

Abstract
Continuous-flow mass spectrometry (CFMS) was used to monitor the products formed during the initial 0.25–20 s of the reactions catalyzed by the flavoprotein N-acetylpolyamine oxidase (PAO) and the pterin-dependent enzymes phenylalanine hydroxylase (PheH) and tyrosine hydroxylase (TyrH). N,N′-Dibenzyl-1,4-diaminobutane (DBDB) is a substrate for PAO for which amine oxidation is rate-limiting. CFMS of the reaction showed formation of an initial imine due to oxidation of an exo-carbon–nitrogen bond. Nonenzymatic hydrolysis of the imine formed benzaldehyde and N-benzyl-1,4-diaminobutane; the subsequent oxidation by PAO of the latter to an additional imine could also be followed. Measurement of the deuterium kinetic isotope effect on DBDB oxidation by CFMS yielded a value of 7.6 ± 0.3, in good agreement with a value of 6.7 ± 0.6 from steady-state kinetic analyses. In the PheH reaction, the transient formation of the 4a-hydroxypterin product was readily detected; tandem mass spectrometry confirmed attachment of the oxygen to C(4a). With wild-type TyrH, the 4a-hydroxypterin was also the product. In contrast, no product other than a dihydropterin could be detected in the reaction of the mutant protein E332A TyrH.
Identification of the product(s) of an enzyme-catalyzed reaction is essential to understanding both the catalytic mechanism of the enzyme and its role in metabolism. In many cases, the initial product is unstable, hindering structural characterization.1−4 Product identification in such cases requires a combination of chemical logic, characterization of stable compounds produced upon further reaction, and/or synthesis of the authentic product or an analogue. Two specific examples occur in the reactions of flavin amine oxidases and pterin-dependent hydroxylases. The former is a large group of enzymes that catalyze the oxidation of a C–N bond in the substrate to an imine;5 the imine typically hydrolyzes to form an aldehyde or ketone and a different amine as the stable products, the products that are routinely detected. The difficulty in detecting the initial imine product because of its instability raised the question of whether the hydrolysis is enzyme-catalyzed. The formation of an l-amino acid during turnover of d-amino acid oxidase in the presence of sodium borohydride provided the initial evidence of an imine as the product released by a flavin amine oxidase;6 this was subsequently confirmed by the use of a cyclic amine so that the imine product was stable.7 Direct detection of an unstable imine product required the use of an organic solvent for the reaction, thereby preventing hydrolysis of the imine.8 However, in the absence of evidence that an imine is always the product of flavoprotein-catalyzed amine oxidation, enzyme-catalyzed hydrolysis of the imine product is still being proposed for flavin amine oxidases.9−11
The pterin-dependent aromatic amino acid hydroxylases catalyze the incorporation of one atom of molecular oxygen into the side chain of the amino acid substrate and the other atom into the pterin substrate to form a 4a-hydroxypterin (4a-HO-pterin) (Scheme 1).12 The latter readily dehydrates in solution to form a quinonoid dihydropterin. The initial demonstration that the pterin product of these enzymes is a hydroxypterin was based on the similarity of the near-UV absorbance spectrum of the pterin product of the phenylalanine hydroxylase (PheH) reaction to that of a synthetic and stable 4a-hydroxy-5-deazapterin;13 further support for the structure of the product came from the 13C nuclear magnetic resonance (NMR) spectrum of the product formed from a specifically labeled tetrahydropterin at cryogenic temperatures.14 Species with similar absorbance spectra were subsequently identified as products in the reactions catalyzed by tyrosine hydroxylase (TyrH) and tryptophan hydroxylase.15,16 While effective with the wild-type enzymes, the use of absorbance spectra to identify the pterin products of these enzymes is less straightforward when it is applied to mutant enzymes in which multiple pterin products are formed. Although deconvolution of product spectra to quantitate the partitioning among different products can be successful,16,17 such an approach is problematic when the partitioning becomes complex. It is also not applicable in addressing the possibility that novel pterin products are formed.18
Scheme 1.

We describe here the use of continuous-flow mass spectrometry (CFMS) to characterize the initial products in the reactions catalyzed by the flavin-dependent amino oxidase N-acetylpolyamine oxidase (PAO) and by the pterin-dependent hydroxylases PheH and TyrH. Structurally, PAO is a member of the monoamine oxidase (MAO) family, a large family of flavoproteins that includes MAO A, MAO B, LSD1, and the l-amino acid oxidases.19 PAO catalyzes the oxidative cleavage of N1-acetylspermine and N1-acetylspermidine, yielding 3-acetamidopropanal and spermidine or putrescine.20 In the case of maize PAO, the reaction has been proposed to involve initial oxidation to an imine followed by its hydrolysis within the enzyme active site to form the final products.9N,N′-Dibenzyl-1,4-diaminobutane (DBDB) (Scheme 2) was selected as a substrate for our studies because oxidation of the amine is the rate-limiting step with it as the substrate for PAO,21 whereas product release is rate-limiting with physiological substrates.22 PheH was selected for the characterization of the product of a pterin-dependent enzyme because it was used in the initial demonstration of the 4a-HO-pterin; in addition, analysis of the TyrH reaction products was conducted with both the wild-type enzyme and a mutant protein in which tetrahydropterin oxidation and amino acid oxidation are uncoupled, so that the 4a-HO-pterin is not the exclusive product.
Scheme 2.

Experimental Procedures
Materials
6-Methyl-5,6,7,8-tetrahydropterin (6MPH4) was purchased from Schircks Laboratories (Jona, Switzerland). Isopropyl β-d-1-thiogalactopyranoside was from Research Products International (Mount Prospect, IL). Diethylenetriaminepentaacetic acid (DTPA), pepstatin A, l-phenylalanine, and l-tyrosine were purchased from Sigma-Aldrich (St. Louis, MO). Leupeptin was from Peptide Institute (Osaka, Japan). N,N′-Dibenzyl-1,4-diaminobutane (DBDB) was purchased from Prime Organics, Inc. (Woburn, MA). N,N′-Bis-perdeuterobenzyl-1,4-diaminobutane (DBDB-d14) was synthesized by the procedure of Henderson Pozzi et al.22
To synthesize ethylenediammonium diacetate (EDDA), acetic acid (71 mL, 2.4 equiv) was added dropwise to ethylenediamine (30.5 g) in 500 mL of dichloromethane at 4 °C. After the addition of acetic acid, solvent and excess acetic acid were removed from the precipitate by rotary evaporation at 30 °C. The off-white solid was washed with dichloromethane and dried under vacuum at room temperature to yield EDDA (97.6 g) as a fluffy, white solid: 1H NMR (600 MHz, D2O) δ 1.87 (6H, s), 3.30 (4H, s); 13C NMR (175 MHz, D2O) δ 22.76, 36.36, 180.78.
Expression and purification of His6-tagged mouse PAO were performed as described by Royo and Fitzpatrick,23 omitting the final chromatography on Sephacryl S-200. Rat phenylalanine hydroxylase lacking the N-terminal regulatory domain (PheH Δ117) was expressed and purified as described by Roberts et al.24 Expression and purification of wild-type and E332A rat TyrH were performed using the procedure of Daubner et al.25 with modifications. Bacterial cells were grown at 37 °C to an OD600 of 0.3; the temperature was then decreased to 20 °C. At an OD600 of 0.7–0.8, isopropyl β-d-1-thiogalactopyranoside was added to induce protein expression. After 21 h at 20 °C, the cells were harvested by centrifugation at 5000g for 30 min or 7500g for 15 min. The cell pellets were stored at −80 °C. Cells were resuspended in buffer containing 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 75 μM diethylenetriaminepentaacetic acid (DTPA), 100 μg/mL phenylmethanesulfonyl fluoride, 10% glycerol, 1 μM leupeptin, 1 μM pepstatin A, and 10% glycerol (pH 7.0) and sonicated. Nucleic acids were precipitated with streptomycin at a final concentration of 1%. The protein was precipitated by addition of ammonium sulfate to 45% saturation and resuspended in the same buffer. The purified protein was obtained by separation on a heparin-Sepharose column using a 0 to 0.8 M sodium chloride gradient. The purified protein was dialyzed against 50 mM HEPES, 100 mM sodium chloride, 20 mM nitrilotriacetic acid, 20 mM DTPA, 1 μM leupeptin, and 1 μM pepstatin A (pH 7.0) to remove bound iron. The chelators were removed by further dialysis against 50 mM HEPES, 100 mM sodium chloride, 1 μM leupeptin, and 1 μM pepstatin A (pH 7.0).
Assays
The oxidation of DBDB by PAO was followed directly in chemical-quench assays by monitoring the concentration of DBDB using a BioLogic (Claix, France) QFM-400 quenched-flow apparatus. Assays were performed by mixing 50 μL of 80 μM DBDB in 10% methanol and 50 mM ammonium acetate (pH 8.5) with 50 μL of 40 μM PAO in 50 mM ammonium acetate (pH 8.5) at 30 °C. N-Methylphenethylamine (MPEA) was included with DBDB at a concentration of 80 μM as an internal standard. Reactions were quenched with 50 μL of 2 M hydrochloric acid. Samples representing t0 were obtained by first mixing DBDB with 2 M hydrochloric acid and then adding PAO to the mixture. The quenched samples were centrifuged to remove protein; the resulting supernatants were diluted 10-fold with 0.1% trifluoroacetic acid in water before being injected onto a Phenomenex Gemini-NX C18 high-performance liquid chromatography (HPLC) column (3.0 μm, 2.0 mm × 150 mm). DBDB was eluted using an isocratic mobile phase of 0.1% trifluoroacetic acid and 17.5% acetonitrile in water and detected by fluorescence, with excitation at 255 nm and emission at 278 nm. The amount of DBDB was determined by comparison with a standard curve.
Steady-state deuterium isotope effects for the oxidation of DBDB by PAO were measured by monitoring oxygen consumption over a range of substrate concentrations using a Yellow Springs Inc. (Yellow Springs, OH) 5300A biological oxygen monitor. Assays contained 2.5–250 μM DBDB or DBDB-d14 and 1.6 μM PAO in 5% methanol and 50 mM ammonium acetate (pH 8.5) at 30 °C; the total reaction volume was 1 mL. Reactions were initiated by the addition of enzyme.
Continuous-Flow Mass Spectrometry
DBDB oxidation was monitored by high-resolution mass spectrometry using a Thermo Scientific (Waltham, MA) LTQ OrbiTrap Discovery mass spectrometer equipped with a custom nanoelectrospray ionization source incorporating a temperature-controlled nanovolume continuous-flow mixer (Eksigent, Dublin, CA). To minimize delay volumes, New Objective (Woburn, MA) SilicaTip emitters with 20 μm inside diameters and 10 μm tips were mounted directly onto the mixing chip. Reactions were performed by mixing the substrate and enzyme solutions at equal flow rates using the same conditions that were used for the chemical-quench assays. MPEA was included with the substrate as an internal standard at a final concentration after mixing of 40 μM. Flow rates ranging from 0.6 to 10 μL/min were used to obtain multiple reaction times. A sample corresponding to a reaction time of 0 s was obtained by mixing DBDB with buffer only. Product isotope measurements were taken using a mixture of 6.7 μM DBDB and 33 μM DBDB-d14. Reaction mixtures were monitored in spectral mode over the range of m/z 85–400. Substrates and reaction products were identified by their respective [M + H]+ ions with a mass error of <2 ppm. Each data point represents an average of at least 200 scans. Ion intensities were normalized against the intensity of MPEA at each flow rate.
For CFMS of pterin oxidation by PheH Δ117, 3 volumes of 25 μM PheH Δ117 and 2.7 mM l-phenylalanine in 50 mM ammonium acetate and 1% methanol (pH 7.0) was mixed with 1 volume of 4.0 mM 6MPH4 in 1 mM HCl and 1% methanol at 5 °C at a flow rate of 5.0 μL/min. Mass scanning of the reaction mixtures was performed over the range of m/z 100–300. Determination of MS/MS spectra of reaction species was performed at a rate of 1.0 μL/min.
CFMS assays of pterin oxidation by wild-type and E332A TyrH were performed by mixing an anaerobic solution of 20 μM enzyme and 500 μM tyrosine in 100 mM EDDA/ethylenediamine (pH 8.0) with an equal volume of an aerobic solution of 100 μM 6MPH4 in 25 mM ammonium chloride/HCl and 10% methanol (pH 3.0) at 5 °C. The enzyme/substrate mixture was prepared in several steps. The stock apoenzyme was exchanged into 100 mM EDDA/ethylenediamine (pH 8.0) using a Sephadex G-25 column (1.0 cm × 17.5 cm) and added to a tonometer containing tyrosine in the same buffer. A side arm containing 1 equiv of ferrous ammonium sulfate in 2 mM HCl was attached. The tonometer was made anaerobic by alternating vacuum and argon for 15 cycles. The contents were then mixed by gently inverting the contents into the side arm several times. Five additional vacuum–argon cycles were applied, and the tonometer was immediately mounted onto the CFMS system. Mass scanning of reactions was performed over the range of m/z 130–400.
Data Analysis
Kinetic data from individual experiments were fit using KaleidaGraph (Synergy Software, Reading, PA). KinTek Explorer26 (KinTek Corp., Austin, TX) was used to perform global analyses of multiple time-dependent data sets through singular-value decomposition to determine observed rate values. The FitSpace Explorer27 package of KinTek Explorer was used to estimate the confidence in the best-fit values by determining the sum square error for all pairs of parameters. The reported confidence intervals are the ranges of values for each parameter for which global fitting gives X2 values no more than 40% greater than the X2 value obtained using the best-fit values; this is termed a X2 threshold of 1.4.
Results and Discussion
Kinetics of Oxidation of DBDB
To utilize CFMS to analyze the kinetics of oxidation of DBDB by PAO, it was necessary to first calibrate the system. For continuous-flow methods, the reaction time at which the analysis occurs is determined by both the flow rate of the reactants and the volume between the mixer and the detector. Either can be varied to analyze the time course of reactions.28,29 In the continuous-flow approach used here, different reaction times were obtained by varying the flow rate. Calculation of the reaction times thus required knowledge of the volume (Vapp) from mixing until the reaction is quenched by desolvation upon electrospray (ESI). The value of Vapp can be determined by monitoring the progress of a reaction with established kinetics at various flow rates (Q), an approach that is commonly used to calibrate rapid-quench systems and determine dead times for stopped-flow instruments. Consequently, the kinetics of oxidation of DBDB by PAO were first analyzed by chemical-quench methods to determine the rate constant for the reaction. To do so, the enzyme was mixed with DBDB and the reaction was quenched at 0.25–8 s. The amount of DBDB remaining at each time was then determined by HPLC with fluorescence detection. Because the concentration of DBDB used was below its KM value under these conditions (50 μM), the time-dependent loss of DBDB (Figure 1) could be fit reasonably well as an exponential decrease (eq 1), yielding a value for the observed rate constant (kobs) for DBDB oxidation of 0.14 ± 0.01 s–1.a
| 1 |
Figure 1.

Time course for the oxidation of DBDB by PAO: (●) chemical-quench assays and (□) continuous-flow mass spectrometry. The reaction conditions were 40 μM DBDB, 20 μM polyamine oxidase, 50 mM ammonium acetate, 5% methanol, pH 8.5, and 30 °C. All concentrations are after mixing. The line indicates the best-fit curve for the chemical-quench data to eq 1.
Next, DBDB and PAO were reacted in the CFMS system under conditions identical to those used for the chemical-quench assays, varying the flow rate to obtain multiple reaction times. As with other mass spectrometry methods, the quantification of ions of interest by CFMS requires normalization against an internal standard. However, quantification using ESI is further complicated by the observation that the intensity of an ion signal varies somewhat with the concentration of the enzyme and the flow rate. In our hands, high concentrations of enzyme decrease the ion intensity, likely because of an increase in the viscosity and surface tension of the reaction mixture, which disrupt the desolvation process of the electrospray. The flow rate dependence of the ion intensity is the result of variations in droplet size and the extent of desolvation.30,31 We have found that this flow-dependent signal variation is ion-specific, such that the signal ratios for two ions may vary significantly at different flow rates. To correct for this, several analogues of DBDB were tested as internal standards, including N,N′-dibenzyl-1,2-diaminoethane, diphenylamine, lysine, and N-methylphenethylamine. N-Methylphenethylamine (MPEA) gave the behavior most consistent with that of DBDB across a range of flow rates, and it is not a substrate for PAO. Consequently, the concentration of DBDB remaining upon detection at each flow rate was determined by normalizing the ion signal against that for MPEA as an internal standard and the known concentration of DBDB at t0.
The reaction time for a continuous-flow analysis is proportional to the inverse of flow rate Q (eq 2).
| 2 |
The value of Vapp can be determined by combining eqs 1 and 2 and substituting in the value for kobs from the chemical-quench experiment (eq 3).
| 3 |
This approach yielded a value for Vapp of 100 ± 20 nL for the mixer used for the experiments described here. Figure 1 shows an overlay of the data from the rapid-quench and MS analyses using this Vapp value to determine the reaction times. A similar analysis conducted with a second mixer yielded a value of 210 ± 20 nL for Vapp, demonstrating the necessity of determining Vapp upon any change in the experimental system.
Products of the Oxidation of DBDB by PAO
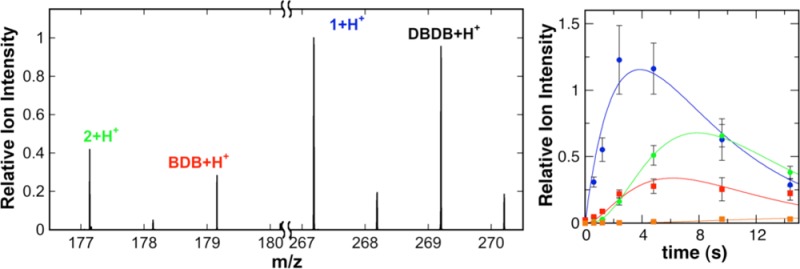
As noted above, a major advantage of CFMS in following reactions is the ability to gain structural insight into transient reactant products. When the oxidation of DBDB was monitored by scanning the range of m/z 85–400, several additional ions were seen during the reaction (Figure 2). The intensities of these ions varied with reaction time in a manner consistent with them being reaction products (Figure 3). Ion 1 was seen at the earliest reaction time (0.6 s) and had an m/z value of 267.1855. This is exactly two hydrogen atoms less than the m/z value for the [M + H]+ ion of DBDB (m/z 269.2012). The simplest possibility for such a species is that it is an imine (m/z 267.1856) formed by abstraction of two protons and two electrons from DBDB (Scheme 3). The magnitude of the signal for 1 increased over the first few seconds of the reaction and then slowly decayed (Figure 3), indicating that 1 is not the final product in the reaction. Consistent with such a conclusion, the magnitude of the signal for an additional species with an m/z value of 179.1540 increased more gradually over the first 5 s of the reaction (Figures 2 and 3). This mass is consistent with the [M + H]+ ion of N-benzyl-1,4-diaminobutane (BDB, m/z 179.1543). As BDB is the expected product in the hydrolysis of an exo-imine formed from DBDB (Scheme 3), 1 can be identified as N-benzyl-N′-benzylidene-1,4-diaminobutane. No significant signal was observed for the ion of benzylamine or N-benzyl-4-aminobutanal, ruling out the endo-imine of DBDB as the major enzymatic product. The intensity of the signal for BDB decayed at later times, and that of an additional ion (2) at m/z 177.1384 increased (Figure 3). The m/z value of the latter ion is exactly two hydrogen atoms less than the signal for BDB, indicating that it represents the [M + H]+ ion (m/z 177.1386) of a dehydrogenation product of BDB. The fact that 2 derives from BDB and not DBDB is supported by the observation of a lag phase in its formation. Similar to 1, 2 can be reasonably assigned as N-benzylidene-1,4-diaminobutane, the exo-imine of BDB (Scheme 3). This establishes that BDB is also a substrate for PAO. As with 1, the magnitude of the signal for 2 decreased with time; this decay was accompanied by the slow increase after several seconds in the magnitude of a signal at m/z 89.1072 (Figure 3). This matches well the [M + H]+ ion of putrescine (m/z 89.1073), consistent with hydrolysis of 2 (Scheme 3). Benzaldehyde must be an additional product in the hydrolyses of 1 and 2 to BDB and putrescine, respectively. However, benzaldehyde is not readily ionized under ESI conditions, and no corresponding ion was observed. Overall, the oxidation of DBDB by PAO can be described by the pathway in Scheme 3. As ESI-MS is sufficiently gentle to keep multimeric protein complexes intact,32−35 reaction products that have not dissociated from the enzyme are not expected to be detectable as individual ions by CFMS. The direct observation of both 1 and 2 in these assays indicates that the imine products are released from the enzyme with their subsequent hydrolysis occurring in solution.
Figure 2.

Representative mass spectrum from the experiment in Figure 1 at a reaction time of 4.8 s. The spectrum represents the average of 250 scans at a flow rate of 1250 nL/min. Ion signals are normalized to the most intense ion ([1 + H]+).
Figure 3.

Time dependence for products of the PAO-catalyzed oxidation of DBDB. The data are reported as the ion intensities for 1 (●), BDB (■), 2 (○), and putrescine (□) relative to that of MPEA. Assay conditions are identical to those described in the legend of Figure 1. The lines are from global fits of the model in Scheme 4 with the values for the rate constants listed in Table 1.
Scheme 3.

To obtain the values of the rate constants for the steps in Scheme 3, the kinetics of formation and decay of the MS signals for DBDB and the different reaction products were analyzed globally using KinTek Explorer.26 A simple four-step model (Scheme 4) was sufficient to fit the complete reaction (Figure 3). In the first step, DBDB is oxidized to 1 by PAO. The second step is the nonenzymatic hydrolysis of the free imine 1 to BDB. The two subsequent steps arise from turnover with BDB as the substrate. The rate constants for the two hydrolysis steps (kH2O) were assumed to be identical in the analysis, because allowing their rate constants to vary independently did not significantly improve the fit and gave similar values for all rate constants. The values from the analysis are listed in Table 1.
Scheme 4.
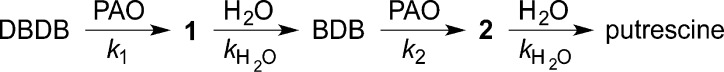
Table 1. Kinetic Parameters for the Kinetic Mechanism in Scheme 4a.
| k1 | 6.0 mM–1 s–1 (4.9–7.1) |
| k2 | 33 mM–1 s–1 (26–62) |
| kH2O | 0.52 s–1 (0.43–0.62) |
Values for the rate constants were determined globally with KinTek Explorer and the data from Figures 1 and 3. The values in parentheses are the confidence intervals reported by FitSpace at a X2 threshold of 1.4, that is, the change in the value that results in an increase in X2 of 40% if all of the other parameters are allowed to vary to improve the fit.
Because these analyses were conducted at a low concentration of DBDB, rate constants k1 and k2 equal the kcat/KM values for DBDB and BDB, respectively. The value for BDB is ∼5-fold greater than that for DBDB, consistent with the former being the better substrate. These results do not allow us to distinguish whether the differences are due to a difference in binding or chemistry. For oxidation of a polyamine, PAO requires that the reacting nitrogen be neutral and that a different nitrogen be charged.22 The primary nitrogen in BDB would have a pKa value higher than that of the benzylic nitrogens in DBDB or BDB, so that more of the latter would be correctly protonated at the pH of these analyses, 8.5. In addition, because the active sites of polyamine oxidases are appropriate for linear saturated substrates,36,37 the absence in BDB of the bulky and rigid second aromatic ring present in DBDB may result in better binding of the former in the active site. As the rates of product release do not contribute to kcat/KM, differences in the rate constants for product release are not responsible for the differences.
Kinetic Isotope Effects for the Oxidation of DBDB
The ability of the CFMS system to directly monitor substrate and products during the oxidation of DBDB by PAO suggested that it should be possible to directly measure the isotope effect on DBDB oxidation. Deuterium isotope effects on steady-state kinetic parameters were first determined by noncompetitive initial rate assays. Rates of oxygen consumption were measured with either DBDB or N,N′-bis-perdeuterobenzyl-1,4-diaminobutane (DBDB-d14) as the substrate for PAO, using conditions similar to those used for the MS-based assays. The data were well fit using eq 4, which applies for an identical isotope effect (Dk) on the kcat and kcat/KDBDB values, yielding an isotope effect of 6.7 ± 0.6.
| 4 |
Alternative analyses assuming different isotope effects on the kcat and kcat/KDBDB values did not yield improved fits. This result is consistent with rate-limiting chemistry with this substrate, as previously concluded.22
The isotope effect was then measured by CFMS. A mixture of DBDB and DBDB-d14 was used as the substrate for PAO, again following the products over time. The initial concentration of DBDB-d14 was ∼5-fold greater than that for the undeuterated substrate to compensate for the decreased rate of formation of the deuterated product due to the large isotope effect measured in the steady-state assays. Figure 4 shows the time dependence for the observed isotope effect on the formation of 1; the value at time zero yields an intrinsic isotope effect of 7.7 ± 0.3. This value is similar to the value for Dk determined in the steady-state assays and more precise. Because of the large isotope effect on imine formation, ion intensities for 2 were too low to perform a similar analysis for the oxidation of BDB in the second turnover.
Figure 4.

Measurement of the deuterium isotope effect for oxidation of DBDB by PAO determined by continuous-flow mass spectrometry. The relative intensities of the ions for deuterated (d13) and undeuterated (h13) 1 were determined at different flow rates. Reaction mixtures contained 6.7 μM DBDB, 33 μM DBDB-d14, and 40 μM PAO in 5% methanol and 50 mM ammonium acetate (pH 8.5) at 30 °C. All concentrations are after mixing. The line indicates the best fit to a single exponential.
Pterin Products of the Aromatic Amino Acid Hydroxylases
The CFMS system was also used to characterize the pterin products produced by wild-type aromatic amino acid hydroxylases and a mutant enzyme. Initial analyses used PheH Δ117, a truncated enzyme that lacks the N-terminal regulatory domain and thus no longer exhibits allostery, simplifying the kinetics.38,39 In reactions with 6MPH4 and l-phenylalanine as substrates, the ion for tyrosine (m/z 182.0811), the amino acid product of the reaction, could be resolved from that of 6MPH4 (m/z 182.1036), despite their very similar masses (Figure 5A, inset). In addition, two ions that could be identified as pterin oxidation products were clearly seen (Figure 5A). One had an m/z value of 180.0877, exactly two hydrogen atoms less than that for 6MPH4, indicating that this species is the [M + H]+ ion of a dihydropterin [6MPH2, m/z 180.0880 (Scheme 1)]. The second ion had an m/z value of 198.0982, greater than that of 6MPH4 by the mass of a single oxygen atom. The likely candidate for this product is the unstable 4a-hydroxy-6-methyltrihydropterin (4a-HO-pterin, m/z 198.0986), formed during the reaction by the incorporation of one atom of molecular oxygen at position 4a of 6MPH4 (Scheme 1).
Figure 5.

(A) Mass spectrum for the reaction of 20 μM PheH Δ117, 2.0 mM l-phenylalanine, and 1.0 mM 6MPH4 at a reaction time of 1.2 s. All concentrations are after mixing. The spectrum is the average of 20 scans. Ion signals are normalized to the most intense ion ([6MPH4 + H]+). The inset is a close-up of the spectrum across the range of m/z 182.07–187.12 showing the resolved peaks for l-tyrosine (Tyr) and 6MPH4. (B) MS/MS spectrum of the 4a-HO-pterin product (m/z 198.0982) from the reaction with PheH Δ117. Reaction conditions were identical to those used for panel A at a reaction time of 6 s. The spectrum is the average of 10 scans. Ion signals are normalized to the most intense ion ([M – CO2]+).
The site of attachment of the hydroxyl moiety in the 4a-hydroxypterin product of the aromatic amino acid hydroxylases was previously established by analysis of the NMR spectrum of the product formed under cryogenic conditions from 6MPH4 that had been specifically labeled with 13C at position 4a.14 The ability to directly detect the HO-pterin by mass spectrometry allowed us to confirm this conclusion with unlabeled substrates at ambient temperatures. The MS/MS spectrum of the putative 4a-HO-pterin (Figure 5B) showed three major product ions at m/z 170.1035, 154.1087, and 86.0348. The first two ions likely result from a fragmentation pathway wherein the pyrimidine ring contracts into an imidazole, resulting in the loss of the carbonyl carbon as CO or CO2 (Scheme 5). The observation that this contraction occurs both with and without loss of the second oxygen atom suggests that the additional oxygen atom is in the proximity of the carbonyl. The third product ion, [M – C5H8N2O]+, is generated by the loss of the methylpiperazine ring and an oxygen atom, indicating that the oxygen atom incorporated during the reaction is bound to the piperazine ring. These observations confirm the incorporation of oxygen at position 4a, as this position is a member of the piperazine ring and adjacent to the carbonyl. Consistent with this conclusion, the same product ion at m/z 86.0348 ([M – C5H8N2]+) was also present in the MS/MS spectrum of 6MPH4 (not shown); in this case, it would be formed by loss of the methylpiperazine alone from the substrate lacking the second oxygen atom.
Scheme 5.

A similar analysis of the reaction of wild-type TyrH with 6MPH4 and l-tyrosine was conducted. Again, two pterin products with m/z values consistent with the dihydropterin and the 4a-HO-pterin were readily detectable (Figure 6A), confirming directly the latter as a product of the reaction catalyzed by this enzyme. The magnitude of the relative signal for 6MPH2 increased at longer reaction times, quickly becoming the dominant signal, while the magnitude of the signal for 6MPH4 decayed as the reaction progressed. In contrast, the magnitude of the signal for the 4a-HO-pterin decreased with time, indicating that this product dehydrates to give the dihydropterin.
Figure 6.

Mass spectrum for the reaction of 10 μM wild-type (A) or E332A (B) TyrH, 250 μM l-tyrosine, and 50 μM 6MPH4 at a reaction time of 1.5 s. All concentrations are after mixing. The spectra are averages of 150 scans. Ion signals are normalized to the most intense ion.
The mutant enzyme E332A TyrH shows almost complete uncoupling of tetrahydropterin oxidation from amino acid hydroxylation.25 Spectroscopic studies of this protein suggested that both the amino acid and the pterin substrate bind normally and convert the iron to the same oxygen-reactive form that is found in the wild-type enzyme18,40 but did not establish the point in the subsequent reaction at which uncoupling occurred. Stopped-flow absorbance spectroscopy was previously used to obtain the absorbance spectrum of the pterin product(s) to clarify the step at which the reaction became unproductive. Comparison of the spectrum with those of known pterins suggested that a pterin distinct from the 4a-HO-pterin was formed in the reaction, possibly a hydroperoxypterin.18 CFMS was used to more definitively identify the pterin product(s) produced by E332A TyrH. The mass spectrum of the reaction mixture did not show evidence of the formation of the 4a-HO-pterin (Figure 6B). Instead, the dihydropterin (6MPH2) was the only pterin product detected. It is not possible to determine from the mass spectrum which tautomer of 6MPH2 forms, but the near-UV absorbance spectrum of the reaction exhibits absorbance between 300 and 400 nm consistent with a significant amount of quinonoid 6MPH2.18 The lack of a signal for a hydroperoxypterin intermediate suggests that it did not form, and that the enzyme oxidizes 6MPH4 directly to 6MPH2. However, we cannot rule out the possibility that the hydroperoxypterin is too unstable to detect even at these short times. The absence of a signal for the 4a-HO-pterin establishes that the E332A TyrH reaction does not catalyze the appropriate heterolytic cleavage of molecular oxygen to generate the Fe(IV)O. Our present understanding of the mechanism of the pterin-dependent hydroxylases is shown in Scheme 6. The altered binding of the pterin due to mutagenesis of Glu332 alters the initial reaction of the enzyme with oxygen, resulting in direct oxidation of the tetrahydropterin, or the mutation prevents the heterolytic cleavage of the oxygen–oxygen bond in the initial intermediate. The previous proposal of Chow et al.18 that Glu332 is responsible for protonation of one of the oxygen atoms to facilitate oxygen bond cleavage would be consistent with the results presented here.
Scheme 6.

Conclusion
The data presented here establish the utility of continuous-flow mass spectrometry for detecting unstable products of enzyme-catalyzed reactions. In contrast to previous methods for isolating imine products of reactions by flavoproteins, which relied on the formation of stable imine products or anhydrous conditions, this approach established directly that an unstable imine is the product of the reaction catalyzed by PAO and allowed measurement of the deuterium kinetic isotope effect for the formation of the imine. It also allowed analysis of the kinetics of further oxidation of the enzymatic products in the reaction. In the reactions of the pterin-dependent enzymes, CFMS definitively identified the unstable 4a-HO-pterin proposed as a product for both PheH and TyrH and ruled out the 4a-HO-pterin as a significant product in the reaction of a mutant TyrH.
Glossary
Abbreviations
- PheH
phenylalanine hydroxylase
- PheH Δ117
phenylalanine hydroxylase lacking the N-terminal regulatory domain
- TyrH
tyrosine hydroxylase
- CFMS
continuous-flow mass spectrometry
- PAO
N-acetylpolyamine oxidase
- MAO
monoamine oxidase
- DBDB
N,N′-dibenzyl-1,4-diaminobutane
- DBDB-d14
N,N′-bis-perdeuterobenzyl-1,4-diaminobutane
- 6MPH4
6-methyl-5,6,7,8-tetrahydropterin
- 6MPH2
6-methyldihydropterin
- 4-HO-pterin
4a-hydroxy-6-methyltrihydropterin
- DTPA
diethylenetriaminepentaacetic acid
- EDDA
ethylenediammonium diacetate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ESI
electrospray ionization
- MPEA
N-methylphenethylamine
- BDB
N-benzyl-1,4-diaminobutane.
The authors declare no competing financial interest.
This work was supported in part by grants from the National Institutes of Health (R01 GM058698) and The Welch Foundation (AQ1245).
Funding Statement
National Institutes of Health, United States
Footnotes
The single-exponential fit is not meant to imply a pseudo-first-order condition of the assay but simply represents a descriptive reflection of the data that can be used as a calibration for the CFMS system.
References
- Stumpe M.; Feussner I. (2006) Formation of oxylipins by CYP74 enzymes. Phytochem. Rev. 5, 347–357. [Google Scholar]
- Fenoll L. G.; Rodríguez-López J. N.; García-Sevilla F.; García-Ruiz P. A.; Varón R.; García-Cánovas F.; Tudela J. (2001) Analysis and interpretation of the action mechanism of mushroom tyrosinase on monophenols and diphenols generating highly unstable o-quinones. Biochim. Biophys. Acta 1548, 1–22. [DOI] [PubMed] [Google Scholar]
- Deery E.; Schroeder S.; Lawrence A. D.; Taylor S. L.; Seyedarabi A.; Waterman J.; Wilson K. S.; Brown D.; Geeves M. A.; Howard M. J.; Pickersgill R. W.; Warren M. J. (2012) An enzyme-trap approach allows isolation of intermediates in cobalamin biosynthesis. Nat. Chem. Biol. 8, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Moore I. F.; Koteva K. P.; Bareich D. C.; Hughes D. W.; Wright G. D. (2004) TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J. Biol. Chem. 279, 52346–52352. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick P. F. (2010) Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 493, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner D. W.; Wellner D. (1971) Demonstration of imino acids as products of the reactions catalyzed by d- and l-amino acid oxidases. Proc. Natl. Acad. Sci. U.S.A. 5, 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick P. F.; Massey V. (1982) Thiazolidine-2-carboxylic acid, an adduct of cysteamine and glyoxylate, as a substrate for d-amino acid oxidase. J. Biol. Chem. 257, 1166–1171. [PubMed] [Google Scholar]
- Woo J. C. G.; Silverman R. B. (1995) Monoamine oxidase B catalysis in low aqueous medium. Direct evidence for an imine product. J. Am. Chem. Soc. 117, 1663–1664. [Google Scholar]
- Binda C.; Coda A.; Angelini R.; Federico R.; Ascenzi P.; Mattevi A. (1999) A 30 Å long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure 7, 265–276. [DOI] [PubMed] [Google Scholar]
- Pawelek P. D.; Cheah J.; Coulombe R.; Macheroux P.; Ghisla S.; Vrielink A. (2000) The structure of l-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J. 19, 4204–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachalova G.; Decker K.; Holt A.; Bartunik H. D. (2011) Crystallographic snapshots of the complete reaction cycle of nicotine degradation by an amine oxidase of the monoamine oxidase (MAO) family. Proc. Natl. Acad. Sci. U.S.A. 108, 4800–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick P. F. (2000) The aromatic amino acid hydroxylases. In Advances in Enzymology and Related Areas of Molecular Biology (Purich D. L., Ed.) pp 235–294, John Wiley & Sons, Inc., New York. [DOI] [PubMed] [Google Scholar]
- Lazarus R. A.; Dietrich R. F.; Wallick D. E.; Benkovic S. J. (1981) On the mechanism of action of phenylalanine hydroxylase. Biochemistry 20, 6834–6841. [DOI] [PubMed] [Google Scholar]
- Lazarus R. A.; DeBrosse C. W.; Benkovic S. J. (1982) Phenylalanine hydroxylase: Structural determination of the tetrahydropterin intermediates by 13C NMR spectroscopy. J. Am. Chem. Soc. 104, 6869–6871. [Google Scholar]
- Dix T. A.; Kuhn D. M.; Benkovic S. J. (1987) Mechanism of oxygen activation by tyrosine hydroxylase. Biochemistry 26, 3354–3361. [DOI] [PubMed] [Google Scholar]
- Moran G. R.; Derecskei-Kovacs A.; Hillas P. J.; Fitzpatrick P. F. (2000) On the catalytic mechanism of tryptophan hydroxylase. J. Am. Chem. Soc. 122, 4535–4541. [Google Scholar]
- Ellis H. R.; Daubner S. C.; Fitzpatrick P. F. (2000) Mutation of serine 395 of tyrosine hydroxylase decouples oxygen-oxygen bond cleavage and tyrosine hydroxylation. Biochemistry 39, 4174–4181. [DOI] [PubMed] [Google Scholar]
- Chow M. S.; Eser B. E.; Wilson S. A.; Hodgson K. O.; Hedman B.; Fitzpatrick P. F.; Solomon E. I. (2009) Spectroscopy and kinetics of wild-type and mutant tyrosine hydroxylase: Mechanistic insight into O2 activation. J. Am. Chem. Soc. 131, 7685–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaweska H.; Fitzpatrick P. F. (2011) Structures and mechanism of the monoamine oxidase family. Biomol. Concepts 2, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; Yankovskaya V.; McIntire W. S. (2003) Cloning, sequencing, and heterologous expression of the murine peroxisomal flavoprotein, N1-acetylated polyamine oxidase. J. Biol. Chem. 278, 20514–20525. [DOI] [PubMed] [Google Scholar]
- Henderson Pozzi M.; Gawandi V.; Fitzpatrick P. F. (2009) pH dependence of a mammalian polyamine oxidase: Insights into substrate specificity and the role of lysine 315. Biochemistry 48, 1508–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson Pozzi M.; Gawandi V.; Fitzpatrick P. F. (2009) Mechanistic Studies of para-Substituted N,N′-Dibenzyl-1,4-diaminobutanes as Substrates for a Mammalian Polyamine Oxidase. Biochemistry 48, 12305–12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo M.; Fitzpatrick P. F. (2005) Mechanistic studies of mouse polyamine oxidase with N1,N12-bisethylspermine as a substrate. Biochemistry 44, 7079–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. M.; Pavon J. A.; Fitzpatrick P. F. (2013) Kinetic Mechanism of Phenylalanine Hydroxylase: Intrinsic Binding and Rate Constants from Single-Turnover Experiments. Biochemistry 52, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubner S. C.; Fitzpatrick P. F. (1999) Site-directed mutants of charged residues in the active site of tyrosine hydroxylase. Biochemistry 38, 4448–4454. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. (2009) Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. (2009) FitSpace Explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41. [DOI] [PubMed] [Google Scholar]
- Zechel D. L.; Konermann L.; Withers S. G.; Douglas D. J. (1998) Pre-steady state kinetic analysis of an enzymatic reaction monitored by time-resolved electrospray ionization mass spectrometry. Biochemistry 37, 7664–7669. [DOI] [PubMed] [Google Scholar]
- Li Z.; Sau A. K.; Furdui C. M.; Anderson K. S. (2005) Probing the role of tightly bound phosphoenolpyruvate in Escherichia coli 3-deoxy-d-manno-octulosonate 8-phosphate synthase catalysis using quantitative time-resolved electrospray ionization mass spectrometry in the millisecond time range. Anal. Biochem. 343, 35–47. [DOI] [PubMed] [Google Scholar]
- Hopfgartner G.; Wachs T.; Bean K.; Henion J. (1993) High-flow ion spray liquid chromatography/mass spectrometry. Anal. Chem. 65, 439–446. [Google Scholar]
- Page J. S.; Kelly R. T.; Tang K.; Smith R. D. (2007) Ionization and transmission efficiency in an electrospray ionization-mass spectrometry interface. J. Am. Soc. Mass Spectrom. 18, 1582–1590. [DOI] [PubMed] [Google Scholar]
- Sobott F.; Robinson C. V. (2004) Characterising electrosprayed biomolecules using tandem-MS: The noncovalent GroEL chaperonin assembly. Int. J. Mass Spectrom. 236, 25–32. [Google Scholar]
- Blackwell A. E.; Dodds E. D.; Bandarian V.; Wysocki V. H. (2011) Revealing the quaternary structure of a heterogeneous noncovalent protein complex through surface-induced dissociation. Anal. Chem. 83, 2862–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgner N.; Montenegro F.; Barrera N. P.; Robinson C. V. (2012) Mass spectrometry: From peripheral proteins to membrane motors. J. Mol. Biol. 423, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R. J.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. (2012) High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods 9, 1084–1086. [DOI] [PubMed] [Google Scholar]
- Binda C.; Angelini R.; Federico R.; Ascenzi P.; Mattevi A. (2001) Structural bases for inhibitor binding and catalysis in polyamine oxidase. Biochemistry 40, 2766–2776. [DOI] [PubMed] [Google Scholar]
- Adachi M. S.; Taylor A. B.; Hart P. J.; Fitzpatrick P. F. (2012) Mechanistic and structural analyses of the roles of active site residues in yeast polyamine oxidase Fms1: Characterization of the N195A and D94N enzymes. Biochemistry 51, 8690–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubner S. C.; Hillas P. J.; Fitzpatrick P. F. (1997) Expression and characterization of the catalytic domain of human phenylalanine hydroxylase. Arch. Biochem. Biophys. 348, 295–302. [DOI] [PubMed] [Google Scholar]
- Daubner S. C.; Hillas P. J.; Fitzpatrick P. F. (1997) Characterization of chimeric pterin-dependent hydroxylases: Contributions of the regulatory domains of tyrosine and phenylalanine hydroxylase to substrate specificity. Biochemistry 36, 11574–11582. [DOI] [PubMed] [Google Scholar]
- Krzyaniak M. D.; Eser B. E.; Ellis H. R.; Fitzpatrick P. F.; McCracken J. (2013) A pulsed EPR study of amino acid and tetrahydropterin binding in a tyrosine hydroxylase nitric oxide complex: Evidence for substrate rearrangements in formation of the oxygen-reactive complex. Biochemistry 52, 8430–8441. [DOI] [PMC free article] [PubMed] [Google Scholar]