

Abstract
Objective
Thymocyte apoptosis is a major event in sepsis; however, how this process is regulated remains poorly understood.
Approach and Results
Septic stress induces glucocorticoids (GC) production which triggers thymocyte apoptosis. Here, we used scavenger receptor BI (SR-BI) null mice, which are completely deficient in inducible GC (iGC) in sepsis, to investigate the regulation of thymocyte apoptosis in sepsis. Cecal ligation and puncture (CLP) induced profound thymocyte apoptosis in SR-BI+/+ mice, but no thymocyte apoptosis in SR-BI−/− mice due to lack of iGC. Unexpectedly, supplementation of GC only partly restored thymocyte apoptosis in SR-BI−/− mice. We demonstrated that HDL is a critical modulator for thymocyte apoptosis. SR-BI+/+ HDL significantly enhanced GC-induced thymocyte apoptosis but SR-BI−/− HDL had no such activity. Further study revealed that SR-BI+/+ HDL modulates GC-induced thymocyte apoptosis via promoting glucocorticoid receptor translocation, but SR-BI−/− HDL loses such regulatory activity. To understand why SR-BI−/− HDL loses its regulatory activity, we analyzed HDL cholesterol contents. There was 3-fold enrichment of unesterified cholesterol in SR-BI−/− HDL compared with SR-BI+/+ HDL. Normalization of unesterified cholesterol in SR-BI−/− HDL by probucol administration or LCAT expression restored GC-induced thymocyte apoptosis, and incorporating unesterified cholesterol into SR-BI+/+ HDL rendered SR-BI+/+ HDL dysfunctional. Using lckCre-GRfl/fl mice in whom thymocytes lack CLP-induced thymocyte apoptosis, we showed that lckCre-GRfl/fl mice were significantly more susceptible to CLP-induced septic death than GRfl/fl control mice, suggesting that GC-induced thymocyte apoptosis is required for protection against sepsis.
Conclusions
The findings in this study reveal a novel regulatory mechanism of thymocyte apoptosis in sepsis by SR-BI and HDL.
Keywords: sepsis, apoptosis, Scarb1, apolipoproteins, lipoproteins
Introduction
Sepsis is a major health issue which claims over 215,000 lives and costs $16.7 billion per year in America alone.1 The death rate of sepsis is high, exceeding 30%, due to the poor understanding of the disease. Sepsis induces profound lymphocyte apoptosis,2 which keeps adaptive immunity in check, but excessive lymphocyte apoptosis causes immunosuppression in the late stage of sepsis, resulting in susceptibility to secondary infections. Thymocyte apoptosis is a major mechanism of lymphocyte depletion in sepsis; however, how this process is regulated remains poorly understood.2–4
Scavenger receptor BI (SR-BI or Scarb1) is a well-characterized HDL receptor mainly expressed in the liver and steroidogenic tissues.5–11 It regulates HDL cholesterol content through reverse cholesterol transport in which SR-BI selectively uptakes cholesteryl ester from HDL9, 10. Despite a two-fold increase in plasma HDL cholesterol concentrations, SR-BI−/− mice are susceptible to atherosclerosis12–16. A number of earlier studies have shown that SR-BI deficiency leads to larger HDL with a profound increase in unesterified cholesterol concentrations on HDL17–20, which may render HDL dysfunctional in SR-BI−/− mice21–24. In support of this notion, an earlier study showed that HDL from SR-BI−/− mice loses activity of selective cholesteryl ester uptake;19 a number of reports suggest that accumulation of unesterified cholesterol on HDL contributes to female infertility and disrupts red blood cell development21–24; we recently reported that HDL from SR-BI−/− mice also loses activity of suppressing lymphocyte proliferation.25 Although it has been recognized for years that HDL has immune regulatory activity, how HDL regulates immunity and the role of SR-BI in the process remain largely unknown.
In this study, we report a novel function of HDL - namely, modulation of thymocyte apoptosis in sepsis. We demonstrate that SR-BI+/+ HDL enhances glucocorticoid (GC)-induced thymocyte apoptosis via promoting glucocorticoid receptor translocation, but SR-BI−/− HDL loses such regulatory activity. We further demonstrate that excessive accumulation of unesterified cholesterol on SR-BI−/− HDL likely renders its dysfunctional. Our findings reveal a novel regulatory mechanism of thymocyte apoptosis by SR-BI and HDL in sepsis.
Materials and Methods
Detailed Materials and Methods are available in the online-only Data Supplement.
Results
SR-BI is a key determinant of thymocyte apoptosis in sepsis
We induced sepsis with CLP in SR-BI+/+ and SR-BI−/− mice and assessed thymocyte apoptosis 18 h following CLP. Compared with sham, CLP caused significant thymic involution in SR-BI+/+ mice, as shown by a 80% reduction in thymus cell number, but only a slight decrease in thymus cell number in SR-BI−/− mice (Figure 1A). Trypan Blue assay revealed a marked thymocyte death in CLP-SR-BI+/+ mice, but much less thymocyte death in CLP-SR-BI−/− mice (Figure 1B). Clear DNA ladders were observed in CLP-SR-BI+/+ mice, but not in CLP-SR-BI−/− mice (Figure 1C). FACS analyses indicated profound thymocyte apoptosis in CLP-SR-BI+/+ mice as shown by 22% TUNEL+ thymocytes, while thymocyte apoptosis was almost completely absent in CLP-SR-BI−/− mice (Figure 1, D and E). H&E staining showed significant apoptotic cell death in the thymic cortex of CLP-SR-BI+/+ mice, but no apoptotic cell death in CLP-SR-BI−/− mice (Figure 1F and Supplemental Figure sI). Taken together, these findings demonstrate SR-BI as a key determinant of thymocyte apoptosis in sepsis.
Figure 1. SR-BI is a key determinant of thymocyte apoptosis in sepsis.

SR-BI+/+ and SR-BI−/− mice were subjected to CLP or sham (22G needle, full ligation) for 18 h and thymocyte apoptosis was analyzed. A) Changes in thymocyte number; B) Trypan Blue assay; C) DNA ladder assay; D and E) TUNEL assay. n = 7–14 per group; F) H&E staining of thymus cortex. Arrows indicate apoptotic cells with fragmented nuclei (representative data. also see Supplemental Fig. sI). **p < 0.01.
SR-BI controls inducible GC (iGC) generation in response to septic stress which is a determinant of thymocyte apoptosis in sepsis
In the early stage of sepsis, adrenals produce high levels of GC that trigger thymocyte apoptosis.2–4 SR-BI is highly expressed in the adrenals. As an HDL receptor, SR-BI mediates the uptake of cholesteryl ester from HDL for GC synthesis, and a number of studies have shown that SR-BI−/− mice have impaired GC production in response to stress 26–28. To understand how SR-BI regulates thymocyte apoptosis, we measured plasma corticosterone concentrations in the early stages of sepsis. As early as 2 h following CLP challenge, SR-BI+/+ mice produced high concentrations of corticosterone, but SR-BI−/− mice failed to upregulate GC in response to septic stress (Figure 2A), which is consistent with our earlier report 26.
Figure 2. SR-BI controls inducible GC generation in response to septic stress, but supplementation of GC only partly restored GC-induced thymocyte apoptosis in SR-BI−/− mice.
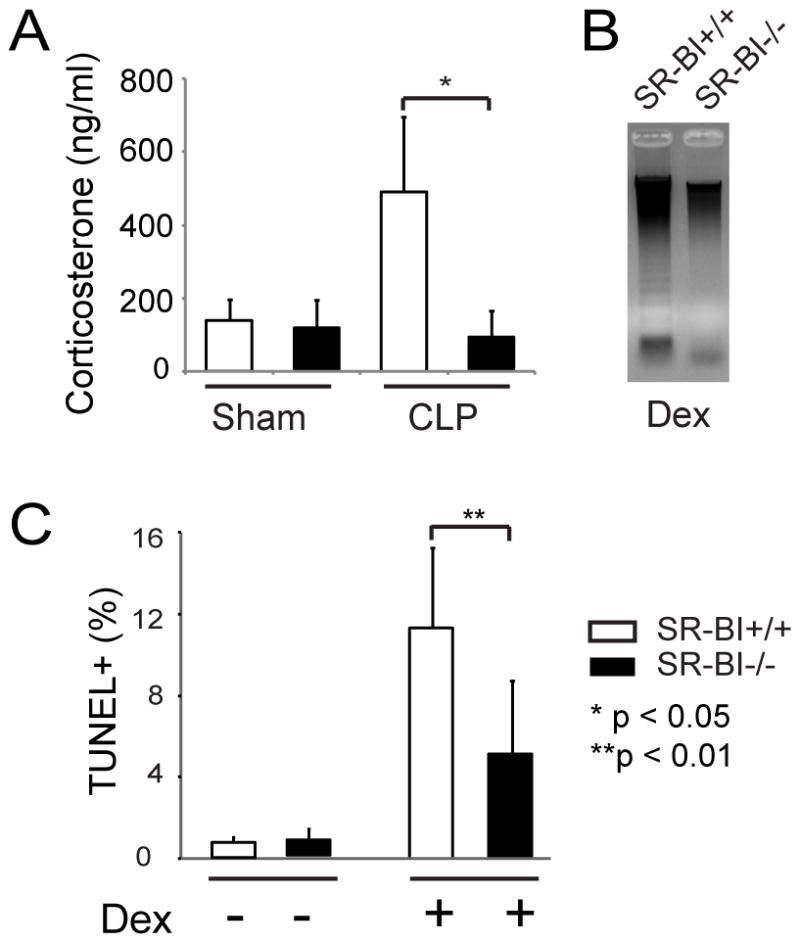
A) Lack of inducible GC production in SR-BI−/− mice in sepsis. SR-BI+/+ and SR-BI−/− mice were subjected to CLP (22G needle, full ligation) for 2 h and the serum corticosterone levels were quantified. n = 6 per group; B and C) Supplementation of dexamethasone only partly restored GC-induced thymocyte apoptosis in SR-BI−/− mice. SR-BI+/+ and SR-BI−/− mice were administered Dex (8mg/kg body weight, i.p.) for18 h. Thymocyte apoptosis was analyzed by DNA ladder assay (B) and by TUNEL assay (C). n = 8–10 per group.
Given the essential role of GC in triggering thymocyte apoptosis 2–4, we reasoned that the lack of SR-BI-mediated iGC generation accounts for the impaired thymocyte apoptosis in sepsis. To test this, we administered dexamethasone (Dex) to SR-BI+/+ and SR-BI−/− mice, expecting that supplementation of GC would restore GC-induced apoptosis in SR-BI−/− mice. Dex administration induced profound thymocyte apoptosis in SR-BI+/+ mice as shown by DNA ladders and a marked increase in TUNEL+ cell percentage (Figure 2, B and C). Unexpectedly, Dex administration only partly restored thymocyte apoptosis in SR-BI−/− mice as shown by a lack of clear DNA ladders and 2-fold fewer TUNEL+ cell percentages in SR-BI−/− mice (Figure 2, B and C). Taken together, these findings indicate that SR-BI controls iGC generation in sepsis, but also imply that SR-BI modulates thymocyte apoptosis via an unidentified mechanism in addition to simply controlling iGC generation.
SR-BI regulates thymocyte apoptosis independent of its expression on thymocytes
SR-BI is moderately expressed in the thymus (Supplemental Figure sIIA). Our earlier study demonstrated that SR-BI induces apoptosis upon serum depletion in vitro 29. This raised the possibility that SR-BI may regulate thymocyte apoptosis intrinsically. To test this, we isolated thymocytes from SR-BI+/+ and SR-BI−/− mice, incubated the cells with corticosterone, and monitored cell death. SR-BI+/+ and SR-BI−/− thymocytes displayed similar cell death in response to corticosterone or Dex (Supplemental Figure sII, B and C). To exclude an intrinsic effect in vivo, we conducted bone marrow transplantation by transferring SR-BI+/+ or SR-BI−/− bone marrow-derived cells into Rag-1−/− mice. Rag-1−/− mice are deficient in CD4+CD8+ (DP), CD4+CD8− and CD4− CD8+ (SP) thymocytes (Supplemental Figure sIIIA). Six weeks following bone marrow transplantation, the Rag-1−/− mice were populated with thymocytes derived from donor mice as shown by the presence of DP and SP thymocytes (Supplemental Figure sIIIA). We then conducted CLP on these mice. As shown in Supplemental Figure sIII, B and C, CLP induced similar thymocyte apoptosis in mice receiving either SR-BI+/+ or SR-BI−/− bone marrow-derived cells, and depleted DP thymocytes equally well in these mice. These findings indicate that SR-BI does not have an intrinsic effect on GC-induced thymocyte apoptosis.
HDL enhances GC-induced thymocyte apoptosis, but HDL from SR-BI−/− mice loses such regulatory activity
SR-BI plays an essential role in HDL metabolism. SR-BI−/− mice display abnormal HDL as shown by larger HDL particles associated with a 2-fold increase in HDL cholesterol content.17–20 We hypothesized that HDL modulates GC-induced thymocyte apoptosis and SR-BI modulates GC-induced thymocyte apoptosis via its effect on HDL. To test this hypothesis, we isolated HDL from SR-BI+/+ and SR-BI−/− mice, and assessed its effect on GC-induced thymocyte apoptosis. We incubated thymocytes isolated from SR-BI+/+ mice with corticosterone in the presence of SR-BI+/+ or SR-BI−/− HDL and quantified thymocyte apoptosis with 7-AAD (7-aminoactinomycin D) staining. As shown in Figure 3, A and B, corticosterone induced significantly thymocyte apoptosis compared to non-corticosterone treatment in the absence of HDL. Interestingly, HDL from SR-BI+/+ mice significantly enhanced corticosterone-induced thymocyte apoptosis, but HDL from SR-BI−/− mice suppressed corticosterone-induced thymocyte apoptosis (Figure 3, A and B). We further tested the dose effect of HDL and found that SR-BI+/+ HDL promotes corticosterone-induced thymocyte apoptosis but SR-BI−/− HDL suppressed corticosterone-induced thymocyte apoptosis in a dose-dependent manner (Supplemental Figure sIV). In vitro cell apoptosis usually undergoes “secondary necrosis” due to lack of phagocyte-mediated engulfment of the apoptotic cells 30, 31. Indeed, we detected a significant increase in necrotic cell death (Figure 3C). We also used Trypan Blue exclusion to test the effect of HDL on thymocyte cell death and obtained similar findings (Figure 3D). These data suggest that normal HDL promotes GC-induced thymocyte apoptosis, but SR-BI−/− HDL loses this regulatory function. Similar results were obtained with Dex incubations (data not shown).
Figure 3. SR-BI+/+HDL enhances GC-induced thymocyte apoptosis through promoting GR translocation, but SR-BI−/− HDL lacks such regulatory activity.

A to D) SR-BI+/+ HDL enhances GC-induced thymocyte apoptosis, but SR-BI−/− HDL lacks such regulatory activity. Thymocytes harvested from wild-type mice were cultured in complete medium and incubated with/without 10 μM corticosterone in the presence/absence of HDL isolated from SR-BI+/+ (20% vol/vol, 0.345±0.011mg/mL by protein) or SR-BI−/− (20% vol/vol, 0.310±0.012 mg/mL by protein) for 18 h and analyzed with 7-AAD staining (A to C) and with Trypan Blue exclusion assays (D). n = 4 with duplicate measurements; Similar data were obtained when SR-BI−/− thymocytes were used (Supplemental Figure sV); E to H) Human HDL enhances GC-induced thymocyte apoptosis. Thymocytes were incubated with/without 10 μM of corticosterone in the presence/absence of human HDL (20% vol/vol, 0.168±0.012 mg/mL by protein) and analyzed with 7-AAD staining (E to G) and with Trypan Blue exclusion assays (H). n = 3 with triplicate measurements; I) SR-BI+/+HDL promoted GC-induced GR translocation but SR-BI−/− HDL lacked such regulatory activity. L929 cells were incubated with/without 100 nM Dex in the presence/absence of SR-BI+/+ or SR-BI−/− HDL for 15 and 30 min. The cytosol and nucleus fractions were isolated and the GR was detected with Western blot. Tubulin and SP1 were used as the markers of cytosol and nucleus, respectively.
Human HDL enhances GC-induced thymocyte apoptosis
We isolated HDL from human serum and tested its effect on corticosterone-induced thymocyte apoptosis. Similar to normal mouse HDL, human HDL significantly enhanced GC-induced thymocyte apoptosis as shown by 7-AAD staining (Figure 3, E to G) and Trypan Blue exclusion (Figure 3H) assays.
HDL enhances GC-induced thymocyte apoptosis via promoting GR translocation, but HDL from SR-BI−/− mice loses such regulatory activity
Upon entering cells, GC binds to GR, resulting in GR translocation from cytosol to the nucleus, where GR functions as transcriptional factor to activate apoptotic signaling. To further understand the molecular mechanisms by which HDL modulates GC-GR apoptotic signaling, we tested the effects of HDL on GR translocation using L929 cells, a cell line widely used for GC-GR signaling 32. As shown in Figure 3I, GR moved rapidly from cytosol to the nucleus in response to Dex incubation. Importantly, HDL from SR-BI+/+ mice markedly enhanced GC-induced GR translocation, but HDL from SR-BI−/− mice suppressed GC-induced GR translocation.
Deficiency of HDL in vivo impairs thymocyte apoptosis in sepsis
ApoAI−/− mice are grossly deficient in HDL (Figure 4A),33 and thus have been employed as an HDL-deficient model to determine the roles of HDL in vivo 34–36. We utilized apoAI−/− mice to elucidate the contribution of HDL to thymocyte apoptosis in vivo. Upon CLP, apoAI−/− mice displayed a 2-fold decrease in the percentage of TUNEL+ thymocytes (Figure 4B), and less depletion in DP thymocytes compared with C57BL/6J mice (Figure 4C). An earlier study indicated that apoAI−/− mice have impaired GC production in response to stress, which raised the possibility that apoAI−/− mice might produce less GC in sepsis 37. To exclude such a possible effect, we induced thymocyte apoptosis by administering Dex to apoAI−/− and C57BL/6J mice. As shown in Figure 4D, Dex-treated apoAI−/− mice had significantly fewer TUNEL+ thymocytes than Dex-treated C57BL/6J mice. Thus, these observations provided in vivo evidence that HDL modulates GC-induced thymocyte apoptosis.
Figure 4. Deficiency of HDL impairs thymocyte apoptosis in sepsis.
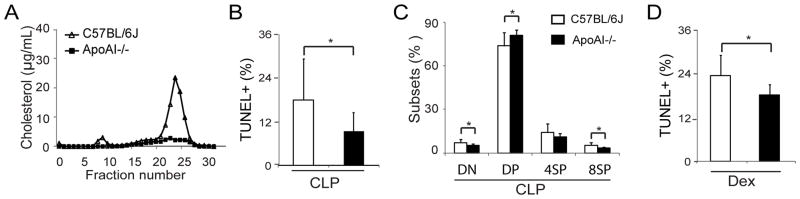
A) Lack of HDL in apoAI−/− mice assessed by FPLC; B and C) Impaired thymocyte apoptosis in apoAI−/− mice in sepsis. ApoAI−/− and control C57BL/6J mice were subjected to CLP (21G needle, half ligation) for 18 h, thymocyte apoptosis was analyzed with TUNEL assay (B) and CD4/CD8 profiles were analyzed (C). n= 11 per group; D) Impaired GC-induced thymocyte apoptosis in apoAI−/− mice. ApoAI−/− and control C57BL/6J mice were administered Dex (8mg/kg body weight, i.p.) for18 h, and thymocyte apoptosis was analyzed with TUNEL assay. n = 5 – 8 per group. *p < 0.05.
Normalization of the unesterified cholesterol content in SR-BI−/− HDL restores its regulatory activity of modulating GC-induced thymocyte apoptosis
SR-BI deficiency leads to larger HDL with a profound increase in unesterified cholesterol ratio of HDL18–20. We analyzed the cholesterol content of these HDL particles. In consistent with the earlier reports18–20, we observed a 3-fold increase in unesterified cholesterol content in HDL, but only marginally increasing in the cholesteryl ester concentrations (Figure 5A, normal diet). The major form of cholesterol in SR-BI+/+ HDL particles is cholesteryl ester, which is located in the inner core of HDL because of its hydrophobic nature. In contrast, unesterified cholesterol is more hydrophilic and polar, and therefore is located on the surface of HDL 22. Given that earlier studies have shown that the accumulation of unesterified cholesterol in circulation causes female infertility and abnormal red blood cell develop in SR-BI−/− mice21–24, it is likely that the massive accumulation of unesterified cholesterol in SR-BI−/− HDL disrupts the structure of HDL, leading to its dysfunction on regulating thymocyte apoptosis. To test this speculation in vivo, we administered SR-BI+/+ and SR-BI−/− mice with probucol, a cholesterol lowering drug that has been shown to normalize unesterified cholesterol levels in SR-BI−/− mice21–24. As shown in Figure 5A (probucol diet), probucol administration normalized the unesterified cholesterol content of SR-BI−/− HDL to almost normal levels. When we administered Dex to the probucol-treated mice, we observed a 2-fold increase in the percentage of TUNEL+ thymocytes in probucol-treated SR-BI−/− mice compared to untreated SR-BI−/− mice (Figure 5B).
Figure 5. Probucol administration normalizes unesterified cholesterol content in SR-BI−/− HDL and restores GC-induced thymocyte apoptosis.

SR-BI+/+ or SR-BI−/− mice were fed diets with/without probucol (0.2% in normal diet) for 3 weeks. A) Probucol administration normalizes unesterified cholesterol content in SR-BI−/− HDL. HDL was isolated by ultracentrifugation and analyzed for cholesterol content. n = 3 – 4 per group with duplicate measurements; B) Probucol administration restores GC-induced thymocyte apoptosis in SR-BI−/− mice. The probucol-treated mice were administered Dex (8 mg/kg body weight, i.p.) for 18 h, and thymocyte apoptosis was analyzed by TUNEL assay. n = 4 – 7 per group; C) SR-BI−/− HDL isolated from probucol-treated mice displays normalized activity on GC-induced thymocyte death. Thymocytes from wild-type mice were cultured in complete medium and incubated with/without 10 μM corticosterone for 18 h in the presence/absence of HDL (20% vol/vol), isolated from the probucol administered mice (SR-BI+/+ HDL, 0.318 mg/mL by protein; SR-BI+/+ HDL probucol diet, 0.181 mg/mL by protein; SR-BI−/− HDL, 0.281 mg/mL by protein; SR-BI−/− HDL probucol diet, 0.296 mg/mL by protein), and the cell death was analyzed by Trypan Blue assay. n = 4 with duplicate measurements.
We then tested the ability of HDL isolated from the probucol-treated mice to modulate corticosterone-induced thymocyte death. As shown in Figure 5C, HDL isolated from probucol-treated SR-BI−/− mice increased the corticosterone-induced thymocyte death by 3-fold compared to HDL isolated from non-probucol-treated SR-BI−/− mice. Thus, both in vivo and in vitro data indicated that correcting the unesterified cholesterol content of SR-BI−/− HDL restores its ability to modulate thymocyte apoptosis in sepsis.
Incorporation of free cholesterol to normal HDL renders the HDL dysfunctional in modulating GC-induced thymocyte apoptosis
Probucol has functions other than lowering plasma cholesterol. To further support our hypothesis, we took an independent approach by incorporating free cholesterol into SR-BI+/+ HDL following a method described by Cooper et al. 38. Compared to control HDL that was treated with dipalmitory lecithin alone, the HDL treated with free cholesterol/dipalmitory lecithin had 3-fold enriched unesterified cholesterol content and a significant increase in the unesterified cholesterol to cholesteryl ester ratio (Figure 6A). We tested the effect of this free cholesterol-enriched HDL on GC-induced thymocyte apoptosis. The control HDL promoted GC-induce thymocyte apoptosis by 2-fold as shown by 7-AAD stainings (Figure 6, B and C). However, the free cholesterol-enriched HDL did not promote GC-induced thymocyte apoptosis, as shown by only 37 % positive 7-AAD stainings in free cholesterol-enriched HDL, versus 70 % positive 7-AAD stainings in control HDL (Figure 6, B and C). Similar findings were obtained with Trypan Blue exclusion assay (Figure 6D). There was no significant difference in GC-induced thymocyte apoptosis between SR-BI+/+ HDL without dipalmitory lecithin treatment and control SR-BI+/+ HDL treated with dipalmitory lecithin (Figures 3 and 6). Of note, we observed that the SR-BI−/− HDL strongly inhibited GC-induced thymocyte apoptosis (Figure 3) but the free cholesterol-enriched HDL neither promote nor inhibit GC-induced thymocyte apoptosis (Figure 6). This difference was likely caused by a higher unesterified cholesterol concentration and higher unesterified cholesterol to cholesteryl ester ratio in SR-BI−/− HDL compared to the free cholesterol-enriched HDL.
Figure 6. Incorporation of free cholesterol into normal HDL impairs HDL’s regulatory activity on GC-induced thymocyte apoptosis.
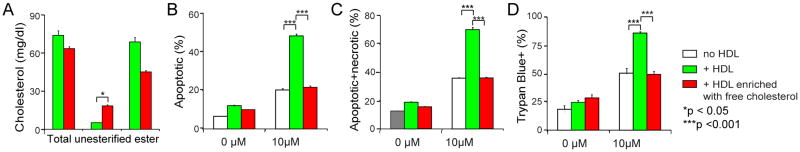
A) Incorporation of free cholesterol into normal HDL. Dipalmitory lecithin was sonicated with/without free cholesterol in saline, and then incubated with mouse serum for 24 h. HDL was isolated by ultracentrifugation and analyzed for cholesterol content. HDL treated with dipalmitory lecithin without free cholesterol addition was used as control HDL. n = 3 per group with duplicate measurements; B) to D). Incorporation of free cholesterol into normal HDL effects loss of HDL activity on GC-induced thymocyte apoptosis. Thymocytes from wild-type mice were cultured in complete medium and incubated with/without 10 μM corticosterone for 18 h in the presence/absence of HDL (20% vol/vol) enriched with free cholesterol (0.149±0.003 mg/mL by protein) or control HDL (0.119±0.010 mg/mL by protein). The cell death was analyzed by 7-AAD staining (B and C) and Trypan Blue assays (D). HDL treated with dipalmitory lecithin without free cholesterol addition was used as control HDL. n = 3 with duplicate measurements.
Lecithin cholesteryl acyltransferase (LCAT) corrects the unesterified cholesterol content of SR-BI−/− HDL and restores its regulatory activity on modulating GC-induced thymocyte apoptosis
An earlier study showed that SR-BI−/− mice have a 90% decrease in endogenous LCAT activity on HDL 20, which may explain why SR-BI−/− HDL has accumulation of unesterified cholesterol. To further test if accumulation of unesterified cholesterol on SR-BI−/− HDL renders its dysfunction, we isolated HDL from LCAT transgenic mice in SR-BI+/+ or SR-BI−/− background (SR-BI−/− LCAT-tg). Over expression of LCAT increased the total cholesterol levels in both SR-BI+/+LCAT-tg HDL and SR-BI−/− LCAT-tg HDL, and markedly normalized the unesterified cholesterol ratio in SR-BI−/− LCAT-tg HDL compared to SR-BI−/− HDL (0.29, SR-BI−/− LCAT-tg HDL versus 0.59, SR-BI−/− HDL. see supplement Table 1). Importantly, SR-BI−/− LCAT-tg HDL displayed much higher activity on GC-induced thymocyte apoptosis compared with SR-BI−/− HDL (Figure 7, A to C).
Figure 7. Lecithin cholesteryl acyltransferase (LCAT) over expression restores SR-BI−/− HDL’s regulatory activity on modulating GC-induced thymocyte apoptosis.
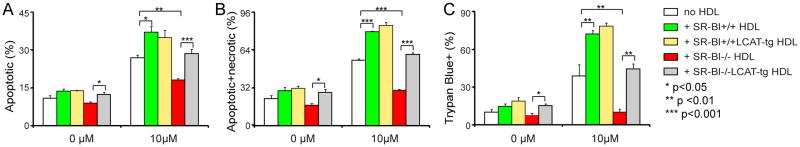
Thymocytes from wild type mice were cultured in complete medium and incubated with/without 10μM corticosterone for 18 h in the presence /absence of HDL (SR-BI+/+ HDL, 0.272±0.013 mg/mL by protein; SR-BI+/+ LCAT-tg HDL, 0.332±0.008 mg/mL by protein; SR-BI−/− HDL, 0.193±0.006 mg/mL by protein; SR-BI−/− LCAT-tg HDL, 0.373±0.003 mg/mL by protein;). The protein concentration of LCAT-tg HDL was adjusted to the same level of non-LCAT-tg HDL. The cell death was analyzed by 7-AAD staining (A and B) and Trypan Blue assays (C). n = 4 with duplicate measurements.
Collectively, these in vivo and in vitro findings indicate that normal HDL enhances GC-induced thymocyte apoptosis through promoting GR translocation, but SR-BI−/− HDL loses this regulatory activity, likely due to disruption of its structure as a result of the accumulation of unesterified cholesterol. To the best of our knowledge, this is the first evidence demonstrating that HDL regulates thymocyte apoptotic signaling in sepsis.
Mice deficient in GC-GR signaling in thymocytes/T cells are susceptible to CLP-induced septic death
We asked whether a deficiency of SR-BI-mediated GC-GR signaling in thymocyte/T cells contributes to septic death. As a deficiency of SR-BI-mediated GC production will affect GC-GR signaling in all types of cells, and GC-GR signaling has been shown to play protective roles in macrophages and endothelial cells in sepsis 39–41, SR-BI−/− mouse model is not suitable for answering this question. Therefore, we utilized lckCre-GRfl/fl mice, a model in which GC-GR signaling is deficient only in thymocytes/T cells. As expected, lckCre-GRfl/fl mice were completely resistant to CLP-induced thymocyte apoptosis (Figure 8, A and B). Importantly, lckCre-GRfl/fl mice were significantly more susceptible to CLP-induced septic death than GRfl/fl control mice (Figure 8C), indicating that the GC-GR signaling in thymocytes/T cells is critical for protection against sepsis.
Figure 8. Mice deficient in GC-GR signaling in thymocytes/T cells are susceptible to CLP-induced septic death.

CLP (25G needle, full ligation) was performed on 8 to 10-week old lckCre-GRfl/fl and GRfl/fl littermates. After 18 h, the apoptotic thymocytes were analyzed with TUNEL staining using FACS (A and B), n = 6 each group. Survival was monitored for 7 days and analyzed with Log-Rank x2. lckCre-GRfl/fl (n = 13) and GRfl/fl (n = 8) littermates,
Discussion
The findings in this study reveal a previously unrecognized regulatory mechanism of thymocyte apoptosis in sepsis. We demonstrate that SR-BI is a key determinant and HDL is a critical modulator of thymocyte apoptosis in sepsis.
SR-BI-mediated iGC production by adrenals is a determinant of thymocyte apoptosis in sepsis
Thymocyte apoptosis is a major event in sepsis; however, how this process is regulated remains poorly understood. GC are potent triggers of thymocyte apoptosis 2–4. Their generation is regulated by three major factors: 1) availability of intracellular cholesterol; 2) transport of cholesterol to mitochondria; and 3) enzymes that convert cholesterol to GC. The organ with the highest SR-BI expression is the adrenal 8. As an HDL receptor, SR-BI takes up cholesteryl ester from HDL to provide cholesterol for GC synthesis in the adrenals 42. The fact that SR-BI−/− mice are completely deficient in iGC production in response to septic stress indicates that the SR-BI-mediated cholesteryl ester uptake from HDL is a rate-limiting step for iGC production. Of note, mouse mainly has HDL but human has both LDL and HDL. This raises a possibility that human may use LDL-LDLr pathway as a source of cholesterol for GC synthesis in addition to HDL-SR-BI pathway. However, patients with homozygous familial hypercholesterolemia (LDLr null) displayed normal iGC production in response to a single dose of ACTH 43. Furthermore, heterozygous carriers of LDLr mutations displayed normal GC production 44, but patients with mutant SR-BI (~ 50% reduction in SR-BI-mediated cholesterol uptake activity) had about 50% reduction in GC production in response to a single dose of ACTH 45. These studies suggest that the SR-BI-mediated cholesteryl ester uptake from HDL is likely a rate-limiting step for iGC production in human in sepsis.
HDL is a critical modulator of GC-induced thymocyte apoptosis
When we administered GC to SR-BI−/− mice, we unexpectedly found that supplementation of Dex only partly restored thymocyte apoptosis in SR-BI−/− mice. This implies that SR-BI modulates thymocyte apoptosis via a secondary mechanism in addition to controlling iGC generation.
By excluding intrinsic effect of SR-BI, we tested our hypothesis that HDL is a modulator of GC-induced thymocyte apoptosis and that SR-BI modulates GC-induced thymocyte apoptosis through HDL. Our findings suggest that HDL profoundly enhances GC-induced thymocyte apoptosis likely through promoting GC-induced GR translocation. Interestingly, HDL from SR-BI−/− mice lost this regulatory function. Importantly, human HDL significantly enhanced GC-induced thymocyte apoptosis. To determine the importance of HDL in modulating thymocyte apoptosis in vivo, we utilized apoAI−/− mice that are grossly deficient in HDL. As expected, mice deficient in HDL displayed impaired thymocyte apoptosis in sepsis, supporting our hypothesis.
Accumulation of unesterified cholesterol on SR-BI−/− HDL renders its dysfunctional
Nascent HDL acquires free cholesterol from peripheral tissues and LCAT catalyzes the free cholesterol to form cholesteryl ester, which is a critical step for HDL metabolism. Interestingly, an earlier study showed that SR-BI−/− mice have a 90% decrease in endogenous LCAT activity in HDL 20. Thus, it is plausible that SR-BI−/− HDL would accumulate unesterified cholesterol due to the lack of LCAT activity. Indeed, in agreement with earlier reports 18–20, we observed a 3-fold increase in unesterified cholesterol contents of SR-BI−/− HDL compared to SR-BI+/+ HDL, but the cholesteryl ester levels of SR-BI−/− HDL were only minimally elevated compared to SR-BI+/+ HDL. Based on the difference in hydrophobic nature between unesterified cholesterol and cholesteryl ester 22, we proposed that SR-BI−/− HDL loses its regulatory activity on GC-induced thymocyte apoptosis due to disruption of HDL structure as a result of the accumulation of unesterified cholesterol on the surface of HDL. To test this hypothesis, we took three independent approaches. First, we administered probucol to SR-BI−/− mice. Probucol normalized the unesterified cholesterol level of SR-BI−/− HDL; importantly, probucol effectively restored GC-induced thymocyte apoptosis both in vivo and in vitro; second, we incorporated free cholesterol to SR-BI+/+ HDL. We found that this free cholesterol-enriched HDL loses its ability to promote GC-induced thymocyte apoptosis; finally we isolated HDL from SR-BI−/− LCAT-tg mice. The over expression of LCAT in SR-BI−/− mice normalized the unesterified cholesterol contents in HDL and partly restored the activity of HDL on GC-induced thymocyte apoptosis.
Although clinical studies have clearly established a negative correlation between plasma HDL cholesterol levels and cardiovascular diseases 46, 47, there is growing appreciation that elevation in HDL cholesterol levels may not always be beneficial and may even be deleterious 48–51. Indeed, SR-BI−/− mice have a two-fold increase in plasma HDL cholesterol concentrations, but are susceptible to atherosclerosis and other cardiovascular diseases 12–16, suggesting that a defect in SR-BI-mediated HDL metabolism may render HDL dysfunctional. Here, using two independent approaches, we demonstrated that the massive accumulation of unesterified cholesterol on SR-BI−/− HDL likely accounts for loss of its regulatory activity on GC-induced thymocyte apoptosis. Just as massive accumulation of unesterified cholesterol on HDL likely disrupts the structure of HDL, it may also impair the other functions of HDL, such as the selective cholesteryl ester uptake. Further study is warranted to test this speculation.
GC-GR signaling in thymocytes/T cells is required for protection against CLP-induced septic death
As opposed to normal physiological conditions in which only about 1% of thymocytes undergo apoptosis, in sepsis, thymocyte apoptosis is increased by 10–20 folds, which indicates a marked increase in thymocyte stress during sepsis. Whether this massive apoptotic cell death is simply a passive response to septic stress or a protective mechanism remains an open question. Using lckCre-GRfl/fl mice in whom thymocytes lack the GR, Mittelstadt et al demonstrated that GC-GR signaling is required for proper selection of thymocytes and absent of GC-GR signaling in thymocytes impairs thymocyte development resulting in generation of functionally compromised T cells 52. Considering that thymocyte apoptosis is a major mechanism of thymocyte selection, we speculate that SR-BI and HDL-regulated GC-GR signaling is required for the removal of the stressed thymocytes and a failure in this process impairs the development of thymocytes resulting in functionally compromised T cells. We conducted CLP on lckCre-GRfl/fl mice. The lckCre-GRfl/fl mice were completely resistant to CLP-induced thymocyte apoptosis and significantly more susceptible to CLP-induced septic death compared with GRfl/fl littermates, which supports our hypothesis.
In summary, this study identifies SR-BI and HDL as key regulators of GC-GR signaling in thymocytes in sepsis. As shown in Supplemental Figure sVI, 1) In adrenals, SR-BI mediates the production of iGC which induces thymocyte apoptosis; 2) in thymus, HDL enhances GC-induced thymocyte apoptosis likely via promoting GR nuclear relocation; 3) lack of SR-BI-mediated reverse cholesterol transport in liver causes accumulation of free cholesterol on HDL that renders it inactive on thymocyte apoptosis; and 4) the SR-BI and HDL regulated GC-GR signaling is likely required for removal of stressed thymocytes, for proper selection of thymocyte and for protection against sepsis
Supplementary Material
Significance.
Thymocyte apoptosis is a major event in sepsis; however, how this process is regulated remains poorly understood. In this study we identify SR-BI and HDL as key regulators in this process. We found that mice deficient in SR-BI are completely resistant to sepsis-induced thymocyte apoptosis. We demonstrated that SR-BI determine thymocyte apoptosis through controlling inducible glucocorticoid production; we further demonstrated that HDL modulates glucocorticoid-induced thymocyte apoptosis through promoting glucocorticoid receptor translocation, but HDL from SR-BI null mice loses such regulatory activity completely. We demonstrated that accumulation of unesterified cholesterol on HDL from SR-BI null mice renders its dysfunction. The SR-BI and HDL regulated GC-GR signaling is likely required for removal of stressed thymocytes, for proper selection of thymocyte and for protection against sepsis.
Acknowledgments
We thank Dr. Marcielle de Beer of the University of Kentucky for protocol of HDL isolation from mouse plasma, Debra Rateri of the University of Kentucky for help in bone marrow transplantation, and Dr. Subbarao Bondada of the University of Kentucky for his invaluable advice.
Funding Sources
This publication was made possible by Grant Number R01GM085231, R01GM085231-2S1 and R01GM085231-5S1 from NIGMS/NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS or NIH. This work was also supported by a grant from the Children’s Miracle Network.
Footnotes
Disclosures: None
References
- 1.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the united states from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: Findings in normal and t- and b-cell-deficient mice. Crit Care Med. 1997;25:1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 3.Ayala A, Herdon CD, Lehman DL, DeMaso CM, Ayala CA, Chaudry IH. The induction of accelerated thymic programmed cell death during polymicrobial sepsis: Control by corticosteroids but not tumor necrosis factor. Shock. 1995;3:259–267. doi: 10.1097/00024382-199504000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: Molecular mechanisms and clinical relevance. Cell Death Differ. 2004;11:S45–S55. doi: 10.1038/sj.cdd.4401456. [DOI] [PubMed] [Google Scholar]
- 5.Landschulz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH. Regulation of scavenger receptor, class b, type i, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest. 1996;98:984–995. doi: 10.1172/JCI118883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calvo D, Vega MA. Identification, primary structure, and distribution of cla-1, a novel member of the cd36/limpii gene family. J Biol Chem. 1993;268:18929–18935. [PubMed] [Google Scholar]
- 7.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of sr-bi, a cd36-related class b scavenger receptor. J Biol Chem. 1994;269:21003–21009. [PubMed] [Google Scholar]
- 8.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor sr-bi as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 9.Holmes RS, LAC Comparative studies of vertebrate scavenger receptor class b type 1: A high-density lipoprotein binding protein. Research and Reports in Biochemistry. 2012;2:9–24. [Google Scholar]
- 10.Mineo C, Shaul PW. Functions of scavenger receptor class b, type i in atherosclerosis. Curr Opin Lipidol. 2012;23:487–493. doi: 10.1097/MOL.0b013e328357ba61. [DOI] [PubMed] [Google Scholar]
- 11.Krieger M. Charting the fate of the “good cholesterol”: Identification and characterization of the high-density lipoprotein receptor sr-bi. Annu Rev Biochem. 1999;68:523–558. doi: 10.1146/annurev.biochem.68.1.523. [DOI] [PubMed] [Google Scholar]
- 12.Arai T, Rinninger F, Varban L, Fairchild-Huntress V, Liang CP, Chen W, Seo T, Deckelbaum R, Huszar D, Tall AR. Decreased selective uptake of high density lipoprotein cholesteryl esters in apolipoprotein e knock-out mice. Proc Natl Acad Sci U S A. 1999;96:12050–12055. doi: 10.1073/pnas.96.21.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M. Loss of sr-bi expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein e-deficient mice. Circ Res. 2002;90:270–276. doi: 10.1161/hh0302.104462. [DOI] [PubMed] [Google Scholar]
- 14.Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Gene transfer and hepatic overexpression of the hdl receptor sr-bi reduces atherosclerosis in the cholesterol-fed ldl receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 2000;20:721–727. doi: 10.1161/01.atv.20.3.721. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Zhang W, Yancey PG, Koury MJ, Zhang Y, Fazio S, Linton MF. Macrophage apolipoprotein e reduces atherosclerosis and prevents premature death in apolipoprotein e and scavenger receptor-class bi double-knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:150–156. doi: 10.1161/01.ATV.0000194096.89476.73. [DOI] [PubMed] [Google Scholar]
- 16.Van Eck M, Twisk J, Hoekstra M, Van Rij BT, Van Der Lans CA, Bos IS, Kruijt JK, Kuipers F, Van Berkel TJ. Differential effects of scavenger receptor bi deficiency on lipid metabolism in cells of the arterial wall and the liver. J Biol Chem. 2003;278:23699–23705. doi: 10.1074/jbc.M211233200. [DOI] [PubMed] [Google Scholar]
- 17.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (hdl) receptor scavenger receptor class b type i reveals its key role in hdl metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brundert M, Ewert A, Heeren J, Behrendt B, Ramakrishnan R, Greten H, Merkel M, Rinninger F. Scavenger receptor class b type i mediates the selective uptake of high-density lipoprotein-associated cholesteryl ester by the liver in mice. Arterioscler Thromb Vasc Biol. 2005;25:143–148. doi: 10.1161/01.ATV.0000149381.16166.c6. [DOI] [PubMed] [Google Scholar]
- 19.Brundert M, Heeren J, Bahar-Bayansar M, Ewert A, Moore KJ, Rinninger F. Selective uptake of hdl cholesteryl esters and cholesterol efflux from mouse peritoneal macrophages independent of sr-bi. J Lipid Res. 2006;47:2408–2421. doi: 10.1194/jlr.M600136-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Ma K, Forte T, Otvos JD, Chan L. Differential additive effects of endothelial lipase and scavenger receptor-class b type i on high-density lipoprotein metabolism in knockout mouse models. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:149–154. doi: 10.1161/01.ATV.0000150414.89591.6a. [DOI] [PubMed] [Google Scholar]
- 21.Miettinen HE, Rayburn H, Krieger M. Abnormal lipoprotein metabolism and reversible female infertility in hdl receptor (sr-bi)-deficient mice. J Clin Invest. 2001;108:1717–1722. doi: 10.1172/JCI13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, Post MJ, Yoerger DM, Picard MH, Krieger JL, Andrews NC, Simons M, Krieger M. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor sr-bi/apolipoprotein e double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–7288. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm TM, Braun A, Trigatti BL, Brugnara C, Sakamoto M, Krieger M, Andrews NC. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor sr-bi. Blood. 2002;99:1817–1824. doi: 10.1182/blood.v99.5.1817. [DOI] [PubMed] [Google Scholar]
- 24.Meurs I, Hoekstra M, van Wanrooij EJ, Hildebrand RB, Kuiper J, Kuipers F, Hardeman MR, Van Berkel TJ, Van Eck M. Hdl cholesterol levels are an important factor for determining the lifespan of erythrocytes. Exp Hematol. 2005;33:1309–1319. doi: 10.1016/j.exphem.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z, Ai J, Foreman O, Daugherty A, Li XA. Deficiency of scavenger receptor bi leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol. 2011;31:2543–2551. doi: 10.1161/ATVBAHA.111.234716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, Bernard P, Daugherty A, Huang B, Li XA. Scavenger receptor bi protects against septic death through its role in modulating inflammatory response. J Biol Chem. 2009;284:19826–19834. doi: 10.1074/jbc.M109.020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoekstra M, Meurs I, Koenders M, Out R, Hildebrand RB, Kruijt JK, Van Eck M, Van Berkel TJ. Absence of hdl cholesteryl ester uptake in mice via sr-bi impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J Lipid Res. 2008;49:738–745. doi: 10.1194/jlr.M700475-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Cai L, Ji A, de Beer FC, Tannock LR, van der Westhuyzen DR. Sr-bi protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J Clin Invest. 2008;118:364–375. doi: 10.1172/JCI31539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XA, Guo L, Dressman JL, Asmis R, Smart EJ. A novel ligand-independent apoptotic pathway induced by scavenger receptor class b, type i and suppressed by endothelial nitric-oxide synthase and high density lipoprotein. J Biol Chem. 2005;280:19087–19096. doi: 10.1074/jbc.M500944200. [DOI] [PubMed] [Google Scholar]
- 30.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: Signals for a good meal. Nat Rev Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 31.Swan R, Chung C-S, Albina J, Cioffi W, Perl M, Ayala A. Polymicrobial sepsis enhances clearance of apoptotic immune cells by splenic macrophages. Surgery. 2007;142:253–261. doi: 10.1016/j.surg.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pariante C, Pearce B, Pisell T, Su C, Miller A. The steroid receptor antagonists ru40555 and ru486 activate glucocorticoid receptor translocation and are not excreted by the steroid hormones transporter in l929 cells. Journal of Endocrinology. 2001;169:309–320. doi: 10.1677/joe.0.1690309. [DOI] [PubMed] [Google Scholar]
- 33.Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein a-i. Proc Natl Acad Sci U S A. 1992;89:7134–7138. doi: 10.1073/pnas.89.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, de Beer FC, Miyake K, Oram JF, Heinecke JW. High-density lipoprotein suppresses the type i interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 2010;122:1919–1927. doi: 10.1161/CIRCULATIONAHA.110.961193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorci-Thomas MG, Zabalawi M, Bharadwaj MS, Wilhelm AJ, Owen JS, Asztalos BF, Bhat S, Thomas MJ. Dysfunctional hdl containing l159r apoa-i leads to exacerbation of atherosclerosis in hyperlipidemic mice. Biochim Biophys Acta. 2012;1821:502–512. doi: 10.1016/j.bbalip.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, Thomas MJ, Sorci-Thomas MG. Apolipoprotein a-i and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29:843–849. doi: 10.1161/ATVBAHA.108.183442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plump AS, Erickson SK, Weng W, Partin JS, Breslow JL, Williams DL. Apolipoprotein a-i is required for cholesteryl ester accumulation in steroidogenic cells and for normal adrenal steroid production. The Journal of Clinical Investigation. 1996;97:2660–2671. doi: 10.1172/JCI118716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper RA, Arner EC, Wiley JS, Shattil SJ. Modification of red cell membrane structure by cholesterol-rich lipid dispersions. A model for the primary spur cell defect. J Clin Invest. 1975;55:115–126. doi: 10.1172/JCI107901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 map kinase. Blood. 2007;109:4313–4319. doi: 10.1182/blood-2006-10-048215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, Bauer M, Schmid W, Schütz G, Libert C, Reichardt HM, Tuckermann JP. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. The FASEB Journal. 2012;26:722–729. doi: 10.1096/fj.11-192112. [DOI] [PubMed] [Google Scholar]
- 41.Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proceedings of the National Academy of Sciences. 2013;110:306–311. doi: 10.1073/pnas.1210200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kraemer FB. Adrenal cholesterol utilization. Mol Cell Endocrinol. 2007;265–266:42–45. doi: 10.1016/j.mce.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Allen JM, Thompson GR, Myant NB. Normal adrenocortical response to adrenocorticotrophic hormone in patients with homozygous familial hypercholesterolaemia. Clin Sci (Lond) 1983;65:99–101. doi: 10.1042/cs0650099. [DOI] [PubMed] [Google Scholar]
- 44.Illingworth DR, Alam NA, Lindsey S. Adrenocortical response to adrenocorticotropin in heterozygous familial hypercholesterolemia. J Clin Endocrinol Metab. 1984;58:206–211. doi: 10.1210/jcem-58-1-206. [DOI] [PubMed] [Google Scholar]
- 45.Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, Hoekstra M, Sierts JA, Dallinga-Thie GM, Motazacker MM, Holleboom AG, Van Berkel TJ, Kastelein JJ, Van Eck M, Kuivenhoven JA. Genetic variant of the scavenger receptor bi in humans. N Engl J Med. 2011;364:136–145. doi: 10.1056/NEJMoa0907687. [DOI] [PubMed] [Google Scholar]
- 46.Barter PJ, Nicholls S, Rye K-A, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of hdl. Circulation Research. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 47.Lewis GF, Rader DJ. New insights into the regulation of hdl metabolism and reverse cholesterol transport. Circulation Research. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 48.Feng H, Li XA. Dysfunctional high-density lipoprotein. Curr Opin Endocrinol Diabetes Obes. 2009;16:156–162. doi: 10.1097/med.0b013e32832922fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith JD. Dysfunctional hdl as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–155. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otocka-Kmiecik A, Mikhailidis DP, Nicholls SJ, Davidson M, Rysz J, Banach M. Dysfunctional hdl: A novel important diagnostic and therapeutic target in cardiovascular disease? Prog Lipid Res. 2012;51:314–324. doi: 10.1016/j.plipres.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Hewing B, Moore KJ, Fisher EA. Hdl and cardiovascular risk. Circulation Research. 2012;111:1117–1120. doi: 10.1161/CIRCRESAHA.112.280958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mittelstadt PR, Monteiro JP, Ashwell JD. Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest. 2012;122:2384–2394. doi: 10.1172/JCI63067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.