

Abstract
Interleukin (IL)-17A is increased both in serum and in kidney biopsies from patients with lupus nephritis, but direct evidence of pathogenicity is less well established. Administration of pristane to genetically intact mice results in the production of autoantibodies and proliferative glomerulonephritis, resembling human lupus nephritis. These studies sought to define the role of IL-17A in experimental lupus induced by pristane administration. Pristane was administered to wild-type (WT) and IL-17A−/− mice. Local and systemic immune responses were assessed after 6 days and 8 weeks, and autoimmunity, glomerular inflammation and renal injury were measured at 7 months. IL-17A production increased significantly 6 days after pristane injection, with innate immune cells, neutrophils (Ly6G+) and macrophages (F4/80+) being the predominant source of IL-17A. After 8 weeks, while systemic IL-17A was still readily detected in WT mice, the levels of proinflammatory cytokines, interferon (IFN)-γ and tumour necrosis factor (TNF) were diminished in the absence of endogenous IL-17A. Seven months after pristane treatment humoral autoimmunity was diminished in the absence of IL-17A, with decreased levels of immunoglobulin (Ig)G and anti-dsDNA antibodies. Renal inflammation and injury was less in the absence of IL-17A. Compared to WT mice, glomerular IgG, complement deposition, glomerular CD4+ T cells and intrarenal expression of T helper type 1 (Th1)-associated proinflammatory mediators were decreased in IL-17A−/− mice. WT mice developed progressive proteinuria, but functional and histological renal injury was attenuated in the absence of IL-17A. Therefore, IL-17A is required for the full development of autoimmunity and lupus nephritis in experimental SLE, and early in the development of autoimmunity, innate immune cells produce IL-17A.
Keywords: glomerulonephritis, interleukin 17A, lupus nephritis, pristane, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is an important autoimmune disease. While SLE has the propensity to involve multiple organs, kidney and central nervous system involvement are associated independently with increased mortality [1]. The production of characteristic autoantibodies is a hallmark of disease, and these autoantibodies induce immune complex deposition and organ injury [2]. Cellular immunity is also involved, and for many years T cells have been implicated in the development of SLE both experimentally [3] and clinically [4]. More recently it has been recognized that dysregulated innate immunity is important in the induction of inflammatory responses and the subsequent development of autoimmunity with organ injury [5].
The pathogenesis of SLE and lupus nephritis involves both genetic predisposition and environmental precipitants. Murine models used to study SLE exploit these factors and include genetically lupus prone mice and C57BL/6 wild-type (WT) mice, which lack genetic susceptibility but develop autoimmunity when administered the hydrocarbon oil, pristane. Administration of pristane to WT mice results in the development of lupus-like humoral and cellular autoimmunity and renal injury, with considerable homology with the human condition [6]. Injury is mediated by humoral autoimmunity, including anti-dsDNA antibodies; however, cellular immunity is also involved. Compared to mice with intact cellular immunity, mice deficient in T cells or mice lacking interferon (IFN)-γ and interleukin (IL)-12p35 (key CD4+ Th1-promoting cytokines) demonstrate significant renal protection [7–9].
IL-17A is produced by both innate immune cells and CD4+ T cells [T helper type 17 (Th17) cells], and promotes autoimmunity, organ inflammation and injury in a number of experimental models [10,11]. Clinical and experimental evidence supports a role for IL-17A in human and experimental lupus nephritis. Results from analyses of human kidney biopsies demonstrated that IL-17A+ cells correlate with clinical disease [12]. Functional roles for IL-17A and IL-23 have been demonstrated in experimental models using genetically prone lupus mice [13,14]. While the potential role of IL-17A and Th17 cells has not been studied in pristine-induced lupus nephritis, we have previously demonstrated increased IL-17A production in C57BL/6 mice which develop lupus nephritis after pristane administration [15]. Others have shown that IL-6, a pleiotropic cytokine involved in the development of Th17 responses, is required for the development of anti-dsDNA antibodies and renal injury in this model [16].
While early focus has been on the role of Th17 cells in driving inflammation, it is recognized increasingly that innate immune cells produce significant amounts of IL-17A and help to direct inflammatory responses and tissue injury [17]. In murine models of infection, early IL-17A production is required for the development of protective Th1 immunity [18]. Previously we have shown that in experimental glomerulonephritis early injurious responses are mediated by IL-17A, while Th1 IFN-γ-producing cells promote the later injury [19,20]. In the current study, we demonstrate that endogenous IL-17A is pathogenic in pristine-induced lupus and lupus nephritis. Pristane-injected C57BL/6 WT mice produce IL-17A early in the course of disease, coming predominantly from innate cells, implicating IL-17A in the subsequent development of autoimmunity, antibody production, renal inflammation and glomerulonephritis.
Materials and methods
Mice and experimental design
WT and IL-17A−/− mice, back-crossed onto a C57BL/6J background and bred at Monash Medical Centre (Melbourne, Australia) as described previously [20], were used for experiments. Studies adhered to the National Health and Medical Research Council of Australia guidelines for animal experimentation. Immune responses and/or renal injury was assessed at three different time-points during the course of disease, using age-and gender-matched, female, WT and IL-17A−/− mice. At 8–10 weeks of age WT and IL-17A−/− mice were injected with 500 μl of pristane (2, 6, 10, 14-tetramethylpentadecane; Sigma-Aldrich, St Louis, MO, USA). We assessed immune responses at an early time-point, 6 days after pristane injection in WT mice and control (untreated) WT mice and at 8 weeks after pristane treatment in WT and IL-17A−/− mice. Renal injury in WT and IL-17A−/− mice was assessed 7 months after pristane injection. Data are expressed as mean ± standard error of the mean (s.e.m.). Groups of data were analysed using an unpaired Student's t-test; when more than two groups were compared, analysis of variance (anova) (GraphPad Software, San Diego, CA, USA) was used. A P-value of <0·05 was considered statistically significant.
Flow cytometric analyses 6 days after pristane administration
Peritoneal cells were isolated 6 days after administration of pristane by lavage and cultured at 1 × 106/ml for 24 h in 10% RPMI-1640 with brefeldin-A (5 ug/ml; Sigma) added for the last 6 h. Cells were first surface-stained using anti-CD11b-allophycocyanin (APC)/cyanin 7 (Cy7) (Biolegend, San Diego, CA, USA), anti-lymphocyte antigen 6G (Ly-6G)-phycoerythrin (PE) (BD Pharmingen, San Diego, CA, USA), anti-F4/80-Alexa647 (in-house), anti-γδT cell receptor (TCR)-APC (eBioscience, San Diego, CA, USA), anti-natural killer (NK)1·1-APC/Cy7 (BD Pharmingen), anti-CD4-PE (BD Pharmingen) and anti-B220-PE (BD Pharmingen), fixed and permeabilized using a fixation/permeabilization kit (BD Pharmingen), then stained intracellularly for IL-17A using fluorescein isothiocyanate (FITC)-anti-IL-17A (eBioscience). Flow cytometric analysis was performed on a MoFlo flow cytometer (Dako, Botany, NSW, Australia). Fluorescence minus one controls were used to ensure specificity in data analysis; doublets were excluded by forward-and side-scatter profile.
Measurement of splenic cytokine production
To assess systemic cytokines after 8 weeks, spleens were removed, a single-cell suspension was obtained and splenocytes (4 × 106 cells/ml per well) were cultured with RPMI-1640/10% fetal calf serum (FCS) at 37°C alone, or with a pure Toll-like receptor (TLR)-4 ligand, highly purified lipopolysaccharide (LPS), (1 μg/ml; Sigma), purified as described previously [21] and a TLR-2 ligand, Pam3-Cys-Ser-Lys4 (Pam3CSK4) (10 μg/ml; Invivogen, San Diego, CA, USA). Both these TLRs are relevant to experimental SLE and to the induction of IL-17A-mediated responses [15,22,23]. Cytokine enzyme-linked immunosorbent assays (ELISAs) were performed on Nunc Maxisorb plates (Nunc, Roskilde, Denmark), as described previously [19,24,25]. Splenocytes from normal mice that did not receive pristane or TLR ligands were also tested to provide baseline levels. The limit of detection for each assay was: IL-17A 7·8 pg/ml, IFN-γ 15·6 pg/ml, TNF 3·9 pg/ml, IL-2 and IL-5 7·8 pg/ml.
Measurement of autoantibodies by ELISA
Total immunoglobulin (Ig)G was detected by incubating ELISA plates with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (κ-chain specific; Southern Biotechnology Associates, Birmingham, AL, USA), as described previously [15]. For the detection of anti-dsDNA antibodies and anti-ribonucleoprotein (RNP) antibodies pre-coated ELISA plates from Alpha Diagnostic International (San Antonio, TX, USA) [26] were used, developed with tetramethylbenzidine (Sigma), and optical density (OD) was read at 450 nm. Results are expressed as OD450 ± s.e.m.
Assessment of functional and histological renal injury
Urinary collections (24 h) were collected at 0, 3 and 5 months and immediately prior to the end of experiments. Albuminuria was measured on 24-h urine collections using a Mouse Albumin ELISA Quantification Kit (Bethyl Laboratories, Montgomery, TX, USA). Glomerular abnormalities were assessed on periodic acid Schiff reagent (PAS)-stained, 3-μm-thick, paraffin-embedded sections on coded slides. The percentage of abnormal glomeruli was determined by scoring ≥50 consecutive glomeruli per mouse for abnormalities according to previously published protocols, using coded slides [15,19]. Abnormalities included glomerular hypercellularity, crescent formation, fibrinoid necrosis, segmental proliferation, hyalinosis and capillary wall thickening.
Glomerular IgG and C3 deposition, leucocyte accumulation and intrarenal cytokine mRNA expression
Glomerular IgG and complement C3 deposition was assessed on 6-μm-thick, frozen sections using FITC-sheep anti-mouse Ig (1 : 100; Silenus, Hawthorn, Victoria, Australia) or goat anti-mouse C3 (1 : 100; Cappell, West Chester, PA, USA). Scores were assigned based on the intensity of IgG/C3 deposition (0–3+), where 0 represents no deposition and 3 denotes intense deposition, as published previously [15,25]. To assess glomerular leucocyte accumulation we used a three-layered immunoperoxidase technique after fixing kidney sections in periodate-lysine-paraformaldehyde. The technique and antibodies were as published previously [19,22]. Results are expressed as cells per glomerular cross-section (c/gcs) after assessing at least 20 glomeruli per mouse.
For measurement of T-bet, retinoic acid-related orphan receptor gamma t (RORγt), GATA-binding protein 3 (GATA-3), forkhead box protein 3 (FoxP3), IFN-γ and CXCL11, RNA was extracted from whole kidney and measured by quantitative polymerase chain reaction (qPCR), as described previously [25,27]. Primer sequences used were as described previously, while expression was standardized to 18S (house-keeping gene) before being expressed as a fold change relative to WT mice treated with pristane.
Results
Innate immune cells produce IL-17A early after pristane treatment
We injected pristane intraperitoneally to WT mice. Six days later experiments ended and peritoneal cells were assessed for IL-17A production by flow cytometry. Treatment with pristane resulted in an increase in the number of peritoneal cells (Fig. 1a) and IL-17A-producing peritoneal cells (Fig. 1b) compared to untreated WT mice not injected with pristane. In pristine-treated mice we assessed the source of IL-17A production. Innate immune cells, including macrophages (F4/80+) and neutrophils (Ly6G+), were the main producers of IL-17A (Fig. 1c). Representative fluorescence activated cell sorter (FACS) plots of peritoneal cells isolated from pristine-treated mice, stained for IL-17A and macrophages and neutrophils (Fig. 1d) are shown. These results demonstrate that pristane treatment induces a local inflammatory infiltrate which produced IL-17A, predominantly from innate immune cells.
Figure 1.
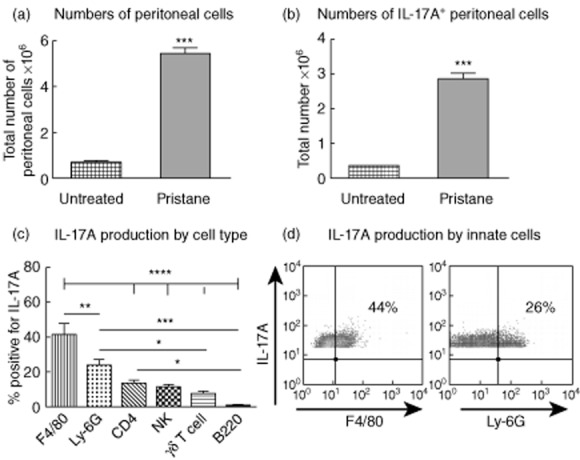
Innate cells are major producers of interleukin (IL)-17A, 6 days after pristane treatment. (a) Compared to ‘untreated’ C57BL/6 wild-type (WT) mice that did not receive pristane (n = 6), administration of pristane to WT mice (n = 8) resulted in an increased number of intraperitoneal cells. (b) In addition, the number of cells within the peritoneum producing IL-17A increased after pristane treatment. Cells from untreated mice were pooled to ensure adequate numbers for analysis. (c) After pristane treatment, the majority of IL-17A-producing cells in the peritoneum were either CD11b+F4/80+ macrophages or CD11b+ lymphocyte antigen 6G (Ly6G+) neutrophils. CD4+ T cells, natural killer T cells and γδ T cells also produced IL-17A. (d) Representative fluorescence activated cell sorter (FACS) plots showing the proportion of peritoneal cells positive for IL-17A and F4/80+ or IL-17A and Ly-6G+ cells after pristane injection. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001.
Systemic immunity in WT and IL-17A−/− mice 8 weeks after pristane treatment
We treated WT and IL-17A−/− mice with pristane and assessed cytokine production from splenocytes at 8 weeks. Spleens were removed and cultured with media alone or with an additional TLR-4 or TLR-2 ligand to assess immune responses when stimulated. Production of IL-17A, the prototypic Th17 cytokine, was measured in untreated WT mice and WT mice receiving pristane. While IL-17A production in splenocytes from mice that did not receive pristane (n = 5) was present only at low levels in some mice, splenocyte IL-17A production was detected readily in mice treated with pristane (Fig. 2a). Compared to splenocytes from WT mice given pristane cultured without ex-vivo stimulation, IL-17A production increased when splenocytes were cultured with either a TLR-4 or a TLR-2 ligand.
Figure 2.
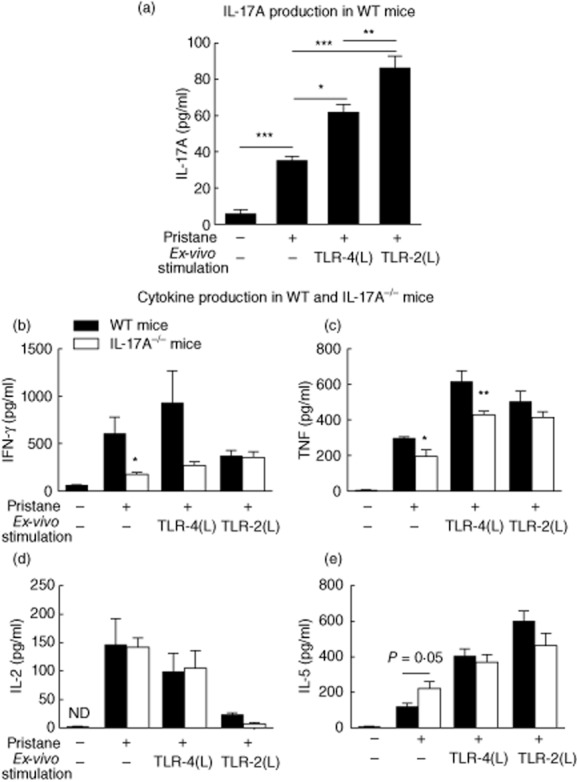
Systemic (spleen) cytokine production in wild-type (WT) and interleukin (IL)-17A−/− mice, 8 weeks after pristane administration. Eight weeks after pristane injection we isolated and cultured splenocytes in media alone, with a Toll-like receptor (TLR)-4 ligand [highly purified lipopolysaccharide (LPS) 1 μg/ml] or a TLR-2 ligand, Pam3CSK4 (10 μg/ml) and measured cytokine production. Cultured splenocytes from pristane injected WT mice exhibited greater production of all cytokines compared to normal WT mice not given pristane (n = 5). (a) Systemic IL-17A, detected only at low levels in splenocytes cultures of WT mice not given pristane, was increased in pristine-treated WT mice (n = 6), and production increased further after culture with a TLR-4 or TLR-2 ligand. (b) Production of interferon (IFN)-γ and (c) tumour necrosis factor (TNF) were increased in WT mice compared to IL-17A−/− mice (n = 7) treated with pristane. (d) There was no difference in IL-2 production between WT and IL-17A−/− mice treated with pristane and (e) a trend towards increased IL-5 production was observed in IL-17A−/− mice. *P < 0·05; **P < 0·01; ***P < 0·001; ND = not detected.
Production of key cytokines by splenocytes was measured after 8 weeks. In WT mice, pristane treatment resulted in increased production of all cytokines. Production of IFN-γ, a Th1-associated cytokine, was increased in unstimulated splenocytes from pristine-treated WT mice compared to unstimulated IL-17A−/− splenocytes (Fig. 2b). TNF was detected readily in unstimulated splenocytes from pristine-treated WT mice and levels were diminished in IL-17A−/− mice (Fig. 2c). Compared to levels detectable in unstimulated splenocytes from pristine-treated mice, levels of IFN-γ (P = 0·07) and TNF (P < 0·001) increased after culture with a TLR-4 ligand in WT mice. IL-2 production from splenocytes isolated from WT or IL-17A−/− mice was similar (Fig. 2d), and there was a trend towards increased IL-5 (a Th2-related cytokine) in splenocytes from IL-17A−/− mice compared to WT mice (Fig. 2e). These results showed that IL-17A is required for full production of IFN-γ and TNF, two key proinflammatory cytokines, 8 weeks after pristane treatment.
Humoral autoimmunity is diminished in IL-17A−/− mice 7 months after pristane treatment
The development of heightened immune responses with hypergammaglobulinaemia and autoantibody production forms the hallmark of experimental and clinical SLE. We measured titres of total IgG and autoantibodies characteristically associated with SLE 7 months after pristane treatment. Compared to WT mice, significantly lower levels of IgG were detected in IL-17A−/− mice at serial dilutions (Fig. 3a). Subsequently we assessed autoantibody levels directed against dsDNA and RNP. Anti-dsDNA levels were detected readily in WT and IL-17A−/− mice, although the autoantibody levels were lower in IL-17A−/− mice (Fig. 3b). There was no difference in antibodies directed against RNP in WT and IL-17A−/− mice (data not shown). These results demonstrate that IL-17A enhanced the development of humoral autoimmunity after pristane treatment.
Figure 3.
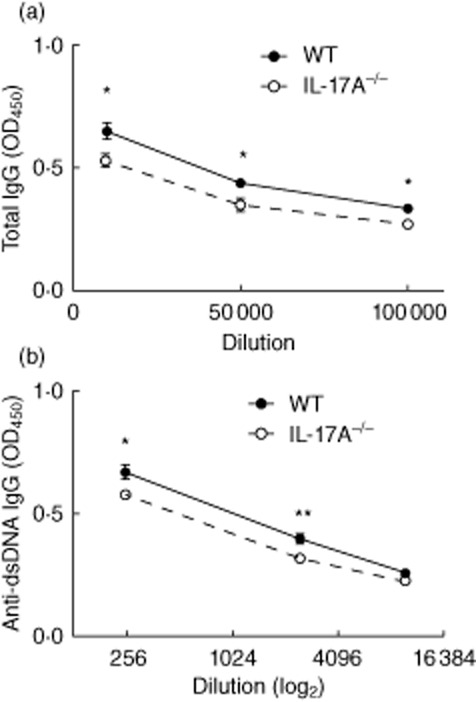
Humoral autoimmunity is decreased in the absence of interleukin (IL)-17A. (a) Seven months after pristane treatment, levels of total immunoglobulin (Ig)G were decreased in IL-17A−/− mice (n = 7) compared to wild-type (WT) mice (n = 9) treated with pristane. (b) Similarly, titres of anti-dsDNA antibodies were decreased in IL-17A−/− mice. *P < 0·05; **P < 0·01.
Glomerular immunoglobulin and C3 deposition is decreased in IL-17A−/− mice
Kidney sections from WT and IL-17A−/− mice were stained for IgG and complement C3 deposition 7 months after pristane administration. IgG was deposited in a granular pattern in WT and IL-17A−/− mice treated with pristane, but was not detectable in age-matched control mice. Compared to the amount of glomerular IgG seen in WT mice, less IgG was detected in glomeruli IL-17A−/− mice (Fig. 4a). Representative glomerular sections from WT and IL-17A−/− mice stained for IgG are shown. Glomerular C3 deposition was observed in all WT and IL-17A−/− mice treated with pristane, but glomerular C3 deposition was decreased in the absence of IL-17A (Fig. 4b). High-power images representative of glomerular C3 deposition in WT and IL-17A−/− mice are shown.
Figure 4.
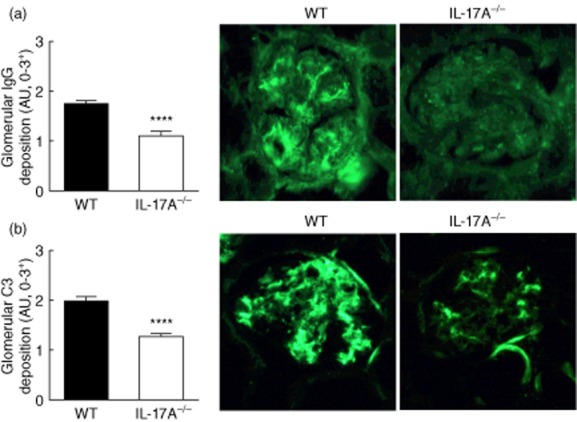
Glomerular immunoglobulin (Ig)G and complement deposition is decreased in interleukin (IL)-17A−/− mice. (a) Glomerular IgG and C3 deposition was assessed 7 months after pristane treatment. Glomerular IgG was detectable in both wild-type (WT) and IL-17A−/− mice, but IL-17A−/− mice had less IgG within glomeruli. Representative photomicrographs (×400) of glomerular IgG deposition in WT and IL-17A−/− mice are shown. (b) Similarly, we found that C3 deposition was decreased in glomeruli of IL-17A−/− mice. Representative images of glomerular C3 deposition in WT and IL-17A−/− mice are shown. ****P < 0·0001. AU = arbitrary units.
Glomerular inflammation is decreased in the absence of IL-17A
Seven months after pristane treatment, kidney mRNA expression of the key Th cell transcription factors, T-bet (Th1), RORγt (Th17) and GATA-3 (Th2), were all decreased (Fig. 5a). There was a trend towards reduced FoxP3 mRNA. In addition, the Th1 cytokine IFN-γ and CXCL11, a key Th1-associated chemokine, were decreased in IL-17A−/− mice (Fig. 5b). While glomerular CD4+ T cells were observed readily in WT mice, CD4+ T cell numbers were decreased in IL-17A−/− mice (Fig. 5c,d). There were no differences in glomerular macrophage or neutrophil recruitment between pristane-injected WT and IL-17A−/− mice (Fig. 5d).
Figure 5.
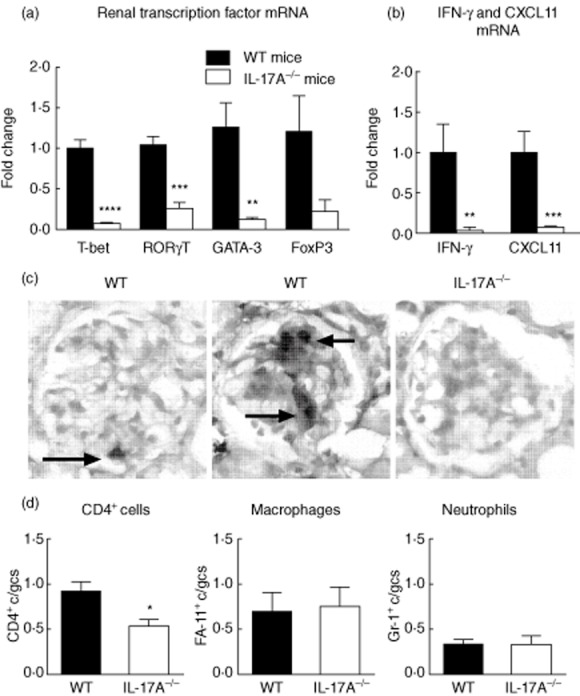
Renal inflammation is diminished in the absence of interleukin (IL)-17A. (a) Seven months after pristane treatment intrarenal mRNA for the T cell transcription factors T-bet, retinoic acid-related orphan receptor gamma t (RORγt) and GATA-binding protein 3 (GATA-3) were decreased in IL-17A−/− mice. The reduction in forkhead box protein 3 (FoxP3) did not reach significance (P = 0·07). (b) mRNA for interferon (IFN)-γ and CXCL11, each a key T helper type 1 (Th1)-associated cytokine and chemokine were decreased in IL-17A−/− mice. (c) Representative images of single and multiple CD4+ T cells in glomeruli of wild-type (WT) mice. Glomerular CD4+ T cells were not observed regularly in IL-17A−/− mice. (d) Compared to WT mice, glomerular CD4+ T cell recruitment was decreased in IL-17A−/− mice. There were no differences in glomerular macrophage or neutrophil recruitment in WT or IL-17A−/− mice. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001. c/gcs = cells per glomerular cross-section.
Functional and histological renal injury is decreased in the absence of IL-17A
Following administration of pristane, we assessed functional injury by sequential measurement of proteinuria and histological injury in WT and IL-17A−/− mice. Previously we have shown that an increase in albuminuria is detectable in WT mice 3 months after pristane treatment [15]. We found that there was a steady increase in urinary albumin excretion in WT mice over the experimental period (Fig. 6a). IL-17A−/− mice were not proteinuric at 3 months and had developed only modest proteinuria by the end of experiments at 7 months. Consistent with the attenuated functional renal injury, histological renal injury measured by the proportion of abnormal glomeruli was attenuated in the absence of IL-17A (Fig. 6b) after 7 months. Representative photomicrographs of the glomerular injury in WT and IL-17A−/− mice are shown (Fig. 6c). Therefore, IL-17A is required for the full expression of functional and histological renal injury in experimental lupus nephritis induced by pristane.
Figure 6.
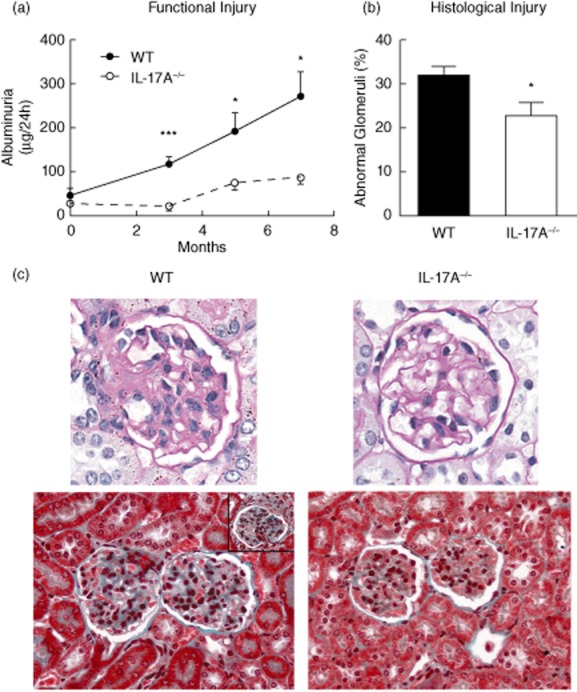
Functional and histological renal injury is diminished in interleukin (IL)-17A−/− mice. (a) Following pristane injection there was a progressive increase in albuminuria. Compared to wild-type (WT, baseline n = 4, disease n = 9) mice treated with pristane, albuminuria was decreased in IL-17A−/− mice (baseline n = 5, disease n = 7). (b) Histological injury was also decreased in the absence of IL-17A, 7 months after pristane treatment. (c) Renal histological injury was attenuated in the absence of IL-17A. Periodic acid Schiff's (PAS) staining of representative images demonstrating glomerular injury with mesangial expansion and hypercellularity in WT mice are shown. In the lower panels, Masson trichrome staining shows accumulation of mesangial matrix and hypercellularity. Crescent formation (insert, WT) was uncommon in WT mice, but was absent in IL-17A−/− mice. *P < 0·05; ***P < 0·001.
Discussion
In these experiments we demonstrate that pristane administration stimulates early IL-17A production, predominantly from innate immune cells. Intact IL-17A responses are required for the full production of IFN-γ and TNF as well as humoral autoimmunity. In association with reduced autoimmunity in IL-17A−/− mice, we found that renal inflammation and injury was diminished. This is an important finding, as therapies targeting IL-17A have been used successfully in clinical practice, in psoriasis, psoriatic arthritis and ankylosing spondylitis [11], and could be used potentially in the treatment of patients with SLE.
Many innate and adaptive immune cells are known to produce IL-17A. In the pristane model the peritoneum harbours an inflammatory infiltrate, which produces inflammatory cytokines including type 1 IFNs, which are linked to the development of both experimental and clinical SLE [28]. Following pristane treatment we found evidence of local inflammation in the peritoneum, with increased numbers of IL-17A-producing cells. The majority of IL-17A-producing cells were innate immune cells, with nearly two-thirds of IL-17A production originating from macrophages or neutrophils. Recently, Bosmann and colleagues have demonstrated that macrophages and in particular peritoneal macrophages produce IL-17A [29]. In experimental endotoxaemia they demonstrated that most IL-17A-producing cells were F4/80+ and pretreatment with anti-F4/80 antibody resulted in diminished circulating levels of IL-17A. Similarly, it has been shown that in response to inhaled environmental toxins alveolar macrophages produce IL-17A [30]. Neutrophils produce IL-17A, including in acute kidney injury [31]. The key role of macrophages and neutrophils in producing IL-17A in these models reflects the importance of local injury in inducing inflammation and driving organ damage. The current studies show that, after pristane administration, other cells besides macrophages and neutrophils produce IL-17A, including CD4+ T cells, NK cells, γδT cells and B cells. These cellular sources of IL-17A are well described in other experimental settings, reviewed recently [17]. Despite being the major source of autoantibodies later in the development of systemic autoimmunity, at this early stage B cells were not major sources of IL-17A.
Both IL-17A and Th17 cells promote renal inflammation. In experimental models of glomerulonephritis, we and others have shown that IL-17A and Th17 cells mediate early glomerular injury [19,20,32]. In patients with SLE, serum IL-17A levels are increased and correlate with disease activity, numbers of Th17 cells are increased in the peripheral blood [33–35] and T cells producing IL-17 have been observed consistently in kidney biopsies from patients with lupus nephritis [12]. IL-17A has been associated with disease in several genetically prone lupus mice. In MRL/lpr mice, SNF1 and BXD2 mice, serum IL-17 levels were increased and IL-17A-producing T cells were found in kidneys [36–38]. By using IL-17A-deficient mice we were able to demonstrate a definite pathogenic role for IL-17A in pristane-induced lupus nephritis. Humoral autoreactivity, including glomerular IgG and C3 deposition, was diminished in the absence of IL-17A and renal injury was decreased significantly, demonstrating that IL-17A is required for nephritogenic immune responses and lupus autoimmunity in this model.
In addition to its effects on humoral autoimmunity, IL-17A also was important in cellular autoimmunity, assessed 8 weeks after pristane administration. There is increasing evidence in murine models of human infection that IL-17A is required for the development of protective Th1 immunity. In experimental Francisella tularensis infection, IL-17A induces IL-12 and IFN-γ secretion from dendritic cells and macrophages and is critical for Th1-mediated bacterial killing [18], with similar results reported in experimental models of Chlamydia muriduram [39], Mycobacterium bovis [40] and viral hepatitis [41]. However, the role of early IL-17A production in Th1 responses in autoimmunity, including autoimmune kidney disease, is not well appreciated. The finding that IFN-γ and TNF production was diminished in the absence of IL-17A highlights the importance of IL-17A in the promotion of these inflammatory pathways, and is consistent with the lack of early IFN-γ production in IL-23p19−/− mice in murine autoimmune anti-glomerular basement membrane disease [42].
To mimic a potential clinical scenario where SLE patients suffer from infections or stresses, which ligate TLRs, we cultured splenocytes with a TLR-2 or TLR-4 ligand. Both TLR-2 [43] and TLR-4 [15] are pathogenic in this model. When WT splenocytes were cultured with a TLR-2 ligand there was a significant increase in IL-17A production, concordant with our findings in experimental vasculitis, where TLR-2 ligation promoted IL-17A responses and subsequent renal injury [22]. TLR-4 ligands promoted an increase in the production of TNF and IFN-γ, Th1-associated cytokines. In addition to decreased Th1 systemic immunity, we found that glomerular CD4+ T cell recruitment was decreased in the absence of IL-17A, with kidney mRNA expression of T-bet, the key Th1-associated transcription factor, and critical Th1-associated cytokine and chemokine levels being decreased in IL-17A−/− mice. Clinical data consistently imply a role for Th1 overactivity in SLE and lupus nephritis [44,45], supported by results from experimental studies and in pristane-induced nephropathy where both IFN-γ−/− and IL-12p35−/− mice were protected [8,9]. The deficit in Th1 immunity and associated proinflammatory cytokines may contribute to the attenuated renal injury observed in IL-17A-deficient mice.
IL-17A-deficient mice exhibited diminished levels of total IgG and anti-dsDNA antibodies. Previously it has been shown that IL-17A is required for the development and maintenance of splenic germinal centres and the generation of dsDNA autoantibodies in BXD2 lupus-prone mice [38]. Similarly, in the ALD-DNA murine model of SLE, induced by exogenous administration of ALD-DNA, treatment with exogenous IL-17A increased dsDNA levels, while neutralizing IL-17A resulted in decreased anti-dsDNA autoantibody levels [26]. However, IL-17A-deficient FcγRIIb−/− lupus-prone mice were protected despite IL-17A deficiency having no effect on autoantibody production [14]. Our results support the concept that IL-17A is important in the full development of humoral immunity in experimental lupus nephritis, with diminished autoantibody levels likely to be contributing to attenuated renal injury.
In conclusion, in lupus nephritis induced in the C57BL/6 mouse strain, with no genetic susceptibility elements, IL-17A is required for the maximal production of humoral and cellular autoimmunity and IL-17A is even produced early in the disease process, predominantly by innate immune cells. IL-17A mediates functional and histological renal injury and represents a promising target for the treatment of SLE and lupus nephritis.
Acknowledgments
These studies were funded by the National Health and Medical Research Council Project grant no. 1010563. Dr Y. Iwakura is thanked for providing the initial breeding pairs of IL-17A−/− mice.
Author contributions
S. A. S. designed and performed the experiments, interpreted the data and wrote the manuscript. D. O. designed and performed the experiments and interpreted the data. M. B. K. performed the experiments. O. M. S. designed the experiments and interpreted the data. Y. Y. performed experiments. S. R. H. designed the experiments and interpreted the data. A. R. K. designed experiments, interpreted the data and wrote the manuscript.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Ward MM, Pyun E, Studenski S. Mortality risks associated with specific clinical manifestations of systemic lupus erythematosus. Arch Intern Med. 1996;156:1337–1344. [PubMed] [Google Scholar]
- 2.Koffler D, Agnello V, Thoburn R, Kunkel HG. Systemic lupus erythematosus: prototype of immune complex nephritis in man. J Exp Med. 1971;134:169s–179. [PubMed] [Google Scholar]
- 3.Wofsy D, Seaman WE. Successful treatment of autoimmunity in NZB/NZW F1 mice with monoclonal antibody to L3T4. J Exp Med. 1985;161:378–391. doi: 10.1084/jem.161.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexopoulos E, Seron D, Hartley RB, Cameron JS. Lupus nephritis: correlation of interstitial cells with glomerular function. Kidney Int. 1990;37:100–109. doi: 10.1038/ki.1990.14. [DOI] [PubMed] [Google Scholar]
- 5.Anders HJ, Schlondorff DO. Innate immune receptors and autophagy: implications for autoimmune kidney injury. Kidney Int. 2010;78:29–37. doi: 10.1038/ki.2010.111. [DOI] [PubMed] [Google Scholar]
- 6.Satoh M, Richards HB, Shaheen VM, et al. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin Exp Immunol. 2000;121:399–405. doi: 10.1046/j.1365-2249.2000.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richards HB, Satoh M, Jennette JC, Okano T, Kanwar YS, Reeves WH. Disparate T cell requirements of two subsets of lupus-specific autoantibodies in pristane-treated mice. Clin Exp Immunol. 1999;115:547–553. doi: 10.1046/j.1365-2249.1999.00825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards HB, Satoh M, Jennette JC, Croker BP, Yoshida H, Reeves WH. Interferon-gamma is required for lupus nephritis in mice treated with the hydrocarbon oil pristane. Kidney Int. 2001;60:2173–2180. doi: 10.1046/j.1523-1755.2001.00045.x. [DOI] [PubMed] [Google Scholar]
- 9.Calvani N, Satoh M, Croker BP, Reeves WH, Richards HB. Nephritogenic autoantibodies but absence of nephritis in Il-12p35-deficient mice with pristane-induced lupus. Kidney Int. 2003;64:897–905. doi: 10.1046/j.1523-1755.2003.00178.x. [DOI] [PubMed] [Google Scholar]
- 10.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 11.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Ito S, Chino Y, et al. Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin Exp Immunol. 2010;159:1–10. doi: 10.1111/j.1365-2249.2009.04031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184:4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pisitkun P, Ha HL, Wang H, et al. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity. 2012;37:1104–1115. doi: 10.1016/j.immuni.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Summers SA, Hoi A, Steinmetz OM, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun. 2010;35:291–298. doi: 10.1016/j.jaut.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–990. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nature Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 18.Lin Y, Ritchea S, Logar A, et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Summers SA, Steinmetz OM, Li M, et al. Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol. 2009;20:2518–2524. doi: 10.1681/ASN.2009030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odobasic D, Gan PY, Summers SA, et al. Interleukin-17A promotes early but attenuates established disease in crescentic glomerulonephritis in mice. Am J Pathol. 2011;179:1188–1198. doi: 10.1016/j.ajpath.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Summers SA, van der Veen BS, O'Sullivan KM, et al. Intrinsic renal cell and leukocyte-derived TLR4 aggravate experimental anti-MPO glomerulonephritis. Kidney Int. 2010;78:1263–1274. doi: 10.1038/ki.2010.327. [DOI] [PubMed] [Google Scholar]
- 22.Summers SA, Steinmetz OM, Gan PY, et al. Toll-like receptor 2 induces Th17 myeloperoxidase autoimmunity while Toll-like receptor 9 drives Th1 autoimmunity in murine vasculitis. Arthritis Rheum. 2011;63:1124–1135. doi: 10.1002/art.30208. [DOI] [PubMed] [Google Scholar]
- 23.Lartigue A, Colliou N, Calbo S, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol. 2009;183:6207–6216. doi: 10.4049/jimmunol.0803219. [DOI] [PubMed] [Google Scholar]
- 24.Summers SA, Chan J, Gan PY, et al. Mast cells mediate acute kidney injury through the production of TNF. J Am Soc Nephrol. 2011;22:2226–2236. doi: 10.1681/ASN.2011020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Summers SA, Steinmetz OM, Ooi JD, et al. Toll-like receptor 9 enhances nephritogenic immunity and glomerular leukocyte recruitment, exacerbating experimental crescentic glomerulonephritis. Am J Pathol. 2010;177:2234–2244. doi: 10.2353/ajpath.2010.100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen Z, Xu L, Xu W, Yin Z, Gao X, Xiong S. Interleukin-17 expression positively correlates with disease severity of lupus nephritis by increasing anti-double-stranded DNA antibody production in a lupus model induced by activated lymphocyte derived DNA. PLOS ONE. 2013;8:e58161. doi: 10.1371/journal.pone.0058161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Summers SA, Phoon RK, Ooi JD, Holdsworth SR, Kitching AR. The IL-27 receptor has biphasic effects in crescentic glomerulonephritis mediated through Th1 responses. Am J Pathol. 2011;178:580–590. doi: 10.1016/j.ajpath.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nacionales DC, Kelly KM, Lee PY, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane) Am J Pathol. 2006;168:1227–1240. doi: 10.2353/ajpath.2006.050125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosmann M, Sarma JV, Atefi G, Zetoune FS, Ward PA. Evidence for anti-inflammatory effects of C5a on the innate IL-17A/IL-23 axis. FASEB J. 2012;26:1640–1651. doi: 10.1096/fj.11-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasahara DI, Kim HY, Williams AS, et al. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol. 2012;188:4558–4567. doi: 10.4049/jimmunol.1102363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Huang L, Vergis AL, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia–reperfusion injury. J Clin Invest. 2010;120:331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paust HJ, Turner JE, Riedel JH, et al. Chemokines play a critical role in the cross-regulation of Th1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. 2012;82:72–83. doi: 10.1038/ki.2012.101. [DOI] [PubMed] [Google Scholar]
- 33.Crispin JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong CK, Ho CY, Li EK, Lam CW. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus. 2000;9:589–593. doi: 10.1191/096120300678828703. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Chu Y, Yang X, et al. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1472–1483. doi: 10.1002/art.24499. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. 2009;183:3160–3169. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang HK, Liu M, Datta SK. Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. J Immunol. 2007;178:7849–7858. doi: 10.4049/jimmunol.178.12.7849. [DOI] [PubMed] [Google Scholar]
- 38.Hsu HC, Yang P, Wang J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 39.Bai H, Cheng J, Gao X, et al. IL-17/Th17 promotes type 1 T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J Immunol. 2009;183:5886–5895. doi: 10.4049/jimmunol.0901584. [DOI] [PubMed] [Google Scholar]
- 40.Gopal R, Lin Y, Obermajer N, et al. IL-23-dependent IL-17 drives Th1-cell responses following Mycobacterium bovis BCG vaccination. Eur J Immunol. 2012;42:364–373. doi: 10.1002/eji.201141569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou L, Jie Z, Desai M, et al. Early IL-17 production by intrahepatic T cells is important for adaptive immune responses in viral hepatitis. J Immunol. 2013;190:621–629. doi: 10.4049/jimmunol.1201970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ooi JD, Phoon RK, Holdsworth SR, Kitching AR. IL-23, not IL-12, directs autoimmunity to the Goodpasture antigen. J Am Soc Nephrol. 2009;20:980–989. doi: 10.1681/ASN.2008080891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urbonaviciute V, Starke C, Pirschel W, et al. Toll-like receptor 2 is required for autoantibody production and development of renal disease in pristane-induced lupus. Arthritis Rheumat. 2013;65:1612–1623. doi: 10.1002/art.37914. [DOI] [PubMed] [Google Scholar]
- 44.Chan RW, Lai FM, Li EK, et al. Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis. Rheumatology. 2006;45:951–957. doi: 10.1093/rheumatology/kel029. [DOI] [PubMed] [Google Scholar]
- 45.Calvani N, Richards HB, Tucci M, Pannarale G, Silvestris F. Up-regulation of IL-18 and predominance of a Th1 immune response is a hallmark of lupus nephritis. Clin Exp Immunol. 2004;138:171–178. doi: 10.1111/j.1365-2249.2004.02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]