

Abstract
Research on child and adolescent mental health problems has burgeoned since the inaugural issue of Development and Psychopathology was published in 1989. In the quarter century since, static models of psychopathology have been abandoned in favor of transactional models, following the agenda set by editor Dante Cicchetti and other proponents of the discipline. The transactional approach, which has been applied to autism, depression, self-injury, and delinquency, (a) specifies vulnerabilities and risk factors across multiple levels of analysis spanning genes to cultures, (b) identifies multifinal and equifinal pathways to psychopathology, and (c) transcends traditional disciplinary boundaries. However, as noted by Rutter and Sroufe (2000), specific mechanisms of continuity, discontinuity, and comorbidity of psychopathology must be identified if we wish to understand etiology fully. In this article, we present a model of early-onset externalizing behavior in which comorbidities and continuities are viewed as ontogenic processes: products of complex longitudinal transactions between interdependent individual-level vulnerabilities (e.g., genetic, epigenetic, allostatic) and equally interdependent contextual risk factors (e.g., coercive parenting, deviant peer group affiliations, neighborhood criminality). Through interactions across levels of analysis, some individuals traverse along the externalizing spectrum, beginning with heritable trait impulsivity in preschool and ending in antisociality in adulthood. In describing our model, we note that (a) the approach outlined in the DSM to subtyping externalizing disorders continues to obscure developmental pathways to antisociality, (b) molecular genetics studies will likely not identify meaningful subtypes of externalizing disorder, and (c) ontogenic trait approaches to psychopathology are much more likely to advance the discipline in upcoming years.
Achenbach's (1974) landmark text, after which the field of developmental psychopathology was named, initiated an upsurge of interest in the study of emerging mental health problems among children and adolescents. At the time of its publication, child and adolescent psychopathology was characterized in much the same way as adult psychopathology, with little attention paid to developmental processes or to transactions between individuals and their environments in shaping maladaptive behavior. Thus, when Achenbach wrote his text, time was ripe for a paradigm shift in research on child (and adult) psychopathology. Dissatisfaction with static formulations of mental illness had been percolating for some time, beginning with specification of diathesis– stress models of schizophrenia (Gottesman & Shields, 1966; Meehl, 1962) and with the related concept of “reaction range” from quantitative behavioral genetics (Gottesman, 1963). Both approaches emphasized the now widely acknowledged supposition that genetic vulnerabilities and potentials give rise to a range of multifinal outcomes, depending on exposure to environmental risk or protection (see, e.g., Cicchetti, 2006; Gottesman & Gould, 2003; Sroufe & Rutter, 1984). The diathesis–stress framework initiated transition away from strict endogenous models of psychopathology, which traced disorder to pathophysiological processes within individuals, and from strict exogenous models of psychopathology, which traced disorder almost exclusively to early adverse experiences and other external events (Cicchetti, 1984; Sroufe, 1997).
Ten years after publication of Achenbach's (1974) text, the field was still emerging. Sroufe and Rutter (1984) defined developmental psychopathology as “the study of the origins and course of individual patterns of behavioral maladaptation, whatever the age of onset, whatever the causes, whatever the transformation in behavioral manifestations, and however complex the course of the developmental pattern may be” (p. 18). This contrasted sharply with traditional child psychiatry, child clinical psychology, and developmental psychology, each of which addressed only part of what developmental psychopathology subsumed (see Beauchaine & Gatzke-Kopp, 2012; Cicchetti, 1984, 1989, 2006). Developmental psychopathologists recognized the need to (a) view genetic and environmental influences as interdependent determinants of behavior, (b) study progressive transformation and reorganization of behavior as developing organisms interact with their environments over time, and (c) acknowledge that stability and change are observed in normal and atypical behavior. Defining features of developmental psychopathology therefore include the study of individual-level (e.g., genetic, neural, hormonal, temperamental) and environmental (e.g., family, peer network, neighborhood, culture) causal processes, developmental continuities and discontinuities in behavior, and multifinal and equifinal outcomes (see Rutter & Sroufe, 2000).
As this brief introductory section implies, the developmental psychopathology perspective was well articulated by the mid-1980s. Nevertheless, its proponents were obligated to publish in journals from preexisting disciplinary traditions that were more restrictive in scope. However, in 1989 Development and Psychopathology, the first and only journal devoted to the new interdisciplinary perspective, was published by Cambridge University Press. This was a watershed event in the evolution of developmental psychopathology for several reasons. Perhaps more than any other event, publication of the new journal established developmental psychopathology as a discipline in its own right, so it could no longer be considered ancillary to developmental psychology, child clinical psychology, or any other branch of knowledge. Editor Dante Cicchetti (1989) invited top scientists from a wide range of theoretical perspectives to submit their work to the Journal, an effort that was immensely successful. Many of these scientists came from disciplines lacking a developmental perspective, and likely would not have published in Development and Psychopathology without Cicchetti's painstaking and consistent editorial leadership. Cicchetti encouraged these scientists to consider the importance of developmental processes in their work and to specify developmental mechanisms of stability and change in behavior and its biological substrates. This may have been the only way to integrate the work of top biological scientists who lacked a developmental perspective into the field.
As a result of these efforts, after only a handful of issues were published, Development and Psychopathology had garnered considerable attention within the scientific community, achieved an impact factor that rivaled those of top developmental and clinical journals, and further legitimized the growing discipline. In years to follow, Cicchetti solicited a series of incisive special issues that shaped the discipline by specifying equifinal and multifinal pathways to psychopathology (Cicchetti & Rogosch, 1996), challenging adevelopmental and anachronistic assumptions about diagnosis and assessment (e.g., Richters & Cicchetti, 1993), ushering advances in research methodology (e.g., Cicchetti & Hinshaw, 2003), and specifying mechanisms of neural plasticity (Cicchetti & Cannon, 1999), among other topics. Some of these special issues were foundational in shaping current and future directions of psychopathology research, with influence extending well beyond developmental psychopathology to child psychopathology, adult psychopathology, neuroscience, and developmental psychology, among other disciplines. For example, special issues devoted to emotion and emotion regulation in psychopathology (Cicchetti, 1996; Cicchetti, Ackerman, & Izard, 1995) continue to influence contemporary research agendas nearly 20 years later.
The first edition of Developmental Psychopathology, a compendium of theoretical, methodological, and empirical works by top scientists in the field, was published in 1995. This two-volume set, which was edited by Cicchetti and Donald Cohen (1995a, 1995b), brought together scientists from various disciplinary perspectives already represented in the Journal. Although the first edition was published before fully articulated multiple levels of analyses models of psychopathology appeared, it provided the first comprehensive interdisciplinary volume in a single outlet, and further defined the field. The second edition of Cicchetti and Cohen (2006a, 2006b, 2006c) was expanded significantly, including new chapters and an additional volume on developmental neuroscience. This was an important and timely addition to the literature given the expanded role of neuroscientific methods, such as magnetic resonance imaging, in developmental psychopathology research. Thus, the second edition of Developmental Psychopathology provided the first fully multiple levels of analysis perspective, laying the groundwork for models such as those represented in this paper.
Sixteen years after publishing their highly influential article in which they defined developmental psychopathology, Rutter and Sroufe (2000) reviewed progress within the field. By 2000, developmental psychopathology encompassed the study of almost all forms of emerging mental illness, including impulse control disorders, autism spectrum disorders, depressive disorders, schizophrenia, pervasive developmental disorders, and personality disorders, to name a few. As foreshadowed by its early proponents (e.g., Achenbach, 1974; Cicchetti, 1984; Sroufe & Rutter, 1984), the field had become “more developmental, contextual, multilevel, dynamic, multidisciplinary, and collaborative” (Masten, 2006, p. 50). Nevertheless, Rutter and Sroufe identified several obstacles that needed to be overcome, and phenomena that needed to be explained, if the discipline was to recognize its full potential. These included improving measurement, especially through use of systematic epidemiological–longitudinal studies; identifying mechanisms of sex differences observed across a variety of disorders; determining how cognitive processes confer risk for various forms of psychopathology; improving our understanding of the interplay between nature and nurture; specifying mechanisms of comorbidity; and studying mechanisms of heterotypic continuity and continuities and discontinuities in normal and atypical development.
Since 2000, progress has been made in several of these areas. Groundbreaking research on gene–environment interdependence has linked specific genetic vulnerabilities to internalizing and externalizing psychopathology, particularly among those exposed to adversity early in life (e.g., Caspi, Hariri, Holmes, Uher, & Moffitt, 2010; Cicchetti, Rogosch, & Thibodeau, 2012; Covault et al., 2007; Gunnar et al., 2012; see also Beauchaine & Gatzke-Kopp, 2013; Rutter, Moffitt, & Caspi, 2006). Furthermore, increasingly sophisticated transactional models of psychopathology have been articulated (Beauchaine, Hinshaw, & Pang, 2010; Beauchaine, Klein, Crowell, Derbidge, & Gatzke-Kopp, 2009; Cicchetti & Toth, 1998; Crowell, Beauchaine, & Linehan, 2009; Dawson, 2008). These models, which are largely unique to developmental psychopathology (see Beauchaine & Gatzke-Kopp, 2012), specify biological vulnerabilities and environmental risk factors that span levels of analysis from genes to cultures, and acknowledge that causal influences operate across these levels of analysis, sometimes changing in direction through internal and external mechanisms (e.g., Cicchetti, 2008; Cicchetti & Blender, 2004; Cicchetti & Dawson, 2002; Cicchetti & Posner, 2005; Ellis, Del Giudice, & Shirtcliff, 2013; Mead, Beauchaine, & Shannon, 2010). Some of these models specify alternative risk/vulnerability mechanisms through which different individuals develop adjustment problems that may be indistinguishable behaviorally (i.e., equifinality; see Davies, Sturge-Apple, Cicchetti, Manning, & Vonhold, 2012; Gatzke-Kopp, 2011; Gatzke-Kopp, Greenberg, Fortunato, & Coccia, 2012) and risk mechanisms through which only some vulnerable individuals develop psychopathology, whereas others do not (i.e., multifinality; see Beauchaine et al., 2009, 2010; Cicchetti, Rogosch,& Thibodeau, 2012). Thus, developmental psychopathology has moved away from mere description of maladaptive behavior and its multiple manifestations and trajectories toward truly integrative mechanistic models. This transition away from description toward explanation reflects maturation of developmental psychopathology as a scientific discipline. The fundamental objective of science is not only to describe but also to identify causal mechanisms of phenomena that were once inexplicable (see Beauchaine, Gatzke-Kopp, & Mead, 2007; Popper, 1985). It is not surprising that Sroufe and Rutter (1984; Rutter & Sroufe, 2000) identified specifying etiologic mechanisms as a principal goal of developmental psychopathology research.
Developmental psychopathologists have therefore placed considerable emphasis on identifying etiologic mechanisms of mental illness by (a) specifying genetic vulnerabilities that predispose to psychopathology (see, e.g., Rutter, 2006), (b) isolating neural and behavioral substrates of genetic vulnerability (i.e., biomarkers and endophenotypes; see, e.g., Beauchaine, 2009), (c) identifying environmental risk factors that potentiate genetic/neural vulnerability (see, e.g., Caspi et al., 2010; Cicchetti et al., 2012), and (d) identifying equifinal pathways to apparently single disorders (see Cicchetti, 2008; Cicchetti & Rogosch, 1996; Gatzke-Kopp, 2011). A core assumption of this approach is that etiology can only be explained through specification of individual-level vulnerabilities, contextual and environmental risk factors, and their complex interactions over time (see Beauchaine & Gatzke-Kopp, 2012, 2013; Rutter et al., 2006). Individual differences in behavior, including emerging psychopathology and its trajectories and comorbidities, must therefore be studied developmentally, across all relevant levels of analysis (Burnett & Cicchetti, 2012; Cicchetti, 2008; Cicchetti & Dawson, 2002).
Two Phenomena for Developmental Psychopathologists to Explain
Despite impressive advances in the disciplinary agenda articulated by Rutter and Sroufe (2000), much work remains. In the program of research conducted in our lab, we are particularly interested in characterizing etiologic mechanisms of two interrelated phenomena identified by Rutter and Sroufe as especially important to understand if we wish to advance the field further in upcoming years. These include (a) homotypic comorbidity (i.e., co-occurrence of multiple externalizing or internalizing disorders within individuals; e.g., Beauchaine et al., 2010; Beauchaine & Gatzke-Kopp, 2012) and (b) heterotypic continuity (i.e., sequential development of different disorders across the life span; see, e.g., Beauchaine et al., 2009, 2010). Although much remains to be learned about these phenomena, considerable research has been conducted since Rutter and Sroufe evaluated progress in the field 13 years ago. Our goals in writing this article are to (a) briefly summarize pertinent literatures addressing these phenomena, (b) present developmental models of comorbidity and continuity in psychopathology that characterize each as ontogenic processes in which neurobiologically rooted vulnerabilities (e.g., trait impulsivity and trait anxiety) interact with environmental risk factors (e.g., coercive parenting, trauma) to canalize maladaptive behavior over time, and (c) demonstrate how characterizing comorbidities and continuities as ontogenic processes can integrate dimensional trait (e.g., research domain criteria [RDoC]; Insel et al., 2010; Sanislow et al., 2010) and traditional categorical approaches to studying and characterizing psychopathology. We focus our discussion on comorbidities and continuities of externalizing behavior disorders. Although a similar approach is also fruitful in the study of internalizing disorders (see, e.g., Cicchetti & Toth, 1998), there is not enough space in a single article to address the internalizing and the externalizing spectra. We begin with a general discussion of comorbidity.
Homotypic Comorbidity
Historical context
Prior to publication of DSM-III-R (American Psychiatric Association [APA], 1987), very little programmatic research on comorbidity had been conducted. Earlier versions of the DSM specified diagnostic hierarchies (i.e., exclusion criteria), which in most cases precluded assigning more than one disorder to any individual (for discussions, see Beauchaine, Klein, Erickson, & Norris, 2013; First, 2005). However, research conducted in the early to mid-1980s suggested that, at least for some disorders, comorbidity was associated with distinct family histories, indicating differential heritability and loss of useful information when one disorder precluded diagnosis of another (e.g., Leckman, Weissman, Merikangas, Pauls, & Prusoff, 1983). Following from these and other findings, almost all diagnostic hierarchies were eliminated from the DSM-III-R, resulting in markedly increased rates of comorbidity (see Klein & Riso, 1993) and significant expansion of comorbidity research (e.g., Angold, Costello, & Erkanli, 1999; Caron & Rutter, 1991; Hinshaw, Lahey, & Hart, 1993). Widespread interest in comorbidity-related phenomena continues to this day, as evidenced by major psychopathology journals devoting special issues and sections to the topic (e.g., Jensen, 2003; Kendall & Drabick, 2010).
Several types of comorbidity have been defined. From a validity standpoint, these can be divided into three overarching categories (see Angold et al., 1999; First, 2005; Klein & Riso, 1993; Lilienfeld, 2003), including artifactual comorbidity (i.e., comorbidity derived by mistakenly splitting one disease entity into multiple diagnoses), spurious comorbidity (e.g., comorbidity resulting from shared diagnostic criteria across distinct disease entities), and true comorbidity (i.e., co-occurrence of separate disease entities within an individual). Disentangling these alternative sources of comorbidity is often impossible without specification of etiology (see First, 2005; Jensen, 2003). Lacking such specification, we are forced to infer psychopathology solely from symptoms, which are often insensitive and nonspecific indicators of disease state (see Beauchaine, Lenzenweger, & Waller, 2008; Meehl, 1995). As a result, etiology-based diagnosis is often a necessary condition for determining whether apparent comorbidity is artifactual, spurious, or true (see Beauchaine & Marsh, 2006; Preskorn & Baker, 2002). For example, even though obsessive–compulsive disorder (OCD) is similar to other DSM-IV-TR (APA, 2000) anxiety disorders phenomenologically, it appears to be distinct etiologically, with differences in longitudinal course, patterns of heritability, and implicated neural circuitry (e.g., Stein et al., 2010). This suggests that there may be advantages to diagnosing OCD independently from other DSM anxiety disorders (APA, 2013; see Hollander, Zohar, Sirovatka, & Regier, 2011; Phillips et al., 2010) and that co-occurrence of OCD with other DSM anxiety disorders reflects true rather than artifactual or spurious comorbidity.
Distinctions between artifactual, spurious, and true comorbidity must be considered when discussing homotypic cooccurrence of psychopathology. Historically, it has been assumed in child psychiatry that (a) different externalizing syndromes such as attention-deficit/hyperactivity disorder (ADHD), oppositional defiant disorder (ODD), conduct disorder (CD), and substance use disorders (SUDs) and (b) different internalizing syndromes such as separation anxiety disorder, other anxiety disorders, depressive disorders, and self-injury reflect distinct forms of psychopathology, despite sometimes substantial overlap in symptoms. This assumption has led to considerable research aimed at identifying dissociable genetic, neural, and other correlates of these disorders. However, as we have outlined elsewhere and describe below, transactional models challenge the assumption that most disorders on either the externalizing or the internalizing spectra are distinct (see Beauchaine & Gatzke-Kopp, 2012; Crowell et al., 2009; Crowell, Derbidge, & Beauchaine, in press; Derbidge & Beauchaine, in press), as do findings from behavioral and molecular genetics studies (e.g., Anney et al., 2008; Burt, Krueger, McGue, & Iacono, 2001; Krueger et al., 2002; Meier, Slutzke, Heath, & Martin, 2011; Tuvblad, Zheng, Raine, & Baker, 2009). Furthermore, many claims of dissociability among homotypic disorders are based on improper use of analysis of covariance and related regression-based statistical control techniques, an issue we and others have commented on elsewhere (Beauchaine et al., 2010; Miller & Chapman, 2001).1 Thus, we must consider the possibility that at least some homotypic comorbidities are artifactual and/or spurious in nature.
In the following sections we discuss likely mechanisms of homotypic comorbidity among externalizing syndromes, and we present a transactional model of externalizing psychopathology in which comorbidity, at least for many individuals, arises not from true co-occurrence of distinct disorders but from developmental changes in the behavioral expression of heritable vulnerability across the life span. According to our model, central nervous system dopamine (DA) dysfunction confers vulnerability to increasingly more intractable externalizing behavior as affected individuals mature (see Beauchaine & Gatzke-Kopp, 2012; Beauchaine et al., 2009). Through recursive feedback mechanisms, high-risk environments amplify preexisting vulnerability over time, thereby facilitating progression along the well-characterized trajectory followed by many antisocial males, beginning with hyperactivity/impulsivity in preschool, followed by delinquency in middle school, and SUDs and antisocial personality disorder (ASPD) in early adulthood (e.g., Loeber & Hay, 1997; Moffitt, 1993; Robins, 1966). The transactional model we present (a) is consistent with the RDoC perspective, which emphasizes the importance of identifying common neurobiological substrates of disorders that have traditionally been considered distinct, and (b) suggests that much comorbidity among externalizing disorders is artifactual (i.e., derived by mistakenly splitting one disease entity into multiple diagnoses) given overlapping etiology.
Latent structure of externalizing spectrum disorders
As traditionally described in the child psychopathology literature, the externalizing spectrum comprises DSM-IV-TR (2000) defined syndromes including ADHD, ODD, and CD, as well as related constructs such as aggression and delinquency (see Achenbach & Edelbrock, 1984; Tackett, 2010). The externalizing spectrum derives from factor analytic studies demonstrating hierarchical latent structure of symptoms in which a single higher order factor (externalizing liability) accounts for much of the covariation among first-order factors (ADHD, ODD, and CD). This latent structure is observed in population-based and twin studies, the latter of which indicate very high heritability coefficients for the externalizing factor (e.g., Dick, Viken, Kapiro, Pulkkinen, & Rose, 2005; Krueger et al., 2002; Krueger, Markon, Patrick, Benning, & Kramer, 2007; Lahey, Van Hulle, Singh, Waldman, & Rathouz, 2011; Tuvblad et al., 2009).
Although the externalizing spectrum was identified originally by child psychopathologists (e.g., Achenbach & Edelbrock, 1984, 1991), the construct has been replicated and extended by adult psychopathologists, who often include SUDs, ASPD, and sometimes psychopathy in their models (e.g., Krueger et al., 2002, 2007; Patrick, Hicks, Krueger, & Lang, 2005). As found in research conducted with children, the factor analytic structure of externalizing behaviors is hierarchal, with a single, heritable, higher order factor accounting for much of the covariation among first-order factors. This general factor structure is illustrated in Figure 1, in which DSM criterion lists are specified at the level of analysis of first-order factors (i.e., behavioral syndromes) and the RDoC approach is specified at the level of analysis of the higher order factor (i.e., cross-cutting vulnerability traits). This characterization suggests that neither the DSM nor the RDoC approach is right or wrong. Rather, each provides information at a different level of analyses, which must be considered in conjunction for a full understanding of etiology. We revisit this theme in later sections.
Figure 1.
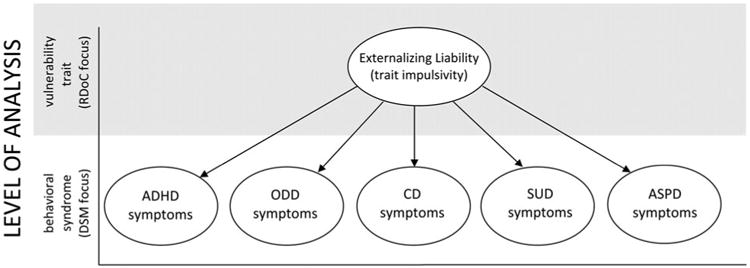
A latent structure of externalizing behavior in which multiple first-order factors (attention-deficit/hyperactivity disorder, oppositional defiant disorder, conduct disorder, substance use disorderss, and antisocial personality disorder) load on a single higher order factor (externalizing liability).
As alluded to above, externalizing spectrum disorders evidence very high rates of comorbidity in nationally representative, cross-cultural, and clinical samples of children, adolescents, and adults (see Beauchaine et al., 2010; Hinshaw, 1987). In a large representative sample of 5- to 15-year-olds, Maughan, Rowe, Messer, Goodman, and Meltzer (2004) reported that 56% of girls and 62% of boys with CD also met criteria for ODD, and that 36% of girls and 46% of boys who met criteria for ODD also met criteria for ADHD and/or CD. In a clinical sample, Gau et al. (2010) reported that children and adolescents with persistent ADHD were 18 times more likely than controls to meet criteria for ODD, and 30 times more likely than controls to meet criteria for CD. In addition, a sizable portion of adolescents who experience conduct problems eventually develop SUDs and/or ASPD (e.g., Kuperman et al., 2001; Myers, Stewart, & Brown, 1998), an issue we return to in later sections. Finally, impulsive personality traits, substance use, and antisocial behavior exhibit high rates of comorbidity among adults (e.g., Kessler, Chiu, Demler, & Walters, 2005; Krueger et al., 2007).
Mechanisms of shared vulnerability
Two important questions emerge from these high rates of comorbidity, and from the consistently replicated factor structure depicted in Figure 1. The first questions is through what mechanism or mechanisms does a common latent trait confer vulnerability to such a wide range of comorbid externalizing syndromes? We have argued that much of this shared liability results from trait impulsivity, conferred trough mesolimbic DA dysfunction and expressed behaviorally as preference for immediate rewards over larger but delayed rewards. Trait impulsivity can be operationalized using DSM-IV (APA, 2000) derived ADHD scales and closely related constructs (e.g., Achenbach & Edelbrock, 1991; Conners, Sitarenios, Parker, & Epstein, 1998), which capture what Sagvolden, Johansen, Aase, and Russell (2005) describe as taking action without forethought and failing to plan ahead—core aspects of personality characteristics such as risk taking, novelty seeking, and sensation seeking (see also Brenner, Beauchaine, & Sylvers, 2005; Hirshfeld-Becker et al., 2002; Neuhaus & Beauchaine, 2013).2
Contemporary neurobiological theories of trait impulsivity all focus at least in part on the mesolimbic DA system and other DA networks (Castellanos & Tannock, 2002; Gatzke-Kopp, 2011; Gatzke-Kopp & Beauchaine, 2007a, Gatzke-Kopp et al., 2009; Kalivas & Nakamura, 1999; Sagvolden et al., 2005). The mesolimbic DA system comprises structures including the ventral tegmental area and its projections to the nucleus accumbens (Swartz, 1999). Mesolimbic theories of trait impulsivity follow from extensive research on incentive motivation, incentive salience, and substance abuse/dependence conducted with rodents, nonhuman primates, and humans. This research demonstrates that (a) electrical and pharmacological stimulation of DA-mediated mesolimbic structures is reinforcing and that trained animals often engage in protracted periods of operant responding to obtain these incentives, often ignoring primary reinforcers such as food and water (see Milner, 1991); (b) mesolimbic neural activity increases during reward seeking, reward anticipation, and after delivery of DA agonists (see Knutson, Fong, Adams, Varner, & Hommer, 2001; Phillips, Blaha, & Fibiger, 1989; Schott et al., 2008); and (c) DA antagonists block the rewarding properties of food, water, and stimulant drugs of abuse (e.g., Rolls et al., 1974). Following from these and other findings, several theories were set forth in the mid-1980s in which impulsivity and related personality constructs such as extraversion, sensation seeking, and novelty seeking were proposed to arise from individual differences in activity/reactivity of mesolimbic DA structures (e.g., Cloninger, 1987; Gray 1987). Soon thereafter, psychopathologists co-opted dopaminergic theories of approach motivation to explain the unrestrained reward-seeking behaviors observed in ADHD, CD, and similar externalizing syndromes (e.g., Fowles, 1988; Quay, 1993).
Although some aspects of these early theories were mistaken (see Beauchaine & Gatzke-Kopp, 2012; Gatzke-Kopp & Beauchaine, 2007a), mesolimbic DA dysfunction is almost certainly an etiological factor in many if not most forms of externalizing psychopathology (Gatzke-Kopp, 2011). Extensive neuroimaging research with humans reveals (a) blunted mesolimbic and/or mesocortical reactivity to incentives among individuals with ADHD (see Bush, Valera, & Seidman, 2005; Carmona et al., 2011; Dickstein, Bannon, Castellanos, & Milham, 2006; Durston, 2003), CD (e.g., Rubia, Smith, et al., 2009), SUDs (see, e.g., Martin-Soelch et al., 2001; Volkow, Fowler, & Wang, 2004), and antisocial traits (e.g., Oberlin et al., 2012); (b) reduced mesolimbic DA transporter, D2 receptor, and/or D3 receptor binding among adults with ADHD (Volkow, Wang, et al., 2009) and alcoholism (e.g., Laine, Ahonen, Räsänen, & Tiihonen, 2001); and (c) compromised functional connectivity between mesolimbic and mesocortical structures among adolescents with ADHD and CD (e.g., Shannon, Sauder, Beauchaine, & Gatzke-Kopp, 2009). This latter finding is of interest because mesocortical structures provide top-down modulatory control over mesolimbic activity and reactivity, especially as individuals mature, an issue we return to below.
The DA dysfunction hypothesis of trait impulsivity is supported by single photon emission computed tomography, positron emission tomography, and functional magnetic resonance imaging studies of children and adults with ADHD. These studies demonstrate that the mechanism of action of DA agonists such as methylphenidate is to increase neural activity in the striatum, located in the mesolimbic reward pathway (e.g., Vles et al., 2003; Volkow, Fowler, Wang, Ding, & Gatley, 2002). Furthermore, methylphenidate normalizes frontocingulate underactivity (Rubia, Halari, Mohammad, Taylor, & Brammer, 2011) and frontostriatal functional connectivity deficits (Rubia, Halari, Cubillo, Mohammad, & Taylor, 2009) observed in children with ADHD. Thus, pharmacologic interventions that increase mesolimbic DA activity and improve functional connectivity by inhibiting reuptake decrease hyperactivity, impulsivity, and related aggressive behaviors (e.g., Hinshaw, Henker, Whalen, Erhardt, & Dunnington, 1989; MTA Cooperative Group, 1999).
Finally, individual differences in DA expression correlate with trait positive affectivity, and infusions of DA into mesolimbic structures produce pleasurable affective states (Ashby, Isen, & Turken, 1999; Berridge, 2003; Berridge & Robindon, 2003; Forbes & Dahl, 2005). In contrast, low levels of striatal DA correspond with trait irritability (Laakso et al., 2003). Children and adults with externalizing disorders including ADHD, ODD, and CD score high on measures of trait irritability and negative affectivity (e.g., Asherson, 2005, Martel & Nigg, 2006). Taken together, these findings provide overwhelming evidence for deficient mesolimbic DA function in the pathophysiology of externalizing behaviors (see also Gatzke-Kopp & Beauchaine, 2007a).3
Given the role of DA in expression of trait impulsivity, it should not be surprising that most genetic association studies of ADHD, ODD, and CD have included genes that affect DA turnover, availability, and/or metabolism. As with almost all psychiatric genetics research, effects sizes for individual genes are small (see Beauchaine & Gatzke-Kopp, 2013). Nevertheless, significant associations have been observed among ADHD, ODD, and/or CD and the DA receptor D4 (DRD4) gene, the DA receptor D5 gene, the DA transporter 1 gene, the monoamine oxidase A (MAOA) gene, and the catechol-O-methyltransferase (COMT) gene (see DeYoung et al., 2010; Faraone & Mick, 2010; Gizer, Ficks, & Waldman, 2009).
Thus, converging sources of evidence derived from experiments conducted with animals, and from neuroimaging and genetics studies conducted with humans, all point toward mesolimbic DA dysfunction as a core neural substrate of trait impulsivity, which predisposes affected individuals to externalizing spectrum disorders (see Beauchaine & Gatzke-Kopp, 2012; Beauchaine et al., 2009, 2010; Gatzke-Kopp, 2011).4 Mesolimbic DA dysfunction is experienced phenomenologically as an aversive, irritable mood state (e.g., Laakso et al., 2003), which affected individuals are motivated to avoid. Reward-seeking and novelty-seeking behaviors function to elevate mood through phasic activation of mesolimbic DA neurons. Unfortunately, any obtained hedonic value is short-lived, leading to searches for larger and more abundant rewards (see Beauchaine et al., 2007; Gatzke-Kopp, 2011; Gatzke-Kopp & Beauchaine, 2007a; Sagvolden et al., 2005). Those with deficient mesolimbic DA function are therefore hyperactive, impulsive, and vulnerable to serious externalizing psychopathology in high-risk environments, a topic we address in detail below.
The second question that emerges from Figure 1, and from the discussion outlined above, concerns differences among externalizing syndromes. If a single, almost entirely heritable higher order externalizing liability factor (expressed neurally as deficient mesolimbic DA function) confers vulnerability to all externalizing spectrum disorders (Krueger et al., 2002; Tuvblad et al., 2009), why do various first-order factors emerge consistently across studies? In addressing this question, we must consider both limitations of factor analysis, and sources of covariation among first-order externalizing syndromes. We discuss these in turn below.
The first-order factor structure of externalizing behaviors is often used as evidence for distinct disorders (i.e., ADHD, ODD, or CD, see Beauchaine et al., 2010). However, this interpretation is mistaken because factor analysis is not suited for identifying subtypes of disorders or people (see Waller & Meehl, 1998). Rather, factor analysis identifies dimensions on which people vary. Consider research on the Big 5 personality dimensions (openness to experience, conscientiousness, extraversion, agreeableness, and neuroticism). Five dimensions of personality in no way suggest five types of personality. Rather, individual differences along five dimensions yield almost unlimited expressions of personality. Similarly, factor analyses of externalizing symptoms do not suggest specific types of disorder. Individuals who score high on one dimension of externalizing conduct usually score high on all others (Hinshaw, 1987), especially if their age confers opportunity to engage in criterion behaviors across syndromes.5
As outlined above, the factor structure depicted in Figure 1 has been replicated consistently across population-based and twin studies of children, adolescents, and adults (e.g., Krueger et al., 2002; Lahey et al., 2011; Tuvblad et al., 2009). However, equally consistent findings are that (a) first-order externalizing syndromes (i.e., ADHD, ODD, CD, SUDs, and ASPD) are highly correlated (e.g., Kessler et al., 2005), (b) first-order externalizing syndromes are considerably less heritable than higher order externalizing liability (e.g., Kreuger et al., 2002), and (c) first-order factors are influenced much more by environment than by externalizing liability (see Burt, 2009; Burt et al., 2001). Given high rates of comorbidity among externalizing syndromes (see above), correlations among first-order factors are not surprising. However, greater environmental influence at the behavioral syndrome level requires elaboration.
Although twin studies indicate that most of the variance in higher order externalizing liability is heritable (see above), environmental factors, especially nonshared, account for considerable variance in specific behavioral syndromes, including ADHD, ODD, CD, SUDs, and ASPD (Krueger et al., 2002; Tuvblad et al., 2009). When combined, shared and nonshared environment often contribute more than heritability to the specific behavioral expression of externalizing liability. At first glance, this may seem counterintuitive. However, consider an individual who is vulnerable to substance dependence by virtue of inherited impulsivity. This person cannot develop a SUD without exposure to alcohol or other drugs of abuse. Similarly, an otherwise vulnerable individual may never engage in criminality or other antisocial behavior if reared in protective familial and cultural environments (see, e.g., Lynam et al., 2000; Meier, Slutske, Arndt, & Cadoret, 2008). Thus, genetic vulnerability is a necessary but insufficient etiological agent in progression from impulsivity early in life (i.e., ADHD) to more serious externalizing disorders.
Interim summary: Heterotypic comorbidity of externalizing syndromes
Externalizing spectrum disorders, including ADHD, ODD, CD, SUDs, and ASPD, are highly comorbid conditions. Most of the covariance among these disorders is accounted for by a single, higher order vulnerability trait, which is almost entirely heritable. Modern genetics and neuroimaging studies point toward mesolimbic DA dysfunction as a neurobiological substrate of inherited vulnerability, which is expressed behaviorally as trait impulsivity. However, even though trait impulsivity is almost entirely heritable, its specific behavioral expression, including whether it advances from ADHD to more serious externalizing pathology, is influenced considerably by environmental factors. Elucidating mechanisms through which environment amplifies or mollifies heritable vulnerability is essential if we wish to understand etiology and prevent lifelong psychopathology for affected individuals (see Beauchaine, Neuhaus, Brenner, & Gatzke-Kopp, 2008). Toward specifying etiology, we must consider developmental continuities and discontinuities in behavior (Rutter & Sroufe, 2000), and extend consideration of causal factors to additional levels of analysis, particularly environmental risk moderators (Beauchaine & Gatzke-Kopp, 2012; Cicchetti, 2008). This leads directly into discussion of heterotypic continuity.
Heterotypic Continuity
Over the past several decades, numerous pathways to delinquency have been described (see, e.g., Crocker, Fryer, & Mattson, 2013; Gatzke-Kopp, 2011; Lynam, 1996; Moffitt, 1993; Shannon Bowen & Gatzke-Kopp, 2013). However, in this article we are concerned with only one externalizing trajectory: that leading from ADHD very early in life to progressively more intractable externalizing behaviors across development. This pathway may account for the majority of individuals who engage in lifelong delinquent behavior (see Beauchaine et al., 2009, 2010; Moffitt, 1993). Since publication of Robins's (1966) landmark text on the development of delinquency, it has been known that antisocial adult males almost invariably follow a developmental trajectory that begins in preschool with severe ADHD, followed in rough temporal sequence by ODD, affiliations with delinquent peers, CD, substance abuse and dependence, ASPD, incarceration, and recidivism (see Beauchaine et al., 2010; Loeber & Hay, 1997; Loeber & Keenan, 1994; Lynam, 1996, 1998). However, no more than half of preschoolers who exhibit ADHD and oppositionality experience more serious conduct problems in later childhood (Campbell, Shaw, & Gilliom, 2000). Thus, ADHD does not determine later delinquency. Any transactional model of externalizing conduct must account for this observation. If vulnerability to externalizing behavior is conferred through a single impulsivity trait, why do some individuals persist to more severe behavioral syndromes as they mature, whereas others continue to suffer only from symptoms of ADHD?6
Addressing this question requires that we take the development component of developmental psychopathology seriously (see Sroufe, 2009). We present in Figure 2 an expanded depiction of the externalizing spectrum in which we add early temperament as a precursor to externalizing syndromes, insert intermittent explosive disorder (IED), which is new in the DSM-5, and denote the developmental trajectory outlined above by placing arrows between disorders. The temperament literature is voluminous and cannot be reviewed here. Nevertheless, it is important to acknowledge that, even though temperament is often assessed earlier in life than are externalizing syndromes, (a) temperamental constructs such as attentional focus, inhibitory control, and effortful control overlap with most definitions of impulsivity (e.g., Foley, McClowry, & Castellanos, 2008); (b) certain aspects of early temperament share genetic underpinnings with externalizing liability (e.g., Schmidt, Fox, Perez-Edgar, & Hamer, 2009); (c) facets of early temperament such as activity level, negative affectivity, and low inhibitory control prospectively predict development of externalizing behavior, especially in high-risk environments (e.g., Stringaris, Maughan, & Goodman, 2010); and (d) temperament is highly heritable (e.g., Saudino, 2009). Thus, even though temperament has not appeared in factor analytic models of the externalizing spectrum, it almost certainly belongs on the developmental pathway depicted in Figure 2. In addition, although very little research has been conducted on IED, we include it because many individuals who meet criteria for a current DSM-IV externalizing spectrum disorder are likely to meet criteria for IED as well and because similar symptoms have been linked to interactions between heritable vulnerability and environmental risk among those with CD (see Beauchaine et al., 2007, 2009), as we describe in later sections.
Figure 2.
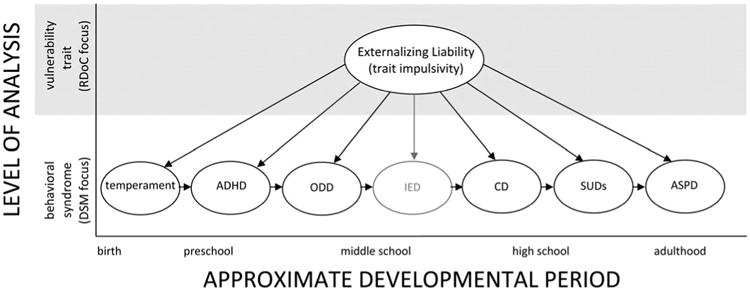
An expanded model in which trait impulsivity serves as a common vulnerability to sequential development of externalizing spectrum disorders across the life span. Temperament and intermittent explosive disorder (IED) have been added. The latter is shaded because it is a new disorder, so its inclusion is based on theoretical rather than empirical grounds.
Although Figure 2 portrays the development of externalizing syndromes in rough temporal sequence for those who progress from temperamental impulsivity to ASPD, it says nothing about mechanisms of continuity or desistance. Understanding such multifinality requires that we consider processes at other levels of analysis in addition to vulnerability traits and behavioral syndromes. Some of these are depicted in Figure 3, where we plot heterotypic development of externalizing syndromes by approximate age along the x axis and levels of analysis including genetic vulnerability (e.g., DRD4 allele), neural/hormonal substrates (e.g., mesolimbic DA function), latent vulnerability traits (e.g., impulsivity), behavioral syndromes (e.g., ADHD), and environmental risk mediators (e.g., parenting quality) down the y axis.
Figure 3.
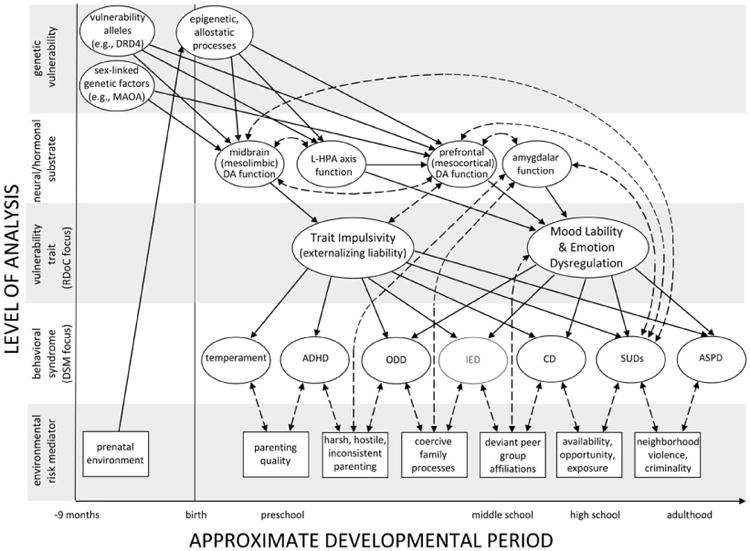
An ontogenic process model of externalizing spectrum behaviors in which levels of analysis are plotted on the y axis and relative age is plotted on the x axis. Heritable trait impulsivity is presumed to be the principal predisposing vulnerability to externalizing spectrum disorders, the syndromal manifestation (e.g., attention-deficit/hyperactivity disorder or conduct disorder) of which is influenced strongly by environmental risk mediators, which change and accrue across development. Trait impulsivity arises from factors specified in the top two panels. However, it is important to note that this heritable vulnerability is exacerbated through recursive feedback loops that span levels of analysis (dashed, bidirectional arrows). Through such mechanisms, high-risk behaviors (e.g., evocative effects on parenting or substance abuse) amplify inherited vulnerability. Emotion dysregulation emerges later in development and is influenced more by environmental influences than by heritability. Despite the daunting complexity of this model, many biological (e.g., head injury, taratogen exposure, serotonergic function) and environmental (e.g., abuse, neglect) etiological factors are left out, as are certain individual level of analysis predictors such as attributional biases and callous–unemotional traits. This illustrates why developmental psychopathology research on any complex trait needs to be conducted across disciplines and levels of analysis if we wish to understand multifinal and equifinal complexities of etiology. Solid arrows represent directional processes, and dashed arrows represent bidirectional processes.
Before describing this model in detail, we acknowledge that some readers will undoubtedly find its complexity bemusing. As Rutter and Sroufe(2000) noted, however, developmental pathways to psychopathology are usually complex, and a primary objective of developmental psychopathology research is to disentangle this complexity. With this goal in mind, Figure 3 illustrates the importance of (a) specifying etiological processes across levels of analysis (Cicchetti, 2008; Cicchetti & Dawson, 2002); (b) considering mechanisms through which processes at one level of analyses (e.g., drug use) interact with, alter functioning of, and feed back to systems at other levels of analysis (e.g., prefrontal DA function), thereby amplifying risk (Beauchaine & Gatzke-Kopp, 2012; Beauchaine, Neuhaus, et al., 2008); and (c) conceptualizing externalizing psychopathology, not as a set of distinct disorders with different causes (a conclusion often reached when we assess static sets of vulnerabilities and risk factors at single time points), but as an ontogenic process through which mechanisms of epigenesis, allostasis, and neural plasticity alter neurobiological and behavioral functioning in some ways that may be reversible, and in other ways that may not be (Beauchaine et al., 2009; Mead et al., 2010; Sroufe, 2009). A corollary of this last point is that two individuals on the same trajectory who are assessed at different developmental time points may exhibit very dissimilar biological and behavioral manifestations of externalizing vulnerability, not because they have different disorders, but because one has progressed much farther into the course of illness than the other.7
With this important point in mind, we now discuss components of the ontogenic process model of heterotypic continuity presented in Figure 3. We note at the outset that a full-length review article could be devoted to each of the following sections, which are necessarily incomplete. We note also that, despite the complexity of our model, a number of biological (e.g., serotonergic function) and environmental (e.g., maltreatment) etiological factors are left out, as are certain individual level of analysis predictors such as attributional biases and callous–unemotional traits. By omitting these influences, we are not suggesting they are unimportant. Each appears to play a significant role in the development of one pathway or another to antisocial behavior (e.g., De Sanctis, Nomura, Newcorn, & Halperin, 2012; Frick & Marsee, 2006; Frick & White, 2008; Lansford, Malone, Dodge, Pettit, & Bates, 2010; Zepf et al., 2008). However, in presenting our model we focus on processes that link specifically to the developmental pathway we describe and/or reduce complexity by limiting the number of levels of analyses and predictors presented. We omit callous–unemotional traits at the individual level of analysis, because such traits may be more important for developmental models of psychopathy (e.g., Frick, Stickle, Dandreaux, Farrell, & Kimonis, 2005). We also omit autonomic nervous system functioning as a level of analysis and child maltreatment as an environmental risk variable. Despite their relevance for the pathway we consider (e.g., Beauchaine et al., 2007; De Sanctis et al., 2012; Matthys, Vanderschuren, & Schutter, 2012), we cannot include all levels of analysis or environmental risk mediators in a single depiction of externalizing spectrum disorder development. In sections to follow, we discuss core components of our model.
Tenets of an ontogenic process model
Psychopathology as an outcome of development
What separates developmental psychopathology from related disciplines including child clinical psychology and psychiatry is its emphasis on complex transactions between individuals and their environments over time (see above). This emphasis follows from the assumption that psychopathology cannot be understood through cross-sectional analyses of associations between variables, regardless of the levels of analysis considered (see Rutter & Sroufe, 2000; Sroufe & Rutter, 1984). We therefore describe the importance of developmental context to our model before describing its constituent parts.
In an incisive paper published in this Journal, Sroufe (1997) emphasized several important points about the developmental psychopathology perspective (see also Sroufe, 2009). First, he noted that certain behavior patterns, although not disordered, render individuals vulnerable to developing psychopathology in the presence of exogenous risk. In the ontogenic process model depicted in Figure 3, this principle is illustrated at the interface between behavioral syndromes and environmental risk mediators. For example, temperamental impulsivity in and of itself is not construed as psychopathology. However, as outlined above, facets of temperament such as activity level, negative affectivity, and low inhibitory control share genetic underpinnings with, and prospectively predict the development of, externalizing behavior, especially in high-risk environments characterized by parental harshness and insensitivity (Muris & Ollendick, 2005; Schmidt et al., 2009; Stringaris et al., 2010). Children with impulsive temperaments are more susceptible to environmental adversity than are their peers (e.g., Bradley & Corwyn, 2008; Kiff, Lengua, & Zalewski, 2011; Kim & Kochanska, 2012; see also Belsky & Pluess, 2009). As a result, parenting mediates links between temperamental impulsivity and development of later ADHD and conduct problems.
Second, when psychopathology is considered at least in part an outcome of development, certain neurobiological processes that might otherwise be construed as causes are better conceptualized as individual differences (Sroufe, 1997, 2009). As outlined above, for example, variation in mesolimbic DA function underlies hereditable trait impulsivity. However, many with such vulnerability may be asymptomatic, and in most cases vulnerability advances beyond ADHD only through interactions with environmental risk (see above and below; Beauchaine & Gatzke-Kopp, 2012; Beauchaine et al., 2009, 2010).8 Thus, although mesolimbic DA function appears to be a neural substrate of individual differences in trait impulsivity, the further along the externalizing spectrum one advances across development, the more complex mediating and moderating pathways from DA function to psychopathology become, and the less any single contributor, including DA hyporesponding, can be interpreted as causal. This is illustrated at the genetic vulnerability level of analysis (upper left corner of Figure 3), where risk alleles for impulsivity are presented. Although genetic factors affect trait impulsivity through midbrain DA responding, there are no direct links from genetic vulnerability to behavior, and indirect links to externalizing syndromes including ADHD, ODD, and ASPD become increasingly complex across development (left to right). When we ignore development, we often draw erroneous and oversimplified causal links between neurobiology and disorder (Sroufe, 2009).
Third, Sroufe (1997, 2009) emphasized the probabilistic rather than the deterministic effects of vulnerabilities, risk factors, and their interactions. Given the overwhelming complexity of influences on behavior across development, including feedback and feedforward mechanisms across levels of analysis (see below), prediction of who will and who will not continue along the externalizing trajectory cannot be accomplished with specificity. Nevertheless, prevention science is advanced enough to offer targeted interventions very early in life to impulsive children who are reared in adversity (see Beauchaine, Gatzke-Kopp, et al., 2013; Webster-Stratton, Reid, & Beauchaine, 2011), with the aim of modifying risk factors such as coercive parenting and deviant peer group affiliations, which increase probabilities of antisocial outcomes (see below; Beauchaine, Neuhaus, et al., 2008).
Impulsivity, the primary source of vulnerability to externalizing spectrum disorders, is a continuously distributed, multi-factorial inherited trait
As reviewed in sections above, considerable evidence points toward trait impulsivity as a principal predisposing vulnerability to externalizing spectrum disorders. Here we emphasize that impulsivity is a multifactorial, continuously distributed individual difference and is therefore influenced by many genetic loci (see above), their interactions with one another, their interactions with other inherited traits (e.g., trait anxiety), and their interactions with the environment (see Neuhaus & Beauchaine, 2013). The importance of such interactions in the phenotypic expression of multifactorial traits (e.g., height) and diseases (e.g., coronary artery disease) has been recognized for decades (see, e.g., Bodmer & Bonilla, 2008). However, in psychiatric genetics we continue to search for genes that are specific to particular disorders, such as ADHD, rather than identifying arrays of genes that confer additive (or multiplicative) vulnerability to traits, such as impulsivity, that cut across disorders. Focusing on disorders assumes implicitly that (a) behavioral syndromes (ADHD, ODD, and CD), as currently defined, represent the proper level of analyses for genetic linkage and association studies and (b) multifactorial inherited traits do not interact with the environment to shape expression of vulnerability (i.e., impulsivity) into a range of phenotypes (i.e., externalizing spectrum disorders). Multifactorial inheritance suggests that no single gene will account for appreciable differences between externalizing syndromes, and that interactions between genetic susceptibility and environmental risk determine specific expression of vulnerability. This is part of the impetus for the RDoC, which focus not on traditional behavioral syndromes such as ADHD, ODD, and CD, but on dimensional traits that cut across traditional diagnostic boundaries (see Figure 3).
Viewing trait impulsivity as a multifactorial inherited trait, the expression of which is determined not by any single gene variant, but by complex interactions between total genetic vulnerability and environmental risk, has significant implications for research aimed at reifying traditional diagnostic boundaries among externalizing syndromes (see Beauchaine et al., 2010). The multifactorial inheritance model implies that genetic differences among those with externalizing spectrum disorders should account for very little variance in behavior within and across syndromes. Research conducted to date is consistent with this supposition. For example, although Caspi et al. (2008) reported that the COMT Val158-Met polymorphism was associated with individual differences in antisocial/aggressive behavior among children with ADHD in three impressively large samples, effect sizes were quite small. Collapsed across samples, the high-risk COMT polymorphism accounted for about 1% of the variance in antisocial behavior. Thus, 99% of the variance in antisocial/aggressive behavior was unaccounted for. This calls into question the authors' assertion that COMT provides a molecular genetic basis for “subtyping” ADHD. Although COMT almost certainly plays a role in the expression of externalizing behavior, it is only one contributor among many (see Waldman & Lahay, 2013).
Furthermore, molecular genetics studies that compare frequencies of candidate gene polymorphisms (e.g., COMT) across subtypes of externalizing disorders (e.g., ADHD vs. CD) often fail to find group differences (e.g., Monuteaux, Biederman, Doyle, Mick, & Faraone, 2009), and genome-wide association studies indicate no added genetic burden for children with ADHD + CD compared with those with ADHD alone (e.g., Anney et al., 2008).
Finally, consistent with the multifactorial inheritance perspective, several recent studies have illustrated the importance of evaluating Gene × Environment interactions in accounting for externalizing conduct. Perhaps the most famous of these was reported by Caspi et al. (2002), who found that the combination of a polymorphism in the MAOA gene and child maltreatment predicted juvenile and adult antisocial behavior. Those exposed to maltreatment as children who also inherited the low MAOA activity genotype were at much higher risk of engaging in antisocial behavior than those who were exposed to maltreatment but did not inherit the low MAOA activity genotype. The MAOA gene encodes for an enzyme that metabolizes DA. The MAOA genotype explained only about 1 % of the variance in antisocial behavior. However, the Maltreatment × Genotype interaction explained about 65%. This illustrates the importance of measuring environment if we wish to gain a full understanding of the direct and indirect effects of genes on behavior.
Taken together, these findings suggest that continued searches for single genes that differentiate between externalizing syndromes may be misguided and that a more fruitful approach will be to determine how multiple vulnerability genes interact with one another and the environment in predicting progression of externalizing behaviors.
Prenatal insults that alter DA function confer vulnerability to externalizing psychopathology through mechanisms of epigenesis and allostasis
The term epigenesis refers to experience-dependent changes in DNA structure (Riggs, Russo, & Martienssen, 1996), whereas allostasis refers to changes in the operating ranges of vital biological systems (Sterling & Eyer, 1988). Allostasis may occur through epigenetic mechanisms or through other neurobiological processes. Sometimes referred to collectively as maternal programming effects, epigenesis and allostasis provide for biological adaptations to environmental adversity (see Mead et al., 2010). As we and others have reviewed elsewhere (Beauchaine et al., 2011; Gatzke-Kopp, 2011; Neuhaus & Beauchaine, 2013), a variety of prenatal risk factors confer vulnerability to later externalizing spectrum disorders through epigenetic and allostatic mechanisms. For example, maternal smoking and second-hand smoke exposure during pregnancy both predict development of later ADHD, CD, and antisocial behavior among offspring, over and above effects of maternal ASPD (e.g., Brennan, Grekin, & Mednick, 1999; Gatzke-Kopp & Beauchaine, 2007b; Wakschlag et al., 1997). This vulnerability appears to be conferred through changes in mid-brain DA function. Rodents exposed to nicotine prenatally exhibit DA hyporeactivity to exogenous stimulation when mature (see Slotkin, 1998). Furthermore, children with high-risk DA transporter and DRD4 polymorphisms are at greatest risk for developing later externalizing disorders when exposed to nicotine prenatally (Becker, El-Faddagh, Schmidt, Esser, & Laucht, 2008; Neuman et al., 2007). In Figure 3, epigenetic/allostatic modulation of midbrain DA activity is indicated by the indirect pathway from prenatal environment, through epigenetic and allostatic processes, to mesolimbic DA function.
Prenatal sensitivity of the mesolimbic DA system to maternal programming effects has profound implications for development of trait impulsivity and vulnerability to psychopathology (see Gatzke-Kopp, 2011).9 As with nicotine, cocaine exposure during gestation elicits downregulation of mesolimbic DA function among rodents and induces permanent structural changes in the developing anterior cingulate cortex, even at low doses (e.g., Minabe, Ashby, Heyser, Spear, & Wang, 1992; Stanwood, Washington, Shumsky, & Levitt, 2001). The anterior cingulate cortex is a DA-rich network critical to self-monitoring and behavior regulation (see Gatzke-Kopp et al., 2009). Although not a direct focus of this paper, maternal substance use and stress exposure during pregnancy also sensitize children's developing limbic–hypothalamic–pituitary–adrenal axis responses to stress in childhood and predict development of ADHD, CD, and aggressive behavior (see Glover, 2011; Hunter, Minnis, & Wilson, 2011). Similarly, exogenous glucocorticoids, which are used prenatally to treat certain medical conditions among mothers and alter DA signaling through epigenetic mechanisms, induce behavioral impulsivity later in life (Kapoor, Petropoulos, & Matthews, 2008).
Circulating cortisol levels play integral roles in the neurodevelopment of DA neurons and in modulating mesolimbic DA system activity and reactivity pre- and postnatally (e.g., Koehl et al., 2001). The hypothalamic–pituitary–adrenal axis appears to modulate sensitivity of midbrain DA neurons to pleasurable effects of strong stimulants such as methamphetamine (e.g., Oswald et al., 2005). Rodent models suggest that through such mechanisms, maternal stress exposure during pregnancy leads to increased sensitivity to stimulant drugs of abuse among adult offspring (e.g., Koehl et al., 2001; Meany, Brake, & Gratton, 2002). These findings are similar to those observed following prenatal exposure to methamphetamine (Bubenikova-Valesova et al., 2009). Thus, as reviewed by Gatzke-Kopp (2011), midbrain DA neurons are exquisitely sensitive during prenatal development to long-term changes in functioning brought about through epigenesis and allostasis, and through mechanisms that damage brain tissue directly (e.g., hypoxia).
Early in life, trait impulsivity is conferred primarily through mesolimbic DA function
In sections above, we reviewed evidence that individual differences in mesolimbic (midbrain) DA function underlie trait impulsivity. However, as most readers are undoubtedly aware, the mesocortical (prefrontal) DA system inhibits impulsive behavior through its roles in decision making, planning, and other executive functions (see, e.g., Floresco & Magyar, 2006). Thus, compromises in the mesocortical DA system and in certain executive function tasks are also associated with impulsivity and conduct problems (see, e.g., Kim & Lee, 2011), and are observed among those with ADHD (e.g., Thorell & Waÿhlstedt, 2006; Willcutt, Doyle, Nigg, Faraone, & Pennington, 2005). As we have noted elsewhere, however (e.g., Neuhaus & Beauchaine, 2013), even though development of executive functions begins in preschool (Garon, Bryson, & Smith, 2008), we do not consider frontal mechanisms of impulsivity to be foundational for most affected children because these brain regions continue to mature into adolescence and early adulthood (e.g., Welsh, Pennington, & Groisser, 1991). We therefore view the mesolimbic DA system as a primary source of trait impulsivity very early in life (see also Halperin & Schulz 2006), with mesocortical contributions increasing across development (see below). For this reason, we place mesolimbic DA function ahead of prefrontal DA function in the temporal sequence depicted in Figure 3. This is not meant to suggest that prefrontal mechanisms of impulsivity are unimportant in the progression of externalizing behaviors. Neurodevelopment of frontal regions may be affected (through mechanisms of neural plasticity, programming, and pruning) by early experiences that are themselves a product of impulsivity (see Beauchaine, Neuhaus, et al., 2008; Sagvolden et al., 2005). Thus, heritable compromises in the early-maturing mesolimbic DA system may alter neurodevelopment in the later-maturing mesocortical DA system, especially in high-risk environments. Specifying such neurodevelopmental sequences is essential if we wish to understand the etiology of psychopathology (see Sroufe, 2009). We therefore return to this point in later sections.
Progression to successively more severe externalizing syndromes occurs through complex, bidirectional transactions between individuals and environments over time
Accumulating evidence suggests that neurobiological vulnerabilities interact with high-risk and protective environments to either promote or inhibit progression along the externalizing trajectory outlined above (for reviews, see Beauchaine et al., 2009, 2010; Beauchaine & Gatzke-Kopp, 2012; Gatzke-Kopp & Beauchaine, 2007a). As a result, children who are impulsive are more likely to engage in delinquent behaviors when reared in environments characterized by hostile and inconsistent parenting (e.g., Drabick, Gadow, & Sprafkin, 2006), maltreatment and neglect (e.g., De Sanctis et al., 2008), neighborhood violence/criminality (e.g., Lynam et al., 2000; Meier et al., 2008), and other forms of adversity. Furthermore, children who are impulsive are more likely than are nonimpulsive children to evoke aversive reactions from their caregivers (O'Connor, Deater-Deckard, Fulker, Rutter, & Plomin, 1998), which may feed back to exacerbate preexisting vulnerabilities (see below).
Bidirectional effects between children's externalizing behaviors and their environments are denoted in Figure 3 by dashed arrows that cross the level-of-analysis boundary between behavioral syndromes and environmental risk mediators. For example, links from ADHD early in life to IED, ODD, and CD operate through a series of environmental risk mediators including overreactive/inconsistent parenting, coercive family processes, and deviant peer group affiliations (see, e.g., Beauchaine & Zalewski, in press; Dishion & Racer, 2013). All of these are associated empirically with progression of externalizing behavior (e.g., Patterson, DeGarmo, & Knutson, 2000; Raudino, Fergusson, Woodward, & Horwood, 2012; Snyder et al., 2005, 2008). Although some have argued that such findings might be explained entirely by active or evocative gene–environment correlations (rGEs),10 rGE cannot account fully for externalizing spectrum progression for at least two reasons. First, research conducted with high-risk samples on links between child difficulty in infancy, hostile parenting in toddlerhood, and later conduct problems in first grade indicates direct effects of maternal hostility, but no effects of child difficulty, and no interactive effects of maternal hostility and child difficulty (Lorber & Egeland, 2011). Thus, parenting appears to affect progression from difficult behaviors in infancy to later conduct problems more than child behavior affects parenting. Second, intervention research reveals that the deviant peer group exposure/affiliation elicits progression of children and adolescents' conduct problems. Among those who exhibit conduct problems and are assigned randomly to group interventions, progression of delinquency is observed over time. Among those assigned randomly to a control condition, delinquency rates remain stable (Dishion, McCord, & Poulin, 1999). These findings cannot be explained by rGE, given random assignment to groups.
However, it is equally clear that children affect their environments in ways that promote progression of delinquency (see Dishion & Racer, 2013). O'Connor et al. (1998) reported an evocative rGE in a sample of children who were both adopted away at birth and at high genetic risk for delinquency. Despite being raised by adoptive parents, these children received more negative parenting than did those in a matched control group. Because the adoptive parents' behaviors could not be explained by shared genetic risk with the child, these data provide strong evidence for an evocative rGE. Neiderhiser et al. (2004) also reported evidence of evocative rGE in a study of parenting by twin mothers.
In addition to evoking aversive reactions from others, impulsive children and adolescents expose themselves to high-risk environments through reward-seeking behaviors, and through associations with deviant peers. Such mechanisms account for age at initiation of nicotine and alcohol use, even though abuse and dependence are determined largely by heritable effects (Boomsma, Koopsman, Van Doormen, & Orlebeke, 1994; Koopsman, Slutzke, Heath, Neale, & Boomsma, 1999; Koopsman, van Doornen, & Boomsma, 1997; McGue, Iacono, Legrand, & Elkins, 2001; Viken, Kaprio, Koskenvuo, & Rose, 1999). Taken together, findings reviewed in this section suggest that continued argument over directions of effect between children and their environments in the progression of externalizing behavior is misplaced and that transactions across levels of analysis are the rule rather than the exception (see also Keijsers, Loeber, Branje, & Meeus, 2011; Pardini, 2008).
Two additional points should be emphasized regarding transactions between vulnerable children/adolescents and their environments. First, although we present environmental risk mediators as if they were phenomenologically and temporally distinct, such distinctions serve only for simplicity of presentation. In reality, environmental risk factors such as inconsistent and coercive parenting are related conceptually, and co-occur (e.g., Arnold, O'Leary, Wolff, & Acker, 1993). Similarly, deviant peer group affiliations and availability/exposure to substances of abuse are highly correlated phenomena (e.g., Fergusson, Swain-Campbell, & Horwood, 2002). As is the case for closely related behavioral syndromes (e.g., ADHD, ODD, and CD; see above), distinguishing among environmental risk mediators, though sometimes useful heuristically, distorts interrelations among influences on externalizing outcomes. This underscores the overwhelming complexity of externalizing spectrum disorder development.
Second, the highly transactional nature of emerging externalizing outcomes among impulsive individuals helps explain why prospective prediction of persistence and escalation is so difficult. One simply cannot know what environmental risk mediators any particular individual will face across his/her lifetime. Emerging evidence suggests that certain environmental risk factors operate cumulatively (e.g., Gerard & Buehler, 2004). On the bright side, this suggests many potential opportunities for desistance in changing environmental contexts, in response to prevention/intervention efforts, or in the presence of individual-level resilience factors (see Beauchaine, Neuhaus, et al., 2008; Rutter, 2012).
Operant reinforcement shapes development of mood lability and emotion dysregulation, which amplify and entrench externalizing behaviors
In several of our previous publications addressing the development of externalizing spectrum disorders, we have advanced the following set of propositions (see Beauchaine et al., 2007, 2009; Beauchaine & Gatzke-Kopp, 2012; Crowell et al., 2009): (a) trait impulsivity is the principal predisposing vulnerability to externalizing disorders; (b) trait impulsivity derives largely from heritable compromises in central DA function; (c) progression of trait impulsivity into more intractable externalizing conduct is facilitated by operant reinforcement of emotional lability within families; and (d) over time, such reinforcement contingencies result in enduring patterns of emotion dysregulation, which predisposes vulnerable individuals to develop ASPD. We have already discussed items (a) and (b) in detail above. Here we briefly summarize mechanisms through which emotional lability and emotion dysregulation are socialized within families, and describe how these mechanisms facilitate progression along the externalizing spectrum.
According to coercion theory (Patterson, 1982; Patterson, DeBaryshe, & Ramsey, 1989), the development of antisocial behavior has roots in aversive dyadic interaction patterns that occur thousands of times between parents and children in high-risk families. During these coercive interactions, aggression and emotional lability are negatively reinforced as children and parents match and oftentimes exceed one another's anger and antagonism levels. This escalation of anger, antagonism, and physiological arousal motivate both parties to terminate the interaction, even if through coercive means, which is reinforcing because it results in escape from the unpleasant interchange (hence the term escape conditioning). Through this mechanism, emotional lability, emotion dysregulation, and physiological reactivity generalize over time, and eventually they become primary means through which affected individuals cope with interpersonal distress, within and outside the family (see Beauchaine & Zalewski, in press). Generalization of coercion and emotion dysregulation may then lead to interpersonal violence, contacts with police, and other adverse sequelae (e.g., Colvin, Cullen, & Vander ven, 2002). Thus, mood lability and emotion dysregulation take on traitlike qualities over time, as indicated in Figure 3.
Evidence for coercive family processes as a mechanism through which antisocial outcomes are shaped and maintained is considerable. In a series of studies using meticulous micro-analytic coding techniques, Snyder and colleagues (e.g., Snyder, Edwards, McGraw, Kilgore, & Holton, 1994; Snyder, Schrepferman, & St. Peter, 1997) demonstrated that in at-risk families, parents often match or exceed aversiveness and arousal levels of their children, who in turn match and exceed aversiveness and arousal levels of their parents. Such exchanges often begin before preschool and continue throughout development, canalizing aversive behaviors and emotional lability (see Beauchaine et al., 2007). Furthermore, impulsive children are more likely than are nonimpulsive children to evoke aversive reactions from their parents, which feeds back to exacerbate their preexisting vulnerability (O'Connor et al., 1998).
At this juncture it is important to reemphasize the transactional nature of our model. Trait impulsivity, which is a heritable vulnerability, is intrinsically insufficient to result in progression from ADHD to more severe externalizing syndromes (Beauchaine et al., 2007, 2009). Rather, it interacts with socialized deficiencies in emotion regulation to amplify risk for ODD, CD, SUDs, and ASPD. Thus, in Figure 3, ODD, CD, SUDs, and ASPD all include directional arrows from trait impulsivity and trait emotion dysregulation, whereas temperament and ADHD are influenced primarily by trait impulsivity. This of course implies that ADHD will not progress to more serious externalizing syndromes in family environments where strong emotion regulation skills are socialized (see Beauchaine et al., 2007, 2009, 2010). Although experimental intervention research indicates that socialization of emotion regulation is possible in young children with ADHD, which reduces conduct problems and aggressive behaviors characteristic of more advanced externalizing syndromes (Beauchaine, Gatzke-Kopp, et al., 2013), it is important to note that, given the high heritability of trait impulsivity, impulsive children are often reared by impulsive parents, who are more likely to react coercively (Patterson, Chamberlain, & Reid, 1982; Patterson et al., 1989, 2000; Patterson, Dishion, & Bank, 1984).
Deficient mood, emotion, and behavior regulation co-develop with compromised mesocortical (prefrontal) brain function
All behavior has neurobiological substrates. Self-regulation, including impulse control and modulation of emotion, is subserved increasingly across development by prefrontal brain regions that mature throughout adolescence into early adulthood (see, e.g., Gogtay et al., 2004; Phillips, Walton, & Jhou, 2007; Thayer, Hansen, Saus-Rose, & Johnsen, 2009). Among typically developing individuals, regulation of reward-related responding, emotion, and mood lability is effected by the prefrontal cortex (PFC), which exerts top-down inhibitory control over subcortical brain regions, including the mesolimbic DA system, the amygdala, the septo-hippocampal system, and their interconnections (see, e.g., Goldsmith, Pollak, & Davidson, 2008; Heatherton, 2011; Heatherton & Wagner, 2011). Such top-down regulatory processes become increasingly important as individuals transition into developmental stages that require endogenous control over their behavior.
Prefrontal influences on trait impulsivity and mood/emotion regulation appear in Figure 3, which indicates a number of complex interrelationships that warrant discussion. Neurodevelopment of the PFC is affected by many influences, including genetic, epigenetic, and allostatic processes (see, e.g., Colantuoni et al., 2011; Lenroot et al., 2007); modulatory effects of other neural and hormonal systems (see, e.g., McCormick & Mathews, 2010); and exogenous factors such as family socialization, trauma, and substance use (see Crews, He, & Hodge, 2007; Hanson et al., 2010; Pollak, 2011). Neurodegenerative effects of stress on the PFC, including those exerted indirectly through the limbic–hypothalamic–adrenal axis, can be structurally extensive, leading to deficiencies in executive functions and impulse control (see Arnsten, 2009; Beauchaine et al., 2011). Thus, heritable vulnerability among children who are impulsive due to compromises in mesolimbic DA function may be amplified in high-stress environments including those characterized by coercion, trauma, neighborhood violence, and criminality. Such environments may alter prefrontal cortical development in ways that potentiate progression of ADHD to more severe externalizing syndromes (see Beauchaine, 2011; Beauchaine, Neuhaus, et al., 2008; Mead et al., 2010). For example, altered patterns of age-related pruning of prefrontal gray matter among those with ADHD and CD, which may be affected by environment influences and normalized via stimulant treatment (see Giedd & Rapoport, 2010), predict risky patterns of substance use and abuse in adolescence (see Bava & Tapert, 2010).
The above paragraph implies that differences in patterns of functional brain activity should be observed among those with ADHD versus those with CD, SUDs, and ASPD. In a recent review of functional magnetic resonance imaging studies comparing children and adolescents with ADHD and CD, Rubia (2011) reported such effects. Consistent with our ontogenic process model, primary neural deficiencies among those with ADHD included reduced activation in striatal (i.e., mesolimbic) brain regions compared with controls. In contrast, children and adolescents with CD showed abnormalities in the ventromedial PFC.11 Like most who conduct group comparisons, Rubia concluded that these differences provide evidence for existing distinctions between ADHD and CD. However, from an ontogenic perspective, an alternative explanation is that CD participants are further along the externalizing trajectory outlined in Figures 2 and 3, and have therefore developed deficiencies in prefrontal function that are not observed among their ADHD-only counterparts. Thus, those who have developed into more advanced stages of the disease process given to interactions between endogenous vulnerabilities and exogenous risk, exhibit more extensive impairment, as reflected in different sets of symptoms. Disentangling these alternative hypotheses cannot be accomplished without painstaking longitudinal research in which effects of environment, brain function, and their interactions are measured repeatedly across development (see Sroufe, 2009).
Functional connections between mesolimbic and mesocortical structures are indicated by a bidirectional dashed arrow in Figure 3. Recent research reveals reduced functional connectivity between mesolimbic and mesocortical brain regions among those with CD and ADHD (e.g., Shannon et al., 2009). Such findings are important given behavior- and emotion-regulatory functions served by feedback and feedforward connections between mesolimbic and mesocortical brain regions (see below). As noted above, the mesocortical DA system inhibits subcortical DA expression in the service of self-regulation. Pharmacologic activation of prefrontal DA levels decreases DA activity in the nucleus accumbens, a mesolimbic structure (Louilot, LeMoal, & Simon, 1989). Conversely, decreasing prefrontal DA increases DA levels in the nucleus accumbens. Disruption in this feedback system, as evidenced by altered functional connectivity, may be one neural substrate of impulsivity (Tisch, Silberstein, Limousin-Dowsey, & Jahanshahi, 2004). Effective top-down modulation of mesolimbic DA activity/reactivity may be especially vulnerable to environmental insults given experience-dependent effects on developing midbrain and cortical DA systems (see above; Arnsten, 2009; Halperin & Schulz, 2006; Spear, 2007; Sullivan & Brake, 2003).
We have also included amygdalar function in Figure 3. Given its roles in processing self-relevant information and generating positive and negative emotional responses (see, e.g., Davis & Whalen, 2001), its developmental sensitivity to environmental programming effects and Gene × Environment interactions (see, e.g., Gillespie, Phifer, Bradley, & Ressler, 2009), and its functional interconnections with other brain regions involved in self-regulation, including the PFC (see above; Kim et al., 2011), any multiple levels of analysis account of externalizing conduct must include amygdalar function.
Few if any studies have found structural or functional abnormalities in the amygda among children or adolescents with ADHD. In contrast, those with CD often exhibit reduced amygdalar volumes and excessive amygdalar reactivity to emotionally evocative stimuli (e.g., Decety, Michalska, Akitsuki, & Lahey, 2009; Fairchild et al., 2011; Sterzera, Stadlerb, Poustkab, & Kleinschmidta, 2007; van Harmelen et al., 2012). Amygdalar reactivity is also associated with individual differences in inhibitory control among adults (e.g., Brown, Manuck, Flory, & Harari, 2006). Furthermore, deficient top-down control of the amygdala by the PFC, and reduced functional connectivity between the amygdala and the PFC, have been implicated in emotional lability and deficient self-control (see, e.g., Churchwell, Morris, Heurtelou, & Kesner, 2009).
An ontogenic process perspective suggests that amygdalar dysfunction and deficient top-down control of the amygdala by the PFC may develop from extensive longitudinal transactions between vulnerable individuals and high-risk environments. Consistent with this supposition, amygdala hyperactivity to angry and fearful faces is observed consistently among those who were abused or mistreated as children (see, e.g., Pollak, 2008; van Harmelen et al., 2013). Although such sensitivity to social cues may be integral to immediate survival in maltreatment contexts, it portends poor social adjustment later in life (Hanson et al., 2010). In turn, poor social adjustment may be one mechanism through which childhood maltreatment facilitates progression of ADHD to more severe conduct problems (see Mead et al., 2010).
Taken together, these findings suggest that among children who are already impulsive, deficiencies in amygdalar function co-develop with deficiencies in prefrontal function and that environmental risk contributes significantly to this process. Deficient top-down control of amygdalar and mesolimbic function by the PFC results in mood lability (including oversensitivity to perceived provocation), emotion dysregulation, and further erosion of impulse control. Through protracted reinforcement and canalization, mood lability and emotion dysregulation assume traitlike qualities (see Figure 3), even though they are far less heritable than impulsivity, at both behavioral and physiological levels of analysis (Goldsmith et al., 2008; Kupper et al., 2004; Sneider, Boomsma, van Doornen, & DeGeus, 1997).
High-risk behaviors amplify deficiencies mesolimbic, mesocortical, and amygdalar brain function, which exacerbates externalizing behavior
In addition to pre- and postnatal effects on neurodevelopment of various stressors, adverse experiences, and exposure to stimulants noted above, many vulnerable individuals engage in high-risk behaviors that compromise brain function further, facilitating progression along the externalizing spectrum. Perhaps the best example of this is substance use, abuse, and dependence, which alter functioning in all of the neural networks discussed previously, often in ways that exacerbate impulsive behavior and contribute to poor behavior and emotion regulation.
Literature on the effects of substance use/dependence on midbrain and forebrain DA systems is voluminous and cannot be reviewed here. Nevertheless, although the neurocircuitry of addiction is complex (see Perry et al., 2011), several important points stand out. First, preexisting mesolimbic and PFC dysfunction, expressed behaviorally as poor self-regulation (see above) places individuals at risk for addiction (see George & Koob, 2010). Second, alcohol and drug abuse and dependence compromise prefrontal/orbitofrontal cortex structure and function further, resulting in more impulsive decision making and susceptibility to relapse (see Schoenbauma & Shahamd, 2008). Third, addiction is facilitated by use-dependent disruption in top-down regulation of mesolimbic reward regions by the PFC, which has additional adverse effects on self-regulation (see Goldstein & Volkow, 2011; Kalivas, 2008). Chronic elevation of DA neural firing in the nucleus accumbens induced by strong stimulant exposure among rodents and nonhuman primates downregulates tonic DA activity, sensitizes phasic DA responding to such stimulants, and suppresses the strength of developing connections between mesolimbic structures and the PFC (see Thomas, Beurrier, Bonci, & Malenka 2001; Vezina, 2004). As noted above, these connections are integral to effective self-regulation.
Given immaturity of the PFC in particular, and to a lesser extent mesocortical structures, adolescents may be especially vulnerable to the reward properties of alcohol and other substances, and to use-dependent alterations in neurodevelopment and self-regulation (e.g., Casey & Jones, 2010). Such alterations include persistent downregulation of DA release in the PFC, with resulting compromises in executive functions (see Volkow, Fowler, Wand, Baler, & Telang, 2009), a well-replicated correlate of early-onset conduct problems and antisocial behavior (e.g., Moffitt, 1993; Pharo, Sim, Graham, Gross, & Hayne, 2011).
Finally, substance-induced alterations in functional projections from the PFC to the amygdala affect extinction of addictive behaviors (see Peters, Kalivas, & Quirk, 2009). Thus, abnormalities in mesolimbic, prefrontal, and amygdalar structure and function confer vulnerability to substance abuse/dependence, are exacerbated by substance abuse/dependence, and amplify preexisting deficiencies in self-regulation to promote progression along the externalizing spectrum to ASPD (see Beauchaine et al., 2009, 2011). Therefore, Figure 3 includes bidirectional arrows between SUDs and mesolimbic, mesocortical, and amygdalar function.
Interim summary: Heterotypic continuity of externalizing syndromes
In traditional conceptualizations of externalizing psychopathology, behavioral syndromes such as ADHD, ODD, CD, SUDs, and ASPD are assumed to be distinct, despite high rates of concurrent comorbidity and heterotypic continuity across development. This has led to a research agenda aimed at identifying discrete genetic susceptibilities, specific neural and affective substrates, and different environmental risk mediators across behavioral syndromes. In contrast to the traditional approach, an ontogenic process perspective views vulnerability as deriving from multifactorial inheritance of impulsivity, which interacts with other vulnerability traits and environmental risk and protective factors to either promote or inhibit progression to increasingly more intractable forms of externalizing behavior across development. According to this approach, homotypic comorbidity and heterotypic continuity cannot be understood without considering effects of complex longitudinal transactions between individuals and their environments on neurodevelopment, including (a) how early experiences, including prenatal insults, can alter brain function in ways that confer vulnerability to later psychopathology through mechanisms of epigenesis and allostasis; (b) how neural mechanisms of impulsivity migrate from primarily subcortical to primarily frontal across development; (c) how brain structure and function are shaped by environmental experience; (d) how mood lability and emotion dysregulation take on traitlike qualities over time through operant reinforcement; and (e) how high-risk behaviors, particularly substance use, abuse, and dependence, compromise functioning in neural networks that are integral to executive functioning and impulse control. None of these processes can be understood by conducting cross-sectional group comparisons of behavior or neurobiological functioning among those with different forms of psychopathology (e.g., ADHD vs. CD). Rather, understanding the emergence of externalizing psychopathology requires that we examine vulnerabilities and their interactions with environmental risk across levels of analysis and time, and that we eschew simplistic main effects explanations of individual differences in behavioral syndromes.
Conclusion
Since the first issue of Development and Psychopathology was published in 1989, great strides have been made in our understanding of the etiology of early-onset externalizing behaviors. Much of this progress follows from seminal papers and special issues published in this Journal (see above). Although considerable energy is still being expended toward reifying traditional adevelopmental boundaries between externalizing disorders (see Beauchaine et al., 2010, 2013), the importance of developmental processes in progression from early life impulsivity to conduct problems, delinquency, and substance use, especially in contexts of adversity and environmental risk, cannot be ignored. When development is taken seriously and the complexity of transactions among vulnerabilities and risk factors is considered across levels of analysis, it becomes clear that externalizing behaviors, at least for a considerable subset of individuals, develop from influences that become increasingly self-reinforcing over time. When viewed from this perspective, the importance of intervening early at all relevant levels of analysis becomes clear (Beauchaine et al., 2013; Beauchaine, Neuhaus, et al., 2008).
Footnotes
This typically involves statistically partialing the effects of one disorder from another when predicting an external criterion. For example, one might examine the relation between ODD and later ASPD, over and above the effects of CD, and vice versa. If one disorder predicts ASPD and the other does not, it may be tempting to consider ODD and CD as distinct. However, the use of analysis of covariance and this interpretation are both inappropriate if ODD and CD are related etiologically. In such cases, analysis of covariance creates statistical entities that do not exist in reality (ODD without liability to CD and CD without liability to ODD), which distorts etiological relations among disorders and obscures patterns of true comorbidity. From a statistical standpoint, covariates and predictors should always be uncorrelated, which avoids these and other sorts of interpretational ambiguities (see, e.g., Pedhazur, 1997).
Although some authors prefer more circumscribed definitions of impulsivity such as errors in maze solving (Porteus, 1965), perseverative errors during set shifting (e.g., Avila, Cuenca, Félix, Parcet, & Miranda, 2004), and performance on continuous performance tasks, ADHD scale scores are much more heritable and explain far more variance in functional outcomes, which suggests greater construct and predictive validity (see Neuhaus & Beauchaine, 2013).
Some (e.g., Rubia, 2011) have suggested different central nervous system substrates for ADHD (frontostriatal) versus CD (ventromedial prefrontal). Although we acknowledge that ventromedial prefrontal cortex dysfunction, a likely neural substrate of emotion dysregulation (e.g., Goldsmith, Pollak, & Davidson, 2008), plays a role in the progression of ADHD to CD, we believe it emerges over time through Person × Environment transactions that can only be understood in developmental context (e.g., Beauchaine & Gatzke-Kopp, 2012; Beauchaine et al., 2007). We address this in later sections.
Other pathways to externalizing vulnerability clearly exist, including those following head injury, teratogen exposure, and hypoxia, among other influences. Readers interested in these alternative pathways, some of which also eventuate in mesolimbic DA dysfunction (see Gatzke-Kopp 2011), are referred to other sources (e.g., Crocker, Fryer, & Mattson, 2013; Shannon Bowen & Gatzke-Kopp, 2013).
A 4-year-old, regardless of his or her externalizing vulnerability, will have difficulty meeting most criteria for CD (e.g., breaking and entering, stealing while confronting a victim, or running away overnight) and cannot possibly meet criteria for ASPD given lack of opportunity.
Research on persistence of ADHD from childhood to adulthood has yielded inconsistent findings (see Barkley, Murphy, & Fischer, 2008). Although our intent is not to review these studies here, several authors have reported such persistence, especially when subthreshold symptoms are accounted for (e.g., Biederman, Petty, Evans, Small, & Faraone, 2010). Persistence into adulthood is of course expected for any highly heritable trait.
Although this notion often receives considerable resistance in psychopathology research, the same holds for heritable medical conditions. For example, vulnerability to type II diabetes is almost entirely heritable (e.g., Medici, Hawa, Ianari, Pyke, & Leslie, 1999), but illness expression advances over time, beginning with mild symptoms such as frequent urination and thirst. As the disease progresses, usually across many years, life-threatening conditions such as renal failure, blindness, and circulatory problems occur. Thus, two affected individuals with type II diabetes will appear very different from one another at the overt symptom level if they are assessed early versus late in the progression of illness. Nevertheless, we would not diagnose them with two disorders. The difference, of course, is that we know the pathophysiology of type II diabetes, so we do not mistake divergent symptom presentations at different developmental epochs for dissociable disorders.
Effect sizes from both functional neuroimaging studies support this point. For example, Scheres, Milham, Knutson, and Castellanos (2007) reported an effect size of d = 1.06 in comparing ventral striatal activation during reward anticipation between adolescents with ADHD and controls. Even though this is a large effect size by Cohen's (1988) standards, it indicates that the distributions overlapped by about 35%. Thus, many adolescents in the control group exhibited striatal responses that were as low or lower than the mean ADHD group response, yet they did not exhibit behavioral symptoms.
At first glance, this may seem incompatible with the assertion that impulsivity is almost entirely heritable. However, because the same prenatal insults are experienced by identical twin pairs, any effects of such insults on behavior are subsumed into the heritability component of a twin study, even though the effects are not genetic in origin (see, e.g., Beauchaine & Gatzke-Kopp, 2013).
Active rGE occurs when a child's heritable vulnerabilities influence his or her selection of environments, whereas evocative rGE occurs when genetically influenced behaviors elicit reactions from others that interact with and exacerbate existing vulnerabilities. For a detailed discussion, see Rutter (2006).
As Rubia (2011) rightly notes, many of these studies included groups with high rates of comorbidity. If one's objective is to differentiate between disorders, this is often viewed as problematic. However, since comorbidity is the rule among externalizing syndromes (see above), recruiting noncomorbid participants is difficult (see Gatzke-Kopp et al., 2009), and any differences between noncomorbid subgroups likely do not reflect the nature of externalizing psychopathology for most affected individuals (see Beauchaine et al., 2010; Miller & Chapman, 2001).
References
- Achenbach T. Developmental psychopathology. New York: Ronald Press; 1974. [Google Scholar]
- Achenbach TM, Edelbrock CS. Psychopathology of childhood. Annual Review of Psychology. 1984;35:227–256. doi: 10.1146/annurev.ps.35.020184.001303. [DOI] [PubMed] [Google Scholar]
- Achenbach TM, Edelbrock CS. Manual for the Child Behavior Checklist/4-18 and 1991 profile. Burlington, VT: University of Vermont, Department of Psychiatry; 1991. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 3rd, rev. Washington, DC: Author; 1987. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th, text rev. Washington, DC: Author; 2000. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th. Washington, DC: Author; 2013. [Google Scholar]
- Angold A, Costello EJ, Erkanli A. Comorbidity. Journal of Child Psychology and Psychiatry. 1999;40:57–87. [PubMed] [Google Scholar]
- Anney RJ, Lasky-Su J, O'Dúshláine C, Kenny E, Neale BM, Mulligan A, et al. Conduct disorder and ADHD: Evaluation of conduct problems as a categorical and quantitative trait in the international multicentre ADHD genetics study. American Journal of Medical Genetics. 2008;147B:1369–1378. doi: 10.1002/ajmg.b.30871. [DOI] [PubMed] [Google Scholar]
- Arnold DS, O'Leary SG, Wolff LS, Acker MM. The Parenting Scale: A measure of dysfunctional parenting in discipline situations. Psychological Assessment. 1993;5:137–144. [Google Scholar]
- Arnsten AF. Stress signaling pathways that impair prefrontal cortex structure and function. Nature Reviews Neuroscience. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby FG, Isen AM, Turken AU. A neuropsychological theory of positive affect and its influence on cognition. Psychological Review. 1999;106:529–550. doi: 10.1037/0033-295x.106.3.529. [DOI] [PubMed] [Google Scholar]
- Asherson P. Clinical assessment and treatment of attention-deficit/hyperactivity disorder in adults. Expert Review of Neurotherapeutics. 2005;5:525–539. doi: 10.1586/14737175.5.4.525. [DOI] [PubMed] [Google Scholar]
- Avila C, Cuenca I, Félix V, Parcet MA, Miranda A. Measuring impulsivity in school-aged boys and examining its relationship with ADHD and ODD ratings. Journal of Abnormal Child Psychology. 2004;32:295–304. doi: 10.1023/b:jacp.0000026143.70832.4b. [DOI] [PubMed] [Google Scholar]
- Barkley RA, Murphy KR, Fischer M. ADHD in adults: What the science says. New York: Guilford Press; 2008. [Google Scholar]
- Bava S, Tapert SF. Adolescent brain development and risk for alcohol and other drug problems. Neuropsychology Review. 2010;20:398–413. doi: 10.1007/s11065-010-9146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP. The role of biomarkers and endophenotypes in prevention and treatment of psychopathological disorders. Biomarkers in Medicine. 2009;3:1–3. doi: 10.2217/17520363.3.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Gatzke-Kopp LM. Instantiating the multiple levels of analysis perspective in a program of study on externalizing behavior. Development and Psychopathology. 2012;24:1003–1018. doi: 10.1017/S0954579412000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Gatzke-Kopp LM. Genetic and environmental influences on behavior. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 111–140. [Google Scholar]
- Beauchaine TP, Gatzke-Kopp L, Mead HK. Polyvagal theory and developmental psychopathology: Emotion dysregulation and conduct problems from preschool to adolescence. Biological Psychology. 2007;74:174–184. doi: 10.1016/j.biopsycho.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Gatzke-Kopp LM, Neuhaus E, Chipman J, Reid MJ, Webster-Stratton C. Sympathetic- and parasympathetic-linked cardiac function and prediction of externalizing behavior, emotion regulation, and prosocial behavior among preschoolers treated for ADHD. Journal of Consulting and Clinical Psychology. 2013;81:481–493. doi: 10.1037/a0032302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Hinshaw SP, Pang K. Comorbidity of attention-deficit/hyperactivity disorder and early-onset conduct disorder: Biological, environmental, and developmental mechanisms. Clinical Psychology: Science and Practice. 2010;17:327–336. [Google Scholar]
- Beauchaine TP, Klein DN, Crowell SE, Derbidge C, Gatzke-Kopp LM. Multifinality in the development of personality disorders: A Biology × Sex × Environment model of antisocial and borderline traits. Development and Psychopathology. 2009;21:735–770. doi: 10.1017/S0954579409000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Klein DN, Erickson NL, Norris AL. Developmental psychopathology and the Diagnostic and Statistical Manual of Mental Disorders. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 29–110. [Google Scholar]
- Beauchaine TP, Lenzenweger MF, Waller NG. Schizotypy, taxometrics, and disconfirming theories in soft science: Comment on Rawlings, Williams, Haslam, and Claridge. Personality and Individual Differences. 2008;44:1652–1662. [Google Scholar]
- Beauchaine TP, Marsh P. Taxometric methods: Enhancing early detection and prevention of psychopathology by identifying latent vulnerability traits. In: Cicchetti D, Cohen D, editors. Developmental psychopathology. 2nd. Hoboken, NJ: Wiley; 2006. pp. 931–967. [Google Scholar]
- Beauchaine TP, Neuhaus E, Brenner SL, Gatzke-Kopp L. Ten good reasons to consider biological processes in prevention and intervention research. Development and Psychopathology. 2008;20:745–774. doi: 10.1017/S0954579408000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Neuhaus E, Zalewski M, Crowell SE, Potapova N. The effects of allostatic load on neural systems subserving motivation, mood regulation, and social affiliation. Development and Psychopathology. 2011;23:975–999. doi: 10.1017/S0954579411000459. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Zalewski M. Physiological and developmental mechanisms of emotional lability in coercive relationships. In: Dishion TJ, Snyder JJ, editors. Oxford handbook of coercive relationship dynamics. New York: Oxford University Press; in press. [Google Scholar]
- Becker K, El-Faddagh M, Schmidt MH, Esser G, Laucht M. Interaction of dopamine transporter genotype with prenatal smoke exposure on ADHD symptoms. Journal of Pediatrics. 2008;152:263–269. doi: 10.1016/j.jpeds.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Belsky J, Pluess M. Beyond diathesis stress: Differential susceptibility to environmental influences. Psychological Bulletin. 2009;135:885–908. doi: 10.1037/a0017376. [DOI] [PubMed] [Google Scholar]
- Berridge KC. Pleasures of the brain. Brain and Cognition. 2003;52:106–128. doi: 10.1016/s0278-2626(03)00014-9. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. Parsing reward. Trends in Neuroscience. 2003;26:507–513. doi: 10.1016/S0166-2236(03)00233-9. [DOI] [PubMed] [Google Scholar]
- Biederman J, Petty CR, Evans M, Small J, Faraone SV. How persistent is ADHD? A controlled 10-year follow-up study of boys with ADHD. Psychiatry Research. 2010;177:299–304. doi: 10.1016/j.psychres.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genetics. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma DI, Koopsman JR, Van Doornen LJ, Orlebeke JF. Genetic and social influences on starting to smoke: A study of Dutch adolescent twins and their parents. Addiction. 1994;89:219–226. doi: 10.1111/j.1360-0443.1994.tb00881.x. [DOI] [PubMed] [Google Scholar]
- Bradley RH, Corwyn RF. Infant temperament, parenting, and externalizing behavior in first grade: A test of the differential susceptibility hypothesis. Journal of Child Psychology and Psychiatry. 2008;49:124–131. doi: 10.1111/j.1469-7610.2007.01829.x. [DOI] [PubMed] [Google Scholar]
- Brennan PA, Grekin ER, Mednick SA. Maternal smoking during pregnancy and adult male criminal outcomes. Archives of General Psychiatry. 1999;56:215–219. doi: 10.1001/archpsyc.56.3.215. [DOI] [PubMed] [Google Scholar]
- Brenner SL, Beauchaine TP, Sylvers PD. A comparison of psychophysiological and self-report measures of BAS and BIS activation. Psychophysiology. 2005;42:108–115. doi: 10.1111/j.1469-8986.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- Brown SM, Manuck SB, Flory JD, Harari AR. Neural basis of individual differences in impulsivity: Contributions of corticolimbic circuits for behavioral arousal and control. Emotion. 2006;6:239–245. doi: 10.1037/1528-3542.6.2.239. [DOI] [PubMed] [Google Scholar]
- Bubenikova-Valesovaa V, Kacerb P, Syslovab K, Rambousekb L, Janovskyc M, Schutovad B, et al. Prenatal methamphetamine exposure affects the mesolimbic dopaminergic system and behavior in adult offspring. International Journal of Developmental Neuroscience. 2009;27:525–530. doi: 10.1016/j.ijdevneu.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Burnett ML, Cicchetti D, editors. Multilevel approaches to understanding antisocial behavior: Current research and future directions. Development and Psychopathology. 2012;24:703–1155. doi: 10.1017/S0954579412000314. Special Issue. [DOI] [PubMed] [Google Scholar]
- Burt SA. Rethinking environmental contributions to child and adolescent psychopathology: A meta-analysis of shared environmental influences. Psychological Bulletin. 2009;135:608–637. doi: 10.1037/a0015702. [DOI] [PubMed] [Google Scholar]
- Burt SA, Krueger RF, McGue M, Iacono WG. Sources of covariation among attention-deficit/hyperactivity disorder, oppositional defiant disorder, and conduct disorder: The importance of shared environment. Journal of Abnormal Psychology. 2001;110:516–525. doi: 10.1037/0021-843X.110.4.516. [DOI] [PubMed] [Google Scholar]
- Bush G, Valera EM, Seidman LJ. Functional neuroimaging of attention-deficit/hyperactivity disorder: A review and suggested future directions. Biological Psychiatry. 2005;57:1273–1284. doi: 10.1016/j.biopsych.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Campbell SB, Shaw DS, Gilliom M. Early externalizing behavior problems: Toddlers and preschoolers at risk for later maladjustment. Development and Psychopathology. 2000;12:467–488. doi: 10.1017/s0954579400003114. [DOI] [PubMed] [Google Scholar]
- Carmona S, Hoekzema E, Ramos-Quiroga JA, Richarte V, Canals C, Bosch R, et al. Response inhibition and reward anticipation in medication-naý¨ve adults with attention-deficit/hyperactivity disorder: A within-subject case-control neuroimaging study. Human Brain Mapping. 2011;33:2350–2361. doi: 10.1002/hbm.21368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron C, Rutter M. Comorbidity in child psychopathology: Concepts, issues and research strategies. Journal of Child Psychology and Psychiatry. 1991;32:1063–1080. doi: 10.1111/j.1469-7610.1991.tb00350.x. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Jones RM. Neurobiology of the adolescent brain and behavior: Implications for substance use disorders. Journal of the American Academy of Child & Adolescent Psychiatry. 2010;49:1189–1201. doi: 10.1016/j.jaac.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: The case of the serotonin transporter gene and its implications for studying complex diseases and traits. American Journal of Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Langley K, Milne B, Moffitt TE, O'Donovan M, Owen MJ, et al. A replicated molecular genetic basis for subtyping antisocial behavior in children with attention-deficit/hyperactivity disorder. Archives of General Psychiatry. 2008;65:203–210. doi: 10.1001/archgenpsychiatry.2007.24. [DOI] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Tannock R. Neuroscience of attention-deficit/hyperactivity disorder: The search for endophenotypes. Nature Reviews Neuroscience. 2002;3:617–628. doi: 10.1038/nrn896. [DOI] [PubMed] [Google Scholar]
- Churchwell JC, Morris AM, Heurtelou NM, Kesner RP. Interactions between the prefrontal cortex and amygdala during delay discounting and reversal. Behavioral Neuroscience. 2009;123:1185–1196. doi: 10.1037/a0017734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti D. The emergence of developmental psychopathology. Child Development. 1984;55:1–7. [PubMed] [Google Scholar]
- Cicchetti D. Developmental psychopathology: Some thoughts on its evolution. Development and Psychopathology. 1989;1:1–4. [Google Scholar]
- Cicchetti D, editor. Regulatory process. Development and Psychopathology. 1996;8:1–305. Special Issue. [Google Scholar]
- Cicchetti D. Development and psychopathology. In: Cicchetti D, Cohen DJ, editors. Developmental psychopathology: vol 1 Theory and method. Hoboken, NJ: Wiley; 2006. pp. 1–23. [Google Scholar]
- Cicchetti D. A multiple-levels-of-analysis perspective on research in developmental psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. Hoboken, NJ: Wiley; 2008. pp. 27–57. [Google Scholar]
- Cicchetti D, Ackerman BP, Izard C, editors. Emotions in developmental psychopathology. Development and Psychopathology. 1995;7:1–226. Special Issue. [Google Scholar]
- Cicchetti D, Blender JA. A multiple-levels-of-analysis approach to the study of developmental processes in maltreated children. Proceedings of the National Academy of Sciences. 2004;101:17325–17326. doi: 10.1073/pnas.0408033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti D, Cannon TD. Neurodevelopmental processes in the ontogenesis and epigenesis of psychopathology. Development and Psychopathology. 1999;11:375–654. doi: 10.1017/s0954579499002114. Special Issue. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Cohen DJ, editors. Developmental psychopathology: vol 1 Theory and method. New York: Wiley; 1995a. [Google Scholar]
- Cicchetti D, Cohen DJ, editors. Developmental psychopathology: vol 2 Risk, disorder, and adaptation. New York: Wiley; 1995b. [Google Scholar]
- Cicchetti D, Cohen DJ, editors. Developmental psychopathology: vol 1 Theory and method. 2nd. New York: Wiley; 2006a. [Google Scholar]
- Cicchetti D, Cohen DJ, editors. Developmental psychopathology: vol 2 Developmental neuroscience. 2nd. New York: Wiley; 2006b. [Google Scholar]
- Cicchetti D, Cohen DJ, editors. Developmental psychopathology: vol 3 Risk, disorder, and adaptation. 2nd. New York: Wiley; 2006c. [Google Scholar]
- Cicchetti D, Dawson G, editors. Multiple levels of analysis. Development and Psychopathology. 2002;14:417–666. doi: 10.1017/s0954579402003012. Special Issue. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Hinshaw SP. Conceptual, methodological, and statistical issues in developmental psychopathology: A Special Issue in honor of Paul E. Meehl. Development and Psychopathology. 2003;15:497–832. doi: 10.1017/s0954579403000269. Special Issue. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Posner M, editors. Integrating cognitive and affective neuroscience and developmental psychopathology. Development and Psychopathology. 2005;17:569–891. doi: 10.1017/S0954579405050273. Special Issue. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Rogosch FA, editors. Developmental pathways: Diversity in process and outcome. Development and Psycho-pathology. 1996;8:597–666. Special Issue. [Google Scholar]
- Cicchetti D, Rogosch FA, Thibodeau EL. The effects of child maltreatment on early signs of antisocial behavior: Genetic moderation by tryptophan hydroxylase, serotonin transporter, and monaoamine oxidase A genes. Development and Psychopathology. 2012;24:907–928. doi: 10.1017/S0954579412000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti D, Toth SL. The development of depression in children and adolescents. American Psychologist. 1998;53:221–241. doi: 10.1037//0003-066x.53.2.221. [DOI] [PubMed] [Google Scholar]
- Cloninger CR. A systematic method for clinical description and classification of personality variants. Archives of General Psychiatry. 1987;44:573–588. doi: 10.1001/archpsyc.1987.01800180093014. [DOI] [PubMed] [Google Scholar]
- Cohen J. Statistical power analysis for the behavioral sciences. 2nd. Hillsdale, NJ: Erlbaum; 1988. [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin M, Cullen FT, Vander ven T. Coercion, social support, and crime: An emerging theoretical consensus. Criminology. 2002;40:19–42. [Google Scholar]
- Conners CK, Sitarenios G, Parker JDA, Epstein JN. The revised Conners' Parent Rating Scale (CPRS–R): Factor structure, reliability, and criterion validity. Journal of Abnormal Child Psychology. 1998;26:257–268. doi: 10.1023/a:1022602400621. [DOI] [PubMed] [Google Scholar]
- Covault J, Tennen H, Armeli S, Conner TS, Herman AI, Cillessen A, et al. Interactive effects of the serotonin transporter 5-HTTLPR polymorphism and stressful life events on college student drinking and drug use. Biological Psychiatry. 2007;61:609–616. doi: 10.1016/j.biopsych.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Crews F, He J, Hodge C. Adolescent cortical development: A critical period of vulnerability for addiction. Pharmacology Biochemistry and Behavior. 2007;86:189–199. doi: 10.1016/j.pbb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker NA, Fryer SL, Mattson SN. Exposure to teratogens as a risk factor for psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 285–316. [Google Scholar]
- Crowell SE, Beauchaine TP, Linehan M. The development of borderline personality: Extending Linehan's theory. Psychological Bulletin. 2009;135:495–510. doi: 10.1037/a0015616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell SE, Derbidge C, Beauchaine TP. Developmental approaches to understanding self-injury and suicidal behaviors. In: Nock MK, editor. Oxford handbook of suicide and self-injury. New York: Oxford University Press; in press. [Google Scholar]
- Davies PT, Sturge-Apple ML, Cicchetti D, Manning LG, Vonhold SE. Pathways and processes of risk in associations among maternal antisocial personality symptoms, interparental aggression, and preschoolers' psychopathology. Development and Psychopathology. 2012;24:807–832. doi: 10.1017/S0954579412000387. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: Vigilance and emotion. Molecular Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Dawson G. Early behavioral intervention, brain plasticity, and the prevention of autism spectrum disorder. Development and Psychopathology. 2008;20:775–803. doi: 10.1017/S0954579408000370. [DOI] [PubMed] [Google Scholar]
- Derbidge C, Beauchaine TP. A developmental model of self-inflicted injury, borderline personality, and suicide risk. In: Lewis M, Rudolph K, editors. Handbook of developmental psychopathology. 3rd. New York: Springer; in press. [Google Scholar]
- Decety J, Michalska KJ, Akitsuki Y, Lahey BB. Atypical empathic responses in adolescents with aggressive conduct disorder: A functional MRI investigation. Biological Psychology. 2009;80:203–211. doi: 10.1016/j.biopsycho.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sanctis VA, Nomura Y, Newcorn JH, Halperin JM. Childhood maltreatment and conduct disorder: Independent predictors of criminal outcomes in ADHD youth. Child Abuse and Neglect. 2012;36:782–789. doi: 10.1016/j.chiabu.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sanctis VA, Trampush JW, Harty SC, Marks DJ, Newcorn JH, Miller CJ, et al. Childhood maltreatment and conduct disorder: Independent predictors of adolescent substance use disorders in youth with attention-deficit/hyperactivity disorder. Journal of Clinical Child and Adolescent Psychology. 2008;37:785–793. doi: 10.1080/15374410802359650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung CG, Getchell M, Koposov RA, Yrigollen CM, Haeffel GJ, Klinteberg B, et al. Variation in the catechol-o-methyltransferase val158 met polymorphism associated with conduct disorder and ADHD symptoms among adolescent male delinquents. Psychiatric Genetics. 2010;20:20–24. doi: 10.1097/YPG.0b013e32833511e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Viken RJ, Kapiro J, Pulkkinen L, Rose RJ. Understanding the covariation among childhood externalizing symptoms: Genetic and environmental influences on conduct disorder, attention-deficit/hyperactivity disorder, and oppositional defiant disorder symptoms. Journal of Abnormal Child Psychology. 2005;33:219–229. doi: 10.1007/s10802-005-1829-8. [DOI] [PubMed] [Google Scholar]
- Dickstein SG, Bannon K, Castellanos XF, Milham MP. The neural correlates of attention-deficit/hyperactivity disorder: An ALE meta-analysis. Journal of Child Psychology and Psychiatry. 2006;47:1051–1062. doi: 10.1111/j.1469-7610.2006.01671.x. [DOI] [PubMed] [Google Scholar]
- Dishion TJ, McCord J, Poulin F. When interventions harm. American Psychologist. 1999;54:755–764. doi: 10.1037//0003-066x.54.9.755. [DOI] [PubMed] [Google Scholar]
- Dishion TJ, Racer KH. Development of adult antisocial behavior. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 453–487. [Google Scholar]
- Drabick DAG, Gadow KD, Sprafkin J. Co-occurrence of conduct disorder and depression in a clinic-based sample of boys with ADHD. Journal of Child Psychology and Psychiatry. 2006;47:766–774. doi: 10.1111/j.1469-7610.2006.01625.x. [DOI] [PubMed] [Google Scholar]
- Durston S. A review of the biological bases of ADHD: What have we learned from imaging studies? Mental Retardation and Developmental Disabilities Reviews. 2003;9:184–195. doi: 10.1002/mrdd.10079. [DOI] [PubMed] [Google Scholar]
- Ellis BJ, Del Giudice M, Shirtcliff EA. Beyond allostatic load: The stress response system as a mechanism of conditional adaptation. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. 2013. pp. 251–284. [Google Scholar]
- Fairchild G, Passamonti L, Hurford G, Hagan CC, von dem Hagen EAH, van Goozen SHM, et al. Brain structure abnormalities in early-onset and adolescent-onset conduct disorder. American Journal of Psychiatry. 2011;168:624–633. doi: 10.1176/appi.ajp.2010.10081184. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Mick E. Molecular genetics of attention-deficit/hyperactivity disorder. Psychiatric Clinics of North America. 2010;33:159–180. doi: 10.1016/j.psc.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergusson DM, Swain-Campbell NR, Horwood LJ. Deviant peer affiliations, crime and substance use: A fixed effects regression analysis. Journal of Abnormal Child Psychology. 2002;30:419–430. doi: 10.1023/a:1015774125952. [DOI] [PubMed] [Google Scholar]
- First MB. Mutually exclusive versus co-occurring diagnostic categories: The challenge of diagnostic comorbidity. Psychopathology. 2005;38:206–210. doi: 10.1159/000086093. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Magyar O. Mesocortical dopamine modulation of executive functions: Beyond working memory. Psychopharmacology. 2006;188:567–585. doi: 10.1007/s00213-006-0404-5. [DOI] [PubMed] [Google Scholar]
- Foley M, McClowry SG, Castellanos FX. The relationship between attention-deficit/hyperactivity disorder and child temperament. Journal of Applied Developmental Psychology. 2008;29:157–169. [Google Scholar]
- Forbes EE, Dahl RE. Neural systems of positive affect: Relevance to understanding child and adolescent depression? Development and Psychopathology. 2005;17:827–850. doi: 10.1017/S095457940505039X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowles DC. Psychophysiology and psychopathology: A motivational approach. Psychophysiology. 1988;25:373–391. doi: 10.1111/j.1469-8986.1988.tb01873.x. [DOI] [PubMed] [Google Scholar]
- Frick PJ, Marsee MA. Psychopathy and developmental pathways to antisocial behavior in youth. In: Patrick CJ, editor. Handbook of psychopathy. New York: Guilford Press; 2006. pp. 353–375. [Google Scholar]
- Frick PJ, Stickle TR, Dandreaux DM, Farrell JM, Kimonis ER. Callous–unemotional traits in predicting the severity and stability of conduct problems and delinquency. Journal of Abnormal Child Psychology. 2005;33:471–487. doi: 10.1007/s10648-005-5728-9. [DOI] [PubMed] [Google Scholar]
- Frick PJ, White SF. Research Review: The importance of callous–unemotional traits for developmental models of aggressive and antisocial behavior. Journal of Child Psychology and Psychiatry. 2008;49:359–375. doi: 10.1111/j.1469-7610.2007.01862.x. [DOI] [PubMed] [Google Scholar]
- Garon N, Bryson SE, Smith IM. Executive function in preschoolers: A review using an integrative framework. Psychological Bulletin. 2008;134:31–60. doi: 10.1037/0033-2909.134.1.31. [DOI] [PubMed] [Google Scholar]
- Gatzke-Kopp LM. The canary in the coalmine: Sensitivity of mesolimbic dopamine to environmental adversity during development. Neuroscience & Biobehavioral Reviews. 2011;35:794–803. doi: 10.1016/j.neubiorev.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Gatzke-Kopp L, Beauchaine TP. Central nervous system substrates of impulsivity: Implications for the development of attention-deficit/hyperactivity disorder and conduct disorder. In: Coch D, Dawson G, Fischer K, editors. Human behavior and the developing brain: Atypical development. New York: Guilford Press; 2007a. pp. 239–263. [Google Scholar]
- Gatzke-Kopp L, Beauchaine TP. Prenatal nicotine exposure and the development of conduct disorder: Direct and passive effects. Child Psychiatry and Human Development. 2007b;38:255–269. doi: 10.1007/s10578-007-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatzke-Kopp LM, Beauchaine TP, Shannon KE, Chipman-Chacon J, Fleming AP, Crowell SE, et al. Neurological correlates of reward responding in adolescents with and without externalizing behavior disorders. Journal of Abnormal Psychology. 2009;118:203–213. doi: 10.1037/a0014378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatzke-Kopp LM, Greenberg MT, Fortunato CK, Coccia MA. Aggression as an equifinal outcome of distinct neurocognitive and neuroaffective processes. Development and Psychopathology. 2012;24:985–1002. doi: 10.1017/S0954579412000491. [DOI] [PubMed] [Google Scholar]
- Gau SSF, Ni HC, Shang CY, Soong WT, Wu YY, Lin LY, et al. Psychiatric comorbidity among children and adolescents with and withpout persistent attention-deficit/hyperactivity disorder. Australian and New Zealand Journal of Psychiatry. 2010;44:135–143. doi: 10.3109/00048670903282733. [DOI] [PubMed] [Google Scholar]
- George O, Koob GF. Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neuroscience & Biobehavioral Reviews. 2010;35:232–247. doi: 10.1016/j.neubiorev.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard JM, Buehler C. Cumulative environmental risk and youth problem behavior. Journal of Marriage and Family. 2004;66:702–720. [Google Scholar]
- Giedd JN, Rapoport JL. Structural MRI of pediatric brain development: What have we learned and where are we going? Neuron. 2010;67:728–734. doi: 10.1016/j.neuron.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CF, Phifer J, Bradley B, Ressler KJ. Risk and resilience: Genetic and environmental influences on development of the stress response. Depression and Anxiety. 2009;26:984–992. doi: 10.1002/da.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: A meta-analytic review. Human Genetics. 2009;126:51–90. doi: 10.1007/s00439-009-0694-x. [DOI] [PubMed] [Google Scholar]
- Glover V. Annual Research Review: Prenatal stress and the origins of psychopathology: An evolutionary perspective. Journal of Child Psychology and Psychiatry. 2011;52:356–367. doi: 10.1111/j.1469-7610.2011.02371.x. [DOI] [PubMed] [Google Scholar]
- Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaituzis AC, et al. Dynamic mapping of human cortical development during childhood through early adulthood. Proceedings of the National Academy of Sciences. 2004;101:8174–8179. doi: 10.1073/pnas.0402680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith HH, Pollak SD, Davidson RJ. Developmental neuroscience perspectives on emotion regulation. Child Development Perspectives. 2008;2:132–140. doi: 10.1111/j.1750-8606.2008.00055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nature Reviews Neuroscience. 2011;12:652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II. Genetic aspects of intelligent behavior. In: Ellis NR, editor. Handbook of mental deficiency. New York: McGraw–Hill; 1963. pp. 253–296. [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Shields J. Schizophrenia in twins: 16 years' consecutive admissions to a psychiatric clinic. British Journal of Psychiatry. 1966;112:809–818. doi: 10.1192/bjp.112.489.809. [DOI] [PubMed] [Google Scholar]
- Gray JA. The neuropsychology of emotion and personality. In: Stahl SM, Iversen SD, Goodman EC, editors. Cognitive neurochemistry. Oxford: Oxford University Press; 1987. pp. 171–190. [Google Scholar]
- Gunnar MR, Wenner JA, Thomas KM, Glatt CE, McKenna MC, Clark AG. The brain-derived neurotrophic factor factor Val66Met polymorphism moderates early deprivation effects on attention problems. Development and Psychopathology. 2012;24:1215–1223. doi: 10.1017/S095457941200065X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin JM, Schulz KP. Revisiting the role of the prefrontal cortex in the patho-physiology of attention-deficit/hyperactivity disorder. Psychological Bulletin. 2006;132:560–581. doi: 10.1037/0033-2909.132.4.560. [DOI] [PubMed] [Google Scholar]
- Hanson JL, Chung MK, Avants BB, Shirtcliff EA, Gee JC, Davidson RJ, et al. Early stress is associated with alterations in the orbitofrontal cortex: A tensor-based morphometry investigation of brain structure and behavioral risk. Journal of Neuroscience. 2010;30:7466–7472. doi: 10.1523/JNEUROSCI.0859-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatherton TF. Neuroscience of self and self-regulation. Annual Review of Psychology. 2011;62:363–390. doi: 10.1146/annurev.psych.121208.131616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatherton TF, Wagner DD. Cognitive neuroscience of self-regulation failure. Trends in Cognitive Sciences. 2011;15:132–139. doi: 10.1016/j.tics.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SP. On the distinction between attention-deficit/hyperactivity and conduct problems/aggression in child psychopathology. Psychological Bulletin. 1987;101:443–463. [PubMed] [Google Scholar]
- Hinshaw SP, Henker B, Whalen CK, Erhardt D, Dunnington RE. Aggressive, prosocial, and nonsocial behavior in hyperactive boys: Dose effects of methylphenidate in naturalistic settings. Journal of Consulting and Clinical Psychology. 1989;57:636–643. doi: 10.1037/0022-006X.57.5.636. [DOI] [PubMed] [Google Scholar]
- Hinshaw SP, Lahey BB, Hart EL. Issues of taxonomy and comorbidity in the development of conduct disorder. Development and Psychopathology. 1993;5:31–49. [Google Scholar]
- Hirshfeld-Becker DR, Biederman J, Faraone SV, Violette H, Wrightsman J, Rosenbaum JF. Temperamental correlates of disruptive behavior disorders in young children: Preliminary findings. Biological Psychiatry. 2002;50:563–574. doi: 10.1016/s0006-3223(01)01299-9. [DOI] [PubMed] [Google Scholar]
- Hollander E, Zohar J, Sirovatka PJ, Regier DA, editors. Obsessive–compulsive spectrum disorders: Refining the research agenda for DSM-V. Washington, DC: American Psychiatric Association; 2011. [Google Scholar]
- Hunter AL, Minnis H, Wilson P. Altered stress responses in children exposed to early adversity: A systematic review of salivary cortisol studies. Stress. 2011;14:614–626. doi: 10.3109/10253890.2011.577848. [DOI] [PubMed] [Google Scholar]
- Insel TR, Cuthbert BN, Garvey MA, Heinssen RK, Pine DS, Quinn KJ, et al. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. American Journal of Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- Jensen P. Comorbidity and child psychopathology: Recommendations for the next decade. Journal of Abnormal Child Psychology. 2003;31:293–300. doi: 10.1023/a:1023281513936. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. Addiction as a pathology in prefrontal cortical regulation of corticostriatal habit circuitry. Neurotoxicity Research. 2008;14:185–189. doi: 10.1007/BF03033809. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Nakamura M. Neural systems for behavioral activation and reward. Current Opinion in Neurobiology. 1999;9:223–227. doi: 10.1016/s0959-4388(99)80031-2. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Petropoulos S, Matthews SG. Fetal programming of hypothalamic–pituitary–adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Research Reviews. 2008;57:586–595. doi: 10.1016/j.brainresrev.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Keijsers L, Loeber R, Branje S, Meeus W. Bidirectional links and concurrent development of parent–child relationships and boys' offending behavior. Journal of Abnormal Psychology. 2011;120:878–889. doi: 10.1037/a0024588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall PC, Drabick DAG, editors. Comorbidity in children's mental health. Clinical Psychology: Science and Practice. 2010;17:265–359. Special Issue. [Google Scholar]
- Kessler RC, Chiu W, Demler O, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiff CJ, Lengua LJ, Zalewski M. Nature and nurturing: Parenting in the context of child temperament. Clinical Child and Family Psychology Review. 2011;14:251–301. doi: 10.1007/s10567-011-0093-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Loucks RA, Palmer AL, Brown AC, Solomon KM, Marchante AN, et al. Structural and functional connectivity of the amygdala: From normal emotion to pathological anxiety. Behavioural Brain Research. 2011;223:403–410. doi: 10.1016/j.bbr.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kochanska G. Child temperament moderates effects of parent–child mutuality on self-regulation: A relationship-based path for emotionally negative infants. Child Development. 2012;83:1275–1289. doi: 10.1111/j.1467-8624.2012.01778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee D. Prefrontal cortex and impulsive decision making. Biological Psychiatry. 2011;69:1140–1146. doi: 10.1016/j.biopsych.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DN, Riso LP. Psychiatric disorders: Problems of boundaries and comorbidity. In: Costello CG, editor. Basic issues in psychopathology. New York: Guilford Press; 1993. pp. 19–66. [Google Scholar]
- Knutson B, Fong GW, Adams CM, Varner JL, Hommer D. Dissociation of reward anticipation and outcome with event-related fMRI. Brain Imaging. 2001;12:3683–3687. doi: 10.1097/00001756-200112040-00016. [DOI] [PubMed] [Google Scholar]
- Koehl M, Lemaire V, Vallee M, Abrous N, Piazza PV, Mayo W, et al. Long-term neurodevelopmental and behavioral effects of perinatal life events in rats. Neurotoxicity Research. 2001;3:65–83. doi: 10.1007/BF03033231. [DOI] [PubMed] [Google Scholar]
- Koopsman JR, Slutzke WS, Heath AC, Neale MC, Boomsma DI. The genetics of smoking initiation and quantity smoked in Dutch adolescent and young adult twins. Behavior Genetics. 1999;29:383–393. doi: 10.1023/a:1021618719735. [DOI] [PubMed] [Google Scholar]
- Koopsman JR, van Doornen LJ, Boomsma DI. Association between alcohol use and smoking in adolescent and young adult twins: A bivariate genetic analysis. Alcoholism: Clinical and Experimental Research. 1997;21:537–546. [PubMed] [Google Scholar]
- Krueger RF, Hicks BM, Patrick CJ, Carlson SR, Iacono WG, McGue M. Etiologic connections among substance dependence, antisocial behavior, and personality: Modeling the externalizing spectrum. Journal of Abnormal Psychology. 2002;111:411–424. [PubMed] [Google Scholar]
- Krueger RF, Markon KE, Patrick CJ, Benning SD, Kramer M. Linking antisocial behavior, substance use, and personality: An integrative quantitative model of the adult externalizing spectrum. Journal of Abnormal Psychology. 2007;116:645–666. doi: 10.1037/0021-843X.116.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman S, Schlosser SS, Kramer JR, Bucholz K, Hesselbrock V, Reich T, et al. Developmental sequence from disruptive behavior diagnosis to adolescent alcohol dependence. American Journal of Psychiatry. 2001;158:2022–2026. doi: 10.1176/appi.ajp.158.12.2022. [DOI] [PubMed] [Google Scholar]
- Kupper NHM, Willemsen G, van den Berg M, de Boer D, Posthuma D, Boomsma DI, et al. Heritability of ambulatory heart rate variability. Circulation. 2004;110:2792–2796. doi: 10.1161/01.CIR.0000146334.96820.6E. [DOI] [PubMed] [Google Scholar]
- Laakso A, Wallius E, Kajander J, Bergman J, Eskola O, Solin O, et al. Personality traits and striatal dopamine synthesis capacity in healthy subjects. American Journal of Psychiatry. 2003;160:904–910. doi: 10.1176/appi.ajp.160.5.904. [DOI] [PubMed] [Google Scholar]
- Lahey BB, Van Hulle CA, Singh AL, Waldman ID, Rathouz PJ. Higher-order genetic and environmental structure of prevalent forms of child and adolescent psychopathology. Archives of General Psychiatry. 2011;68:181–189. doi: 10.1001/archgenpsychiatry.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine TPJ, Ahonen A, Räsänen P, Tiihonen J. Dopamine transporter density and novelty seeking among alcoholics. Journal of Addictive Disease. 2001;20:95–100. doi: 10.1300/j069v20n04_08. [DOI] [PubMed] [Google Scholar]
- Lansford JE, Malone PS, Dodge KA, Pettit GS, Bates JE. Developmental cascades of peer rejection, social information processing biases, and aggression during middle childhood. Development and Psychopathology. 2010;22:593–602. doi: 10.1017/S0954579410000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckman JF, Weissman MM, Merikangas KR, Pauls DL, Prusoff BA. Panic disorder and major depression: Increased risk of depression, alcoholism, panic, and phobic disorders in families of depressed probands with panic disorder. Archives of General Psychiatry. 1983;40:1055–1060. doi: 10.1001/archpsyc.1983.01790090017002. [DOI] [PubMed] [Google Scholar]
- Lenroot RK, Schmitt JE, Ordaz SJ, Wallace GL, Neale MC, Lerch JP, et al. Differences in genetic and environmental influences on the human cerebral cortex associated with development during childhood and adolescence. Human Brain Mapping. 2007;30:163–174. doi: 10.1002/hbm.20494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienfeld SO. Comorbidity between and within childhood externalizing and internalizing disorders: Reflections and directions. Journal of Abnormal Child Psychology. 2003;31:285–291. doi: 10.1023/a:1023229529866. [DOI] [PubMed] [Google Scholar]
- Loeber R, Hay D. Key issues in the development of aggression and violence from childhood to early adulthood. Annual Review of Psychology. 1997;48:371–410. doi: 10.1146/annurev.psych.48.1.371. [DOI] [PubMed] [Google Scholar]
- Loeber R, Keenan K. Interaction between conduct disorder and its comorbid conditions: Effects of age and gender. Clinical Psychology Review. 1994;14:497–523. [Google Scholar]
- Lorber MF, Egeland B. Parenting and infant difficulty: Testing a mutual exacerbation hypothesis to predict early onset conduct problems. Child Development. 2011;82:2006–2020. doi: 10.1111/j.1467-8624.2011.01652.x. [DOI] [PubMed] [Google Scholar]
- Louilot A, LeMoal M, Simon H. Opposite influences of dopaminergic pathways to the prefrontal cortex or the septum on the dopaminergic transmission in the nucleus accumbens: An in vivo voltammetric study. Neuroscience. 1989;29:45–56. doi: 10.1016/0306-4522(89)90331-x. [DOI] [PubMed] [Google Scholar]
- Lynam DR. The early identification of chronic offenders: Who is the fledgling psychopath? Psychological Bulletin. 1996;120:209–234. doi: 10.1037/0033-2909.120.2.209. [DOI] [PubMed] [Google Scholar]
- Lynam DR. Early identification of the fledgling psychopath: Locating the psychopathic child in the current nomenclature. Journal of Abnormal Psychology. 1998;107:566–575. doi: 10.1037//0021-843x.107.4.566. [DOI] [PubMed] [Google Scholar]
- Lynam DR, Caspi A, Moffitt TE, Wikstro¨m PH, Loeber R, Novak S. The interaction between impulsivity and neighborhood context in offending: The effects of impulsivity are stronger in poorer neighborhoods. Journal of Abnormal Psychology. 2000;109:563–574. doi: 10.1037//0021-843x.109.4.563. [DOI] [PubMed] [Google Scholar]
- Martel MN, Nigg JT. Child ADHD and personality/temperament traits of reactive and effortful control, resiliency, and emotionality. Journal of Child Psychology and Psychiatry. 2006;47:1175–1183. doi: 10.1111/j.1469-7610.2006.01629.x. [DOI] [PubMed] [Google Scholar]
- Martin-Soelch C, Leenders KL, Chevalley AF, Missimer J, Kunig S, Magyar A, et al. Reward mechanisms in the brain and their role in dependence: Evidence from neurophysiological and neuroimaging studies. Brain Research Reviews. 2001;36:139–149. doi: 10.1016/s0165-0173(01)00089-3. [DOI] [PubMed] [Google Scholar]
- Masten AS. Developmental psychopathology: Pathways to the future. International Journal of Behavioral Development. 2006;30:47–54. [Google Scholar]
- Matthys W, Vanderschuren LJMJ, Schutter DJLG. The neurobiology of oppositional defiant disorder and conduct disorder: Altered functioning in three mental domains. Development and Psychopathology. 2012;25 doi: 10.1017/S0954579412000272. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Maughan B, Rowe R, Messer J, Goodman R, Meltzer H. Conduct disorder and oppositional defiant disorder in a national sample: Developmental epidemiology. Journal of Child Psychology and Psychiatry. 2004;45:606–621. doi: 10.1111/j.1469-7610.2004.00250.x. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Mathews IZ. Adolescent development, hypothalamic–pituitary–adrenal function, and programming of adult learning and memory. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2010;34:756–765. doi: 10.1016/j.pnpbp.2009.09.019. [DOI] [PubMed] [Google Scholar]
- McGue M, Iacono WG, Legrand LN, Elkins I. Origins and consequences of age at first drink: II. Familial risk and heritability. Alcoholism: Clinical and Experimental Research. 2001;25:1166–1173. [PubMed] [Google Scholar]
- Mead HK, Beauchaine TP, Shannon KE. Neurobiological adaptations to violence across development. Development and Psychopathology. 2010;22:1–22. doi: 10.1017/S0954579409990228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Brake W, Gratton A. Environmental regulation of the development of mesolimbic dopamine systems: A neurobiological mechanism for vulnerability to drug use? Psychoneuroendocrinology. 2002;27:127–138. doi: 10.1016/s0306-4530(01)00040-3. [DOI] [PubMed] [Google Scholar]
- Medici F, Hawa M, Ianari A, Pyke DA, Leslie RD. Concordance rate for type II diabetes mellitus in monozygotic twins: Actuarial analysis. Diabetologia. 1999;42:146–150. doi: 10.1007/s001250051132. [DOI] [PubMed] [Google Scholar]
- Meehl PE. Schizotaxia, schizotypy, schizophrenia. American Psychologist. 1962;17:827–838. [Google Scholar]
- Meehl PE. Bootstraps taxometrics: Solving the classification problem in psychopathology. American Psychologist. 1995;50:266–275. doi: 10.1037//0003-066x.50.4.266. [DOI] [PubMed] [Google Scholar]
- Meier MH, Slutske WS, Arndt S, Cadoret RJ. Impulsive and callous traits are more strongly associated with delinquent behavior in higher risk neighborhoods among boys and girls. Journal of Consulting and Clinical Psychology. 2008;117:377–385. doi: 10.1037/0021-843X.117.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier MH, Slutzke WS, Heath AC, Martin NG. Sex differences in genetic and environmental influences on childhood conduct disorder and adult antisocial behavior. Journal of Abnormal Psychology. 2011;120:377–388. doi: 10.1037/a0022303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chapman JP. Misunderstanding analysis of covariance. Journal of Abnormal Psychology. 2001;110:40–48. doi: 10.1037//0021-843x.110.1.40. [DOI] [PubMed] [Google Scholar]
- Milner PM. Brain stimulation reward: A review. Canadian Journal of Psychology. 1991;45:1–36. doi: 10.1037/h0084275. [DOI] [PubMed] [Google Scholar]
- Minabe Y, Ashby CR, Heyser C, Spear LP, Wang RY. The effects of prenatal cocaine exposure on spontaneously active midbrain dopamine neurons in adult male offspring: An electrophysiological study. Brain Research. 1992;586:152–156. doi: 10.1016/0006-8993(92)91387-t. [DOI] [PubMed] [Google Scholar]
- Moffitt TE. Adolescence-limited and life-course-persistent antisocial behavior: A developmental taxonomy. Psychological Review. 1993;100:674–701. [PubMed] [Google Scholar]
- Monuteaux MC, Biederman J, Doyle AE, Mick E, Faraone SV. Genetic risk for conduct disorder symptom subtypes in an ADHD sample: Specificity to aggressive symptoms. Journal of the American Academy of Child & Adolescent Psychiatry. 2009;48:757–764. doi: 10.1097/CHI.0b013e3181a5661b. [DOI] [PubMed] [Google Scholar]
- MTA Cooperative Group. A 14-month randomized clinical trial of treatment strategies for attention-deficit/hyperactivity disorder. Archives of General Psychiatry. 1999;56:1073–1086. doi: 10.1001/archpsyc.56.12.1073. [DOI] [PubMed] [Google Scholar]
- Muris P, Ollendick TH. The role of temperament in the etiology of child psychopathology. Clinical Child and Family Psychology Review. 2005;8:271–289. doi: 10.1007/s10567-005-8809-y. [DOI] [PubMed] [Google Scholar]
- Myers MG, Stewart DG, Brown SA. Progression from conduct disorder to antisocial personality disorder following treatment for adolescent substance use. American Journal of Psychiatry. 1998;155:479–485. doi: 10.1176/ajp.155.4.479. [DOI] [PubMed] [Google Scholar]
- Neiderhiser JM, Reiss D, Pedersen NL, Lichtenstein P, Spotts EL, Hansson K, et al. Genetic and environmental influences on mothering of adolescents: A comparison of two samples. Developmental Psychology. 2004;40:335–351. doi: 10.1037/0012-1649.40.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus E, Beauchaine TP. Impulsivity and vulnerability to psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 197–226. [Google Scholar]
- Neuman RJ, Lobos E, Reich W, Henderson CA, Sun LW, Todd RD. Prenatal smoking exposure and dopaminergic genotypes interact to cause a severe ADHD subtype. Biological Psychiatry. 2007;61:1320–1328. doi: 10.1016/j.biopsych.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Oberlin BG, Dzemidzic M, Bragulat V, Lehigh CA, Talavage T, O'Connor SJ, et al. Limbic responses to reward cues correlate with antisocial trait density in heavy drinkers. NeuroImage. 2012;60:644–652. doi: 10.1016/j.neuroimage.2011.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor TG, Deater-Deckard K, Fulker D, Rutter M, Plomin R. Genotype–environment correlations in late childhood and adolescence: Antisocial behavior problems and coercive parenting. Developmental Psychology. 1998;34:970–981. doi: 10.1037//0012-1649.34.5.970. [DOI] [PubMed] [Google Scholar]
- Oswald LM, Wong DF, McCaul M, Zhou Y, Kuwabara H, Choi L, et al. Relationships among ventral striatal dopamine release, cortisol secretion, and subjective responses to amphetamine. Neuropsychopharmacology. 2005;30:821–832. doi: 10.1038/sj.npp.1300667. [DOI] [PubMed] [Google Scholar]
- Pardini D. Novel insights into longstanding theories of bidirectional parent–child influences: Introduction to the Special Section. Journal of Abnormal Child Psychology. 2008;36:627–631. doi: 10.1007/s10802-008-9231-y. [DOI] [PubMed] [Google Scholar]
- Patrick CJ, Hicks BM, Krueger RF, Lang AR. Relations between psychopathy facets and externalizing in a criminal offender sample. Journal of Personality Disorders. 2005;19:339–356. doi: 10.1521/pedi.2005.19.4.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GR. Coercive family process. Eugene, OR: Castalia; 1982. [Google Scholar]
- Patterson GR, Chamberlain P, Reid JB. A comparative evaluation of parent training procedures. Behavior Therapy. 1982;13:638–650. doi: 10.1016/j.beth.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Patterson GR, DeBaryshe BD, Ramsey E. A developmental perspective on antisocial behavior. American Psychologist. 1989;44:329–335. doi: 10.1037//0003-066x.44.2.329. [DOI] [PubMed] [Google Scholar]
- Patterson GR, DeGarmo DS, Knutson NM. Hyperactive and antisocial behaviors: Comorbid or two points in the same process? Development and Psychopathology. 2000;12:91–107. doi: 10.1017/s0954579400001061. [DOI] [PubMed] [Google Scholar]
- Patterson GR, Dishion TJ, Bank L. Family interaction: A process model of deviancy training. Aggressive Behavior. 1984;10:253–267. [Google Scholar]
- Pedhazur E. Multiple regression in behavioral research. 3rd. New York: Harcourt Brace; 1997. [Google Scholar]
- Perry JL, Joseph JE, Jiang Y, Zimmerman RS, Kelly TH, Darna M, et al. Prefrontal cortex and drug abuse vulnerability: Translation to prevention and treatment interventions. Brain Research Reviews. 2011;65:124–149. doi: 10.1016/j.brainresrev.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learning and Memory. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharo H, Sim C, Graham M, Gross J, Hayne H. Risky business: Executive function, personality, and reckless behavior during adolescence and emerging adulthood. Behavioral Neuroscience. 2011;125:970–978. doi: 10.1037/a0025768. [DOI] [PubMed] [Google Scholar]
- Phillips AG, Blaha CD, Fibiger HC. Neurochemical correlates of brain-stimulation reward measured by ex vivo and in vivo analyses. Neuroscience & Biobehavioral Reviews. 1989;13:99–104. doi: 10.1016/s0149-7634(89)80017-x. [DOI] [PubMed] [Google Scholar]
- Phillips KA, Stein DJ, Rauch SL, Hollander E, Fallon BA, Barsky A, et al. Should an obsessive–compulsive spectrum grouping of disorders be included in DSM-V? Depression and Anxiety. 2010;27:528–555. doi: 10.1002/da.20705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PE, Walton ME, Jhou TC. Calculating utility: Pre-clinical evidence for cost-benefit analysis by mesolimbic dopamine. Psychopharmacology. 2007;191:483–495. doi: 10.1007/s00213-006-0626-6. [DOI] [PubMed] [Google Scholar]
- Pollak SD. Mechanisms linking early experience and the emergence of emotions: Illustrations from the study of maltreated children. Current Directions in Psychological Science. 2008;17:370–375. doi: 10.1111/j.1467-8721.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak SD. Early social experience and the ontogenesis of emotion regulatory behavior in children. Developments in Primatology: Progress and Prospects. 2011;36:333–341. [Google Scholar]
- Popper KR. The aim of science. In: Miller D, editor. Popper selections. Princeton, NJ: Princeton University Press; 1985. pp. 162–170. Original work published 1957. [Google Scholar]
- Porteus SD. Porteus maze tests: Fifty years application. Palo Alto, CA: Pacific Books; 1965. [Google Scholar]
- Preskorn SH, Baker B. The overlap of DSM-IV syndromes: Potential implications for the practice of polypsychopharmacology, psychiatric drug development, and the human genome project. Journal of Psychiatric Practice. 2002;8:170–177. doi: 10.1097/00131746-200205000-00006. [DOI] [PubMed] [Google Scholar]
- Quay HC. The psychobiology of undersocialized aggressive conduct disorder: A theoretical perspective. Development and Psychopathology. 1993;5:165–180. [Google Scholar]
- Raudino A, Fergusson DM, Woodward LJ, Horwood LJ. The intergenerational transmission of conduct problems. Social Psychiatry and Psychiatric Epidemiology. 2012 doi: 10.1007/s00127-012-0547-0. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Richters JE, Cicchetti D. Toward a developmental perspective on conduct disorder. Development and Psychopathology. 1993;5:1–4. [Google Scholar]
- Riggs AD, Russo VEA, Martienssen RA. Epigenetic mechanisms of gene regulation. Plainview, NY: Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- Robins LN. Deviant children grown up. Baltimore, MD: Williams & Wilkins; 1966. [Google Scholar]
- Rolls ET, Rolls BJ, Kelly PH, Shaw SG, Wood RJ, Dale R. The relative attenuation of self-stimulation, eating, and drinking produced by dopamine receptor blockade. Psychopharmacologia. 1974;38:219–230. doi: 10.1007/BF00421374. [DOI] [PubMed] [Google Scholar]
- Rubia K. “Cool” inferior frontostriatal dysfunction in attention-deficit/hyperactivity disorder versus “hot” vetromedial orbitofrontal-limbic dysfunction in conduct disorder: A review. Biological Psychiatry. 2011;69:e69–e87. doi: 10.1016/j.biopsych.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Rubia K, Halari R, Cubillo A, Mohammad M, Taylor E. Methylphenidate normalises activation and functional connectivity deficits in attention and motivation networks in medication-na¨ýve children with ADHD during a rewarded continuous performance task. Neuropharmacology. 2009;57:640–652. doi: 10.1016/j.neuropharm.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Rubia K, Halari R, Mohammad M, Taylor E, Brammer M. Methylphenidate normalizes frontocingulate underactivation during error processing in attention-deficit/hyperactivity disorder. Biological Psychiatry. 2011;70:255–262. doi: 10.1016/j.biopsych.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubia K, Smith A, Halari R, Matukura F, Mohammad M, Taylor E, et al. Disorder-specific dissociation of orbitofrontal dysfunction in boys with pure conduct disorder during reward and ventrolateral prefrontal dysfunction in boys with pure attention-deficit/hyperactivity disorder during sustained attention. American Journal of Psychiatry. 2009;166:83–94. doi: 10.1176/appi.ajp.2008.08020212. [DOI] [PubMed] [Google Scholar]
- Rutter M. Genes and behavior: Nature–nurture interplay explained. Oxford: Blackwell; 2006. [Google Scholar]
- Rutter M. Annual research review: Resilience: Clinical implications. Journal of Child Psychology and Psychiatry. 2012 doi: 10.1111/j.1469-7610.2012.02615.x. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Rutter M, Moffitt TE, Caspi A. Gene–environment interplay and psychopathology: Multiple varieties but real effects. Journal of Child Psychology and Psychiatry. 2006;47:226–261. doi: 10.1111/j.1469-7610.2005.01557.x. [DOI] [PubMed] [Google Scholar]
- Rutter M, Sroufe LA. Developmental psychopathology: Concepts and challenges. Development and Psychopathology. 2000;12:265–296. doi: 10.1017/s0954579400003023. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Johansen EB, Aase H, Russell VA. A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behavioral and Brain Sciences. 2005;28:397–468. doi: 10.1017/S0140525X05000075. [DOI] [PubMed] [Google Scholar]
- Sanislow CA, Pine DS, Quinn KJ, Kozak MJ, Garvey MA, Heinssen RK, et al. Developing constructs for psychopathology research: Research domain criteria. Journal of Abnormal Psychology. 2010;119:631–639. doi: 10.1037/a0020909. [DOI] [PubMed] [Google Scholar]
- Saudino LJ. The development of temperament from a behavioral genetics perspective. Advances in Child Development and Behavior. 2009;37:201–231. doi: 10.1016/s0065-2407(09)03705-7. [DOI] [PubMed] [Google Scholar]
- Scheres A, Milham MP, Knutson B, Castellanos FX. Ventral striatal hyporesponsiveness during reward anticipation in attention-deficit/hyperactivity disorder. Biological Psychiatry. 2007;61:720–724. doi: 10.1016/j.biopsych.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Schmidt LA, Fox NA, Perez-Edgar K, Hamer DH. Linking gene, brain, and behavior: DRD4, frontal asymmetry, and temperament. Psychological Science. 2009;20:831–837. doi: 10.1111/j.1467-9280.2009.02374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbauma G, Shahamd Y. The role of orbitofrontal cortex in drug addiction: A review of preclinical studies. Biological Psychiatry. 2008;63:256–262. doi: 10.1016/j.biopsych.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott BH, Minuzzi L, Krebs RM, Elmenhorst E, Lang M, Winz OH, et al. Mesolimbic functional magnetic resonance imaging activations during reward anticipation correlate with reward-related ventral striatal dopamine release. Journal of Neuroscience. 2008;28:14311–14319. doi: 10.1523/JNEUROSCI.2058-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon KE, Sauder C, Beauchaine TP, Gatzke-Kopp L. Disrupted effective connectivity between the medial frontal cortex and the caudate in adolescent boys with externalizing behavior disorders. Criminal Justice and Behavior. 2009;36:1141–1157. [Google Scholar]
- Shannon Bowen KE, Gatzke-Kopp LM. Brain injury as a risk factor for psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 317–340. [Google Scholar]
- Slotkin TA. Fetal nicotine or cocaine exposure: Which one is worse? Journal of Pharmacology and Experimental Therapeutics. 1998;285:931–945. [PubMed] [Google Scholar]
- Sneider H, Boomsma DI, van Doornen LJP, DeGeus EJC. Heritability of respiratory sinus arrhythmia: Dependency on task and respiration rate. Psychophysiology. 1997;34:317–328. doi: 10.1111/j.1469-8986.1997.tb02402.x. [DOI] [PubMed] [Google Scholar]
- Snyder J, Edwards P, McGraw K, Kilgore K, Holton A. Escalation and reinforcement in mother–child conflict: Social processes associated with the development of physical aggression. Developmental and Psychopathology. 1994;6:305–321. [Google Scholar]
- Snyder J, Schrepferman L, McEachern A, Barner S, Johnson K, Provines J. Peer deviancy training and peer coercion: Dual processes associated with early-onset conduct problems. Child Development. 2008;79:252–268. doi: 10.1111/j.1467-8624.2007.01124.x. [DOI] [PubMed] [Google Scholar]
- Snyder J, Schrepferman L, Oeser J, Patterson G, Stoolmiller M, Johnson K, et al. Deviancy training and association with deviant peers in young children: Occurrence and contribution to early-onset conduct problems. Development and Psychopathology. 2005;17:397–413. doi: 10.1017/s0954579405050194. [DOI] [PubMed] [Google Scholar]
- Snyder J, Schrepferman L, St Peter C. Origins of antisocial behavior: Negative reinforcement and affect dysregulation of behavior as socialization mechanisms in family interaction. Behavior Modification. 1997;21:187–215. doi: 10.1177/01454455970212004. [DOI] [PubMed] [Google Scholar]
- Spear LP. Assessment of adolescent neurotoxicity: Rationale and methodological considerations. Neurotoxicology and Teratology. 2007;29:1–9. doi: 10.1016/j.ntt.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroufe LA. Psychopathology as an outcome of development. Development and Psychopathology. 1997;9:251–268. doi: 10.1017/s0954579497002046. [DOI] [PubMed] [Google Scholar]
- Sroufe LA. The concept of development in developmental psychopathology. Child Development Perspectives. 2009;3:178–183. doi: 10.1111/j.1750-8606.2009.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroufe LA, Rutter M. The domain of developmental psychopathology. Child Development. 1984;55:17–29. [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Shumsky JS, Levitt P. Prenatal cocaine exposure produces consistent developmental alterations in dopamine-rich regions of the cerebral cortex. Neuroscience. 2001;106:5–14. doi: 10.1016/s0306-4522(01)00256-1. [DOI] [PubMed] [Google Scholar]
- Stein DJ, Fineberg NA, Bienvenu OJ, Denys D, Lochner C, Nestadt G, et al. Should OCD be classified as an anxiety disorder in DSM-V? Depression and Anxiety. 2010;27:495–506. doi: 10.1002/da.20699. [DOI] [PubMed] [Google Scholar]
- Sterling P, Eyer J. Allostasis: A new paradigm to explain arousal pathology. In: Fisher S, Reason J, editors. Handbook of life stress, cognition and health. New York: Wiley; 1988. pp. 629–649. [Google Scholar]
- Sterzera P, Stadlerb C, Poustkab F, Kleinschmidta A. A structural neural deficit in adolescents with conduct disorder and its association with lack of empathy. NeuroImage. 2007;37:335–342. doi: 10.1016/j.neuroimage.2007.04.043. [DOI] [PubMed] [Google Scholar]
- Stringaris A, Maughan B, Goodman R. What's in a disruptive disorder? Temperamental antecedents of oppositional defiant disorder: Findings from the Avon Longitudinal Study. Journal of the American Academy of Child & Adolescent Psychiatry. 2010;49:474–483. doi: 10.1097/00004583-201005000-00008. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Brake WG. What the rodent prefrontal cortex can teach us about attention-deficit/hyperactivity disorder: The critical role of early developmental events on prefrontal function. Behavior and Brain Research. 2003;146:43–55. doi: 10.1016/j.bbr.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Swartz JR. Dopamine projections and frontal systems function. In: Miller BL, Cummings JL, editors. The human frontal lobes: Functions and disorders. New York: Guilford Press; 1999. pp. 159–173. [Google Scholar]
- Tackett JL. Toward an externalizing spectrum in DSM-V: Incorporating developmental concerns. Child Development Perspectives. 2010;4:161–167. [Google Scholar]
- Thayer J, Hansen AL, Saus-Rose E, Johnsen BH. Heart rate variability, prefrontal neural function, and cognitive performance: The neurovisceral integration perspective on self-regulation, adaptation, and health. Annals of Behavioral Medicine. 2009;37:141–153. doi: 10.1007/s12160-009-9101-z. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: A neural correlate of behavioral sensitization to cocaine. Nature Neuroscience. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Thorell LB, Waÿhlstedt C. Executive functioning deficits in relation to symptoms of ADHD and/or ODD in preschool children. Infant and Child Development. 2006;15:503–518. [Google Scholar]
- Tisch S, Silberstein P, Limousin-Dowsey P, Jahanshahi M. The basal ganglia: Anatomy, physiology, and pharmacology. Psychiatric Clinics of North America. 2004;27:757–759. doi: 10.1016/j.psc.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Tuvblad C, Zheng M, Raine A, Baker LA. A common genetic factor explains the covariation among ADHD, ODD, and CD symptoms in 9–10-year-old boys and girls. Journal of Abnormal Child Psychology. 2009;37:153–167. doi: 10.1007/s10802-008-9278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harmelen AL, van Tol MJ, Demenescu LR, van der Wee NJA, Veltman DJ, Aleman A, et al. Enhanced amygdala reactivity to emotional faces in adults reporting childhood emotional maltreatment. Social Cognitive and Affective Neuroscience. 2013;8:362–369. doi: 10.1093/scan/nss007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P. Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neuroscience & Biobehavioral Reviews. 2004;27:827–839. doi: 10.1016/j.neubiorev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Viken RJ, Kaprio J, Koskenvuo M, Rose RJ. Longitudinal analyses of the determinants of drinking and of drinking to intoxication in adolescent twins. Behavior Genetics. 1999;29:455–461. doi: 10.1023/a:1021631122461. [DOI] [PubMed] [Google Scholar]
- Vles J, Feron F, Hendriksen J, Jolles J, van Kroonenburgh M, Weber W. Methylphenidate down-regulates the dopamine receptor and transporter system in children with attention deficit hyperkinetic disorder. Neuropediatrics. 2003;34:77–80. doi: 10.1055/s-2003-39602. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. The addicted human brain viewed in light of imaging studies: Brain circuits and treatment strategies. Neuropharmacology. 2004;47:3–13. doi: 10.1016/j.neuropharm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine's role in drug abuse and addiction. Neuropharmacology. 2009;56(Suppl. 1):3–8. doi: 10.1016/j.neuropharm.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang G, Ding Y, Gatley SJ. Mechanism of action of methylphenidate: Insights from PET imaging studies. Journal of Attention Disorders. 2002;6:S31–S43. doi: 10.1177/070674370200601s05. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Kollins SH, Wigal TL, Newcorn JH, Telang F, et al. Evaluating dopamine reward pathway in ADHD: Clinical implications. Journal of the American Medical Association. 2009;302:1084–1091. doi: 10.1001/jama.2009.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakschlag LS, Lahey BB, Loeber R, Green SM, Gordon RA, Leventhal BL. Maternal smoking during pregnancy and the risk of conduct disorder in boys. Archives of General Psychiatry. 1997;54:670–676. doi: 10.1001/archpsyc.1997.01830190098010. [DOI] [PubMed] [Google Scholar]
- Waldman ID, Lahey BB. Oppositional defiant disorder, conduct disorder, and juvenile delinquency. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. 2nd. Hoboken, NJ: Wiley; 2013. pp. 411–452. [Google Scholar]
- Waller NG, Meehl PE. Multivariate taxometric procedures. Thousand Oaks, CA: Sage; 1998. [Google Scholar]
- Webster-Stratton C, Reid MJ, Beauchaine TP. Combining parent and child training for young children with attention-deficit/hyperactivity disorder. Journal of Clinical Child and Adolescent Psychology. 2011;40:191–203. doi: 10.1080/15374416.2011.546044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh MC, Pennington BF, Groisser DB. A normative-developmental study of executive function: A window on prefrontal function in children. Developmental Neuropsychology. 1991;7:131–149. [Google Scholar]
- Willcutt EG, Doyle AE, Nigg JT, Faraone SV, Pennington BF. Validity of the executive function theory of attention-deficit/hyperactivity disorder: A meta-analytic review. Biological Psychiatry. 2005;57:1336–1346. doi: 10.1016/j.biopsych.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Zepf FD, Holtmann M, Stadler C, Demisch L, Schmitt M, Wöckel L, et al. Diminished serotonergic functioning in hostile children with ADHD: Tryptophan depletion increases behavioural inhibition. Pharmacopsychiatry. 2008;41:60–65. doi: 10.1055/s-2007-1004593. [DOI] [PubMed] [Google Scholar]