

Abstract
Context:
Thyroid cancer is the most common form of endocrine cancer, and it is a disease whose incidence is rapidly rising. Well-differentiated epithelial thyroid cancer can be divided into papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC). Although FTC is less common, patients with this condition have more frequent metastasis and a poorer prognosis than those with PTC.
Objective:
The objective of this study was to characterize the molecular mechanisms contributing to the development and metastasis of FTC.
Design:
We developed and characterized mice carrying thyroid-specific double knockout of the Prkar1a and Pten tumor suppressor genes and compared signaling alterations observed in the mouse FTC to the corresponding human tumors.
Setting:
The study was conducted at an academic research laboratory. Human samples were obtained from academic hospitals.
Patients:
Deidentified, formalin-fixed, paraffin-embedded (FFPE) samples were analyzed from 10 control thyroids, 30 PTC cases, five follicular variant PTC cases, and 10 FTC cases.
Interventions:
There were no interventions.
Main outcome measures:
Mouse and patient samples were analyzed for expression of activated cAMP response element binding protein, AKT, ERK, and mammalian target of rapamycin (mTOR). Murine FTCs were analyzed for differential gene expression to identify genes associated with metastatic progression.
Results:
Double Prkar1a-Pten thyroid knockout mice develop FTC and recapitulate the histology and metastatic phenotype of the human disease. Analysis of signaling pathways in FTC showed that both human and mouse tumors exhibited strong activation of protein kinase A and mTOR. The development of metastatic disease was associated with the overexpression of genes required for cell movement
Conclusions:
These data imply that the protein kinase A and mTOR signaling cascades are important for the development of follicular thyroid carcinogenesis and may suggest new targets for therapeutic intervention. Mouse models paralleling the development of the stages of human FTC should provide important new tools for understanding the mechanisms of FTC development and progression and for evaluating new therapeutics.
Thyroid cancer is the most common endocrine malignancy, with most patients exhibiting well-differentiated epithelial thyroid cancer, either papillary thyroid cancer (PTC) or follicular thyroid cancer (FTC). Although most patients with thyroid cancer have excellent long-term survival, FTC is more likely to produce distant metastases, leading to a poorer prognosis for FTC patients than those with PTC (1). Thus, identifying the mechanisms leading to the increased aggressiveness of FTC remains an important area for investigation.
Individuals with the inherited tumor predispositions Carney complex (CNC; Online Mendelian Inheritance in Man [OMIM] #160980) and Cowden syndrome (CS; OMIM # 158350) have an increased incidence of thyroid cancer (2, 3). The cancers in both CNC and CS are a mixture of FTC and PTC, although there is an overrepresentation of FTC in these patients (4, 5). We have previously shown that mice heterozygous for the CNC gene, Prkar1a, recapitulate many of the features of CNC, including thyroid cancer in 10% of aged animals (6). PRKAR1A, the type 1A regulatory subunit of the cAMP-dependent protein kinase (protein kinase A, or PKA), is the most ubiquitous of the PKA regulatory subunits, and inactivation of PRKAR1A leads to enhanced PKA activity (7). More recently we have shown that deletion of Prkar1a in the mouse thyroid leads to locally invasive FTC without distant metastases (8).
Similarly, mice heterozygous for Pten mutations develop a variably penetrant thyroid phenotype, possibly reflecting strain effects (9). Thyroid-specific deletion of Pten results mainly in follicular adenomas [(10) and Pringle, D.R., and L.S. Kirschner, unpublished observations], although analysis of mice up to 2 years of age showed the development of FTC at advanced ages (11).
To evaluate how the combination of PKA and Akt activation interact to affect thyroid growth in vivo, we generated mice with thyroid-specific knockout of both Prkar1a and Pten. We report that these double Prkar1a-Pten mice (DRP-TpoKO mice) develop aggressive FTC with 100% penetrance and frequently develop well-differentiated lung metastases. Analysis of signaling pathways demonstrated consistent activation of the PKA and mammalian target of rapamycin (mTOR) pathways, a molecular signature that was also observed in a cohort of human FTCs. Thus, the DRP-TpoKO mice represent a new and highly relevant model of metastatic FTC and indicate that PKA-mTOR crosstalk may be a mechanism to foment FTC metastatic progression. This potential PKA-mTOR crosstalk could have significant implications for developing new targeted therapies for the treatment of FTC patients.
Materials and Methods
Animal studies
Mice were maintained in a sterile environment under an Institutional Animal Care and Use Committee-approved protocol. Prkar1aloxP/loxP, PtenloxP/loxP, and Thyroid Peroxidase-cre (TPO-cre) mice have been previously described (6, 34, 35). All mice were of mixed background, with predominant contributions of 129/Sv126 and FVB/N. Serum hormone levels were analyzed by the National Hormone and Peptide Program (Harbor-UCLA Medical Center, Torrance, California).
Mouse histology
Immunohistochemistry (IHC)/immunofluorescence (IF) experiments were performed as described previously (36) with the following antibodies: Cell Signaling Technology, pCREBSer133 (9198), pAktSer473 (3787), and pErkThr202/Tyr204 (9101); AbCam, pmTORSer2448 (ab51044) and phospho-p70S6KThr389/412 (ab129230).
Microarray
RNA was isolated from using the QIAGEN miRNeasy kit and hybridized to GeneChip Mouse Exon 1.0 ST array (Affymetrix). The heat map of genes with linear trend effects was generated by filtering out genes with high variance and subjecting them to hierarchical clustering using Pearson's correlation with MeV, part of the TM4 Microarray Software Suite (37). Functional annotation was performed using Database for Annotation, Visualization, and Integrated Discovery (DAVID) (38).
Human samples
Sporadic FTC samples were obtained from the Ukrainian Center of Endocrine Surgery (Kiev) under an institutional review board approved protocol at the Uniformed Services University of the Health Sciences. IHC was performed as previously described (32), with the following antibodies: Cell Signaling Technologies, pCREBSer133 (9198), pAktSer473 (9271), pErkThr202/Tyr204 (9101), and phospho-p70S6KThr421/Ser424 (9204). Scoring criteria were as follows: 0, no staining; 1, low intensity focal staining; and 2, intense staining in greater than 50% of the cells.
Statistics
For analysis of repeated measurements of mouse weight over time, a linear model was used, and adjusted P values were obtained using Holm's method, values of P < .05 considered significant. Microarray signals were subject to background correction and quantile normalization and gene expression summarized using Robust Multi-array Average (39). A linear model was used to detect differentially expressed genes. Estimates of variability and statistical tests for differential expression were performed as described using moderated t statistics with variance smoothing (40). Linear trend was assessed for gene expression among Pten-, R1a-, and DRP-TpoKO genotypes and was normalized to matched controls. Linear model and t test were used to obtain linear trend significance.
Results
Ablation of Prkar1a and Pten in the thyroid leads to hyperthyroidism and metastatic FTC
To study how interactions of PKA and Akt could affect thyroid cancer development, we generated mice harboring heterozygous deletion of both Prkar1a and Pten. Although these mice showed reduced survival compared with single heterozygotes (Supplemental Figure 1A, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org), no increased incidence of thyroid cancer was observed. To verify specificity of the Prkar1a-Pten interaction, we also crossed Prkar1a+/− mice with mice heterozygous for the endocrine tumor suppressor genes Lkb1 or Men1. Neither Prkar1a+/−; Lkb1+/− nor Prkar1a+/−;Men1+/− mice showed altered survival (Supplemental Figure 1, B and C). To better model thyroid cancer in the context of Prkar1a and Pten mutations, we generated mice with thyroid-specific deletion of both Prkar1a and Pten, referred to as DRP-TpoKO (Double R1a-Pten knockout) mice. Although control mice exhibited no significant morbidity up to 12 months, median age for mortality or euthanasia due to morbidity (labored breathing and/or weight loss) for DRP-TpoKO animals was 6 months (n = 97) (Supplemental Figure 2). This survival was significantly less that mice carrying thyroid-specific KO of Prkar1a or Pten alone (R1a-TpoKO and Pten-TpoKO, respectively).
Serum was collected from 28 DRP-TpoKO animals and 10 WT littermates for measurement of thyroid function (Supplemental Figure 3, A and B). Free T4 levels were markedly increased (28.75 ng/dL in DRP-TpoKO, 3.2 ng/dL in WT, P < .0001). TSH was measured from the cohort, although the poor low-end sensitivity of the assay (similar to first generation human TSH assays) did not enable detection of the expected TSH suppression in DRP-TpoKO animals. To assess for physiological hyperthyroidism, male WT and DRP-TpoKO animals were weighed monthly until 6 months or euthanasia. DRP-TpoKO animals had lower body weight beginning at 1 month of age, which became statistically significant at 5 months (Supplemental Figure 3C). This reduced weight was associated with reduced sc and visceral adipose tissue. These data confirm that DRP-TpoKO animals are physiologically hyperthyroid.
Grossly, DRP-TpoKO thyroids were enlarged compared with wild type (WT) and showed obvious nodular growths (Figure 1, A and C). Histological examination demonstrated that 100% of these mice (n = 56) developed FTC by 6 months of age, based on the presence of capsular and/or vascular invasion (Figure 1, E and F). Notably, analysis of mice (n = 9) at 8 weeks of age also demonstrated 100% penetrance of FTC, making this model both rapid and highly reproducible.
Figure 1.
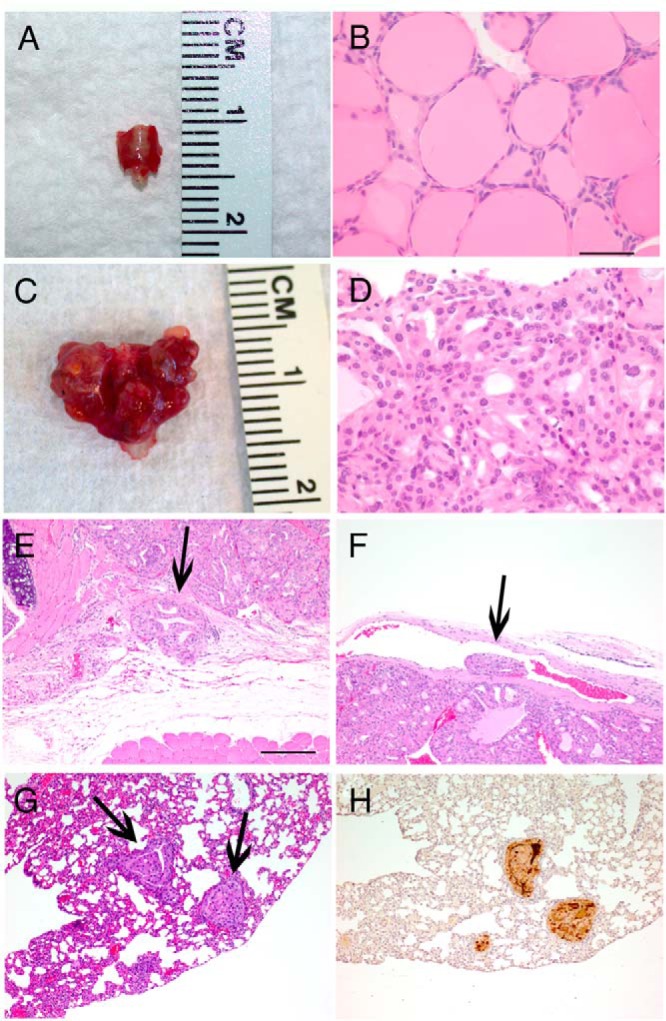
DRP-TpoKO tumors show consistent features of aggressive FTC and develop FTC-derived lung metastases. Macroscopic images of representative WT (A) and DRP-TpoKO (C) thyroids. Representative hematoxylin and eosin staining of high-magnification images of WT (B) and DRP-TpoKO (D) thyroids. Evidence of capsular (E, arrow) and vascular (F, arrow) invasion in DRP-TpoKO tumors. Representative photomicrographs of DRP-TpoKO follicular thyroid carcinoma lung metastases (arrows) stained with hematoxylin and eosin (F) and thyroglobulin (H). Scale bar (B, applies to D), 125 μm, (E, applies to F, G, and H), 500 μm.
Metastatic FTC was detected in the lungs of 27% of these animals (15 of 56). The metastases were well differentiated, including maintaining follicular structure with colloid (Figure 1G), and staining for thyroglobulin (Figure 1H). Focused examination of the liver and long bones of mice with metastatic disease as well as survey necropsy of other organs did not detect the presence of other metastases.
Multiple oncogenic pathways are activated in DRP-TpoKO cancers
The effects of deletion of Prkar1a and Pten on thyroid oncogenesis were first studied by analyzing thyrocyte proliferation in DRP-TpoKO thyroids, along with Pten-TpoKO (benign neoplasia only) and R1a-TpoKO thyroids (locally invasive FTC) (Supplemental Figure 4). Surprisingly, R1a-TpoKO tumors exhibited the highest proliferation rate, with Pten-TpoKO and DRP-TpoKO showing only mild increases (Supplemental Figure 5).
To evaluate oncogenic signaling pathways, thyroids were assessed by immunohistochemistry. WT thyroids exhibited little to no phosphorylation (activation) of cAMP response element binding protein (CREB), Akt, ERK (p42/44), p70S6K, and mTOR (Figure 2, A–E). In contrast, Pten-, R1a-, and DRP-TpoKO tumors exhibited increased activation of at least two of these pathways. In Pten-TpoKO thyroids (Figure 2, F–J), there was widespread phospho-Akt staining as expected as well as increases in phosphorylated (p) mTOR and phospho-p70S6K, consistent with their roles as pathways downstream from Akt activation in the thyroid, as previously demonstrated (12). In R1a-TpoKO mice (Figure 2, K–O), there was strong nuclear staining for pCREB as well as an unexpected increase in pmTOR/pp70S6K. Lastly, DRP-TpoKO tumors (Figure 2, P–T) also demonstrated strong nuclear pCREB and diffuse pmTOR and pp70S6K. Patchy pERK staining was observed, although there was no localization to tumor leading edges, as has been observed in human PTC (13). Intriguingly, pAkt staining in DRP-TpoKO tumors exhibited membranous localization (Figure 2Q, inset), in contrast to the diffuse nuclear-cytoplasmic staining seen in Pten-TpoKO thyroids (Figure 2G, inset). Localization to the membrane is a required step in Akt activation (14), but sustained membranous pAkt is uncommon. Membranous localization of pAkt has been described in a prostate cancer model and is increased in castrated animals (15). However, membranous pAkt was observed, regardless of sex in our tumors, suggesting sex hormones are likely not involved in this observation. Because the function and localization of Akt may be affected by binding to multiple other proteins (16), it is conceivable that PKA may affect Akt localization indirectly in this manner; efforts to understand this phenomenon and its implications are currently underway.
Figure 2.

Activation of FTC-related pathways in DRP-TpoKO, R1a-TpoKO, and Pten-TpoKO tumors. Immunohistochemical staining of WT thyroids (A–E), Pten-TpoKO tumors (F–J), R1a-TpoKO tumors (K–O), and DRP-TpoKO tumors (P–T) for pCREB (A, F, K, and P), pAkt (B, G, L, and Q), pErk (C, H, M, and R), phospho-p70S6k (D, I, N, and S), and pmTOR (E, J, O, and T). Scale bar (A, applies to all), 500 μm.
Human sporadic FTCs show a molecular signature similar to DRP-TpoKO tumors
To determine if signaling alterations in DRP-TpoKO tumors correlated to those observed in human FTC, a panel of thyroid cancer subtypes was stained for pCREB, pAKT, pERK, and pp70S6K (Figure 3). Included in this panel were normal thyroid, PTC, follicular variant of PTC (FVPTC), and FTC. Similar to the mouse cancers, human FTC exhibited strong staining for nuclear pCREB as well as diffuse staining of pp70S6K as a marker of mTOR activation. As shown in Table 1, 70% and 80% of the FTC cancers stained strongly for pCREB and pp70S6K, respectively, whereas strong staining for pAKT and pERK were seen in only 30%–40%. As expected, PTCs tend to exhibit activation of ERK and AKT. We conclude that signaling in the DRP-TpoKO model closely mimics the signaling changes associated with human FTC.
Figure 3.

Activation of cancer pathways in human FTC. Immunohistochemical staining of human normal thyroid (A–D), PTC (E–H), FVPTC (I–L), and FTC (M–P) for pCREB (A, E, I, and M), pERK (B, F, J, and N), pAKT (C, G, K, and O), and phospho-p70S6K (D, H, L, and P). Scale bar (A, applies to all), 500 μm.
Table 1.
Percentage of Cases With a Score of 2.
| pCREB | pAkt | pERK | pp70S6K | |
|---|---|---|---|---|
| Normal thyroid (n = 10) | 0 | 0 | 0 | 0 |
| PTC (n = 30) | 16.6 | 56.6 | 36.6 | 33.3 |
| FVPTC (n = 5) | 60 | 0 | 0 | 40 |
| FTC (n = 10) | 70 | 30 | 40 | 80 |
Thyroid cancers from CNC and CS patients show activation of both PKA and phosphatidylinositol 3-kinase
CNC and CS are rare syndromes with thyroid cancer rates of 2.5% and approximately 35%, respectively (2, 3). We were able to obtain one CNC-associated PTC and one CS-associated FTC for testing. Despite the differing genetic backgrounds, both cancers exhibited activation of both CREB and AKT (Supplemental Figure 6), similar to the observations in the DRP-TpoKO thyroids. Unfortunately, due to limited amounts of sample, further analysis on these samples was not possible.
PKA and mTOR are active in the same cells in DRP-TpoKO tumors
To verify that PKA and mTOR crosstalk was related to FTC, we sought to determine whether these proteins were activated in the same cells. IHC analysis (Figure 4, A–C) demonstrated that pmTOR and pCREB were observed in the same tumor regions. To verify this observation at the cellular level, we used IF to stain DRP-TpoKO tumors for pmTOR and pCREB (Figure 4, D and E). Colocalization of the fluorescent signals confirmed that these molecules were activated in the same cells.
Figure 4.
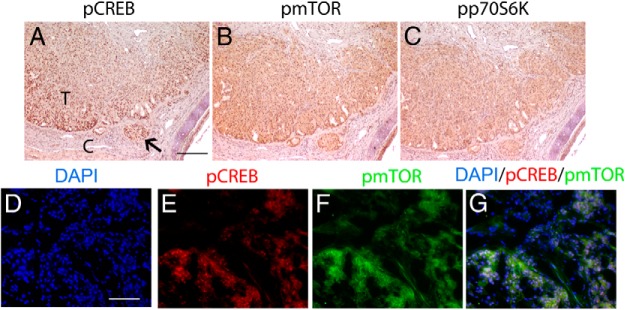
Activation of PKA and mTOR occurs in the same cells in DRP-TpoKO tumors. Immunohistochemical staining of pCREB (A), pmTOR (B), and phospho-p70S6K (C) in DRP-TpoKO tumors is shown. IF staining of 4′,6′-diamino-2-phenylindole (DAPI; D), pCREB (E, red), pmTOR (F, green) in DRP-TpoKO tumors (G is the merged image of D–F). T, FTC; C, capsule. Arrow indicates capsular invasion. Scale bar (A, applies to B and C), 125 μm; (D, applies to E–G), 250 μm.
Microarray analyses confirm activation of networks related to mTOR function
To further characterize the molecular changes associated with cancer development in DRP-TpoKO mice, we performed expression microarray analyses comparing DRP-TpoKO tumors to WT thyroids. The resulting gene list (Supplemental Table 1) was analyzed using Ingenuity pathways analysis (IPA; www.ingenuity.com), which revealed that two of the top five networks were related to known functions of mTOR (Supplemental Table 2), including protein degradation, protein synthesis, and metabolic diseases (Supplemental Figure 7, A and B). Although we do not observe alterations in mTOR expression, we hypothesize that aberrant mTOR activation is responsible for the alteration of these gene sets. Additionally, IPA analysis identified a network related to proliferation as well as endocrine system function and development (network 3, Supplemental Table 2). This network (Supplemental Figure 7C) includes genes associated with altered PKA signaling, including Creb and CyclinD1 (Ccnd1), previously shown to be responsive to PKA activity (17). Comparison of the mouse microarray data with a three-gene signature (PLAB/GDF15, PCSK2, and CCDN2) proposed to differentiate human FTA from FTC (18) demonstrated excellent correlation with alterations in Plab (up-regulated 5.04-fold, P = 6.38E-11) and Pcsk2 (down-regulated 28.4-fold, P = 1.18E-12). Although alterations in Ccnd2 were not observed, we observed up-regulation of Ccnd1̧ suggesting an up-regulation of the same process through a different cyclin D family member. It is unclear whether the use of Ccnd1 (in mouse) vs CCND2 (in human) reflects a difference between our model and human FTC or reflects a more general difference between cyclin usage in mouse and humans. These data may also indicate that genes other than CCND2 may be more appropriate targets in the human data.
In addition to data from R1a- (8) and DRP-TpoKO mice, we also generated microarray data from our own colony of Pten-TpoKO mice, which exhibit benign follicular thyroid hyperplasia (Supplemental Table 3). These three mouse data sets were used to perform principal component (PC) analysis to compress and then identify trends in the data. After discarding age (PC1), a plot of PC2 vs PC3 revealed an excellent discrimination between WT and neoplastic thyroids (Figure 5A). Furthermore, the Pten-, R1a-, and DRP-TpoKO tumors segregate from left to right, indicating a progression of gene expression changes as the tumors become more aggressive.
Figure 5.

Microarray data comparing Pten-, R1a-, and DRP-TpoKO tumors. Principal Component Analysis plot of all tumor microarray data (A) is shown. Circles group each of the tumor genotypes. Heat map of genes identified as having significant linear trend effect between the three data sets (B) is shown. Red bar denotes linear trend genes down-regulated in DRP-TpoKO tumors, yellow bar denotes those down-regulated in malignant tumors, green bar denotes those up-regulated in DRP-TpoKO tumors, and the blue bar denotes those up-regulated in malignant tumors. DRP WT, R1a WT, and Pten WT denote the WT littermates used for comparison for each model.
We next performed a linear-trend analysis on the data to identify the most relevant mRNA changes (Supplemental Table 4). A heat map of these genes (Figure 5B) identified four clusters that showed differential regulation between benign and malignant tumors as well as between locally invasive and metastatic cancers. Given our interest in the metastatic phenotype, we focused on the set of genes up-regulated only in the DRP-TpoKO thyroids (Figure 5B, green bar). These 67 genes were analyzed by IPA, which revealed that the top predicted function of these genes was cellular movement, with 22 of the 67 genes associated with this function. Examples of genes in this cluster include Icam1, Mmp9, and Mmp13, genes that control cell adhesion. DAVID functional annotation of the altered transcripts also detected alterations related to cell surface functionality, including transmembrane proteins/domains and glycoproteins/domains. Thus, genes associated with cellular movement and cell surface regulation are altered in the DRP-TpoKO tumors suggesting mechanisms of metastasis which lessen cell-cell adhesion to promote invasion.
Discussion
Although patients with PTC typically exhibit activation of the Ras-Raf-Mek-ERK pathway (eg, the activating BRAFV600E mutation), the signaling pathways that contribute to FTC are less well defined. A number of genetic lesions have been identified in human FTC, including mutations in RAS, PTEN, and the PPARG-PAX8 fusion gene, but the connection between these mutations and carcinogenesis is less clear, especially because mouse models carrying these mutations singly either do not generate FTC (Ras and Pparg-Pax8) or generate it only unreliably (Pten deletion). In contrast, we recently demonstrated that thyroid-specific KO of Prkar1a leads to FTC in more than 40% of mice by 1 year of age (8). However, these tumors did not exhibit hematogenous metastasis, a key feature of human FTC.
Although phosphatidylinositol 3-kinase signaling is a key pathway leading to thyroid carcinogenesis, the interaction of this pathway with the TSH/cAMP/PKA pathway is somewhat controversial, with data in Wistar rat thyroid cells showing that TSH stimulates Akt activation (19), whereas similar studies in PCCL3 rat thyroid cells showing the opposite (20). To study the effects of activation of both pathways on thyroid carcinogenesis, we introduced into our R1a-TpoKO model conditional Pten alleles to generate DRP-TpoKO mice. These mice exhibited aggressive FTC, with 100% penetrance by 8 weeks of age. Furthermore, these mice exhibited well-differentiated lung metastases in a good analogy to human FTC. Given the size and volume of lung metastases in this model, it was disappointing that we were not able to detect metastases in other tissues. However, it is worthwhile to note that the thyroid tumors in the DRP-TpoKO mice are locally aggressive, leading to compressive symptoms necessitating the killing of the mice. Because metastatic disease to other sites tend to be a later complication of human FTC, it seems possible that the incidence of metastasis might be increased if the primary tumors could be ablated and the mice kept alive longer.
The most striking observation in this study is that both mouse and human FTC exhibit high levels of both nuclear pCREB and activation of mTOR. This observation was made in the absence of mutation data for the human tumors, suggesting that these may be common downstream endpoints regardless of the initiating mutation.
In our model, we believe that nuclear pCREB is a direct effect of excess PKA activity (due to Prkar1a KO), although CREB can also be phosphorylated by non-PKA-dependent mechanisms (21). We have previously demonstrated that other pathways leading to CREB phosphorylation (eg, ERK, p38) are not active in our tumors. Likewise, analysis of human FTC has detected activation of ERK only rarely (Table 1), suggesting that CREB phosphorylation is likely mediated by PKA. This concept is consistent with the epidemiological observation of Haymart et al (22) that elevated TSH (with presumed downstream activation of PKA) predisposes to the development of thyroid cancer, and with the mouse data of the Fagin group showing a role for TSH in enhancing B-raf-mediated thyroid carcinogenesis (23).
In addition to the activation of CREB, mouse FTC exhibited significant activation of mTOR with strong staining for both activated mTOR and its downstream target p70S6K. It has previously been suggested that mTOR may be activated by deletion of Prkar1a and may directly interact with Prkar1a (24), although these data are somewhat controversial (25). Additionally, mTOR activation by TSH has been suggested to be, at least partly, due to PKA phosphorylation of the target of rapamycin complex 1 complex member PRAS40 (26). In the DRP-TpoKO tumors, activation of mTOR may be partly a consequence of Akt activation, although the fact that mTOR is activated in R1a-TpoKO tumors indicates that PKA independently plays a role in this process. It has previously been demonstrated that Prkar1a silencing enhances ribosomal protein S6 kinase 1 activation (27), which may also modulate mTOR activation.
It is also worth noting that mice made hypothyroid by methimazole treatment exhibited marked elevation of TSH and mTOR activation in the absence of Akt, indicating that mTOR was the driver of thyrocyte proliferation (28). In contrast to our findings, this manuscript reported reduced phosphorylation of CREB and suggested that PKA may not contribute to the proliferation phenotype. However, it has been shown in vivo that chronic cAMP stimulation can reduce PKA activity and CREB phosphorylation (29), indicating that the correlation of cAMP/PKA activation with pCREB is not straightforward.
Although we considered the possibility that PKA-mediated mTOR activation might occur through intercellular crosstalk (eg, via the tumor microenvironment), our data clearly indicate that dual PKA-mTOR activation occurs within the same cells. Even in tumors with a predisposing activation of PKA (ie, CNC) or AKT (ie, CS), we observe activation of both pathways. Finally, it is worth noting that PKA is not known to directly phosphorylate mTOR, and the mechanism by which this interaction occurs remains under study.
One issue we have not yet addressed in this model is the role of hyperthyroidism in promoting and/or modulating the tumor phenotype. It is worth noting that single R1a-TpoKO mice are hyperthyroid and develop thyroid cancer, although it is not metastatic (8). In contrast, as just mentioned, elevation of TSH by methimazole causes enhanced proliferation in the hypothyroid state, suggesting that elevation of T4/T3 is not necessary for thyrocyte proliferation (28).
Analysis of microarray data from the three mouse models suggests an overlapping genetic signature, which is a good recapitulation of the cancer progression seen in vivo (Figure 5A). Linear trend effects across the three models suggest that metastasis in the DRP-TpoKO animals is achieved via up-regulation of genes associated with cellular movement and alteration of cell-cell adhesions. Among the transcripts identified in this manner were a number of members of the matrix metalloproteinase (MMP) gene family. These proteins, which are also up-regulated in human FTC (18), are known to play multiple roles in carcinogenesis and metastasis (30).
Another interesting finding in the mouse model is the membranous localization of pAkt (Figure 2R, inset). The mechanisms and consequences of this membranous localization of pAkt are unclear. However, with previous data showing Akt's importance in FTC invasion and metastasis in vivo in the TRβPV/PV model (31) and its activation of p21-activated kinases, which have been shown to be important for PTC cell migration in vitro (32), we hypothesize that this pattern of localization may be important for cell motility and invasion, but this remains to be proven.
Over the past few years, we have developed mouse models that recapitulate the progression of human FTC from benign follicular adenoma (at one year of age) in the Pten-TpoKO (10) to locally invasive FTC as in the R1a-TpoKO (8) and finally to widely invasive and distantly metastatic FTC seen in the mice described here. Using these model systems, we identified activation of PKA and mTOR as key signaling nodes and showed that mTOR activation can occur independently of Akt. Although unexpected, this coactivation of PKA and mTOR and was observed in human FTC and suggests that activation of PKA (including or through pCREB) leads to the activation of mTOR/p70S6K, resulting in thyroid cancer. It is interesting to note that opposite effects have been reported in mouse embryonic fibroblasts and HEK293T cells (33), highlighting the highly cell type-specific nature of PKA signaling.
Together our data from the R1a- and DRP-TpoKO animals indicates that dysregulation of PKA is a key regulator of FTC development in both mice and humans. Additionally, these data could lead to insight, not only into the context of how PKA and mTOR may interact in FTC progression but also as to how to exploit the interaction of these signaling pathways to better treat patients with aggressive FTC.
Acknowledgments
We acknowledge Alan Flechtner, HTL (ASCP), and Lisa Rawahneh for their excellent assistance with processing and sectioning tissues. We also thank Drs Constantine Stratakis and Charis Eng for sharing their valuable patient samples.
This work was supported by National Institutes of Health Grants CA112268 (to L.S.K.), PO1CA124570 (to D.R.P., L.Y., M.S., D.J., K.M.D.L.P., M.D.R., and L.S.K.), and CA16058 (to the Ohio State University Comprehensive Cancer Center). D.R.P. was supported by a Jeffery J. Seilhamer Memorial Fellowship and a Pelotonia Graduate Fellowship.
Current address for D.R.P.: Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, CA 94158.
Disclosure Summary: The authors declare no conflicts of interest.
Footnotes
- CNC
- Carney complex
- CREB
- cAMP response element binding protein
- CS
- Cowden syndrome
- DAVID
- Database for Annotation, Visualization, and Integrated Discovery
- DRP-TpoKO
- double Prkar1a-Pten mice
- FTC
- follicular thyroid cancer
- FVPTC
- follicular variant of PTC
- IF
- immunofluorescence
- IHC
- Immunohistochemistry
- IPA
- Ingenuity pathways analysis
- KO
- knockout
- MMP
- matrix metalloproteinase
- mTOR
- mammalian target of rapamycin
- p
- phosphorylated
- PC
- principal component
- PKA
- protein kinase A
- PTC
- papillary thyroid cancer
- Pten-TpoKO
- Pten KO mice
- R1a-TpoKO
- Prkar1a KO mice
- WT
- wild type.
References
- 1. Sugino K, Ito K, Nagahama M, et al. Prognosis and prognostic factors for distant metastases and tumor mortality in follicular thyroid carcinoma. Thyroid. 2011;21:751–757 [DOI] [PubMed] [Google Scholar]
- 2. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009;94:2085–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab. 2011;96:E2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sandrini F, Matyakhina L, Sarlis NJ, et al. Regulatory subunit type I-α of protein kinase A (PRKAR1A): a tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer. 2002;35:182–192 [DOI] [PubMed] [Google Scholar]
- 6. Kirschner LS, Kusewitt DF, Matyakhina L, et al. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005;65:4506–4514 [DOI] [PubMed] [Google Scholar]
- 7. Kirschner LS. Use of mouse models to understand the molecular basis of tissue-specific tumorigenesis in the Carney complex. J Intern Med. 2009;266:60–68 [DOI] [PubMed] [Google Scholar]
- 8. Pringle DR, Yin Z, Lee AA, et al. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr Relat Cancer. 2012;19:435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones GN, Manchanda PK, Pringle DR, Zhang M, Kirschner LS. Mouse models of endocrine tumours. Best Pract Res Clin Endocrinol Metab. 2010;24:451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007;67:959–966 [DOI] [PubMed] [Google Scholar]
- 11. Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene. 2010;29:5678–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yeager N, Brewer C, Cai KQ, Xu XX, Di Cristofano A. Mammalian target of rapamycin is the key effector of phosphatidylinositol-3-OH-initiated proliferative signals in the thyroid follicular epithelium. Cancer Res. 2008;68:444–449 [DOI] [PubMed] [Google Scholar]
- 13. Vasko V, Saji M, Hardy E, et al. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet. 2004;41:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Testa JR, Bellacosa A. Membrane translocation and activation of the Akt kinase in growth factor-stimulated hematopoietic cells. Leuk Res. 1997;21:1027–1031 [DOI] [PubMed] [Google Scholar]
- 15. Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24:7401–7409 [DOI] [PubMed] [Google Scholar]
- 17. Nadella KS, Kirschner LS. Disruption of protein kinase a regulation causes immortalization and dysregulation of D-type cyclins. Cancer Res. 2005;65:10307–10315 [DOI] [PubMed] [Google Scholar]
- 18. Weber F, Shen L, Aldred MA, et al. Genetic classification of benign and malignant thyroid follicular neoplasia based on a three-gene combination. J Clin Endocrinol Metab. 2005;90:2512–2521 [DOI] [PubMed] [Google Scholar]
- 19. Tsygankova OM, Saavedra A, Rebhun JF, Quilliam LA, Meinkoth JL. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol Cell Biol. 2001;21:1921–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lou L, Urbani J, Ribeiro-Neto F, Altschuler DL. cAMP inhibition of Akt is mediated by activated and phosphorylated Rap1b. J Biol Chem. 2002;277:32799–32806 [DOI] [PubMed] [Google Scholar]
- 21. Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861 [DOI] [PubMed] [Google Scholar]
- 22. Haymart MR, Repplinger DJ, Leverson GE, et al. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J Clin Endocrinol Metab. 2008;93:809–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franco AT, Malaguarnera R, Refetoff S, et al. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proc Natl Acad Sci USA. 2011;108:1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mavrakis M, Lippincott-Schwartz J, Stratakis CA, Bossis I. mTOR kinase and the regulatory subunit of protein kinase A (PRKAR1A) spatially and functionally interact during autophagosome maturation. Autophagy. 2007;3:151–153 [DOI] [PubMed] [Google Scholar]
- 25. Day ME, Gaietta GM, Sastri M, et al. Isoform-specific targeting of PKA to multivesicular bodies. J Cell Biol. 2011;193:347–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blancquaert S, Wang L, Paternot S, et al. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol Endocrinol. 2010;24:1453–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chaturvedi D, Cohen MS, Taunton J, Patel TB. The PKARIα subunit of protein kinase A modulates the activation of p90RSK1 and its function. J Biol Chem. 2009;284:23670–23681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res. 2007;67:8002–8006 [DOI] [PubMed] [Google Scholar]
- 29. Aroor AR, Jackson DE, Shukla SD. Dysregulated phosphorylation and nuclear translocation of cyclic AMP response element binding protein (CREB) in rat liver after chronic ethanol binge. Eur J Pharmacol. 2012;679:101–108 [DOI] [PubMed] [Google Scholar]
- 30. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27 [DOI] [PubMed] [Google Scholar]
- 31. Saji M, Narahara K, McCarty SK, et al. Akt1 deficiency delays tumor progression, vascular invasion, and distant metastasis in a murine model of thyroid cancer. Oncogene. 2011;30:4307–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCarty SK, Saji M, Zhang X, et al. Group I p21-activated kinases regulate thyroid cancer cell migration and are overexpressed and activated in thyroid cancer invasion. Endocr Relat Cancer. 2010;17:989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xie J, Ponuwei GA, Moore CE, Willars GB, Tee AR, Herbert TP. cAMP inhibits mammalian target of rapamycin complex-1 and -2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cell Signal. 2011;23:1927–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kusakabe T, Kawaguchi A, Kawaguchi R, Feigenbaum L, Kimura S. Thyrocyte-specific expression of Cre recombinase in transgenic mice. Genesis. 2004;39:212–216 [DOI] [PubMed] [Google Scholar]
- 35. Trimboli AJ, Cantemir-Stone CZ, Li F, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones GN, Tep C, Towns WH, 2nd, et al. Tissue-specific ablation of Prkar1a causes schwannomas by suppressing neurofibromatosis protein production. Neoplasia. 2008;10:1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378 [DOI] [PubMed] [Google Scholar]
- 38. Huang da W, Sherman BT, Zheng X, et al. Extracting biological meaning from large gene lists with DAVID. Curr Protoc Bioinformatics. 2009;Chapter 13:Unit 13.11 [DOI] [PubMed] [Google Scholar]
- 39. Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264 [DOI] [PubMed] [Google Scholar]
- 40. Yu L, Gulati P, Fernandez S, Pennell M, Kirschner L, Jarjoura D. Fully moderated T-statistic for small sample size gene expression arrays. Stat Appl Genet Mol Biol. 2011;10. [DOI] [PMC free article] [PubMed] [Google Scholar]