 2b. This possibility highlights the importance of identifying the roles of virus and mutation in the loss of diversity.
Conclusion
Ultimately, the loss in TCR diversity is only relevant to the extent to which it impacts health. While extremely low diversity would not allow for recognition of most pathogens and hence lead to poor health due to lack of immune protection,60 the shape of the functional relationship between increasing diversity and increasing immune robustness remains unclear. Indirectly, oligioclonal expansions (memory inflation) in elderly humans have been associated with shorter life expectancies,61 and a controlled study in mice has suggested a similar association.42 Diversity and protection need to be more directly linked.
Quantitative population models show that thymic involution cannot, by itself, explain the observed loss in diversity. However, these models suggest that virus- and mutation-induced oligoclonal expansions could cause such a loss. While these mechanisms produce similar dynamics, they have sharply contrasting implications for treatment to halt or reverse the loss in diversity. With the future production of larger amounts of quantitative data, such mathematical models coupled with experimental data will allow us to understand the mechanisms underlying aging and suggest strategies to rejuvenate the immune repertoire in old age.
Acknowledgments
This research was supported in part by NIH grants K99 GM104158 to PJ, U01 GM070749 and R01 AI110720 to RA, U19 AI090019 and R01 AG045799 to JG.
In this paper, we use the term ‘mutation’ broadly to refer to any heritable genetic or epigenetic change.
Disclosures
The authors declare no conflicts of interest.
References
- 1.de Boer RJ, Perelson AS. How diverse should the immune system be? Proc Biol Sci. 1993;252:171–5. doi: 10.1098/rspb.1993.0062. [DOI] [PubMed] [Google Scholar]
- 2.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human αβ T cell receptor diversity. Science. 1999;286:958–61. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 3.Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murugan A, Mora T, Walczak AM, Callan CG. Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc Natl Acad Sci U S A. 2012;109:16161–6. doi: 10.1073/pnas.1212755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 6.Venturi V, Quigley MF, Greenaway HY, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol. 2011;186:4285–94. doi: 10.4049/jimmunol.1003898. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Ye C, Ji G, Han J. Determinants of public T cell responses. Cell Res. 2012;22:33–42. doi: 10.1038/cr.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. 1985;22:563–75. doi: 10.1111/j.1365-3083.1985.tb01916.x. [DOI] [PubMed] [Google Scholar]
- 9.Bains I, Thiebaut R, Yates AJ, Callard R. Quantifying thymic export: combining models of naive T cell proliferation and TCR excision circle dynamics gives an explicit measure of thymic output. J Immunol. 2009;183:4329–36. doi: 10.4049/jimmunol.0900743. [DOI] [PubMed] [Google Scholar]
- 10.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–64. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moon JJ, Dash P, Oguin TH, McClaren JL, Chu HH, Thomas PG, Jenkins MK. Quantitative impact of thymic selection on Foxp3+ and Foxp3− subsets of self-peptide/MHC class II-specific CD4+ T cells. Proc Natl Acad Sci U S A. 2011;108:14602–7. doi: 10.1073/pnas.1109806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akondy RS, Monson ND, Miller JD, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183:7919–30. doi: 10.4049/jimmunol.0803903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-specific CD4+ memory-phenotype T cells are abundant in unexposed adults. Immunity. 2013;38:373–83. doi: 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanchot C, Lemonnier FA, Pérarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naïve or memory T cells. Science. 1997;276:2057–62. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 15.Tanchot C, Rocha B. The peripheral T cell repertoire: independent homeostatic regulation of virgin and activated CD8+ T cell pools. Eur J Immunol. 1995;25:2127–36. doi: 10.1002/eji.1830250802. [DOI] [PubMed] [Google Scholar]
- 16.Freitas AA, Rocha B. Population biology of lymphocytes: the flight for survival. Annu Rev Immunol. 2000;18:83–111. doi: 10.1146/annurev.immunol.18.1.83. [DOI] [PubMed] [Google Scholar]
- 17.Vezys V, Yates A, Casey KA, Lanier G, Ahmed R, Antia R, Masopust D. Memory CD8 T-cell compartment grows in size with immunological experience. Nature. 2009;457:196–9. doi: 10.1038/nature07486. [DOI] [PubMed] [Google Scholar]
- 18.Sansoni P, Cossarizza A, Brianti V, et al. Lymphocyte subsets and natural killer cell activity in healthy old people and centenarians. Blood. 1993;82:2767–73. [PubMed] [Google Scholar]
- 19.Macallan DC, Wallace D, Zhang Y, et al. Rapid turnover of effector-memory CD4+ T cells in healthy humans. J Exp Med. 2004;200:255–60. doi: 10.1084/jem.20040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyman O, Krieg C, Homann D, Sprent J. Homeostatic maintenance of T cells and natural killer cells. Cell Mol Life Sci. 2012;69:1597–608. doi: 10.1007/s00018-012-0968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takada K, Jameson SC. Naive T cell homeostasis: from awareness of space to a sense of place. Nat Rev Immunol. 2009;9:823–32. doi: 10.1038/nri2657. [DOI] [PubMed] [Google Scholar]
- 22.Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, Ahmed R. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. 2002;195:1541–8. doi: 10.1084/jem.20020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choo DK, Murali-Krishna K, Antia R, Ahmed R. Homeostatic turnover of virus-specific memory CD8 T cells occurs stochastically and is independent of CD4 T cell help. J Immunol. 2010;185:3436–44. doi: 10.4049/jimmunol.1001421. [DOI] [PubMed] [Google Scholar]
- 24.Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–81. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- 25.Grossman Z, Paul WE. Adaptive cellular interactions in the immune system: the tunable activation threshold and the significance of subthreshold responses. Proc Natl Acad Sci U S A. 1992;89:10365–9. doi: 10.1073/pnas.89.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 2006;312:114–6. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed M, Lanzer KG, Yager EJ, Adams PS, Johnson LL, Blackman MA. Clonal expansions and loss of receptor diversity in the naive CD8 T cell repertoire of aged mice. J Immunol. 2009;182:784–92. doi: 10.4049/jimmunol.182.2.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudd BD, Venturi V, Davenport MP, Nikolich-Zugich J. Evolution of the antigen-specific CD8+ TCR repertoire across the life span: evidence for clonal homogenization of the old TCR repertoire. J Immunol. 2011;186:2056–64. doi: 10.4049/jimmunol.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudd BD, Venturi V, Li G, Samadder P, Ertelt JM, Way SS, Davenport MP, Nikolich-Zugich J. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc Natl Acad Sci U S A. 2011;108:13694–9. doi: 10.1073/pnas.1107594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cicin-Šain L, Messaoudi I, Park B, et al. Dramatic increase in naive T cell turnover is linked to loss of naive T cells from old primates. Proc Natl Acad Sci U S A. 2007;104:19960–5. doi: 10.1073/pnas.0705905104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naylor K, Li G, Vallejo AN, et al. The influence of age on T cell generation and TCR diversity. J Immunol. 2005;174:7446–52. doi: 10.4049/jimmunol.174.11.7446. [DOI] [PubMed] [Google Scholar]
- 32.Czesnikiewicz-Guzik M, Lee W-W, Cui D, et al. T cell subset-specific susceptibility to aging. Clin Immunol. 2008;127:107–18. doi: 10.1016/j.clim.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Selin LK, Vergilis K, Welsh RM, Nahill SR. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infections. J Exp Med. 1996;183:2489–99. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Messaoudi I, Lemaoult J, Guevara-Patino JA, Metzner BM, Nikolich-Zugich J. Age-related CD8 T cell clonal expansions constrict CD8 T cell repertoire and have the potential to impair immune defense. J Exp Med. 2004;200:1347–58. doi: 10.1084/jem.20040437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sylwester AW, Mitchell BL, Edgar JB, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–85. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pita-Lopez ML, Gayoso I, DelaRosa O, Casado JG, Alonso C, Muñoz-Gomariz E, Tarazona R, Solana R. Effect of ageing on CMV-specific CD8 T cells from CMV seropositive healthy donors. Immun Ageing. 2009;6:11. doi: 10.1186/1742-4933-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R, Schönitzer D, Grubeck-Loebenstein B. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005;79:3675–83. doi: 10.1128/JVI.79.6.3675-3683.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–52. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolich-Žugich J, Li G, Uhrlaub JL, Renkema KR, Smithey MJ. Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin Immunol. 2012;24:356–64. doi: 10.1016/j.smim.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson PLF, Yates AJ, Goronzy JJ, Antia R. Peripheral selection rather than thymic involution explains sudden contraction in naive CD4 T-cell diversity with age. Proc Natl Acad Sci U S A. 2012;109:21432–7. doi: 10.1073/pnas.1209283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikolich-Žugich J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat Rev Immunol. 2008;8:512–22. doi: 10.1038/nri2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smithey MJ, Li G, Venturi V, Davenport MP, Nikolich-Žugich J. Lifelong persistent viral infection alters the naive T cell pool, impairing CD8 T cell immunity in late life. J Immunol. 2012;189:5356–66. doi: 10.4049/jimmunol.1201867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moolgavkar SH, Knudson AG., Jr Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 1981;66:1037–52. doi: 10.1093/jnci/66.6.1037. [DOI] [PubMed] [Google Scholar]
- 45.Buchholz VR, Flossdorf M, Hensel I, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340:630–5. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 46.Gerlach C, Rohr JC, Perié L, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340:635–9. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 47.Chang JT, Palanivel VR, Kinjyo I, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–91. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 48.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor β-chain repertoire by massively parallel sequencing. Genome Res. 2009;19:1817–24. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C, Sanders CM, Yang Q, et al. High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proc Natl Acad Sci U S A. 2010;107:1518–23. doi: 10.1073/pnas.0913939107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simpson EH. Measurement of diversity. Nature. 1949;163:688. [Google Scholar]
- 51.DeKosky BJ, Ippolito GC, Deschner RP, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. 2013;31:166–9. doi: 10.1038/nbt.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40:237–64. [Google Scholar]
- 53.Daley T, Smith AD. Predicting the molecular complexity of sequencing libraries. Nat Methods. 2013;10:325–7. doi: 10.1038/nmeth.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.den Braber I, Mugwagwa T, Vrisekoop N, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. 2012;36:288–97. doi: 10.1016/j.immuni.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Zhang SL, Bhandoola A. Losing TREC with age. Immunity. 2012;36:163–5. doi: 10.1016/j.immuni.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Storek J, Lalovic BB, Rupert K, Dawson MA, Shen DD, Maloney DG. Kinetics of B, CD4 T, and CD8 T cells infused into humans: estimates of intravascular:extravascular ratios and total body counts. Clin Immunol. 2002;102:249–57. doi: 10.1006/clim.2001.5174. [DOI] [PubMed] [Google Scholar]
- 57.Kohler S, Wagner U, Pierer M, et al. Post-thymic in vivo proliferation of naive CD4+ T cells constrains the TCR repertoire in healthy human adults. Eur J Immunol. 2005;35:1987–94. doi: 10.1002/eji.200526181. [DOI] [PubMed] [Google Scholar]
- 58.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9:57–62. doi: 10.1038/nri2471. [DOI] [PubMed] [Google Scholar]
- 59.Lynch HE, Goldberg GL, Chidgey A, Van den Brink MR, Boyd R, Sempowski GD. Thymic involution and immune reconstitution. Trends Immunol. 2009;30:366–73. doi: 10.1016/j.it.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nikolich-Žugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4:123–32. doi: 10.1038/nri1292. [DOI] [PubMed] [Google Scholar]
- 61.Hadrup SR, Strindhall J, Køllgaard T, Seremet T, Johansson B, Pawelec G, thor Straten P, Wikby A. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol. 2006;176:2645–53. doi: 10.4049/jimmunol.176.4.2645. [DOI] [PubMed] [Google Scholar]