

Abstract
High Density Lipoprotein (HDL) mediates reverse cholesterol transport and it is known to be protective against atherosclerosis. In addition, HDL has potent anti-inflammatory properties that may be critical for protection against other inflammatory diseases. The molecular mechanisms of how HDL can modulate inflammation, particularly in immune cells such as macrophages, remain poorly understood. Here we identify the transcriptional repressor ATF3, as an HDL-inducible target gene in macrophages that down-regulates the expression of Toll-like receptor (TLR)-induced pro-inflammatory cytokines. The protective effects of HDL against TLR-induced inflammation were fully dependent on ATF3 in vitro and in vivo. Our findings may explain the broad anti-inflammatory and metabolic actions of HDL and provide the basis for predicting the success of novel HDL-based therapies.
The innate immune system utilizes several families of signaling receptors to detect invading pathogens or tissue damage1. While an appropriate inflammatory response is critical for clearing pathogens and maintaining tissue integrity, excessive or prolonged inflammation can lead to harmful systemic inflammation and organ dysfunction. However, multiple regulatory mechanisms have evolved to limit the extent and duration of the inflammatory response. For instance, while Toll-like receptor (TLR) activation leads to the expression of pro-inflammatory cytokines it also induces the expression of many negative regulators, acting to limit signal transduction, mRNA transcription or translation2. Of these, Activating Transcription Factor 3 (ATF3) is a key inducible transcriptional repressor, which binds to specific CREB-ATF elements within the promoter loci of specific target genes resulting in chromatin remodeling and subsequent inhibition of transcription factor binding3. ATF3 is induced by TLR stimulation as well as a multitude of other stimuli4 and acts in a negative feedback system to limit excessive production of pro-inflammatory cytokines including TNF, IL-6 and IL-12p403, 5. Understanding how these regulatory pathways work in the context of chronic inflammatory disease is of great interest and may lead to the design of beneficial therapeutics.
Atherosclerosis is the chronic inflammatory condition underlying coronary artery disease and is driven in part via the recognition of metabolic danger signals by innate immune receptors on macrophages6, 7. Elevated levels of HDL cholesterol are associated with reduced atherothrombotic events8. Furthermore, animal studies have shown HDL to be beneficial on the key cell types and processes in atherosclerosis, in part by mediating the removal of cholesterol from lipid-laden macrophages (known as foam cells)9. However, therapies based on simply increasing HDL cholesterol levels have been largely unsuccessful10,11, prompting the suggestion that the functionality of HDL may be more important to disease outcome than the quantity of HDL itself12. Indeed, cholesterol efflux capacity is better correlated with disease risk than total HDL cholesterol levels13. HDL also has anti-oxidant, anti-thrombotic and anti-inflammatory properties, which may also protect against atherogenesis14–16. The anti-inflammatory nature of HDL could also be beneficial in other inflammatory diseases: it can lower endothelial cell vascular cell adhesion molecule 1 (VCAM1) expression, increase endothelial nitric oxide synthase (eNOS) production and reduce monocyte CD11b expression and migration along an monocyte chemotactic protein-1 (MCP-1) gradient17. However, the molecular mechanisms by which HDL mediates its anti-inflammatory functions, especially in macrophages and outside of the context of atherosclerosis, remain poorly understood.
We therefore investigated the effects of HDL on macrophage activation by TLRs and in macrophage-dependent models of inflammation under normo-cholesterolemic conditions. Here we show that HDL inhibits TLR-induced production of pro-inflammatory cytokines from macrophages. These effects were independent of HDL binding TLR ligands and did not involve modulation of TLR signaling events, or disruption of cytokine secretory processes. Rather, in a time-dependent manner, HDL suppressed the ability of TLRs to induce the expression of pro-inflammatory cytokines (such as IL-6, IL-12p40 and TNF) on the transcriptional level. Using a systems biology approach we identified ATF3 as an HDL-inducible negative regulator of macrophage activation. Transcriptome analysis revealed that HDL regulated many genes in an ATF3-dependent manner, and ATF3 ChIP sequencing confirmed that several key pro-inflammatory genes were directly targeted by ATF3 following its induction by HDL. Furthermore, the ability of HDL to reduce TLR responses was found to be fully dependent on ATF3 expression both in vitro and in vivo, demonstrating the critical role of this transcriptional repressor in the anti-inflammatory effects of HDL.
RESULTS
HDL is protective against TLR-induced inflammation
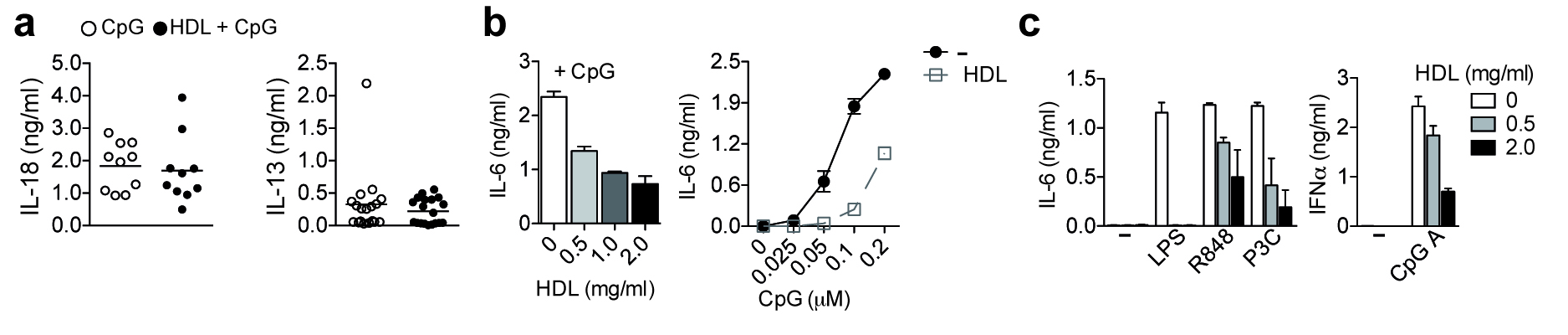
HDL has multiple functions beyond promoting the efflux of cholesterol from lipid-laden cells. To study the anti-inflammatory effects of HDL under normo-cholesterolemic conditions, we utilized a well-established in vivo model of acute inflammation, in which the systemic release of pro-inflammatory cytokines from TLR activated macrophages leads to liver damage18, 19. Mice were injected with HDL freshly isolated from patients (native HDL) or with purified human Apolipoprotein A1 (ApoAI; the major protein component of HDL) reconstituted with phospholipids (referred to hereafter as HDL) and subsequently challenged with TLR agonists. Pre-treatment with either native HDL or HDL significantly reduced liver damage induced by the TLR9 activator CpG-DNA (CpG), as measured by alanine aminotransferase (ALT) release (Fig. 1a,b) or hepatocyte cell death (Fig. 1c and data not shown). In addition, mice injected with CpG following pre-treatment with native HDL displayed significantly reduced serum levels of the pro-inflammatory cytokines IL-6 and IL-12p40 (Fig. 1a). Similarly, mice pre-treated with HDL showed reduced serum concentrations of CpG-induced IL-6, IL-12p40, TNF, MCP-1 and IL-10 (Fig. 1b), whereas IL-18 and IL-13 remained unaffected (Supplementary Fig. 1a). In addition, HDL pre-treatment significantly reduced liver damage and inflammation mediated by the TLR1-2 ligand, Pam3CysK4 (P3C) (Fig. 1d). We further investigated the anti-inflammatory effect of HDL in vitro using bone marrow-derived macrophages (BMDMs). Pre-treatment of BMDMs with HDL substantially reduced IL-6, IL-12p40 and TNF cytokine output in response to a variety of TLR ligands (Fig. 1e), without impacting cell viability (Fig. 1f). HDL reduced TLR-mediated cytokine production in a dose-dependent manner (Supplementary Fig. 1b). Comparable decreases in TLR-induced IL-6 production were also observed following pre-treatment of BMDMs with native HDL (Fig. 1g). HDL’s ability to decrease TLR-induced inflammation was also evident in human peripheral blood mononuclear cells (PBMCs) (Supplementary Fig. 1c). Collectively, these data suggest HDL is protective against TLR-induced inflammation by down modulating the production of pro-inflammatory cytokines from monocytes and macrophages.
Figure 1.

HDL inhibits TLR-induced cytokine production from macrophages in vivo and in vitro. (a–d) Mice were injected intraperitoneally (i.p.) with 2 mg HDL for 6 h before i.p. injection of TLR ligand (CpG: 20 μg, P3C 2 μg) and D-galactosamine (10 mg). Serum cytokines were measured after 1 h and serum alanine aminotransferase (ALT) and liver histology after 10 h. a C3H/HeJ mice injected with native HDL and CpG: ALT release (n=12) and serum cytokines (CpG n=28, native HDL+CpG n= 27) were measured. b,c C57BL/6 mice injected with HDL and CpG: ALT release (n=9); serum cytokines (CpG n=18, HDL+CpG n=19) (b), and liver histology by hemotoxylin and eosin staining shows hepatic cell death. (c). d, C57BL/6 injected with HDL and P3C: ALT release (n=15) and serum cytokines (n=15) were measured. e, ELISA of BMDMs treated with 2 mg/ml HDL for 6 h followed by overnight stimulation with the TLR4 ligand, LPS (100 ng/ml); TLR9 ligand, CpG (100 nM); TLR7/8 ligand, R848 (5 ng/ml); or TLR2/1 ligand P3C (50 ng/ml). f, Cell viability was measured with CellTitre- Blue® reagent. g, BMDMs were pre-treated with recombinant or native HDL (2 mg/ml) for 6 h and stimulated overnight with CpG (100 nM) or P3C (50 ng/ml) and IL-6 cytokine secretion measured by ELISA. a,b,d Data are presented as mean values ±S.E.M, CpG versus HDLs+CpG *p<0.05, **p<0.01, ***p<0.005, c, images are representative of mice with median ALT concentration; scale bars, 100 μm, e,f,g data is shown as mean ±S.D. and is representative of at least three independent experiments.
HDL regulates inflammation at the transcriptional level
HDL can directly neutralize the inflammatory activity of LPS20–22. To examine whether HDL neutralizes other TLR activators by direct interaction, we analyzed if HDL can change the size exclusion profile of fluorescent TLR agonists. As expected, HDL led to a shift of LPS fluorescence into the higher molecular weight HDL fractions (Fig. 2a), demonstrating LPS binding to HDL. In contrast, HDL did not change the elution profile of fluorescent CpG or P3C (Fig. 2a and data not shown), indicating that HDL does not directly interact with these TLR ligands. Furthermore, direct binding by HDL should abolish or reduce the ability of TLR ligands to activate their downstream signaling pathways. Consistent with our size exclusion data, HDL did not block CpG- or P3C-induced p38 mitogen-activated protein kinase (MAPK) phosphorylation (p-p38) in BMDMs, indicating successful interaction of the ligands with their receptors (Fig. 2b). In contrast, HDL co-treatment abolished LPS induced p38 activation (Fig. 2b). These data suggest that while HDL can attenuate LPS-induced inflammation by sequestration, they do not explain the anti-inflammatory effects of HDL on other TLR ligands, including CpG and P3C.
Figure 2.

HDL inhibits TLR-induced pro-inflammatory cytokine transcription. a, Bodipy-labeled LPS, Alexa 647-labeled CpG 1826 or HDL were run over an S200 size exclusion column separately (a, upper panel) or together (a, lower panel) and absorbance profiles examined. b, LPS (200 ng/ml), CpG (100 nM) or P3C (50 ng/ml) were incubated with HDL (2 mg/ml), and BMDMs stimulated for 30 min. Whole cell lysates were analyzed for p38 phosphorylation (p-p38) relative to total β-actin. c,d BMDMs were pre-treated with HDL (2 mg/ml) for 6 h before stimulation with CpG (100 nM) for indicated times: total cellular cholesterol (c) and phosphorylation of p38, JNK, NF-κB p65 and IκBα degradation (d). e,f BMDMs were pre-treated with HDL as before, and stimulated with CpG (100 nM) for 30 min: subcellular localization of NF-κB p65 (β-tubulin: cytoplasmic loading control; poly ADP ribose polymerase (PARP): nuclear loading control) (e) and NF-κB binding to a target probe as analysed by EMSA (f). g,h BMDMs were pre-treated with HDL as before: CpG-induced phospho-NF-κB p65 and intracellular IL-6 and IL-1β were measured in whole cell extracts, (g); secreted IL-6 measured by ELISA (h). mRNA expression of BMDMs pre-treated with HDL as before, and stimulated with 100 nM CpG for 4 h (i). j, Liver mRNA profile 1 h after C57BL/6 mice were injected with CpG following 6 h HDL pre-treatment (n=8). a, Data is representative of at least three independent experiments. b, Representative immunoblot and densitometric analysis combined from three independent experiments (mean ±S.E.M, each ligand; no HDL versus HDL incubation ***p<0.005). c–i, Representative data from at least three independent experiments (mean ±S.D). j, Mean values ±S.E.M, CpG versus HDL+CpG *p<0.05.
HDL promotes the efflux of cholesterol from macrophages and thus, subsequent TLR activation might be inhibited via changes to lipid raft cholesterol content23, 24. However, while pre-treatment of BMDMs with HDL decreased cellular cholesterol concentration (Fig. 2c and Supplementary Fig. 2a), it did not inhibit CpG-induced signaling, including activation of p38 MAPK, JNK, NF-κB p65, or the degradation of IκBα (Fig. 2d). Furthermore, HDL pre-treatment had no effect on the nuclear translocation of NF-κB (Fig. 2e), nor did it prevent NF-κB binding to target DNA sequences (Fig. 2f). Together these data exclude the possibility of interference with early TLR signaling mechanisms. To assess whether HDL affects cytokine secretion, we measured cytokines in intracellular compartments. Although HDL pre-treatment had no effect on the activation of NF-κB p65 in response to TLR stimulation, intracellular cytokine expression was diminished in HDL pre-treated BMDMs (Fig. 2g), as was the amount of secreted cytokines (Fig. 2h and Supplementary Fig. 2b,c). These data suggest that HDL interferes with cytokine production but not its secretion. Consistent with this hypothesis, relative mRNA expression of pro-inflammatory cytokines was notably reduced when BMDMs were treated with HDL or native HDL prior to CpG stimulation (Fig. 2i and Supplementary Fig. 2d). Cytokine mRNA expression was also decreased in the livers of mice injected with HDL or native HDL prior to CpG (Fig. 2j and Supplementary Fig. 2e). We next assessed if HDL was affecting the half-life of TLR-induced cytokine mRNA or protein. HDL pre-treatment of BMDMs did not alter the stability of IL-6 mRNA transcripts (Supplementary Fig. 2f) nor the stability of IL-1β protein (Supplementary Fig. 2g). Together these data indicate that HDL may regulate anti-inflammatory effects in macrophages by down modulating pro-inflammatory gene transcription.
ATF3 is identified as a candidate transcription factor
We noted that macrophages exposed to HDL for longer periods of time displayed a more pronounced reduction in cytokine production (Fig. 3a), which persisted up to 24 h after removal of HDL (Fig. 3b). This suggested that the underlying molecular mechanism might involve de novo transcription of an inflammatory repressor. We therefore used genome-wide RNA profiling to assess whether HDL affected the transcriptional response to TLR stimulation, by comparing resting BMDMs with HDL pre-treated BMDMs subsequently stimulated with CpG. Principle component analysis and hierarchical clustering indicated that HDL induced significant transcriptional changes in resting and CpG-treated BMDMs (Supplementary Fig. 3a,b). As expected, HDL treatment alone induced the gene expression of several enzymes involved in the cholesterol biosynthesis pathway (Supplementary Fig. 3c). This finding correlated with an increased appearance of cholesterol precursors following HDL treatment as identified by mass spectrometry (Supplementary Fig. 3d).
Figure 3.
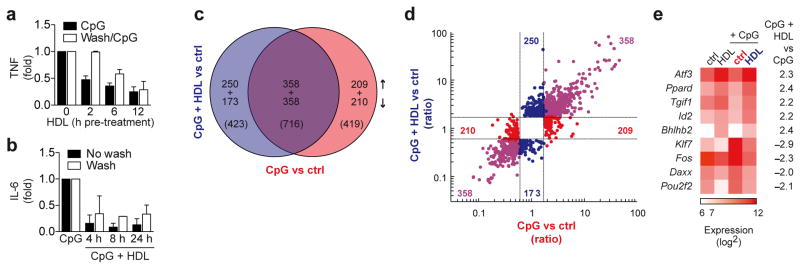
Microarray analysis identifies ATF3 as a candidate gene for the anti-inflammatory function of HDL. a, Immortalized-BMDMs were pre-treated with HDL (2 mg/ml) for indicated times, then either washed twice in serum-free DMEM, or left unwashed, before overnight stimulation with CpG (100 nM). TNF secretion was measured by ELISA and normalized to CpG treatment alone. b, BMDMs were pre-treated with HDL (2 mg/ml) for 12 h, then either washed twice in serum-free DMEM, or left unwashed, before stimulation with CpG (100 nM) for indicated times. IL-6 was measured in culture supernatants and normalized to CpG treatment alone. c–f, Microarray analysis of BMDMs pre-treated for 6 h with HDL (2 mg/ml) then stimulated with CpG (100 nM) for 4 h. c, Venn diagram shows genes with differential expression (vs control) and the overlap in these genes (Fold Change limit 1.8, False discovery rate p<0.05). d, A fold change/fold change plot shows directional gene expression resulting from CpG treatment (red), CpG and HDL treatment (blue) or genes that are co-regulated in both treatments (purple). e, Expression of transcription factors following HDL, CpG or combined treatment as described in the methods and presented as a heat map. TFs are ranked according change in expression across treatments, while color intensity shows the mean expression value per condition. a,b, Representative data from at least three independent experiments (mean ±S.D) c–e, At least three biological replicates per condition were generated.
Stimulation of BMDMs with CpG resulted in regulation of 1135 genes in total; 419 of which were not influenced by HDL pre-treatment (Fig. 3c,d). The remaining 716 CpG regulated genes (Fig. 3c, purple overlap) were also regulated by CpG in the presence of HDL relative to resting to cells. However, HDL further enhanced or reduced the expression of many of these genes (Figure 3d, purple). HDL treatment also modulated the expression of 423 genes (250 induced and 173 repressed) independently of CpG treatment (Fig. 3c,d blue). To identify a potential transcription factor that might be responsible for the anti-inflammatory effects of HDL, we interrogated our transcriptome data using several bioinformatics approaches. Firstly, we performed Gene Ontology Enrichment Analysis (GOEA) using the genes that were regulated by CpG and further modified by HDL, and visualized this as a Gene Ontology (GO) – term network (Supplementary Fig. 3e). This analysis identified one subnet, based on genes down-regulated by HDL (blue edges and nodes), that could be summarized as ‘innate immune response’-related processes, while another large subnet, based on genes induced by HDL (red edges and nodes), was linked to cholesterol and lipid metabolism. However, the most significant enrichment scores were linked to a central dense subnet best summarized as ‘regulation of metabolic processes’. Based on this pronounced enrichment of regulatory processes we searched for candidates of transcriptional regulation in this subnet and identified the transcriptional repressor ATF3 in most of the subnet’s GO-terms (yellow area underlying the network in Supplementary Fig. 3e). Secondly, using the transcription factor (TF) catalogue database (TFCat database)25, we identified ATF3 as the TF with the highest expression levels in the presence of HDL (Fig. 3e). Finally, we performed TF promoter-binding prediction using the genes most significantly repressed by HDL as bait (fold change (FC)<-3, p<0.05, false discovery rate (FDR), n=33, (Methods and Supplementary Fig. 3f). Among the 143 TF candidates potentially binding to these promoters, only ATF3 (FC 2.3) was up-regulated by HDL. TF module analysis validated these findings and identified potential ATF3 binding sites in 28 out of 33 input genes (Methods and Supplementary Table 1). Collectively, these analyses strongly support a model in which HDL leads to the repression of CpG-induced genes by increasing the expression of the transcriptional repressor ATF3.
ATF3 binds target gene promoters following HDL induction
Consistent with our transcriptome analysis, BMDMs treated with HDL displayed increased ATF3 mRNA (Fig. 4a) and protein (Fig. 4b) expression in a time-dependent manner. The induction of ATF3 mRNA and protein by HDL was further potentiated by subsequent CpG or P3C stimulation (Fig. 4c,d). Native HDL also induced ATF3 mRNA expression to comparable levels in BMDMs (Fig. 4e). Additionally, HDL treatment increased ATF3 mRNA and protein in human monocytes (Fig. 4f,g). In vivo, HDL injection increased hepatic ATF3 mRNA expression in C57BL/6 mice (Fig. 4h), specifically within Kupffer cells (the resident macrophages of the liver) (Fig. 4i). To analyze whether HDL treatment would modulate ATF3 binding to target gene promoters, we performed ATF3 ChIP sequencing (ChIP-Seq) on wild type (WT) and Atf3-deficient BMDMs. We used signals from Atf3-deficient BMDMs to subtract background non-specific binding of the ATF3 antibody. HDL treatment, either alone or in combination with CpG, dramatically increased ATF3 binding across the genome, while CpG treatment alone reduced ATF3 binding (Fig. 5a). Global assessment of ATF3 peak localization relative to transcription start sites revealed that ATF3 was specifically enriched in promoter regions of genes (Fig. 5b). Consistent with our findings that HDL treatment results in reduced production of pro-inflammatory cytokines, our ChIP-Seq analysis demonstrated significant ATF3-binding at the promoters of IL-6, IL-12p40 and TNF in the presence of HDL or HDL+CpG, but not CpG alone (Fig. 5c). No enrichment of ATF3 was seen at the promoters of non-HDL regulated cytokines that we identified in vivo (Supplementary Fig. 1a) such as, IL-18 and IL-13 (Supplementary Fig. 4a). Together these data demonstrate that following induction by HDL, ATF3 is specifically enriched at the promoters of cytokines found to be regulated by HDL both in vivo and in vitro, strongly suggesting that the anti-inflammatory effects of HDL are mediated via this mechanism.
Figure 4.

HDL induces ATF3 expression. a,b, BMDMs were treated with HDL (2 mg/ml) for the indicated time points or LPS (200 ng/ml) for 6 h, and ATF3 mRNA expression (a) or protein (*indicates a non-specific band) relative to total β-actin was analyzed (b) c,d, BMDMs were pre-treated with HDL (2 mg/ml) for 6 h and stimulated with CpG (100 nM) or P3C (50 ng/ml) for 4 h : ATF3 mRNA (c) and ATF3 protein (d). e, BMDMs were treated with HDL or native HDL (2 mg/ml) or LPS for 10 h and ATF3 mRNA expression was measured by qPCR (e). f,g CD14+ monocytes were pre-treated with HDL as before, and stimulated with P3C (1 μg/ml) for 4 h; ATF3 mRNA expression (f) or protein (g). h,i, qPCR analysis of ATF3 mRNA expression in the livers (n=9) (h) or Kupffer cells (n=6) (i) of C57BL/6 mice 10 h after 2 mg i.p. HDL injection. a,c,e One of at least three independent experiments shown (mean ±S.D.) b, Representative immunoblot and densitometric analysis combined from four independent experiments (mean ±S.E.M, untreated versus HDL treated *p<0.05). d,g, Representative blot of at least two separate experiments. f, Data is represented as the mean from four individual donors (one-tailed student t test, untreated versus HDL treated *p<0.05) h,i Data are shown as the mean ±S.E.M, PBS versus HDL injected mice *p<0.05.
Figure 5.

ATF3 is active following induction by HDL. a–c, ATF3 ChIP sequencing of BMDMs pre-treated with HDL (2 mg/ml) for 6 h followed by stimulation with CpG (100 nM) for 4 h. Distribution of ATF3 binding within the genome (upper) and promoter regions (lower panel) (a) and global ATF3 peak localization relative to transcription start site (TSS); normalized average of tags per peak per bp from −1kb to +1kb (b). Genomic loci of Il6, Il12p40 and Tnf with ChIP-Seq signals for ATF3 binding under the stimulation conditions, red bars indicate significant peaks (c). d,e qPCR analysis of ATF3, IL-6, IL-12p40 or Ch25h mRNA expression in livers from Apoe-deficient mice fed on a high fat diet diet and injected intravenously (i.v.) with PBS or HDL (100 mg/kg) (n=5). a–c, At least three biological replicates per condition were generated. d,e Data are shown as the mean ±S.E.M, PBS versus HDL **p<0.01, ***p<0.001.
Functions of HDL are most intimately associated with processes related to atherosclerosis. Thus, we next examined whether ATF3 was induced following HDL treatment in an animal model of atherosclerosis. We examined the livers of atherosclerosis-prone Apoe-deficient mice fed on a high fat diet and observed that HDL-injected mice displayed significantly greater hepatic ATF3 mRNA expression compared with PBS control mice (Fig 5d). Induction of ATF3 correlated with a substantial reduction in the levels of the direct ATF3 target genes IL-6, IL-12p40 and cholesterol 25-hydroxylase (Ch25h) (Fig. 5e). Notably, within the livers of HDL injected Apoe-deficient mice, a significant induction of ATF3 mRNA was observed in Kupffer cells, while hepatocytes showed 10-fold less expression (Supplementary Fig. 4b). This data demonstrates that in an animal model of atherosclerosis HDL induces the expression of ATF3 within macrophages, which may act to repress the expression of inflammatory target genes.
HDL’s anti-inflammatory effects are mediated via ATF3
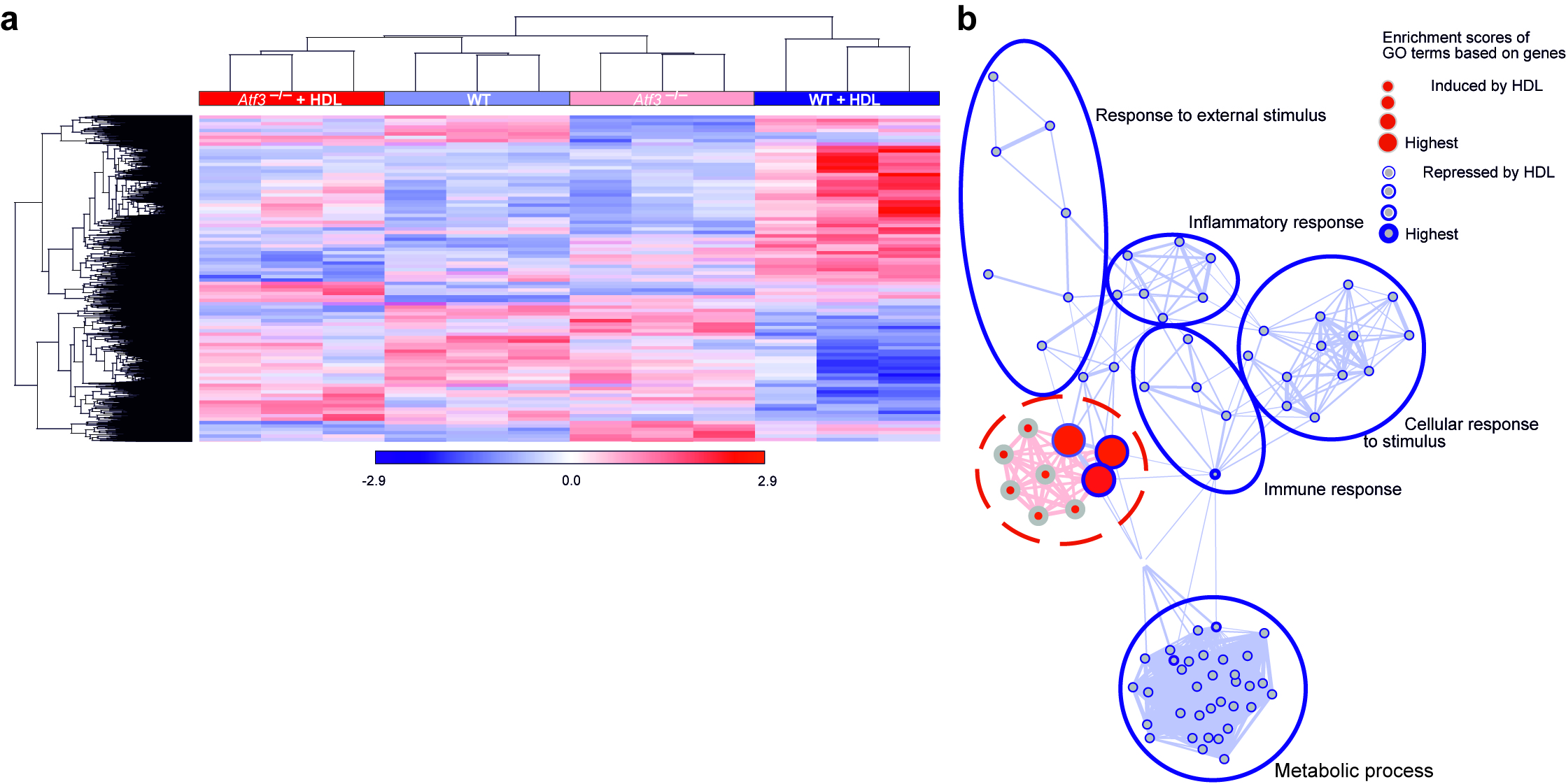
To further assess the role of ATF3 in mediating the anti-inflammatory effects of HDL we performed genome-wide transcriptome analysis on WT and Atf3-deficient BMDMs pre-treated with HDL and subsequently stimulated with CpG or P3C. Using the model presented in Figure 6a, we observed that ATF3 deficiency affected a large set of TLR-driven, HDL-modulated genes (n=224, Supplementary Table 2a). Transcripts induced or repressed in WT BMDMs by CpG or P3C alone (green dots) and counter regulated by HDL pre-treatment (black dots) were no longer modified in the absence of ATF3 (red dots) (Fig. 6b, Supplementary Fig. 5a and Supplementary Table 2b–e). We observed substantially overlapping gene sets whether CpG or P3C stimulation was used (Supplementary Fig. 5b). Furthermore, GOEA network visualization of TLR-regulated genes demonstrated that immune response related functions modified by HDL were ATF3-dependent, while lipid metabolism terms were ATF3-independent (Supplementary Fig. 5c). In fact, the majority of genes within the cholesterol biosynthesis pathway showed ATF3-independent regulation by HDL (Supplementary Fig. 5d). Accordingly, we did not detect differential gene expression between WT and Atf3-deficient BMDMs for Srebf226 or Lxr target genes27 (data not shown), indicating that ATF3 is not involved in their regulation in response to HDL. Of the genes we identified to be regulated by HDL and CpG in WT cells but not in Atf3-deficient cells (n=130), approximately 20 % were confirmed as direct ATF3 target genes (n=27) by ChIP-Seq, and GO-term analysis of these direct ATF3 targets identified significant enrichment of genes involved in inflammatory processes (Supplementary Fig. 5e).
Figure 6.
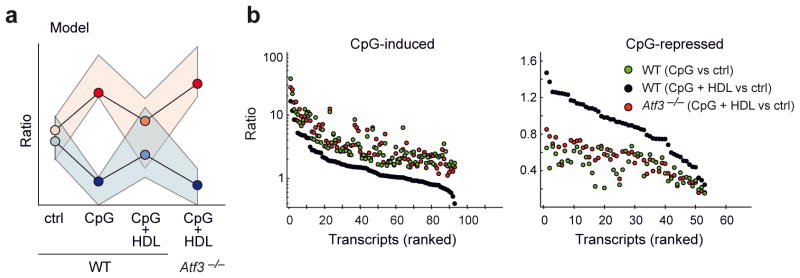
ATF3 mediates much of the transcriptional response to HDL treatment of macrophages. a–b, Microarray and ChIP-Seq analysis of WT and Atf3-deficient BMDMs pre-treated with HDL (2 mg/ml) for 6 h and subsequently stimulated for 4 h with CpG (100 nM) or P3C (50 ng/ml). a Schematic representation of the model used to identify ATF3 target genes (also see Methods). b Visualization of transcripts induced (left panel) or repressed (right panel) by CpG. At least three biological replicates per condition were generated.
To confirm that ATF3 was required for HDL mediated inhibition of TLR-induced cytokine secretion, we compared the effects of HDL pre-treatment on subsequent TLR responses in WT and Atf3-deficient BMDMs. As expected, HDL significantly reduced CpG-induced IL-6 and IL-12p40 production from WT BMDMs, but not from Atf3-deficient BMDMs (Fig. 7a). Additionally, we assessed the relevance of ATF3 induction by HDL in vivo by comparing WT and Atf3-deficient mice subjected to inflammation and liver injury induced by injection of CpG. Strikingly, HDL was also no longer protective against CpG-induced liver damage in Atf3-deficient mice, as observed by comparable ALT release from Atf3-deficient animals injected with or without HDL (Fig. 7b). Accordingly, unlike WT mice, Atf3-deficient mice injected with HDL showed no significant reductions in serum concentration of IL-12p40 and TNF, or in hepatic TNF protein expression in response to CpG (Fig. 7b). These data demonstrate that repression of pro-inflammatory cytokine responses by HDL both in vitro and in vivo are mediated by the induction of ATF3.
Figure 7.

ATF3 is required for the anti-inflammatory effect of HDL in vitro and in vivo. a, ELISA from WT or Atf3-deficient BMDMs pre-treated with HDL (2 mg/ml) for 6 h prior to overnight stimulation with CpG (indicated doses). b, WT or Atf3-deficient mice injected i.p with HDL (2 mg) 6 h before subsequent injection with CpG (30 μg) and D-gal (10 mg) for a further 10 h. Serum ALT, serum TNF and hepatic TNF (WT n=10, Atf3−/− n=8,), and serum IL-12p40 (n=6) were measured. c,d, HDL increased re-endothelialization after carotid artery injury in WT, but not Atf3-deficient mice. Carotid injury was performed on WT or Atf3-deficient mice, 3 h later PBS or HDL (20 mg/kg) was injected i.v. Re-endothelialization was evaluated 3 days following carotid injury (WT PBS n=8, WT HDL n=7, Atf3−/− PBS n=7, Atf3−/− HDL n=9). e Gene set enrichment analysis using the macrophage/carotid injury overlapping gene set applied to the carotid injury dataset assessing gene enrichment in HDL treated samples derived from WT versus Atf3−/− mice. a, Combined data from three independent experiments are shown as the mean ±S.E.M (WT versus Atf3−/− *p<0.05, **p<0.01). b, Data are presented as mean values ±S.E.M, CpG versus HDL+CpG (per genotype) *p<0.05, ***p<0.001. c, Data are presented as mean values ±S.E.M, PBS vs HDL (per genotype) **p<0.01. d, Images are representative of each group in each genotype. e, At least three biological replicates per condition were generated.
The protective functions of HDL are best documented in the context of atherosclerosis. Cardiovascular risk factors including hypertension, toxins from cigarette smoke, high blood sugar and turbulent blood flow can lead to localized and recurrent endothelial damage, a process thought to be critical in atherogenesis28. Endothelial damage is followed by an inflammatory response of macrophages that modulates the rate and extent of re-endothelialisation29,30. Since HDL is known to accelerate vascular wound healing and re-endothelialisation31–33, we next assessed whether ATF3 is also critical for HDL’s protective function in an atherosclerosis relevant model of vascular repair. While HDL effectively increased the amount of re-endothelialization after carotid artery injury in WT mice, no change was observed in Atf3-deficient mice (Fig. 7c,d). These data suggest that HDL can dampen the local inflammation that occurs during vascular injury and that ATF3 thereby contributes to the vascular repair process. To broadly assess HDL’s effects in the vascular wall after injury, we performed transcriptional profiling of RNA isolated from the injured carotid artery segments from WT and Atf3-deficient mice that were injected with HDL or PBS. Hierarchical clustering of genes and samples showed that the replicate samples within the four groups clustered as four individual groups (Supplementary Fig. 6a). HDL treatment strongly modulated genes in WT samples but not Atf3-deficient samples, implying that the effects of HDL treatment were dependent on the presence of ATF3 (Supplementary Fig. 6a). We next characterized the genes regulated by HDL and ATF3 in carotid artery samples and overlapped them with those genes earlier identified as regulated by HDL via ATF3 in macrophages in vitro (Fig. 6 and Supplementary Fig. 5). We performed GOEA analysis with these genes and visualized the changes of GO-terms induced by HDL in WT but not ATF3 deficient samples using BINGO networks. Consistent with our in vitro macrophage analysis, HDL led to an ATF3-dependent reduction of the inflammatory responses in carotid artery samples (Supplementary Fig. 6b). We validated these approaches using Gene Set Enrichment Analysis (GSEA) on the macrophage-carotid injury overlapping gene set. GSEA ranks RNA expression results based on their correlation with the phenotype and significant enrichment. These analyses revealed a highly significant enrichment of these ATF3 target gene sets in HDL treated samples from WT mice but not Atf3-deficient mice (Fig. 7e). This data indicates that HDL’s beneficial effects on carotid artery re-endothelialization are for the most part driven by ATF3 and the reduction of macrophage inflammation
DISCUSSION
Epidemiological studies have consistently documented that low concentrations of HDL cholesterol in the blood represent a strong, independent risk factor for cardiovascular disease. The protective effect of HDL in cardiovascular and possibly other inflammatory diseases is thought to be the result of several beneficial functions. HDL relieves cells from excessive amounts of cholesterol, has strong anti-oxidative effects, inhibits platelet aggregation and exerts several vasoprotective effects34. Most notably, HDL also has the remarkable ability to modulate the inflammatory response in various cell types. HDL isolated from healthy individuals or reconstituted from purified ApoA1 and phospholipids is strongly anti-inflammatory. However, during an acute phase response or in chronic inflammatory states HDL can be modified to become dysfunctional33.
The mechanisms by which HDL reduces inflammatory responses, particularly in macrophages, are not well understood. While it is known that HDL can sequester LPS and thereby prevent cellular activation through TLR4, this mechanism does not explain why HDL can block a broad spectrum of TLR-mediated inflammatory responses in macrophages. In contrast to the current understanding that HDL can impair TLR signaling through interference with raft cholesterol levels, we found that TLR signaling and subsequent nuclear translocation of the pro-inflammatory transcription factor NF-κB remained unaffected by HDL. We identified that the induction of ATF3, a key transcriptional repressor of innate immune response genes, is the main mechanism by which HDL mediates its anti-inflammatory activities in macrophages. ATF3 can be induced by TLR activation and acts as part of an important negative feedback loop to limit TLR-driven inflammatory responses in macrophages. Hence, by inducing ATF3, HDL utilizes an ancient regulatory feedback system and can broadly and directly modify the inflammatory response of macrophages towards TLR stimulation.
This discovery has broad implications as TLR activation triggers macrophage responses that contribute to a large number of inflammatory pathologies. For example, targeted deletion of TLRs or components of their signaling pathways in mice confers protection against atherosclerosis35–37. Furthermore, TLR-inducible pro-inflammatory cytokines such as IL-6, IL-12p40 and TNF all contribute to atherosclerosis development38. Of note, ATF3 can also directly regulate cholesterol metabolism pathways by repressing Ch25h expression, an enzyme required for 25-hydroxycholesterol mediated foam cell formation. Indeed, mice deficient in both ATF3 and ApoE have enhanced atherosclerotic lesions, which was attributed to increased expression of Ch25h39. Consistently, we observed that injection of HDL in Apoe-deficient mice induced ATF3 expression and reduced the expression of Ch25h, as well as the pro-inflammatory cytokines, IL-6 and IL-12p40. Hence, HDL appears to modulate both foam cell formation and inflammation via ATF3, which could contribute to HDL’s protective nature in atherosclerosis. However, HDL is known to exert other effects that can beneficially influence atherosclerosis that are likely independent of ATF3. For example, HDL’s anti-oxidative function can prevent LDL oxidation, which itself contributes to atherogenesis17. Modulation of cellular cholesterol by HDL may also affect antigen presentation and the induction of an adaptive immune response16. Furthermore, HDL reverses hyper-proliferation of hematopoietic stem cell precursors, which can exaggerate atherosclerosis40. Similarly, HDL affects the activation of platelets which do not have a nucleus41, and it reduces the integrin-mediated adhesion of leukocytes too quickly to involve transcription42. Therefore, the contribution of ATF3 to HDL’s protective effects against atherosclerosis in vivo may be difficult to separate from the rest of its protective functions.
Our ChIP-Seq analysis in macrophages and RNA profiling studies in macrophages and injured carotid arteries revealed that HDL regulates an exceptionally broad set of target genes via ATF3 under normo-cholesterolemic conditions. This could be explained by ATF3’s ability to recruit co-repressors such as histone deacetylase 1 (HDAC1). HDAC1 recruitment by ATF3 is thought to result in less histone acetylation at target gene promoters, which is typically associated with a closed chromatin and inactivation of genes3, 39. Thus, via this mechanism, HDL could influence epigenetic modification of target genes. Indeed, we observed that ATF3 was constitutively bound across target gene promoters in resting macrophages, which was lost upon CpG stimulation, consistent with transcriptional activation of these genes. However, HDL treatment further increased ATF3 binding across the genome, presumably remodeling chromatin structure to prevent transcriptional activation of these genes. These results provide a long sought mechanistic link between HDL and its anti-inflammatory function.
While HDL’s protective role is most well characterized in atherosclerosis, its ability to broadly modulate TLR responses suggests that HDL may be beneficial in other chronic inflammatory diseases. Our findings raise the possibility of using HDL-based therapeutics in treating inflammatory disorders not directly linked to hypercholesterolemia. Furthermore, our results imply that fine-tuning the negative regulators of inflammation, such as ATF3, may provide a therapeutic avenue for the treatment of a range of inflammatory pathologies.
METHODS
Reagents
Ultrapure LPS (E. coli 0111:B4), Pam3CK4 (referred to as P3C in the text) and R848 were from Invivogen. Phosphorothioate backbone modified CpG DNA oligonucleotides (referred to as CpG in the text) were synthesized by Metabion. Murine TLR9 stimulatory CpG DNA type B 1826 (5′-TCCATGACGTTCCTGACGTT-3′) and human TLR9 stimulatory CpG DNA type A 2216 (5′-GGGGGACGATCGTCGGGGGG-3′) were used. Anti-phospho-p65 NF-κB (3033), anti-phospho-p38 MAPK (4511), anti-phospho-SAPK/JNK (4668) and anti-IκB-α (4814) antibodies were from Cell Signaling Technology. Anti-IL-1β (BAF401) and anti-IL-6 (BAF406) antibodies were from R & D systems. Anti-ATF3 (C19; sc-188) was from Santa Cruz and anti-PARP (C2-10; 4338) was from Trevigen. Anti-β-tubulin (926-42211), anti-α-tubulin (926-42213), anti-β-actin (926-42210) and secondary antibodies; anti-mouse (926-32212), anti-rabbit (926-32213) and anti-goat (926-3214), were from LI-COR Biosciences. Bovine Serum Albumin (BSA), Actinomycin-D and cyclohexamide were purchased from Sigma.
Generation of reconstituted HDL
A reconstituted form of HDL (CSL-111, referred to as HDL in the text) with a protein to phospholipid ratio of 1:160, was generated as described previously43 and kindly provided by CSL Behring.
Generation of native HDL from human donors
Native HDL from sera of human donors was purified using a stepwise ultracentrifugation within a density of 1.063–1.21 g/ml as described previously44. The distribution and chemical composition of separated lipoproteins in human serum was followed by dialysis against PBS. Alternatively, native HDL was purchased from Meridian Life Science.
Animal models
TLR-induced inflammation and liver injury mouse model
Eight-week old C3H/HeJ female mice were injected intraperitoneally (i.p.) with 2 mg of native HDL or 500 μl control filtrate 6 hours before i.p. injection of 20 μg CpG DNA and 10 mg D-galactosamine (D-gal). C3H/HeJ mice (TLR4 mutant) were used to avoid the effects of endotoxin carried in native HDL. Six- to eight-week old C57BL/6 female mice were injected i.p. with 2 mg reconstituted HDL or 100 μl PBS for 6 h before i.p. injection of 20 μg CpG DNA and 10 mg D-gal. Six- to eight-week old Atf3-deficient mice and matched wild type (WT) mice were injected i.p. with 2 mg reconstituted HDL or 100 μl PBS for 6 h before i.p. injection of 30 μg CpG DNA and 10 mg D-gal. Blood was collected 1 h following CpG DNA/D-gal injection and mice were sacrificed after 10 h. At sacrifice livers were analyzed by haematoxylin and eosin staining and serum analyzed for aminotransferase (ALT) with DRI-CHEM4000 from Fujifilm. Cytokines were measured by Bio-Plex assay from BIO-RAD. These animal experiments were performed in accordance with the Teikyo University animal experiments committee of the Teikyo University School of Medicine and also in accordance with the animal ethics committee of Monash University.
Atherosclerosis mouse model
Eight-week old Apoe-deficient mice were fed a high fat diet (1.25% Cholesterol) for 8 weeks. During week 8, mice were injected intravenously (i.v.) two times with HDL (100 mg/kg) or PBS 4 days apart before the animals were sacrificed and livers collected for RNA isolation and mRNA analysis by qPCR as described below. These animal experiments were performed with approval from the Landesamt für Natur, Umwelt und Verbraucherschutz NRW and in accordance with German animal protection law.
Carotid injury mouse model
For carotid artery injury approximately 12-week old male WT and Atf3-deficient mice were anesthetized with i.p. injection of 150 mg/kg ketaminehydrochloride (Ketanest, Pharmacia) and 0.1 mg/kg xylazinehydrochloride (Rompun 2%, Bayer). A small incision from the cranial apex of the sternum to just below the mandible was made. After careful preparation of an approximately 6 mm long segment proximal of the bifurcation, the common carotid artery was electrically denuded. A 4 mm long lesion was made by applying two serial 5 second bursts of 2 Watt using 2 mm wide forceps. The skin was then sutured and the mice allowed to recover in individual cages before returning to their littermates. Three hours later the mice received a single 200 μl i.v. injection of 20 mg/kg HDL or PBS. After three days, 50 μl Evan’s blue solution was injected i.v. and allowed to circulate for 5 min. The mice were then sacrificed and both common carotid arteries fully excised. The arteries were rinsed in 0.9 % NaCl solution and the residual connective tissue carefully removed. Images were taken, and the total lesion area (4 mm) and remaining denuded area (stained blue) measured using AxioVision version 4.5.0 software (Zeiss). The percentage of re-endothelialization was calculated as the difference of blue stained area compared to the total injured area. These animal experiments were performed with approval from the Landesamt für Natur, Umwelt und Verbraucherschutz NRW and in accordance with German animal protection law.
Cells
Bone marrow-derived macrophages (BMDMs) were obtained by culturing bone marrow cells from 6- to 8-week old WT C57BL/6 mice in DMEM supplemented with 10% fetal calf serum (FCS), 10 μg/ml Ciprobay-500 and 40 ng/ml M-CSF (R & D Systems). Six days later, BMDMs were harvested and plated.. Age- and sex-matched WT control mice were used to derive BMDMs for comparative experiments with Atf3-deficient BMDMs. Immortalized BMDMs were cultured in DMEM supplemented with 10% FCS and 10 μg/ml Ciprobay-500. Human peripheral blood mononuclear cells (PBMCs) were purified from whole blood over Ficoll density gradient (GE Healthcare); erythrocytes were lysed in red cell lysis buffer (Miltenyi Biotec) and seeded in RPMI supplemented with 10% FCS and 10 μg/ml Ciprobay-500. Human CD14+ monocytes were isolated from PBMCs using CD14 microbeads (Miltenyi Biotec).
Isolation of Kupffer Cells and hepatocytes
Livers were perfused via the portal vein with a solution of 0.05% collagenase (Sigma-Aldrich), then mechanically disrupted, filtered through mesh (300 μm) and centrifuged at 300 rpm for 5 min. The hepatocyte cell pellet and the supernatant containing nonparenchymal liver cells were separated and washed once with Hank’s Balanced Salt Solution. Nonparenchymal liver cells were labeled with anti-CD11b antibody-labeled MACS microbeads and purified using immunomagnetic separation (Miltenyi).
Measurement of secreted cytokine levels by ELISA
Cytokines in cell-culture supernatants were measured by ELISA as per the manufacturer’s instructions. Murine IL-6, IL-12p40 and TNF, and human IL-6 and TNF ELISA kits were from R & D Systems; the human IFN-α ELISA kit was from eBiosciences.
Cell viability assay
Cell viability of BMDMs determined using CellTitre-Blue® reagent (Promega) according to the manufacturer’s instructions.
Fast Performance Liquid Chromatography (FPLC)
Approximately 2 nM of 3′ Alexa-647 labeled CpG DNA 1826 (Eurofins MWG Operon) or 40 μg of Bodipy-FL labeled LPS (Invitrogen), were incubated with either PBS or HDL (600 μg) for 2 h at 37°C. Samples were loaded onto a Superdex-200 size exclusion column (GE Healthcare) and absorbance was read at 260 nm (RNA/DNA), 280 nm (protein). Fluorescence absorbance was recorded at 505 nm and 647 nm for LPS and CpG, respectively.
Quantification of cellular cholesterol
BMDMs were lysed in 2x RIPA buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate) and cholesterol content was measured using the Amplex® Red Cholesterol Assay Kit (Invitrogen) according to manufacturer’s instructions.
Cell lysis and immunoblotting
Up to 1.5 × 106 BMDMs or CD14+ human monocytes were lysed on ice for 30 min with 1x RIPA buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate) supplemented with 0.1 μM PMSF, cOmplete protease inhibitors and PhosSTOP (Roche Biochemicals). Lysates were clarified by centrifugation at 13,000 × g for 10 min at 4°C and protein concentration measured by BCA assay (Pierce). Equal amount of protein per sample were then run on 4–12% pre-cast SDS-PAGE gels (Novex, Invitrogen) with MES or MOPS buffer (Novex, Invitrogen). Separated proteins were transferred onto PVDF membranes (Millipore) and blocked in 3% BSA in Tris Buffered Saline with Tween-20 before overnight incubation with specific primary antibodies. Membranes were then washed and incubated with the appropriate secondary antibodies, and immunoreactivity was observed by near-infrared detection (Odyssey, LI-COR).
Preparation of nuclear and cytoplasmic extracts
Up to 3 × 106 BMDMs were lysed in cytoplasmic lysis buffer (10 mM HEPES pH 7.4, 1.5 M MgCl2 and 10 mM KCl with 0.1% NP-40) supplemented with 0.1 μM PMSF and cOmplete protease inhibitors (Roche) before intact nuclei were separated from cytoplasmic content by centrifugation at 2000 × g for 10 min at 4°C. Supernatants were harvested as the ‘cytoplasmic extract’. Nuclei were washed with cytoplasmic lysis buffer, before being lysed in nuclear lysis buffer (20 mM HEPES pH 7.8, 420 mM NaCl, 20% glycerol, 0.2 mM EDTA and 1.5 mM MgCl2) and then centrifuged at >13,000 × g for 15 min at 4°C. The supernatant was then harvested as the ‘nuclear extract’. Protein content was determined by BCA assay (Pierce). Protein was then subjected to immunoblotting as described above.
Electromobility Shift Assay (EMSA)
Up to 3 × 106 BMDMs were gently lysed in buffer I (10 mM Tris pH 7.8, 5 mM MgCl2, 10 mM KCl, 1 mM EGTA pH7, 5% Sucrose with DTT, cOmplete protease inhibitors, 0.6% NP-40) and centrifuged at 2000 × g for 10 min at 4°C. The nuclei were then lysed in buffer II (20 mM Tris pH7.8, 5 mM MgCl2, 330 mM KCl and 200 μM EGTA pH7, 25% glycerol with DTT and protease inhibitors), centrifuged at >13,000 × g for 15 min at 4°C. The supernatant was then used as ‘nuclear extract’. Protein content of the nuclear extract was determined by BCA assay (Pierce) and 5 μg of nuclear extract was incubated with the IRDye-700 NF-κB consensus oligonucleotide (LI-COR) with Poly dI:dC, DTT and NP-40 from the Odyssey EMSA buffer kit as per the instructions. Samples were run on a 4% TBE native polyacrylamide gel and bands observed by near-infrared detection (Odyssey, LI-COR).
Quantification of sterols by mass spectrometry
Immortalized BMDMs grown in DMEM supplemented with 0.5% FCS and 10 μg/ml Ciprobay-500 were treated with HDL (2 mg/ml) for up to 6 h. Culture supernatants and cells were then subjected to Gas chromatography-mass spectrometry-selected ion monitoring (GC-MS-SIM) to determine cholesterol and cholesterol precursor levels as described elsewhere45, 46.
RNA isolation and generation of cDNA for analysis by SYBR Green qPCR
Up to 3 × 106 BMDMs, isolated Kupffer cells, hepatocytes or homogenised tissue samples were lysed and RNA isolated with RNeasy or miRNeasy mini kits according to the manufacturer’s protocol (Qiagen). Approximately 1 μg of isolated RNA from each sample was synthesized into cDNA using an oligo dT(18) primer and SuperScript™ III Reverse Transcriptase (Invitrogen). cDNA abundance was measured by quantitative real time PCR (qPCR) using the Maxima™ SYBR Green/ROX qPCR Master Mix (Fermentas) on a 7900T thermocycler (Applied Biosystems). The primer sequences were as follows:
| Gene: | Forward primer (5′ to 3′): | Reverse primer (5′ to 3′): |
|---|---|---|
| murine HPRT | TGAAGTACTCATTATAGTCAAGGGCA | CTGGTGAAAAGGACCTCTCG |
| human HRPT | TCAGGCAGTATAATCCAAAGATGGT | AGTCTGGCTTATATCCAACACTTCG |
| murine ATF3 | GAGCTGAGATTCGCCATCCA | CCGCCTCCTTTTCCTCTCAT |
| human ATF3 | CCAACCATGCCTTGAGGATAA | GGCAAGGTGCTGAAAATCCTT |
| murine IL-6 | CCAGAAACCGCTATGAAGTTCC | CGGACTTGTGAAGTAGGGAAGG |
| murine IL-12p40 | GGAAGCACGGCAGCAGAATA | AACTTGAGGGAGAAGTAGGAATGG |
| murine IL-1β | TTGACGGACCCCAAAAGATG | CAGCTTCTCCACAGCCACAA |
| murine Ch25h | TGACCTTCTTCGACGTGCTG | AGCCAAAGGGCACAAGTCTG |
| Murine TNF | TATGGCCCAGACCCTCACA | GGAGTAGACAAGGTACAACCCATC |
Primer specificity was checked by melt curve analysis. Expression of target genes was normalized to the expression of the housekeeping gene, HPRT. For fold change analysis data was transformed using the ‘Relative Standard Curve Method and Comparative CT Method (ΔΔCt)’ as described by Applied Biosystems.
RNA isolation for microarray
Up to 107 cells were lysed in 1 ml of TRIZOL reagent and stored at −80°C until further RNA isolation. A total of 0.2 ml chloroform was added to 1 ml of TRIZOL. The upper colorless aqueous phase was transferred into a new tube after centrifugation. RNA was precipitated by addition of 0.5 ml of isopropyl alcohol with a subsequent washing step in at least 1 ml of 75% ethanol. The precipitated RNA was dissolved in RNase free water. The RNA quality was assessed by measuring the ratio of absorbance at 260 nm and 280 nm using a Nanodrop 2000 Spectrometer (Thermo Scientific), as well as by visualization of the integrity of the 28S and 18S band on an agarose gel.
Genome-wide transcriptome assessment by microarray
Total RNA was purified using the MinElute Reaction Cleanup Kit (Qiagen) prior to array based gene expression profiling. By using the TargetAmp™ Nano-g™ Biotin-aRNA Labeling Kit for the Illumina System (Epicentre), biotin-labeled cRNA was generated. The biotin-labeled cRNA (1.5 μg) was hybridized to MouseWG-6 v2.0 Beadchips (Illumina) and scanned on an Illumina iScan system.
Data generation and bioinformatics of microarray data
Processing of raw intensity data was performed by BeadStudio 3.1.1.0 (Illumina). All experiments performed for this study are listed in Supplementary Table 3. Data were exported using the Partek report builder and imported as non-normalized data into Partek Genomics Suite V6.6 (PGS) (Partek). Data were then quantile normalized prior to further analysis. Differentially expressed (DE) genes between the different conditions, as well as transcripts with variable expression within the dataset were calculated using two- or three-way ANOVA models including batch correction. DE genes were defined by a fold change (FC) greater than 2, and a false discovery rate (FDR)-corrected p-value<0.05 unless stated otherwise. Principle component analysis (PCA) using all transcripts and default settings in PGS was utilized for visualization of sample relationships. Similarly, default settings within PGS were used for hierarchical clustering to visualize the sample and transcript relationships of the 1,000 most variable genes based on FDR-corrected p-values within the dataset. To link differential gene expression to biological processes involved, gene ontology- (GO-) and pathway-enrichment analysis were performed in PGS. Furthermore, for visualization of GO-enrichment, BINGO (http://www.psb.ugent.be/cbd/papers/BINGO/Home.html) combined with Enrichment Map (http://baderlab.org/Software/EnrichmentMap) were utilized as plugins implemented in Cytoscape (http://www.cytoscape.org). Heatmaps based on log2-transformed mean expression values were generated using MayDay (http://www-ps.informatik.uni-tuebingen.de/mayday/wp/). To identify potential transcriptional regulators, transcripts differentially expressed between CpG-stimulated BMDMs in the presence or absence of HDL were filtered using the TFCat database47 and their expression values (log2) visualized as a gene-ranked heatmap.
To predict genes directly targeted by ATF3 following HDL treatment, gene expression experiments were also performed in Atf3-deficient BMDMs. This allowed the identification of genes differentially regulated by HDL in WT but not in Atf3-deficient BMDMs, as potential direct targets of ATF3 downstream of HDL (see Figure 6a). In this model, genes were defined as ATF3 targets if they met the following three criteria; 1) TLR-modified genes being elevated (FC >1.5) or reduced (FC < −1.5) in expression by stimulation with either CpG or P3C, 2) TLR-modified genes showing reduced differences (up or down towards control) in the presence of HDL by at least 30%, and 3) these same genes showing a loss of the HDL effect in Atf3-deficient BMDMs.
Gene Set Enrichment Analysis (GSEA; http://www.broadinstitute.org/gsea/index.jsp) was used to determine whether the ATF3 target gene set is statistically significantly enriched in the carotid injury model from HDL treated wild-type mice (Enrichment score: 0.81; p-value 0.0; FDR: 0.0)48.
Transcription factor binding prediction
Transcription factor (TF) binding prediction was performed using the Genomatix software suite (http://www.genomatix.de) (Supplementary Figure 5b). Briefly, gene symbols of bait genes (most significantly downregulated genes (FC<-3; FDR.p<0.05) in BMDMs treated with CpG in the presence of HDL) were uploaded into the Genomatix module ‘Gene2Promoter’ to determine transcripts and promoters of these gene loci. Using standard settings, promoter sequences were compiled as a new sequencing file and further interrogated with the Genomatix module ‘common TF sites’ to determine TFs with enriched binding sites within the promoters of the chosen gene loci. For the 33 genes 108 promoter models (104 sequences, 70,162 bp) were identified by Genomatix search for common TF sites in multiple sequences and chosen for further analysis. Nine TF families representing 143 TF candidates revealed TF binding sites in at least 80% of the included promoter models. TF candidates were interrogated for expression as well as differential expression using the comparison of the transcriptome data (131/143 TFs covered) derived from BMDMs treated with CpG either in the presence or absence of HDL. Next, the percentage of expressed genes within the transcriptome dataset (23/131, 17.5%) was determined. Among those TFs, ATF3 was the only TF upregulated (FC 2.3) in macrophages stimulated with CpG in the presence of HDL (Figure 3f). As a second approach, TF modules on the 108 promoter models were computed using Genomatix FrameWorker, which is designed to extract a common framework of elements from a set of TF binding sites. As search criteria, TF modules containing CREB (containing ATF3) or ETSF (containing PU.1) binding sites were given. A total of 80 sequences, from 30 gene loci with 51,851 bp were interrogated. Significantly enriched (p<0.03) module hits (n=27) were identified covering 26/30 genes. The Genomatix module ‘Model inspector’ was utilized to assess the location of the generated TF modules. The large majority of hits contained combined CREB-ETFS (69%) modules, with the ETSF family containing the macrophage lineage TF PU.1 (Sfpi1). Calculation of the percentage of gene loci covering CREB-ETSF TF modules was performed in EXCEL.
ATF3 ChIP Sequencing (ChIP-seq)
ChIP for ATF3 was performed using the EZ-Magna ChIP G Kit (Millipore) according to the manufacturer’s instructions. In brief, three biological replicates of BMDMs from WT and Atf3-deficient mice were treated with HDL, CpG, HDL then CpG or left untreated. Cells were then cross-linked with 1% paraformaldehyde, before being lysed and chromatin sheared by sonication for 30 min with the Covaris S220. Immunoprecipitation was performed over night at 4°C using 2.5 μg of rabbit polyclonal anti-ATF3 antibody (C-19X, Santa Cruz). After washing, eluted samples were reverse cross-linked and RNaseA- and proteinase-K-treated as per the manufacturer’s recommendations. ChIP fragments were ligated to NEXTflex ChIP-Seq barcode adaptors using the NEXTflex ChIP-Seq Kit (both BioO Scientific). Adaptor ligated DNA fragments were size selected (150–250 bp), PCR amplified, further size selected (150–250 bp), pooled, and sequenced on an Illumina HiSeq-1000 sequencer according to the manufacturer’s instructions.
ChIP-Seq analysis
Reads were aligned to the UCSC mm9 reference mouse genome, using Bowtie 0.12.849. After ChIP-Seq quality control using HOMER v4.2 (http://biowhat.ucsd.edu/homer)50, reads of biological replicates were pooled, and peak identification was performed using MACS 1.4.2 with the samples from Atf3-deficient BMDMs set as unspecific background to identify specific ATF3 binding51. Afterwards, HOMER v4.2 was used to annotate the genomic location of identified peaks. To generate histograms for the distribution of tag densities, position-corrected tags in 5 bp windows were normalized using Java-genomics-toolkit (http://palpant.us/java-genomics-toolkit/) and the Atf3-deficient tags subtracted from the WT tags. For visualization, only enrichment in WT cells is depicted. Tag coverage of HDL-specific peaks relative to genomic TSS sites was calculated for WT and Atf3-deficient BMDMs for all conditions using HOMER v4.2.
Statistics
Data are typically presented as mean values ±S.E.M, where a p value of <0.05 was considered significant as determined by a two-tailed Student’s t test; or as described in individual figure legends. Analyses were performed with Excel (Microsoft), Prism (GraphPad Software, Inc.), or for microarray data with Partek Genomics Suite (Partek) using ANOVA-models.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We acknowledge Christoph Thiele (University of Bonn) for helpful discussions and Juan-Carlos Hernandez (University of Medellin) for help with experiments. We also thank Christine M. De Nardo for critical reading of the manuscript. The work was funded by grants from the NIH (1R01HL093262 to E.L., 1R01HL112661 to E.L. and M.L.F), the German Research foundation (SFB670) and Excellence Cluster ImmunoSensation to E.L. and J.L.S.
Footnotes
ACCESSION NUMBERS
All microarray data of this study where uploaded onto Gene Expression Omnibus (GEO) and can be accessed under GSE44034.
AUTHOR CONTRIBUTIONS
D.D, L.I.L, H.K, R.S, designed and performed experiments and analyzed data, S.V.S, D.X. F.A.S, J.V, A.K, M.Kr, N.B, A.G, C.L, S.Z, N.H performed experiments, S.V.S, M.B, T.U, W.K, and J.L.S, analyzed transcriptome and ChIP sequencing data, M.Kn. and M.R. provided the Atf3-deficient and matched WT control mice, D.L, M.L.F, B.W, P.K and S.D.W analyzed data and provided critical suggestions and discussions throughout the study, D.D, L.I.L, J.L.S and E.L. designed the study and along with S.D.W. wrote the paper.
COMPETING FINANCIAL INTERESTS
S.D.W is a paid employee of CSL Behring.
References
- 1.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 3.Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 4.Hai T, Wolford CC, Chang YS. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr. 2010;15:1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitmore MM, et al. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- 6.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seneviratne AN, Sivagurunathan B, Monaco C. Toll-like receptors and macrophage activation in atherosclerosis. Clin Chim Acta. 2012;413:3–14. doi: 10.1016/j.cca.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 9.Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. 2012;111:1079–1090. doi: 10.1161/CIRCRESAHA.111.258673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz GG, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 11.Boden WE, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 12.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 13.Khera AV, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 15.Murphy AJ, Westerterp M, Yvan-Charvet L, Tall AR. Anti-atherogenic mechanisms of high density lipoprotein: effects on myeloid cells. Biochim Biophys Acta. 2012;1821:513–521. doi: 10.1016/j.bbalip.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norata GD, Pirillo A, Ammirati E, Catapano AL. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis. 2012;220:11–21. doi: 10.1016/j.atherosclerosis.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 17.Barter PJ, et al. Antiinflammatory properties of HDL. Circ Res. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 18.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 19.Sparwasser T, et al. Macrophages sense pathogens via DNA motifs: induction of tumor necrosis factor-alpha-mediated shock. Eur J Immunol. 1997;27:1671–1679. doi: 10.1002/eji.1830270712. [DOI] [PubMed] [Google Scholar]
- 20.Ulevitch RJ, Johnston AR. The modification of biophysical and endotoxic properties of bacterial lipopolysaccharides by serum. J Clin Invest. 1978;62:1313–1324. doi: 10.1172/JCI109252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci U S A. 1993;90:12040–12044. doi: 10.1073/pnas.90.24.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wurfel MM, Hailman E, Wright SD. Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J Exp Med. 1995;181:1743–1754. doi: 10.1084/jem.181.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–3206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulton DL, et al. TFCat: the curated catalog of mouse and human transcription factors. Genome Biol. 2009;10:R29. doi: 10.1186/gb-2009-10-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horton JD, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimarino M, et al. Optical coherence tomography accurately identifies intermediate atherosclerotic lesions--an in vivo evaluation in the rabbit carotid artery. Atherosclerosis. 2007;193:94–101. doi: 10.1016/j.atherosclerosis.2006.08.047. [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P, et al. Vascular wound healing and neointima formation induced by perivascular electric injury in mice. Am J Pathol. 1997;150:761–776. [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmer S, et al. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ Res. 2011;108:1358–1366. doi: 10.1161/CIRCRESAHA.111.243246. [DOI] [PubMed] [Google Scholar]
- 31.Seetharam D, et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 32.Petoumenos V, Nickenig G, Werner N. High-density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J Cell Mol Med. 2009;13:4623–4635. doi: 10.1111/j.1582-4934.2008.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speer T, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity. 2013;38:754–768. doi: 10.1016/j.immuni.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Saemann MD, et al. The versatility of HDL: a crucial anti-inflammatory regulator. Eur J Clin Invest. 2010;40:1131–1143. doi: 10.1111/j.1365-2362.2010.02361.x. [DOI] [PubMed] [Google Scholar]
- 35.Michelsen KS, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim TW, et al. The critical role of IL-1 receptor-associated kinase 4-mediated NF-kappaB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J Immunol. 2011;186:2871–2880. doi: 10.4049/jimmunol.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gold ES, et al. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med. 2012;209:807–817. doi: 10.1084/jem.20111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yvan-Charvet L, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lerch PG, Spycher MO, Doran JE. Reconstituted high density lipoprotein (rHDL) modulates platelet activity in vitro and ex vivo. Thromb Haemost. 1998;80:316–320. [PubMed] [Google Scholar]
- 42.Murphy AJ, et al. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. 2008;28:2071–2077. doi: 10.1161/ATVBAHA.108.168690. [DOI] [PubMed] [Google Scholar]
- 43.Lerch PG, Fortsch V, Hodler G, Bolli R. Production and characterization of a reconstituted high density lipoprotein for therapeutic applications. Vox Sang. 1996;71:155–164. doi: 10.1046/j.1423-0410.1996.7130155.x. [DOI] [PubMed] [Google Scholar]
- 44.Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thelen KM, Laaksonen R, Paiva H, Lehtimaki T, Lutjohann D. High-dose statin treatment does not alter plasma marker for brain cholesterol metabolism in patients with moderately elevated plasma cholesterol levels. J Clin Pharmacol. 2006;46:812–816. doi: 10.1177/0091270006289851. [DOI] [PubMed] [Google Scholar]
- 46.Crameri A, et al. The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J. 2006;25:432–443. doi: 10.1038/sj.emboj.7600938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fulton DL, et al. TFCat: the curated catalog of mouse and human transcription factors. Genome Biol. 2009;10:R29. doi: 10.1186/gb-2009-10-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.