

1. Introduction
Nitrogen is an essential element contained in many biomolecules necessary to sustain life.1,2 This element is abundantly available in Earth’s atmosphere in the form of dinitrogen (N2) gas, yet most organisms are unable to metabolize N2 because it is relatively inert.3,4 Instead most organisms must obtain their N from “fixed” forms such as ammonia (NH3) or nitrate (NO3–).5−7 Because fixed forms of N are continuously sequestered into sediments, rendering them unavailable for metabolism, and because they are also continuously converted to N2 through the combined processes of nitrification and denitrification, life can only be sustained by conversion of N2 to NH3.6,7 This latter process is known as N2 fixation8 and is a critical step in the biogeochemical N cycle.5,7,9 N2 fixation occurs in three different ways: (i) through geochemical processes such as lightning,9 (ii) biologically through the action of the enzyme, nitrogenase,10,11 found only in a select group of microorganisms,12,13 and (iii) industrially through the Haber–Bosch process.2,14,15 From the evolution of nitrogenase, approximately two billion years ago16 until the widespread use of the Haber–Bosch process in the 1950s, all life derived N from biological nitrogen fixation, with geochemical processes representing a minor contributor to the supply of fixed nitrogen.2,7 Since the increase in use of the Haber–Bosch process, the biological and industrial processes contribute comparably to N2 fixation.5,7,9
Nitrogen fixation has a profound agronomic, economic, and ecological impact owing to the fact that the availability of fixed nitrogen represents the factor that most frequently limits agricultural production throughout the world.2 Indeed, nearly half of the existing human population could not exist without application of the Haber–Bosch process for production of nitrogen fertilizers.2,5 Given that over half of the fixed nitrogen input that sustains Earth’s population is supplied biologically, there has been intense interest in understanding how the nitrogenase enzyme accomplishes the difficult task of N2 fixation at ambient temperature and pressure.17,18 An understanding of biological N2 fixation may further serve as the foundation for achieving two highly desirable, although so far unmet, goals: genetically endowing higher plants with the capacity to fix their own nitrogen,19−21 and developing improved synthetic catalysts based on the biological mechanism.3,4,22−25
It has been over 150 years since Jodin first suggested that microbes could “fix” N2,26 and more than a century since the first isolation of N2-fixing bacteria around 1900. In 1934, Burk coined the term “nitrogenase”10,11 for the enzyme that catalyzes the conversion of N2 to a bioaccessible form of nitrogen, and initiated the first meaningful studies of nitrogenase in living cells. Methods for extracting nitrogenase in an active form were developed in the early 1960s,27−29 opening the way for serious mechanistic investigations. The next 35 years witnessed intensive efforts by numerous investigators to reveal the structure and catalytic function of nitrogenase.30−34 These developments were summarized in the magisterial review by Burgess and Lowe in 1996.17 Key advances in understanding nitrogenase structure and function during those intervening years included the following: (i) It was determined that nitrogenase is a two-component system35−37 composed of the MoFe protein (also called dinitrogenase or component I) and the electron-transfer Fe protein (also called dinitrogenase reductase or component II).34,38−41 (ii) A reducing source and MgATP are required for catalysis.42−45 (iii) Fe protein and MoFe protein associate and dissociate in a catalytic cycle involving single electron transfer and MgATP hydrolysis.38 (iv) It was discovered that the MoFe protein contains two metal clusters: the iron–molybdenum cofactor (FeMo-co),30,46 which provides the active site for substrate binding and reduction, and P-cluster, involved in electron transfer from the Fe protein to FeMo-co.39,47−50 (v) Crystallographic structures were solved for both Fe51 and MoFe32,48,52−54 proteins. (vi) Also, the alternative V- and Fe-type nitrogenases, in which the Mo of FeMo-co is replaced by V or Fe, were discovered.18 Despite this accumulation of functional and structural information, the catalytic mechanism remained elusive.
The years since the Burgess and Lowe review17 have seen profound advances in understanding many aspects of nitrogenase structure and function. For example, the solutions of a number of high-resolution X-ray structures of the nitrogenase component proteins55−69 have provided insights into the nature of the active site FeMo-cofactor, most recently identifying the presence of an interstitial C atom,70−77 while structures of the two proteins in the complex78−81 have identified their binding interface (Figures 1 and 2) and its alterations with the state of the bound nucleotide.67 Likewise, great strides have been made in understanding the biosynthesis and insertion of the metal clusters of nitrogenase to form the mature proteins,21,82−89 and the properties of the V-type nitrogenase.90−98 Recent studies have begun to shed light on the order of events during the catalytic cycle,99−103 including the nature of electron transfer between the metal clusters62,104−111 and the roles of ATP binding and hydrolysis in these processes.55,68,99,112−121 Considerable progress has been made in the application of theoretical methods to various aspects of the nitrogenase mechanism.122−140 Finally, progress has been made in expanding in the substrates of nitrogenases93,141−149 to include CO95,96,98,150,151 and CO2.148,152
Figure 1.

Molybdenum nitrogenase. (A) One catalytic half of the Fe protein:MoFe protein complex with the Fe protein homodimer shown in tan, the MoFe protein α subunit in green, and the β subunit in cyan. (B) Space filling and stick models for the 4Fe–4S cluster (F), P-cluster (P), and FeMo-co (M). Made with Pymol and ChemDraw using PDB:2AFK.
Figure 2.

FeMo-cofactor and the side chains of selected amino acid residues of the MoFe protein. Numbering of iron atoms is according to the structure PDB coordinate 2AFK. Iron is shown in rust, molybdenum in magenta, nitrogen in blue, sulfur in yellow, carbon in gray, and oxygen in red.
The present narrative focuses on recent progress in understanding the mechanism of N2 activation and reduction to ammonia by Mo-nitrogenase. The discussion begins with a short reminder of the kinetic scheme that describes nitrogenase catalysis.33,103 It then turns to the successes in trapping catalytic intermediates of the MoFe protein by rapid freezing of turnover mixtures of Fe protein and of MoFe proteins, both wild-type and variants containing selected amino acid substitutions as a means to modulating reactivity.146,148,153,154 The use of EPR/ENDOR/ESEEM spectroscopic techniques applied to isotopically substituted trapped intermediates has allowed the identification and characterization of key intermediates along the N2 reduction pathway.154−156 This led to the formulation of a reaction mechanism based on the properties of catalytic intermediates and grounded in the reaction of hydrides associated with FeMo-co.156 The mechanism not only satisfies all constraints on the mechanism provided by earlier studies, but has suggested and passed a stringent test.157 This report recounts these advances, and expands on them.
2. Background
Two issues require consideration as a basis for discussion of recent advances in nitrogenase mechanism.155,156 The first is the kinetic model that has been developed to describe the multistep reduction of N2 to two NH3, and its implications for the stoichiometry of this reaction,33,103 implications that were mutually supported by experiment.158 The second is the strategies and procedure that at last enabled the trapping of catalytic intermediates whose characterization by advanced paramagnetic resonance techniques underlies the progress in mechanism described here.
2.1. Kinetics and Stoichiometry
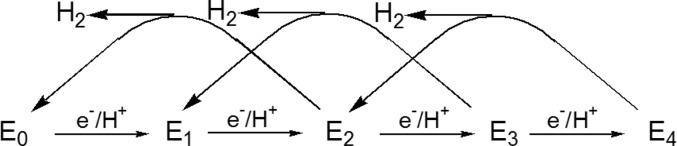
A “kinetic” foundation for a nitrogenase mechanism was developed by extensive studies in the 1970s and 1980s by many groups, especially Lowe and Thorneley and their co-workers.17,33,103 The culmination of these extensive kinetic studies, which involved steady-state, stopped-flow, and freeze–quench kinetics measurements, was the Lowe–Thorneley (LT) kinetic model for nitrogenase function,17,33,103 which describes the kinetics of transformations among catalytic intermediates (denoted En) where n is the number of steps of electrons/protons delivery to MoFe protein, Figure 3. Electron transfer from Fe protein to MoFe protein is driven by the binding and hydrolysis of two MgATP species within the Fe protein;99 the release of the Fe protein after delivery of its electron is the rate-limiting step of catalysis.33
Figure 3.

Simplified LT kinetic scheme that highlights correlated electron/proton delivery in eight steps. Although in the full LT scheme N2 binds at either the E3 or E4 levels, the pathway through E3 is de-emphasized here. LT also denotes the protons bound to FeMo-co (e.g., E1H1); for clarity we have omitted these protons in this scheme.
A central consequence of the kinetic measurements and defining feature of this scheme, Figure 3, is that the limiting enzymatic stoichiometry for enzyme-catalyzed nitrogen fixation is not what would be given by the simple balanced equation for reduction of N2 to two NH3 by six electrons/protons, but is given by eq 1
| 1 |
This is a conclusion that is in agreement with stoichiometric experiments by Simpson and Burris.158 This equation highlights several key aspects of the nitrogenase mechanism, including the involvement of ATP hydrolysis in substrate reduction and the obligatory formation of 1 mol of H2 per mole of N2 reduced, an apparent “waste” of two reducing equivalents and four ATP per N2 reduced.17,33
Although the close of the previous millennium saw the accumulation of a vast breadth and depth of information about the reduction of N2, H+, and a variety of other nonphysiological substrates,17 it was not until recently that studies have succeeded in characterizing En intermediate states beyond the resting-state E0.154−156 Thus, the early studies provided little direct experimental evidence regarding a reaction pathway, and hence, there was no possibility of integrating a reaction pathway and kinetic scheme, as is central to development of a mechanism based on the properties of catalytic intermediates.156
2.2. Trapping and Characterization of Substrates
The first 40 years of study of purified nitrogenase did not see the definitive characterization of any intermediates associated with the binding and reduction of N2,159 leaving the identity of the reaction pathway unresolved. The way forward was provided by studies of nitrogenases with individual amino acid substitutions, which revealed that the residue at position α-70 within the MoFe protein, a valine, acts as a “gatekeeper” that sterically controls the access of substrate to the active site FeMo-co (Figure 2).146,154 The side chain of this amino acid residue is located over one FeS face of FeMo-cofactor (that includes Fe atoms 2, 3, 6, and 7) thereby also implicating Fe as the site of substrate binding, while the α-195His was inferred to be involved in proton delivery (Figure 2).160−165
Use of MoFe protein substituted at one or both of these residues enabled freeze–quench trapping of a number of nitrogenase turnover intermediates, almost all of which show an EPR signal arising from an S = 1/2 state of FeMo-co, rather than the S = 3/2 state of resting-state FeMo-co.153,154 The procedures developed with these variants even enabled N2−intermediate trapping with enzyme.166 The first fruit of this approach was the trapping of a state during reduction of the alkyne propargyl alcohol to the corresponding alkene.145,167 An intermediate trapped using MoFe protein variants was shown by ENDOR studies to be a wholly novel bio-organometallic structure in which the alkene product of alkyne reduction by nitrogenase binds as a π-complex/ferracycle to a single Fe ion of FeMo-cofactor, presumed to be Fe6.168 This was followed by characterization of intermediates formed during the reduction of H+ under Ar,169 and, finally, identification of four associated with N2 reduction itself.147,153,160,166,170,171
Paramagnetic resonance methods have proven to be uniquely advantageous for characterization of trapped nitrogenase intermediates.155 At the most basic level, FeMo-co in the E0 resting-state of MoFe protein is an odd-electron (“Kramers”; half-integer spin, S = 3/2172), EPR-active cluster, and therefore, intermediate states that have accumulated an even number of electrons also will be EPR-active. Focusing on nitrogen fixation, FeMo-co then will be EPR-active in the En states, n = 2, 4, 6, 8, formed along the pathway for accumulation of the stoichiometrically required eight [e–/H+], eq 1. In contrast, En states n = 1, 3, 5, 7 will be even-electron, and FeMo-co will either be diamagnetic or in an integer-spin (“non-Kramers” spin-state)173 cluster, which also can be EPR-active under appropriate conditions.173,174
As will be illustrated below, electron–nuclear double-resonance (ENDOR) spectroscopy,175,176 supported by related techniques ESEEM and HYSCORE,177 is uniquely suited for the study of freeze–quench trapped intermediates. These techniques give NMR-like spectra of nuclei that are hyperfine-coupled to the electron spin of an EPR-active cluster. The importance of the techniques rests on several aspects. ENDOR is broad-banded: with isotopic enrichment it can monitor every atom in a metalloenzyme active site. Thus, when interpreted in the context of the X-ray structure of the resting-state, it can reveal the electronic and metrical structure of a catalytic intermediate. It is selective: it interrogates only EPR-active states. It is high-resolution: it can resolve and interrogate the signals from multiple distinct EPR-active centers. It is sensitive: we have successfully analyzed the properties of intermediates present in ∼20% abundance in a sample containing ∼100 μM MoFe protein. Viewed another way, ENDOR is capable of selecting and characterizing a small fraction of the MoFe protein in a sample. In contrast, for example, Mössbauer and X-ray absorption techniques, which have made enormous contributions to the study of resting-state nitrogenase, interrogate all FeMo-co in a sample, and if the state of interest is a small minority, its signal is buried and lost. Recently, however, an X-ray spectroscopic study has given information about a freeze–quenched nitrogenase intermediate.178
3. Intermediates of Nitrogenase Activation
According to the simplified LT kinetic scheme of Figure 3, the first four of the eight [e–/H+] of nitrogen fixation accumulate prior to N2 binding, which occurs at the E4 stage. The complete scheme17,33,103 allows for N2 binding at E3 as well, but E4 uniquely places the enzyme on the pathway to N2 hydrogenation.
3.1. E1–E3
The E1 state contains one-electron reduced cofactor, and has been assigned as an integer-spin species on the basis of Mössbauer studies of MoFe protein trapped during turnover under N2.179,180
High-spin EPR signals (S = 3/2), denoted as 1b and 1c, thought to be associated with En states, n ≤ 4, were first observed 35 years ago for samples of wild-type nitrogenase trapped during turnover using a variety of conditions,181 and more recently were studied by rapid freeze–quench EPR.182 The kinetics of appearance of 1b and 1c demonstrated that they must be assigned to reduced states of cofactor, n > 1, rather than just as conformers of the FeMo-co resting-state. However, the kinetics of appearance of the stronger 1b signal was a puzzle: they were best described by assigning 1b to the E3 state, which would seem to require that FeMo-co be in an integer-spin (non-Kramers) state, contrary to observation. Most likely, this apparent contradiction reflects uncertainties in the rate constants used in the kinetics analysis, and 1b represents an E2 state. During cryoannealing experiments183 discussed below, we definitively observed that FeMo-co of E2 is in a high-spin (S = 3/2) state, but at least in the α-70Ile variant its g-values were distinct from those of 1b. The spectrum of the 1c species is weaker in intensity. It may represent a conformer of the resting or 1b states, or may correspond to even more reduced states, as its effective formation requires a high molar ratio of Fe protein to MoFe protein, corresponding to higher electron flux.
3.2. E4: The “Janus Intermediate”
In this subsection we describe the trapping and EPR/ENDOR characterization of the E4 intermediate as activated by the accumulation of four [e–/H+] for binding and reduction of N2. The structure of E4 as determined by ENDOR spectroscopy, and integrated into the LT kinetic scheme, has been the key to recognizing the central role of hydrides in the mechanism for nitrogen fixation.156 We then discuss the E1–E4 states associated with electron accumulation by MoFe protein; subsequent sections discuss the trapped states associated with the N2 reduction pathway following N2 binding.
Early in the search for intermediates,146 the α-70Val→Ile substitution in the MoFe protein was shown to deny access of all substrates to the active site, except protons.169,184 Samples of this substituted MoFe protein freeze–quenched during turnover under Ar exhibited a new S = 1/2 EPR signal,169 which also can be observed at lower concentrations during turnover of wild-type MoFe protein under Ar.181,1851,2H ENDOR spectroscopic analysis of this trapped state169 revealed the presence of two strongly hyperfine-coupled, metal-bridging hydrides [M–H–M′]: (i) The finding that the bound hydrides have a large isotropic hyperfine coupling, aiso ≈ 24 MHz, led to their assignment as hydrides bound to metal ion(s) of the core. (ii) The anisotropic hyperfine contribution, T = [−13.3, 0.7, 12.7] MHz, exhibits almost complete rhombicity, as defined by the form Trh ≈ [t, 0, −t]. This form rules out terminal hydrides, which would have a roughly axial T,186 and is precisely the form first predicted187 and then confirmed188 to be associated with a hydride bridging two paramagnetic metal ions, namely as [Fe–H–Fe] and/or [Mo–H–Fe] fragments.
95Mo ENDOR measurements subsequently established that both hydrides bridge two Fe ions, forming two [Fe–H–Fe] fragments (Figure 4), as follows.189 Equations for the anisotropic hyperfine interaction matrix, T, of a nucleus that undergoes through-space dipolar interactions to two spin-coupled metal ions187 were generalized to describe an arbitrary [M1–H–M2] fragment of a spin-coupled cluster. The components of T are a function of the [M1–H–M2] geometry and of the coefficients [K1, K2] that describe the projection of the total cluster spin on the two local M-ion spins. The 95Mo ENDOR measurements of the intermediate showed a very small isotropic hyperfine coupling, aiso(95Mo) ∼ 4 MHz, which indicated that KMo is too small to yield the rhombic dipolar coupling, Trh, observed in this intermediate.189 The model for E4 displayed in Figure 4 is completed by placement on sulfurs of the two protons190,191 that form part of the delivery of 4[e–/H+] (Figure 3). The protons are so placed because they must be near to the negative charge density associated with the hydrides in order to obtain the electrostatic stabilization implicit in the required accumulation of one proton for each electron delivered to MoFe protein;17 other arrangements are possible, such as putting both protons on doubly bridging sulfur, but see below.
Figure 4.
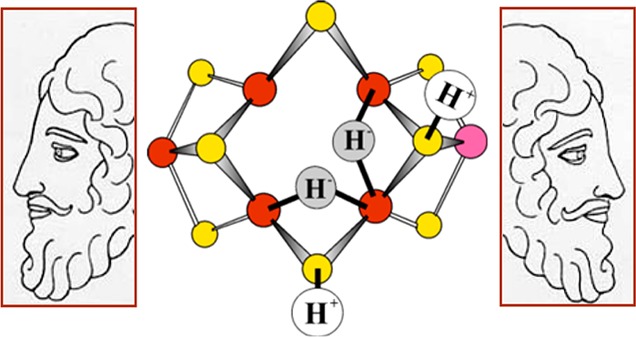
Depiction of E4 as containing two [Fe–H–Fe] moieties, emphasizing the essential role of this key “Janus intermediate”, which comes at the halfway point in the LT scheme, having accumulated four [e–/H+], and whose properties have implications for the first and second halves of the scheme. Janus image adapted from http://www.plotinus.com/janus_copy2.htm. Figure adapted with permission from ref (156). Copyright 2013 American Chemical Society.
Cryoannealing this “dihydride” intermediate in the frozen state at −20 °C, which prevents further delivery of electrons from the Fe protein, showed that it relaxes to the resting FeMo-co state by the successive loss of two H2 molecules.183 According to the LT scheme, only E4 would undergo this two-step relaxation process (Scheme 1), with the first relaxation step of E4 yielding H2 and the E2 state, the second step returning FeMo-co to the E0 stage with loss of a second H2, and the production of H2 being revealed by solvent kinetic isotope effects in both stages. This relaxation protocol thus revealed that the trapped intermediate is the E4 state, which has accumulated n = 4 electrons and protons.183 As the relaxation measurements involved tracking the kinetically linked conversion of E4 into E2, and the conversion of E2 into resting-state E0, the measurements further allowed an unambiguous identification of the EPR signal associated with E2 (see above).
Scheme 1.
Examination of the simplified version of the LT scheme of Figure 3 reveals that E4 is a key stage in the process of N2 reduction.33,103 Indeed, we have denoted it as the “Janus” intermediate, referring to the Roman God of transitions who is represented with two faces, one looking to the past and one looking to the future (Figure 4).156 Looking “back” from E4 to the steps by which it is formed, E4 is the culmination of one-half of the electron/proton deliveries during N2 fixation: four of the eight reducing equivalents are accumulated in E4, before N2 even becomes involved. Looking “forward”, toward NH3 formation, E4 is the state at which N2 hydrogenation begins, and it is involved in one of the biggest puzzles in N2 fixation: “why” and “how” H2 is lost upon N2 binding.
To date, we have visualized E4 by placing its two hydrides on the Fe2, 3, 6, 7 face of resting-state FeMo-co and sharing a common vertex at Fe6, Figure 4. Although the hydrides may well exhibit fluxionality at ambient temperature, their ability to adopt a configuration with a common vertex is required by the reductive elimination (re) mechanism of reversible H2 release upon N2 binding (section 7), and Fe6 is favored from earlier kinetic studies on MoFe protein variants.69,144,145,154,166,168 However, this model is only one of four possible configurations based on the resting structure that have two hydrides sharing an Fe6 vertex. To visualize these structures we have built the bound hydrides onto the crystal structure of resting-state FeMo-co using Fe–H distances from model complexes,188,192 Figure 5.
Figure 5.
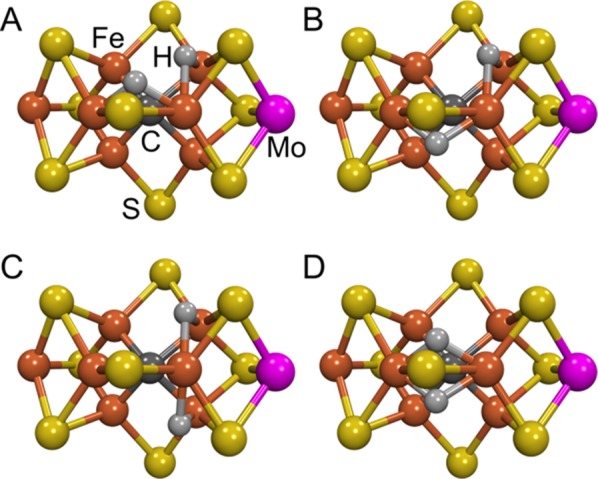
Mockups of the “Janus” E4 intermediate in which the two bridging hydrides [Fe–H–Fe] revealed by ENDOR spectroscopy are built onto the resting-state crystal structure. These models of FeMo-co have Fe6 as a “vertex” for the two bridging hydrides to facilitate reductive elimination. The figure was generated using the coordinate file PDB:2AFK. Iron is shown in rust, molybdenum in magenta, sulfur in yellow, carbon in dark gray, and hydrogen in light gray.
Quantum chemical computations will test these alternatives. However, the experimentally determined relative orientation of the hyperfine tensors of the two hydrides provides a significant constraint on their placement within E4. Given the stability of the FeMo-co structure that is likely imparted by the interstitial carbide, it seemed plausible to us that consideration of the constructed models of Figure 5 would allow us to test these alternative hydride distributions, even though it is beyond doubt that the structure of FeMo-co will distort upon substrate binding. This exercise (see Supporting Information) provides support for the topology of hydride binding pictured for the Janus E4 intermediate in Figure 4, with hydrides bridging Fe2/Fe6 and Fe6/Fe7 (Figure 5A,B), as opposed to Figure 5C,D, but does not discriminate between the structures of Figure 5A,B. In discussions below, we retain the placement of the E4 hydrides shown in Figure 4 (Figure 5A) as being more readily visualized in discussions of mechanism.
The characterization of the hyperfine interactions of the metal-ion core of E4 that began with the 95Mo ENDOR measurements189 was completed by an ENDOR study of the 57Fe atoms of the E4 FeMo-co through use of a suite of advanced ENDOR methods.193 The determination of hyperfine interactions for two ligand hydrides and all eight metal ions of FeMo-cofactor in this state will provide the experimental test that guides future computational studies that seek to characterize the geometric and electronic structure of E4.
Storage of the reducing equivalents accumulated in the E4 state as bridging hydrides has major consequences. A bridging hydride is less susceptible to protonation than a terminal hydride, and thus bridging hydride(s) diminish the tendency to lose reducing equivalents through the formation of H2 (Scheme 1), thereby facilitating the accumulation of reducing equivalents by FeMo-co. This mode also lowers the ability of the hydrides to undergo exchange with protons in the environment, a characteristic that is shown to be of central importance below. However, the bridging mode also lowers hydride reactivity toward substrate hydrogenation, relative to that of terminal hydrides.194,195 As a result, substrate hydrogenation most probably incorporates the conversion of hydrides from bridging to terminal binding modes.196 We next discuss how the structure found for E4 guides assignment of structures for the E1–E3 states. Subsequently, we show how the E4 structure defined possible mechanisms for coupling H2 loss to N2 binding.
3.3. Redox Behavior and Hydride Chemistry of E1–E3: Why Such a Big Catalytic Cluster?
Given that the four accumulated electrons of E4 reside not on the metal ions but, instead, are formally assigned to the hydrides of the two Fe-bridging hydrides, what then are the proper descriptions of E1–E3? The addition of one electron/proton to the MoFe protein results in the E1 state, and a Mössbauer study of nitrogenase trapped during turnover under N2180 suggested that this state contains the reduced metal-ion core of FeMo-co, denoted M– in Figure 6A. The presence in E4 of two bridging hydrides/two protons led us to propose that upon delivery of the second electron/proton to form E2 the metal–sulfur core of the FeMo-cofactor “shuttles” both electrons onto one proton to form an [Fe–H–Fe] hydride, leaving the second proton bound to sulfur for electrostatic stabilization and the core formally at the resting-state, M0, redox level (also commonly referred to as, MN),197 Figure 6A. A subsequent, analogous, two-stage process would then yield the E4 state, with its two [Fe–H–Fe] hydrides, two sulfur-bound protons, and the core at the resting-state, M0, redox level.193
Figure 6.
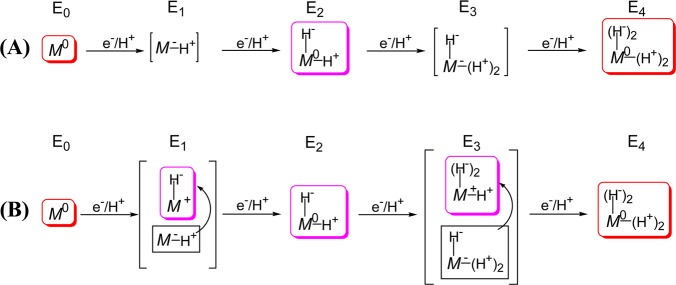
Formulations of E1–E4 derived from consideration of E4 as containing two bound hydrides and two protons. (A) Assuming reduction of the core in n = 1, 3 states. (B) Alternative formulation of E1–E4 under the assumption of hydride formation at every stage, in which case the core is formally oxidized for En, n = 1, 3. Symbols: M represents FeMo-co core; superscripts are charge difference between core and that of resting-state (commonly denoted MN); the number of bound protons/hydrides are indicated. Adapted with permission from ref (156). Copyright 2013 American Chemical Society.
Such a process of acquiring the four reducing equivalents of E4 involves only a single redox couple connecting two formal redox levels of the FeMo-co core of eight metal ions; M0 the resting-state, and M– the one-electron reduced state of the core, Figure 6A.193 Indeed, comparisons of the 57Fe ENDOR results for the E4 intermediate with earlier 57Fe ENDOR studies and “electron inventory analyses”155,198 of nitrogenase intermediates led us to the remarkable suggestion that, throughout the nitrogenase catalytic cycle, the FeMo-cofactor would cycle through only two formal redox levels of the metal-ion core. On reflection, it seems obvious that only by “storing” the equivalents as hydrides is it possible to accumulate so much reducing power at the constant potential of the Fe protein. We further proposed that such “simple” redox behavior of a complex metal center might apply to other FeS enzymes carrying out multielectron substrate reductions.193
Considering the critical role of hydrides in storing reducing equivalents, we also suggested that the E1 and E3 states, respectively, might well contain one and two bridging hydrides bound to a formally oxidized metal-ion core (Figure 6B),100 in which case the single redox couple accessed would formally be that between M0 and M+. In section 9, below we adopt this “oxidative” formulation of the E1–E3 structures. We emphasize that a third formulation of E1(E3), with hydride(s) bound to M0 and the presence of oxidized P-cluster, is ruled out by the absence of EPR signals from P+ in samples trapped under turnover conditions.
If the FeMo-cofactor does not utilize more than one redox couple during catalysis, then why is it constructed from so many metal ions? As discussed above, the hydrides of E4 bind to at least two, and plausibly three Fe atoms of a 4-Fe face of FeMo-co, as shown in Figures 4 and 5. It is further possible that catalysis is modulated by the linkage of Fe ion(s) to the anionic atom C that is centrally located within the metal–sulfur core of the FeMo-cofactor.70,71 Formation of such a 4Fe face and the incorporation of C is not likely with less than a trigonal prism of six Fe ions linked by sulfides to generate these structural features. In this view, the trigonal prismatic FeMo-cofactor core of six Fe ions plus C generates the catalytically active 4Fe face. This prism is capped, and its properties are likely “tuned”, by two “anchor” ions, one Fe plus a Mo, or a V or Fe in the alternative nitrogenases.
Finally, and far from least, as we have consistently noted (see section 7), there is good reason to imagine N2 and/or the N2Hx reduction intermediates may interact with multiple Fe ions on a FeMo-co face.
3.4. Why Does Nitrogenase Not React with H2/D2/T2 in the Absence of N2?
The following question is commonly raised: If electrons accumulated in En intermediates, n = 2–4, can relax to En-2 through formation and release of H2 during turnover, as captured in the partial LT scheme, Scheme 1, why does the enzyme not exhibit the reverse of this reaction, and react with H2/D2 in what might appear to be the “microscopic reverse” of H2 release? We have proposed that H2 formation involves protonation of an [Fe–H–Fe], and at a basic level, all three relaxation processes of Scheme 1 should have much the same characteristics. For simplicity in addressing this issue, we focus on the “first” of these, the E2 → E0 relaxation, and ask why E0 is not reduced by H2 to form E2, eq 2
| 2 |
A logical answer to this question begins with the recognition that the LT kinetic scheme for N2 fixation, Figure 3 (also denoted the “MoFe protein cycle”), and the segment presented in Scheme 1, omit the reactions of the Fe protein for clarity; these are treated as a separate “Fe-protein cycle”.17,33,103,154 A stoichiometrically correct scheme that merges the Fe protein and MoFe protein cycles is given in Figure 7. It reminds us that E2 is formed by two steps of Fe → MoFe protein ET, with each step involving hydrolysis of two ATP molecules to drive a reaction that is highly “uphill” energetically.
Figure 7.

Formation and relaxation of E2. In-line: The “on-path” two-step, ATP-dependent addition of two H+/e– to MoFe protein to form E2. Off-line: Representation of the exergonic (free energy, +|ΔGh|) “off-path” relaxation of E2, liberating H2 and directly regenerating E0 without intervention of Fe protein, and of the energetically (free energy, +|ΔGh|) and kinetically forbidden reverse of this process; E0′ is a putative intermediate state that causes the reaction of E0 not to be the microscopic reverse of the release of H2 from E2 (see text).
Clearly the E2 → E0 relaxation with accompanying loss of H2 is not the “reverse” of the turnover formation of E2 from E0; neither Fe protein reduction nor ATP formation is involved. Instead, it is a side-reaction of E2. Indeed, it is even quite unlikely that a direct reaction of H2 with E0 to form E2 (eq 2) would be the microscopic reverse of the E2 → E0 relaxation with accompanying loss of H2. Moreover, the steric congestion caused by the sulfurs at the six tetrahedral [FeS3C] sites of the FeMo-co “waist” requires that the core must relax for the Fe to bind any ligand; in particular it is probable that the structure of the [Fe7S9MoC] core of FeMo-cofactor of E0 (denoted MN) is altered during the reduction of E0 by two [e–/H+] to form E2 (also see section 9.2). In this case, as illustrated in Figure 7, the relaxation of E2 with loss of H2 to form the resting E0 state would be a 2-step process. The loss of H2 by E2 would be expected to form a state (denoted here E0′) that contains FeMo-cofactor in a conformation approximating that of E2, corresponding to a metastable conformation of its resting redox level (denoted MN′); this conformer would in turn undergo a MN′ → MN structural relaxation associated with E0′ → E0 relaxation. The reduction of E0 by H2 (eq 2) by the microscopic reverse of this two-step relaxation would correspondingly take place in two steps, Figure 7, with the initial E0 → E0′ thermal activation associated with the conformational change, MN → MN′, adding an activation free energy (denoted |ΔG†|) and kinetic barrier to the endergonic reduction of E0′ → E2 by H2 (free energy denoted |ΔGh′|).
What would be the free energy for reduction of nitrogenase by H2, as in eq 2? An upper bound for the free energy change for this reaction, ΔGh2, would be 4 times the negative of the free energy change for the hydrolysis of ATP to form ADP and Pi (−ΔGHyd ∼ +7 kcal/mol; total, endergonic by ∼ +28 kcal/mol), that is required for the formation of E2 through the delivery of reducing equivalents by Fe protein; roughly compatible with that, oxidative addition of H2 to an Fe–S center (hydrogenase), corresponding to |ΔGh′|, is uphill by at least +20 kcal/mol,199,200 to which must be added the conformational free energy |ΔG†|. Given the strongly endergonic nature of eq 2, coupled with the kinetic penalty associated with the activation of E0 to E0′, it becomes clear why H2 is not observed to reduce FeMo-cofactor.
4. “Dueling” N2 Reduction Pathways
Researchers have long considered two competing proposals for the second half of the LT kinetic scheme, the reaction pathway for N2 reduction that begins with the Janus E4 state.17,139,155 These invoke distinctly different intermediates, Figure 8, and computations suggest they likely involve different metal-ion sites on FeMo-co.139 The “distal” (D) pathway is associated with the Chatt4,201 or Chatt–Schrock cycle3 because it is utilized by inorganic Mo complexes discovered by these investigators to cleave N2 (Chatt and co-workers202,203) and, most dramatically, to catalytically fix N2 (Schrock and co-workers24,204−206). In this cycle, which has been suggested to apply to nitrogen fixation by nitrogenase with Mo as the active site,139 a single N of N2 is hydrogenated in three steps until the first NH3 is liberated, and then the remaining nitrido-N is hydrogenated three more times to yield the second NH3. In the “alternating” (A) pathway that has been suggested to apply to catalysis at Fe of FeMo-co,25,131 the first two hydrogenations generate a diazene-level intermediate, the next two form hydrazine, and the first NH3 is liberated only by the fifth hydrogenation (Figure 8). As one can imagine alternative structures for the intermediates, the figure focuses on the defining difference between D and A pathways as being the release of the first NH3 in the D as occurring after three hydrogenations of substrate, the addition of three [e–/H+] to substrate, but only after five hydrogenations in A.
Figure 8.

Comparison of distal (D) and alternating (A) pathways for N2 hydrogenation, highlighting the stages that best distinguish them, most especially noting the different stages at which NH3(1) is released.
Simple arguments can be made for both pathways and for either Fe and Mo as the active site.17,154,155,207 For example, the A route is suggested by the fact that hydrazine is both a substrate of nitrogenase and is released upon acid or base hydrolysis of the enzyme under turnover,17,208−211 and is favored in computations with reaction at Fe,131 while the D route was suggested by the fact that until recently the only inorganic complexes that catalytically fix N2 employ Mo and function via the D route,24 which is computationally favored for reaction at Mo.139 Interestingly, this argument is somewhat weakened by a recent study that reported small W clusters fix N2 by the A pathway.212 More significantly, the argument based on N2 cleavage and catalytic N2 fixation by Mo model complexes has lost ground by the quite recent discovery of Fe model complexes that cleave N2 (Holland and covworkers22,213) and indeed that also catalytically fix N2 (Peters and co-workers214).
Further support for the A pathway is provided by considerations of the alternative nitrogenases. It is most economical to suggest that both the Mo-dependent nitrogenase studied here and the V-type nitrogenase reduce N2 by the same pathway. As V-nitrogenase produces traces of N2H4 while reducing N2 to NH3,215 then according to Figure 8 this enzyme can be concluded to function via the A pathway, implying the same is true for Mo-nitrogenase.
5. Intermediates of N2 Reduction: En, n ≥ 4
As can be seen in Figure 8, characterization of catalytic intermediates formed during the reduction of N2 could distinguish between the D and A pathways. However, such intermediates had long eluded capture until four intermediates associated with N2 fixation were freeze-trapped and characterized by ENDOR spectroscopic studies.154,155 These four states were generated under the hypothesis that intermediates associated with different reduction stages could be trapped using N2 or semireduced forms of N2 or their analogues: N2; NH=NH; NH=N–CH3; H2N–NH2.17,153,154 These included a proposed early (e) stage of the reduction of N2, e(N2), obtained from wild-type (WT) MoFe protein with N2 as substrate;166,170 two putative “midstage” intermediates, m(NH=N–CH3), obtained from α-195Gln MoFe protein with CH3–N=NH as substrate170,171 and m(NH=NH), obtained from the doubly substituted, α-70Ala/α-195Gln MoFe protein during turnover with in-situ-generated NH=NH;147 and a “late” stage, l(N2H4), from the α-70Ala/α-195Gln MoFe protein during turnover with H2N-NH2160,170 as substrate. Both hydrazine and diazene are substrates of wild-type nitrogenase that, like N2, are reduced to ammonia.17,147,160,211,216
5.1. Intermediate I
A combination of X/Q-band EPR and 15N,1,2H ENDOR measurements on the intermediates formed with the three semireduced substrates during turnover of the α-70Val→Ala/α-195His→Gln MoFe protein subsequently showed that in fact they all correspond to a common intermediate (here denoted I) in which FeMo-co binds a substrate-derived [NxHy] moiety (Figure 9).154−156,207 Thus, both the diazenes and hydrazine enter and “flow through” the normal N2-reduction pathway (Figure 8), and the diazene reduction must have “caught up” with the “later” hydrazine reaction.
Figure 9.
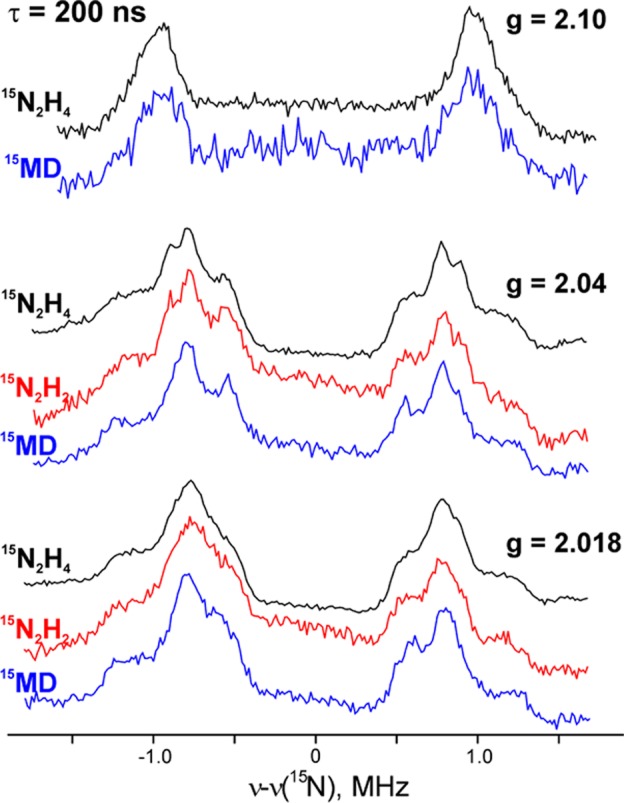
Comparison of 35 GHz ReMims pulsed 15N ENDOR spectra of intermediates trapped during turnover of the α-70Ala/α-195Gln MoFe protein with 15N2H4, 15N2H2, and 15NH=N—CH3 (denoted 15MD). Adapted with permission from ref (207). Copyright 2011 American Chemical Society.
1,2H and 15N 35 GHz CW and pulsed ENDOR measurements next showed that I exists in two conformers, each with metal ion(s) in FeMo-co having bound a single nitrogen from a substrate-derived [NxHy] fragment.154,155 Subsequent high-resolution 35 GHz pulsed ENDOR spectra and X-band HYSCORE measurements showed no response from a second nitrogen atom, and when I was trapped during turnover with the selectively labeled CH3—15N=NH, 13CH3—N=NH, or C2H3—N=NH, no signal was seen from the isotopic labels.207 From these results we concluded the N–N bond had been cleaved in forming I, which thus represents a late stage of nitrogen fixation, after the first ammonia molecule already has been released and only a [NHx] (x = 2 or 3) fragment of substrate is bound to FeMo-co.207
5.2. Nitrogenase Reaction Pathway: D versus A
Given that states that could correspond to I are reached by both A and D pathways (Figure 8), the identity of this [NHx] moiety need not in itself distinguish between pathways. However, the spectroscopic findings about I, in conjunction with a variety of additional considerations, led us to propose that nitrogenase functions via the A reaction pathway of Figure 8 for reduction of N2.207 As one example, to explain how nitrogenase could reduce each of the substrates, N2, N2H2, and N2H4, to two NH3 molecules via a common A reaction pathway, one need only postulate that each substrate “joins” the pathway at the appropriate stage of reduction, binding to FeMo-co that has been “activated” by accumulation of a sufficient number of electrons (possibly with FeMo-co reorganization), and then proceeds along that pathway. Energetic considerations,139 in combination with the strong influence of α-70Val substitutions of MoFe protein without modification of FeMo-co reactivity, then implicate Fe, rather than Mo, as the site of binding and reactivity.146,154,217
5.3. Intermediate H
When nitrogenase is freeze–quenched during turnover, the EPR signals from trapped intermediates in odd-electron FeMo-co states (Kramers states; S = 1/2, 3/2,...; En, n = even),154,155 plus the signals from residual resting-state FeMo-co, never quantitate to the total FeMo-co present, indicating that EPR-silent states of FeMo-co must also exist. These silent MoFe protein states must contain FeMo-co with an even number of electrons, and thus correspond to En, n = odd (n = 2m +1, m = 0–3) intermediates in the LT scheme. As noted above, such states may contain diamagnetic FeMo-co, or FeMo-co in integer-spin (S = 1, 2, ...), “non-Kramers (NK)” states,179,180,218 but no EPR signal from an integer-spin form of FeMo-co had been detected until careful examination of samples that contain intermediate I(154−156) revealed an additional broad EPR signal at low field in Q-band spectra that arises from an integer-spin system with a ground-state non-Kramers doublet with spin S ≥ 2 (Figure 10).219
Figure 10.

2K Q-band CW EPR spectrum of α-70Val→Ala, α-195His→Gln MoFe protein in resting-state (S = 3/2) and trapped during turnover with 14N2H4. Kramers intermediate I and non-Kramers intermediate, H, are noted in the turnover spectrum. Adapted with permission from ref (219). Copyright 2012 National Academy of Sciences.
Earlier work showed how to characterize a non-Kramers doublet with ESEEM spectroscopy (NK-ESEEM),173,174 so NK-ESEEM time-waves were collected for the NK intermediates trapped during turnover with: 14N and 15N isotopologs of N2H2 and N2H4 substrates; 95Mo-enriched α-70Val→Ala/α-195His→Gln MoFe protein; H—14N=14N—CH3, H—15N=14N—CH3, and H—14N=14N—CD3. Figure 11 presents representative 35 GHz (2 K) three-pulse NK-ESEEM time-waves collected at several relatively low fields from the nitrogenase NK intermediates generated with isotopologs of the three substrates. The NK-ESEEM time-waves for the intermediates trapped during turnover with the corresponding 14N and 15N isotopologues of N2H2, N2H4, and HN2CH3 substrates are identical at all fields, indicating that they are associated with a common intermediate, denoted H, trapped during turnover with all three substrates. 95Mo enrichment of α-70Val→Ala, α-195His→Gln MoFe protein produces a significant change of the NK-ESEEM time-wave. This analysis established that the NK-EPR signal of H arises from the Mo-containing FeMo-co in an integer-spin-state with S ≥ 2, and not the all-iron electron-transfer P cluster, also present in the MoFe protein, or even the [4Fe–4S] cluster of the Fe protein.219
Figure 11.
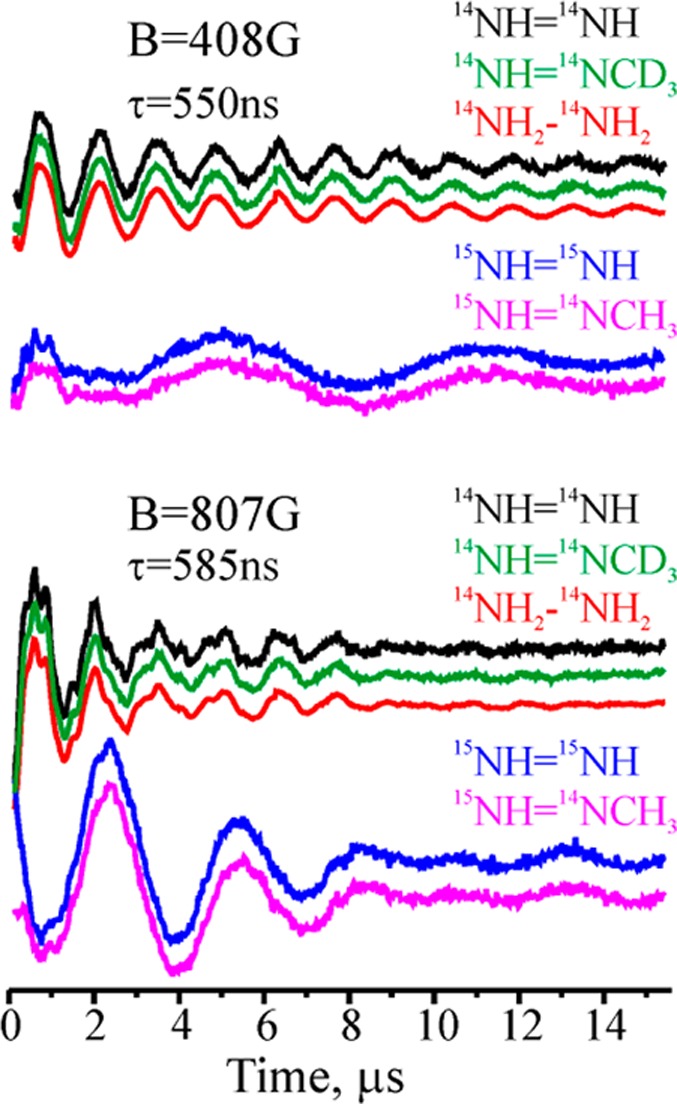
Three-pulse ESEEM traces after decay-baseline subtraction for NK intermediate H of α-70Val→Ala, α-195His→Gln MoFe protein trapped during turnover with 14NH=14NH, 14NH=14NCD3, 14NH2—14NH2, 15NH=15NH, 15NH=14NCH3. Adapted with permission from ref (219). Copyright 2012 National Academy of Sciences.
Comparison of the 14N/15N NK-ESEEM of H in Figure 11 indicates that a nitrogenous ligand derived from substrate is directly bound to FeMo-co of α-70Val→Ala/α-195His→Gln MoFe protein. Modulation is absent from the second 14N that would be present if the N–N bond of substrate remained intact, as shown by comparison of the time-waves for the H prepared with H—15N=15N—H versus H—15N=14N—CH3, as is modulation from 2H of H–14N=14N-CD3. This indicates that H contains an NHx fragment that remains bound to FeMo-co after cleavage of the N–N bond and loss of NH3. Quadrupole coupling parameters for the NHx fragment indicated it is not NH3, and that H has bound [−NH2].219
6. Unification of the Nitrogenase Reaction Pathway with the LT Kinetic Scheme
The H and I intermediates provide “anchor-points” that allow assignment of the complete set of En intermediates that follow E4, 5 ≤ n ≤ 8. As illustrated in Figure 12, the loss of two reducing equivalents and two protons as H2 (eq 1) upon N2 binding to the FeMo-co of E4 leaves FeMo-co activated by two reducing equivalents and two protons. We argued that when N2 binds to FeMo-co it is “nailed down” by prompt hydrogenation, Figure 12, with N2 binding, H2 loss, and reduction to the diazene level, all occurring at the E4 kinetic stage of the LT scheme.219 The identification of H with its En stage is achieved as follows. (i) As the same intermediate H is formed during turnover with the two diazenes and with hydrazine, the diazenes must have catalytically “caught up” to hydrazine, and H must occur at or after the appearance of a hydrazine-bound intermediate. (ii) As noted above, H contains FeMo-co in an integer-spin (NK) state, and thus corresponds to an En state with n = odd. As H is a common intermediate that contains a bound fragment of substrate, it must, therefore, correspond to E5 or E7, and analysis of the pathway alternatives in the light of the EPR/ESEEM measurements indicated that H corresponds to the [NH2]−-bound intermediate formed subsequent to N–N bond cleavage and NH3 release at the E7 stage of the P–A pathway.
Figure 12.

Integration of LT kinetic scheme with “prompt” (P) alternating (A) pathway for N2 reduction. The ? represents the product of N2 binding with H2 release, whose identity is discussed below. Also shown is how diazene and hydrazine join the N2 reduction pathway. Note: M denotes FeMo-co in its entirety, and substrate-derived species are drawn to indicate stoichiometry only, not mode of substrate binding. En states, n = even, are Kramers states; n = odd are non-Kramers. MN denotes resting-state FeMo-co. Individual charges on M and a substrate fragment, not shown, sum to the charge on resting FeMo-co. Adapted with permission from ref (156) with corrections based on the re mechanism for H2 loss upon N2 binding discussed below. Copyright 2013 American Chemical Society.
By parallel arguments, the only possible assignment for the S = 1/2 state I, which we showed earlier to occur after N–N bond cleavage,207 is as E8: I must correspond to the final state in the catalytic process (Figure 12), in which the NH3 product is bound to FeMo-co at its resting redox state, prior to release and regeneration of the resting-state form of the cofactor. The trapping of a product-bound intermediate I is analogous to the trapping of a bio-organometallic intermediate during turnover of the α-70Val→Ala MoFe protein with the alkyne, propargyl alcohol; this intermediate was shown to be the allyl alcohol alkene product of reduction.168 With assignments of E4, E7, and E8, then filling in the LT “boxes” for E5, E6 of Figure 3 is straightforward, thus unifying the reaction pathway for N2 reduction with the LT scheme.
Figure 12 adopts a “prompt” (P)–alternating pathway for the stages following N2 binding and H2 loss, which offers explanations for how the hydrogenated reaction intermediates, diazene and hydrazine, join the N2 reduction pathway. Key to this issue was the finding that H2 inhibits the reduction of diazene,147 but not hydrazine.211 We took the simplest view, that under turnover, diazene and hydrazine each joins the N2 reduction pathway at its own characteristic entry point, and each then proceeds to generate both H and I. As shown in Figure 12, diazene binds to E2 with the release of H2, thereby entering the N2 pathway as the “final” interconverting form of the E4 state. N2H4 instead binds to E1 (as proposed for another two-electron substrate, C2H217,33,103,220), joining the N2 pathway with the release of NH3 to form a stage corresponding to E7 in the N2 reduction scheme.156
7. Obligatory Evolution of H2 in Nitrogen Fixation: Reductive Elimination of H2
The En assignments of Figure 6 plus those of Figure 12 give proposed structures to all En states of the LT kinetic scheme (Figure 3), but the assignments have been developed through independent analyses of the two four-electron halves of the eight-electron catalytic cycle (eq 1). In the first half (part I) of the pathway, accumulation of four electrons/protons activates FeMo-co, generating E4; in the second half (part II), bound N2 is hydrogenated by two of those electrons/protons plus an additional four electrons/protons. However, the assignments are silent about the mechanism by which the E4 Janus intermediate, Figure 4, connects these two halves: the obligatory production of an H2 molecule upon N2 binding, as shown in Figure 12.33,156,158 Why nitrogenase should “waste” fully 25% of the ATP required for nitrogen fixation through H2 generation (eq 1) has remained a mystery, and indeed is not even accepted uniformly.23,221
Consideration of the finding that E4 stores its four reducing equivalents as two bridging hydrides (Figure 4) within the context of the well-known organometallic chemistries of hydrides194,222 and dihydrogen223 led us to examine the two alternative mechanisms by which this state might bind and activate N2 with release of H2, and proceed to the prompt formation of FeMo-co with a bound diazene-level species (N2H2) without additional accumulation of [e–/H+], as featured in the P–A reaction pathway, Figure 12. In one, H2 is formed by hydride protonation (hp mechanism), Figure 13, upper; the other forms H2 through reductive elimination (re),195 Figure 13, lower. We first describe these two mechanisms, and then show that the re mechanism is operative.
Figure 13.

Visualization of hp and re mechanisms for H2 release upon N2 (blue) binding to E4. The following is shown: the Fe-2,3,6,7 face of resting FeMo-co; the structure of FeMo-co must distort in different stages of catalysis. The Fe that binds N2 is presumed to be Fe6, as indicated by studies of α-70Val variants; when bold, red, Fe6 is formally reduced by two equivalents (see text). The bridging hydrides of E4 (green) are positioned to share an Fe “vertex”, as suggested by re mechanism of H2 release upon N2 binding. Alternative binding modes for N2-derived species can be envisaged.
7.1. Hydride Protonation (hp) Mechanism
In the hp scheme (Figure 13, upper), N2 binding is accompanied by the activation of one bridging hydride to the terminal form and protonation of this hydride by a sulfide-bound proton to form and release H2. Such a mechanism for H2 formation is invoked in discussions of hydrogenases,223,224 and there is strong precedence for replacement of a metal-bound H2 with N2. In this context, by analogy to the mechanism for the (much less demanding) reduction of alkynes/alkenes one might propose that transient terminalization of the “second” hydride would then lead to hydrogenation and protonation of the bound N2, to form FeMo-co bound N2H2 (see Figure 13, upper, below). For reasons that will become clear below, the hydrogenation of N2 to form metal bound-N2H2 must be reversible.
7.2. Reductive Elimination (re) Mechanism
The second mechanism for H2 loss upon N2 binding begins with transient terminalization of both E4 hydrides, Figure 13, lower. This is followed by reductive elimination of H2 as N2 binds, steps with considerable precedence.4,194,222,223,225 Of key importance, the departing H2 carries away only two of the four reducing equivalents stored in E4, while the Fe that binds N2 becomes highly activated through formal reduction by two equivalents; for example a formal redox state of Fe(II) would be reduced to Fe(0). This delivery of two reducing equivalents to the FeMo-co core which otherwise is reduced by at most one equivalent during electron/proton activation (Figures 6 and 7) would poise the cofactor to deliver the two activating electrons to N2, whose π acidity could be further enhanced by electrostatic interactions with the two sulfur-bound protons: combined delivery of the two electrons and protons would directly yield cofactor-bound N2H2. This amounts to a “push–pull” mechanism for the hydrogenation of N2, in which the “push” of electrons from the doubly reduced cofactor onto N2 is enhanced by the electrostatic “pull” of the protons bound to sulfur. As discussed in section 9, below, models of E4(N2) constructed by placing N2 and two protons on the doubly reduced FeMo-co core modeled with its resting-state structure provide a convincing illustration of this mechanism (and other insights, as well). The diminished electron donation to Fe by protonated sulfides would not only facilitate reductive elimination, but also would act to localize the added electrons on the Fe involved, limiting charge delocalization over the rest of the cofactor. This mechanism provides a compelling rationale for obligatory H2 formation during N2 reduction: the transient formation of a state in which an electrostatically activated N2 is bound to a highly activated, doubly reduced site, thereby generating a state optimally activated to carry out the initial hydrogenations of N2, the most difficult process in N2 fixation.
7.3. Mechanistic Constraints Reveal That Nitrogenase Follows the re Mechanisms
A clear choice between hp and re mechanisms is achieved by testing them against the numerous constraints that are associated with the reaction of D2 with the diazene-level E4(N2/N2H2) state formed when N2 binds to the cofactor and is reduced. The three principal constraints are listed in Chart 1. The first test that they provide for a mechanism is that it must accommodate the finding that when nitrogenase turns over in the presence of both N2 and D2, then two HD are formed through D2 cleavage and solvent-proton reduction, with the stoichiometry summarized as constraint i of Chart 1.17,226−228 Such HD formation only occurs in the presence of N2, and not during reduction of H+ or any other substrate.226,229,230
Chart 1.

The second key constraint and mechanistic test was revealed by Burgess and co-workers 30 years ago; the absence of exchange into solvent of D+/T+ derived from D2/T2 gas, Chart 1, constraint ii.226 When nitrogenase turns over under a mixture of N2 and T2, HT is formed with stoichiometry corresponding to Chart 1, constraint i, but during this process only a negligible amount of T+ is released to solvent (∼2%). The third constraint is provided by a later study of α-195His- and α-191Gln-substituted MoFe proteins.161 It provided persuasive evidence that HD formation under N2/D2 requires that the enzyme be at least at the E4 redox level, with a FeMo-co-bound N–N species at the reduction level of N2H2 or beyond, corresponding to the third constraint, Chart 1, iii.161 Constraint iii, plus the stoichiometry of HD formation according to constraint i implies a process described as
| 3 |
Thus, N2H2 formation is reversible, as shown in Figure 13.
Figure 14, upper, shows that the characteristics of HD formation during turnover under N2/D2cannot be reconciled with the hp mechanism. In the reverse of this mechanism, D2 binding and N2 release would generate an E4 state that has one deuteride bridge, which is deactivated for exchange with solvent. However, it carries the other deuteron in the form of D+ bound to sulfur (or a protein residue), which likely would be solvent exchangeable. Exchange of that D+ would violate the stoichiometric constraint, of eq 3 (line i, Chart 1), as relaxation of E4 to E2 within the reverse-hp mechanism would generate only one HD per D2, not two as required. Correspondingly, replacement of D2 by T2 in Figure 14, upper, with exchange of T+ bound to sulfide would lose roughly one T+ per T2 to solvent, contrary to the few percent loss observed by Burgess et al. (constraint ii).226 The possibility that the proton-bearing site is “shielded” from exchange seems implausible for a catalytic cluster that depends on proton delivery for its catalytic function, and in any case solvent exchange need not be fast; the rate-limiting step in nitrogenase turnover is the off-rate for Fe protein after it has delivered its electron to MoFe,33,99 and this process is quite slow, with a rate constant of ∼6 s–1.
Figure 14.
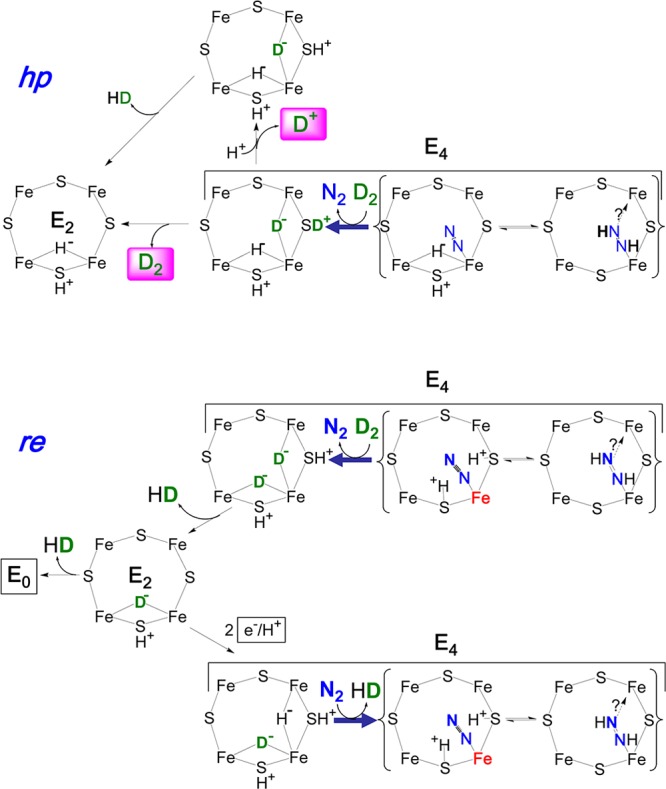
Reversal of hp and re mechanisms upon D2 binding. Details as in Figure 13. Bold arrows replace equilibrium arrows to emphasize the relaxation process.
If this proton were nonetheless shielded from exchange, relaxation to E2 would occur with regeneration of D2, without the generation of HD, in disagreement with the stoichiometric constraint of Chart 1. This objection would be overcome if at ambient temperatures the hydrides/protons can “migrate” over the FeMo-co face, but this instead would require multiple sites to be “shielded” for slow exchange, while FeMo-co is accessible to rapid proton delivery. Overall, we conclude that the hp process fails to satisfy the constraints of Chart 1, as the reverse hp process satisfies neither the stoichiometry of eq 3 nor the constraint that T+ is not released to solvent (Chart 1).
In contrast, in the reverse of the re mechanism, shown in Figure 14, lower, D2 binding and N2 release would generate E4(2D), the E4 isotopomer in which both atoms of D2 exist as deuteride bridges. This state would relax with loss of HD to E2(D), and then to E0 with loss of the second HD, thus satisfying the stoichiometry of eq 3. If the reaction were carried out under T2, essentially no T+ would be lost to solvent because the bridging deuterides are deactivated for exchange with the protein environment and solvent, thus satisfying the “T+ exchange” constraint, Chart 1.
One alternative fate of the E4(2D) formed by D2 replacement of N2 would be to rebind an N2, but this would merely release the D2 that had started the reverse process, creating a cycle invisible to detection. As a second alternative, E2(D) could acquire two additional electrons/protons to achieve the monodeutero E4 state. However, as shown in Figure 14, lower, if this state then bound N2 it would release the second HD, again without solvent exchange, whereas if it ultimately relaxed to E0 it would release the second HD along with an H2. Thus, the re mechanism for N2 binding and H2 release not only has the compelling chemical rationale discussed above, but also satisfies the three critical HD constraints for the various alternatives that arise when it is run in reverse, Chart 1.
In short, the re mechanism, Figure 13, lower, satisfies the constraints summarized in Chart 1, as visualized in Figure 14, lower; to the best of our knowledge, it likewise satisfies all other constraints on the mechanism provided by earlier studies, most of which are not directly tied to D2 binding.
8. Test of the re Mechanism
Subsequent to formulation of the re mechanism for the activation of FeMo-cofactor to reduce N2 (Figure 13, lower),156 we noted that addition of C2H2 to a N2/D2 reaction mixture should offer a rigorous test of the mechanism. The test is founded on a defining characteristic of nitrogenase catalysis, an exact distinction between hydrons (H/D/T) associated with the gaseous diatomics, H2/D2/T2, and those derived from solvent water. Thus, when nitrogenase in protic buffer is turned over under N2/D2, gaseous D2 can displace N2 from the E4(N2/N2H2) state (Figure 14, lower), stoichiometrically yielding two HD.226−228 This and other observations clearly show that diatomic H2/D2 is not used to reduce N2 during turnover under N2//H2/D2 (in particular, T incorporated into the ammonia product of N2 fixation would exchange with solvent).17 Likewise, as demonstrated below, when C2H2 is reduced in the presence of D2, no deuterated ethylenes are generated.
8.1. Predictions
With this foundation, we recognized that the re mechanism predicts that turnover under C2H2/D2/N2 should not only incorporate H from solvent to generate C2H4 by the normal reduction process, but through the agency of the added N2 also should breach the separation of gaseous D2 from solvent protons by generating both C2H3D and C2H2D2 (Figure 15). According to the re mechanism, when turnover is carried out under N2/D2, D2 can react with E4(N2/N2H2), replacing the N2 and undergoing oxidative addition to generate E4(2D). We recognized that this state in fact might be expected to react with C2H2 to form C2H2D2 through the idealized mechanism (Figure 16A) involving terminalization of an [Fe–D–Fe] bridge of E4, and migratory insertion of bound C2H2 into the Fe–D bond to form an Fe-alkenyl intermediate, followed by reductive elimination of C2H2D2.195,231 Previous studies17,103 could not distinguish reaction at the E4 state from reaction at the E2 state when C2H2 is reduced in the absence of N2, as N2 is required to enable gaseous D2 to enter the nitrogenase catalytic process. The possibility that acetylene can access different nitrogenase redox states, however, had been suggested on the basis of experiments using a nitrogenase variant that exhibits N2 reduction that is resistant to inhibition by acetylene.232,233
Figure 15.

Formation of deuterated acetylenes during turnover under N2/D2/C2H2 as predicted according to re mechanism. Cartoons again depict the Fe2,3,6,7 face of resting-state FeMo-co, with no attempt to incorporate likely structural modifications. Figure shows that the “reverse” of re mechanism through displacement of N2 by D2 produces, successively, E4(2D) and E2(D), further showing potential reaction channels for capture of E4(2D) and E2(D) intermediates with C2H2.
Figure 16.

Schematic mechanism for reaction of C2H2 with E4(2D) and E2(D). (A) Formation of C2H2D2, which follows Scheme 15.20 of Hartwig:231 mi = migratory insertion; re = reductive elimination. In braces: Possible alternative reaction channel that leads to formation of C2H3D, ap = alkenyl protonolysis. (B) Schematic mechanism for formation of C2H3D from reaction of C2H2 with E2(D). (C) Illustration of possibility that C2H2 displaces D2 formed by reductive elimination of the E4(2D) deuterides, leading to direct formation of C2H4 without D incorporation.
The E4(2D) state also would relax through the loss of HD to form E2(D), an E2 state whose unique isotopic composition can be generated in no other way. Interception of the E2(D) state by C2H2 would then generate C2H3D, with Figure 16B presenting a plausible mechanism: deuteride terminalization and insertion, followed by alkenyl protonolysis.195,231 This reaction also might occur through an alternative reaction channel of E4(2D), as noted in Figure 15.
8.2. Testing the Predictions
We tested the predictions based on the re mechanism of an unprecedented involvement of gaseous D2 in substrate reduction by use of C2H2 reduction under N2/D2/C2H2 gas mixtures to intercept the E4(2D) and E2(D) states. As expected, the control reaction of turnover under D2/C2H2 generates only C2H4, without incorporation of D from gaseous D2 to generate either C2H3D or C2H2D2 (Figure 17). In dramatic contrast, C2H2 reduction by nitrogenase under a N2/D2/C2H2 gas mixture in fact produces readily measured amounts of C2H2D2 and even greater amounts of C2H3D (Figure 17).157
Figure 17.

Time-dependent formation of 13C2H3D and 13C2H2D2, catalyzed by nitrogenase reduction of 13C2H2. 13C2H3D determined by GC/MS monitoring of m/z = 31 for a reaction mixture containing 13C2H2 and including D2 and N2 (■), just D2 (x inside □), or H2 and N2 (□). Inset: 13C2H2D2, m/z = 32, formation starting with 13C2H2/D2/N2 (●), just D2 (◊), or H2/N2 (○). Partial pressures of 0.02 atm 13C2H2, 0.25 atm N2, and 0.7 atm H2/D2, where present. The molar ratio of Fe protein to MoFe protein was 2:1. All assays incubated at 30 °C. Adapted with permission from ref (157). Copyright 2013 National Academy of Sciences.
On reflection, the success of this test for the formation of E4(2D) is a consequence of the greater reactivity of C2H2 compared to that of N2 and/or of the difference in the likely ways that these two substrates bind to FeMo-co: side-on for C2H2, end-on for N2. Otherwise, in a process analogous to that for N2H2 formation in the re mechanism (Figure 13, lower), C2H2 might in principle displace D2 formed by reductive elimination of the E4(2D) deuterides, leading to direct formation of C2H4 without D incorporation, Figure 16C. The yield of C2H2D2 may be less than that of C2H3D, because the contribution from this reaction channel diminishes the yield of the former, but it is perhaps more likely that the binding and reduction of C2H2 by E4(2D) is substantially less likely than the relaxation of E4(2D) to E2(D) through loss of HD, and the reduction of C2H2 by E2(D) (Figure 15).
These observations are enriched by consideration of the dependences of the yields of C2H3D and C2H2D2 on the partial pressures of C2H2, D2, N2, and electron flux, all of which are understandable in terms of the production of the E4(2D) and E2(D) states under these turnover conditions, as predicted by the re mechanism for FeMo-cofactor activation for N2 binding and reduction.157 For example, reduction of acetylene and N2 are mutually exclusive, with complicated inhibition kinetics between these two substrates.217,234 Therefore, it was of interest to determine the effect of varying the N2 partial pressure on the formation of C2H3D and C2H2D2 at fixed C2H2 and D2 pressures. The yields of C2H3D and C2H2D2 increase in parallel with increasing partial pressure of N2 (Figure 18). This can be explained by enhanced formation of E4[N2/N2H2] by reaction of N2 with E4. Increased formation of E4[N2/N2H2] in turn would enhance reaction with D2 to form E4(2D), which can be intercepted by acetylene to form deuterated ethylenes (Figure 15).157
Figure 18.

Deuterated ethylene formation as a function of N2 partial pressure. The partial pressure of C2H2 was 0.02 atm and D2 was 0.6 atm. The molar ratio of Fe protein to MoFe protein was 4:1. Assay conditions as in Figure 17. Adapted with permission from ref (157). Copyright 2013 National Academy of Sciences.
It is of interest to note that the reduction of C2H2 to C2H3D by reaction with E2(D) formally corresponds to the reduction of C2H2 by the HD that otherwise would form during relaxation of E2(D) to E0, a perspective that highlights the contrast between this result, achieved in the presence of N2, with the failure of nitrogenase to use H2/D2 to reduce any substrate in the absence of N2. As an eleboration on this perspective, the formation of HD during turnover under N2/D2, with stoichiometry (eq 3, above),17 can be seen to correspond to the nitrogenase-catalyzed reduction of protons by D2 and electrons with N2 as cocatalyst, eq 3′
| 3′ |
as the reaction neither proceeds without N2 nor consumes N2. Likewise, although C2H2D2 is well-known to form during nitrogenase reduction of C2D2 in H2O buffer (or C2H2 in D2O buffer),148 formation of this species during turnover under C2H2/D2/N2 corresponds to the previously unobserved reduction of C2H2 by gaseous D2 with N2 as cocatalyst (eq 4).
| 4 |
Correspondingly, the formation of C2H3D involves incorporation of D– derived from D2 along with H+ from solvent with N2 as cocatalyst.
9. Completing the Mechanism of Nitrogen Fixation
Figure 12, above, presents a formal integration of the reaction pathway for nitrogen fixation (intermediates E4–E8) with the LT kinetic scheme, the key to the resulting mechanism being N2 binding and H2 release through the re mechanism, Figure 13, lower. This mechanism is built on the structure of the E4 intermediate and its implication that hydride chemistry is central to nitrogen fixation by nitrogenase (section 3). As a corresponding implication, we further offered the two alternative sets of proposed structures for the “early”, E1–E3, intermediates (Figure 6). We now discuss in greater depth the E5–E8 intermediates of nitrogen fixation, proposing in Figure 19, II not only more detailed structures for the stages following the formation of N2H2-bound FeMo-co, the E4(N2H2) state, written as binding diazene itself, but also the nature of the chemical transformations that link these stages during the delivery of the ‘second half’ of the eight [e–/H+] that comprise the stoichiometry of nitrogen fixation, eq 1. The analysis further leads us to provisionally assign the early, “first half” intermediates to the alternative described in Figure 6B, now visualized in Figure 19, I. When combined with the reductive elimination (re) mechanism for the binding N2 and release of H2, Figure 13, lower, the result, Figure 19, is a self-consistent proposal for the structures of all intermediates in the nitrogen fixation mechanism and a formal description of the transformations that convert each stage to the subsequent one: a complete, though of course still simplified, mechanism for nitrogen fixation by nitrogenase.
Figure 19.

Proposed mechanism displaying structures of all intermediates in nitrogen fixation, inspired by the assumption of primacy of hydride chemistry associated with the Fe2,3,6,7 face of FeMo-co, and containing a formal description of the transformations that convert each stage to the subsequent one. In I the mechanism tentatively adopts and visualizes the view of En states n = 1–4 presented in Figure 6B; in II it visualizes bridging hydrides by analogy, without evidence for or against terminal hydrides for n = 5–7. Likewise, the structure of the N2H2 species as end-on bound diazene is suggestive, not definitive, etc. I and II are connected by the re mechanism, Figure 13, lower. Formal charges are included as useful to help guide the reader.
Figure 19, II, is constructed on two assumptions that (i) the formation and reactions of hydrides is key; (ii) beginning with N2H2, the hydrogenation of reduced forms of N2 involves migratory insertion into Fe–H bonds. These assumptions lead to the conclusion that [e–/H+] transfer to FeMo-co of the E4(N2H2) and E6 states creates E5 and E7 that each contain an [Fe–H–Fe] bridging hydride moiety bound to an oxidized FeMo-co, Figure 19, II, in correspondence with the analogous [e–/H+] transfer to E0 and E2, shown in the cartoon of Figure 6B, and now visualized in Figure 19, I. In the case of E5, an accompanying migratory insertion of the N2H2 into an Fe–H bond (presumably formed by terminalization of the bridge) forms the [N2H3]− moiety bound to the oxidized cluster; in the case of E7, migratory insertion leads to N–N bond cleavage and formation of [NH2]− bound to the formally oxidized cofactor (Figure 19, II). The follow-up [e–/H+] transfer to E5, E7 generates the E6 and E8 states, respectively. This mechanistic picture is anchored by the final stages, E7 and E8, whose structures match those proposed in the EPR/ENDOR/ESEEM studies of intermediates H, assigned to E7, and I, assigned to E8, Figure 12.
The proposal completes a mechanism in which the stoichiometrically required delivery of all 8 [e–/H+] to FeMo-co is controlled by the hydride chemistry of the cofactor. The clearly understandable differences between the first “half” of the catalytic cycle, visualized in Figure 19, I, and the “second half”, Figure 19, II, arise because the former involves accumulation of reducing equivalents while the latter involves delivery of reducing equivalents to substrate.
The two halves are similar in that addition of [e–/H+] to form an n = odd intermediate (n = 1, 3, first half; n = 5, 7, second half) generates an [Fe–H–Fe] bridging hydride attached to a formally oxidized FeMo-co core. They differ in that the hydride is “stored” in n = 1, 3, but is promptly transferred to substrate in n = 5, 7 to form a (formally) anionic reduced substrate. Upon addition of [e–/H+] to any one of these four n = odd intermediates, to form the subsequent n = even intermediate, the electron formally reduces the core to the resting-state redox level. In the first half (n = 2, 4), the H+ is delivered to a sulfur and its charge balances that on a hydride; in the second half (n = 6, 8), the proton neutralizes the anionic nitrogenous ligand, to form the neutral, N2H4 of E6, NH3 of E8.
The two halves of the nitrogen fixation mechanism are joined at the E4 stage, as described above and displayed as Figure 19, re: the E4(2H) intermediate formed by accumulation of four [e–/H+] and containing two bridging hydrides undergoes reductive elimination as it binds N2 and releases the two “sacrificial” reducing equivalents as H2. Figure 19 thus represents a complete mechanism for nitrogen fixation by nitrogenase that invokes the primacy of the hydride chemistry of FeMo-co.
9.1. Uniqueness of N2 and Nitrogenase
The mechanistic proposal of Figure 19 invokes the primacy of hydride chemistry associated with a 4Fe face of FeMo-co, a structural feature made possible only with a cluster of at least six metal ions. The hydrogenations of reduced forms of N2, starting with N2H2, involve migratory insertion of substrate into Fe–H bonds, one at a time. This is the same mechanism visualized for the “normal” reduction of C2H2 at the E2 stage, and even for the rare trapping of E4 by C2H2, Figure 16; we suggest migratory insertions are likely to be involved in the hydrogenation of all other substrates.
But N2 is not reactive to hydride insertion. So nitrogenase adopts a different “strategy” for attacking its physiological substrate. It is forced to accumulate four reducing equivalents as two Fe hydrides, which requires a 4-Fe face, and thus the large cluster is “held together” by the carbide at its core. We have concluded that this cluster can only become activated for N2 hydrogenation through reductive elimination of two of those equivalents in the form of H2.156,157 The “push” of the doubly reduced metal-ion core of the cluster, compounded by the electrostatic “pull” of sulfur-bound protons, is required to overcome the high barrier to the initial hydrogenation of N2, directly to N2H2, Figure 19.
9.2. Structure of the E4(N2) Intermediate: Some Implications
As an exercise to illustrate four points worth noting, we have modeled alternative structures of the E4(N2) intermediate by building the bound substrate onto the crystal structure of resting-state FeMo-co using structural information from model complexes.235−240 It seems most likely, on the basis of the structures of model complexes, that N2 binds end-on, rather than bridging. As illustrated in Figure 20, and emphasized over the years,137,241,242 end-on bound N2 can bind to FeMo-co in two basic, alternative modes: endo, with the N2 “nestled” in the pocket above the Fe2,3,6,7 face; exo, with N2 pointed away from that face. The first point is as follows. According to our mechanism, E4(N2) contains doubly reduced metal-ion core with two protons bound to sulfur. There are multiple potential dispositions of the H+ on different sulfurs, but distance measurements with the mockups show that the atoms of N2 and protons can indeed be in close enough proximity to support the electrostatic “pull” postulated above.
Figure 20.

Models for the two alternative modes for N2 binding at Fe6 of FeMo-cofactor in the E4(N2) state, with two protons bound to two adjacent sulfides as in Figure 4: (A) endo mode; (B) exo mode. The side chains of selected amino acid residues are shown as sticks. The figure was generated in Pymol by building N2 onto the resting-state of FeMo-co using the coordinate file PDB:2AFK. Iron is shown in rust, molybdenum in magenta, sulfur in yellow, carbon in dark gray, hydrogen in light gray, nitrogen in blue, and oxygen in red.
Second, this mockup demonstrates the commonly understood need for the FeMo-co core to “relax” upon substrate binding. In the resting-state the Fe ions are roughly tetrahedral, and without such relaxation, the N2–S distances would be far too short. The normal assumption would be that Fe6 roughly forms a plane with three S atoms, with a major contribution to the relaxation being an elongation of the bond trans to N2.
The third issue is the resulting structural/electronic-structure consequences of the identity of the trans ligand in exo versus endo N2 binding, and it does not appear to have been widely discussed. The modulation of metal-ion reactivity by variations in the trans ligand (the “trans effect”) is well-known,231 and recently, a series of trigonal Fe complexes that are biomimetic of nitrogenase have shown that the trans ligands to a terminal Fe–N2 can regulate the ability of the complex to catalytically reduce N2.214 In the exo binding mode, the interstitial carbide is trans to N2. This mode would favor the idea that carbide modulates the properties of Fe6 through the trans effect, and may well act as a hemilabile ligand. However, in the endo mode, which has been favored by some computations, the trans ligand is now a S that bridges to Mo. As C is (roughly) an “in-plane” ligand, not trans, its influence on reactivity would be different than for endo binding, in which case modulation of Fe6 reactivity by the trans effect would involve [S–Mo] being “axial” ligand.
There is a corollary to considerations of the endo binding mode. It is widely assumed that the catalytic centers of the alternative nitrogenases have the same structure as FeMo-co, with the heterometal atom Mo being replaced by V or Fe.18 Thus, if N2 does bind endo to Fe6 of a FeMo-co-like structure in all three systems, its reactivity would be modulated by differences in the axial −S–M “ligand” caused by differences in the properties of Mo, V, and Fe.
The fourth point is the possible importance of interactions of substrate with adjacent amino acid residues. In the endo binding mode the N2 is nestled within a binding pocket capped by the side-chain of α-70Val; in the exo mode, the N2 is pointed to a pocket surrounded by α-191Gln and the homocitrate ligand of Mo. The present mockups suggest that the protein environment in either binding mode could readily accommodate, and even stabilize, N2 with no more than minor conformational rearrangements.
10. Summary of Mechanistic Insights
10.1. Catalytic Intermediates of N2 Fixation
Two major points can be made regarding intermediates trapped: (1) Characterization of the E4 “Janus” intermediate as bearing four reducing equivalents in the form of two [Fe–H–Fe] bridging hydrides has provided the foundation for proposals that the FeMo-co core is never oxidized or reduced by more than one equivalent relative to the resting-state, and that the oxidative couple in fact is operative, Figure 19, I. (2) The characterization of the common intermediates H and I, trapped during turnover with nitrogenous substrates, led to the proposed unification of kinetic scheme and A reaction pathway, Figure 12.
10.2. re Mechanism
Reductive elimination of two hydrides upon N2 binding (re mechanism) provides an explanation for the nitrogenase stoichiometry (eq 1) and for the obligatory formation of H2 upon N2 binding. This mechanism for H2 production upon N2 binding to E4, Figure 13, lower, satisfies both the stoichiometric constraint of HD formation (Chart 1, line i) and the “T+” constraint against exchange of gas-derived hydrons with solvent (Chart 1, line ii), whereas the hp mechanism (Figure 13, upper) satisfies neither. The re mechanism further involves D2 binding to a state at the “diazene level” of reduction, as required by the constraint of eq 3 and Chart 1, line iii. Finally, to the best of our knowledge, all other constraints on the mechanism, most of which are not directly tied to D2 binding, are satisfied, as well.
This mechanism answers the following long-standing and oft-repeated question: Why does nature “waste” four ATP/two reducing equivalents through an obligatory loss of H2 when N2 binds? The answer follows: reductive elimination of H2 upon binding of N2 to FeMo-co of the E4 state generates a state in which highly reduced FeMo-co binds N2, which likely is activated for reduction through electrostatic interactions with the remaining two sulfur-bound protons. Transfer of the two reducing equivalents generated by the reductive elimination, combined with transfer of the two activating protons, then forms N2H2, Figure 13, lower, in keeping with the P–A scheme of Figure 12. It appears that only through this activation is the enzyme able to hydrogenate N2.
10.3. Turnover under N2/D2/C2H2 as a Test of the re Mechanism
This mechanism has been supported by a rigorous test which provided experiments in which C2H2 is added to an N2/D2 reaction mixture. Although diatomic D2 does not reduce nitrogenase C2H2 in the absence of N2, the re mechanism successfully predicted that turnover under C2H2/D2/N2 would breach the separation of gaseous D2 from solvent protons by generating both C2H3D and C2H2D2.
The conclusions regarding H2 formation upon N2 binding reached from this study are as follows. (i) The unprecedented incorporation of D from D2 into the nitrogenase reduction products C2H2D2 and C2H3D during turnover under C2H2/D2/N2 in H2O demonstrates the presence of the E4(2D) and E2(D) states under these conditions. In our view any model that fails to incorporate obligatory H2 loss as a fundamental aspect of N2 activation is unlikely to provide a robust description of the chemistry associated with the biological process.242 (ii) This incorporation provides a very clear demonstration of the essential mechanistic role for obligatory, reversible loss of H2 upon N2 binding and thus of the eight-electron stoichiometry for nitrogen fixation by nitrogenase embodied in eq 1. Until now, the data indicating that some H2 must be evolved during N2 reduction has been viewed as being much more compelling than the data indicating an obligatory evolution of one H2 for every N2 reduced, leading to the stoichiometry of eq 1.17 (iii) The formation of E4(2D) and E2(D) during turnover under D2/N2 in H2O is predicted by the re mechanism for the activation of FeMo-cofactor for reduction of N2, and the interception of these intermediates by C2H2 thus provides direct experimental evidence in support of this mechanism (Figure 17). (iv) The well-known reduction of protons by D2 to form 2HD during turnover under D2/N2 in H2O and the newly discovered reductions of C2H2 by D2/N2 should be viewed as being catalyzed by nitrogenase with N2 as cocatalyst. (v) This review has proposed an explanation of the inability of H2/D2 to reduce nitrogenase and/or catalyze substrate reduction in the absence of N2.
11. Conclusions
The trapping and characterization of five nitrogenase catalytic intermediates, which correspond to three of the five stages involved in binding and reduction of nitrogen (Figure 3), most especially the “Janus intermediate”, E4, and including the nitrogenous intermediate states H (E7) and I (E8), have identified the “prompt–alternating (P–A)” pathway of Figure 12, carried out on a four-Fe face of FeMo-co, as most likely operative for nitrogenase and led to the unification of the nitrogenase reaction pathway and the LT kinetic scheme.
The recognition of the central role played by conversion of accumulated [e–/H+] into metal hydrides has led to the proposal that this most complex of biological catalytic clusters, and by extension perhaps all biological clusters involved in multielectron substrate hydrogenation, function through a limited set of redox couples, and indeed most likely through a single couple, with multiple reducing equivalents being stored as hydrides rather than as reduced metal ions, Figures 6, 19. Only in this way can a cluster accumulate equivalents delivered at a constant potential set by its biological partners. These considerations provide part of the reason why such a large cluster is required for nitrogenase catalysis.
Simple energetic considerations have further illuminated the heretofore puzzling observation that states of nitrogenase activated by the accumulation of multiple [e–/H+] can relax through release of H2 (Scheme 1), but H2 cannot reduce nitrogenase in what appears to be the reverse process: the answer is that the processes are not microscopic reverses.
Perhaps the central question of nitrogen fixation by nitrogenase has been that of stoichiometry: “Why does (or even, does) nature ‘waste’ four ATP/two reducing equivalents through an obligatory loss of H2 when N2 binds?” An answer has been proposed on the basis of further consideration of hydride chemistry exhibited by E4: the enzyme exhibits the stoichiometry of eq 1 because reductive elimination (re) of two [e–/H+] in the form of H2 activates FeMo-co for hydrogenation of N2 to N2H2 via a “push–pull” mechanism, Figure 13, lower. A test of the mechanism involving turnover under N2/D2/C2H2, as in Figure 15, validated the re mechanism, and in so doing confirmed the stoichiometry of nitrogen fixation, eq 1, as requiring eight [e–/H+].
The test reaction further highlighted the role of N2 as cocatalyst in reductions catalyzed by nitrogenase that would not occur in the absence of N2. We have further noted some issues regarding the uniqueness of N2 and nitrogenase as the catalyst for its hydrogenation, and of the implications of alternative structures of the N2 complex.
The result of these efforts is the mechanism for nitrogen fixation presented in Figure 19 for further tests, both experimental and theoretical.
Acknowledgments
This work was supported by the NIH (HL 13531, B.M.H.; GM59087, L.C.S. and D.R.D.), NSF (MCB 0723330, B.M.H.), and DOE (DE-SC0010687 to L.C.S. and D.R.D.). We gratefully acknowledge many illuminating discussions with friends and colleagues, most especially noting Professors Richard H. Holm, Pat Holland, and Jonas Peters. Thanks to Dr. K. Danyal for assistance with Figure 1.
Biographies

Brian M. Hoffman was an undergraduate at the University of Chicago, received his Ph.D. from Caltech, and spent a postdoctoral year at MIT. From there, he went to Northwestern University, where he is the Charles E. and Emma H. Morrison Professor in the Departments of Chemistry and of Molecular Biosciences. He is a member of the National Academy of Sciences.

Dmitriy Lukoyanov received a M.S. degree and a Ph.D. from Kazan State University. He is a postdoctoral fellow at Northwestern University.

Zhi-Yong Yang received a B.S. degree in chemistry and a Ph.D. degree in organic chemistry from Nankai University in Tianjin, China. Following a position at Shanghai ChemPartner as an organic scientist, he joined the Ph.D. program in biochemistry at Utah State University, where he recently completed his degree. During the past 12 years, his research has covered biomimetic chemistry of hydrogenases, and the mechanism of nitrogenase. His research interest is in the activation and formation of chemical bonds catalyzed by biological and synthetic transition-metal catalysts.

Dennis R. Dean received a B.A. from Wabash College and a Ph.D. from Purdue University. He is currently a University Distinguished Professor at Virginia Tech where he also serves as the Director of the Fralin Life Science Institute and the Virginia Bioinformatics Institute.

Lance C. Seefeldt received a B.S. degree in chemistry from the University of Redlands in California and a Ph.D. in biochemistry from the University of California at Riverside. He was a postdoctoral fellow at the Center for Metalloenzyme Studies at the University of Georgia before joining the faculty of the Chemistry and Biochemistry Department at Utah State University, where he is now Professor. He was recently awarded the D. Wynne Thorne Career Research Award and elected a Fellow of the American Association for the Advancement of Science. Over the past 25 years, his research has focused on elucidating the mechanism of nitrogenase.
Supporting Information Available
Analysis of the relative orientations of bridging hydrides of E4 intermediate. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ferguson S. J. Curr. Opin. Chem. Biol. 1998, 2, 182. [DOI] [PubMed] [Google Scholar]
- Smil V.Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; MIT Press: Cambridge, MA, 2004. [Google Scholar]
- Jia H.-P.; Quadrelli E. A. Chem. Soc. Rev. 2014, 43, 547. [DOI] [PubMed] [Google Scholar]
- MacKay B. A.; Fryzuk M. D. Chem. Rev. 2004, 104, 385. [DOI] [PubMed] [Google Scholar]
- Canfield D. E.; Glazer A. N.; Falkowski P. G. Science 2010, 330, 192. [DOI] [PubMed] [Google Scholar]
- Cheng Q. J. Integr. Plant Biol. 2008, 50, 786. [DOI] [PubMed] [Google Scholar]
- Thamdrup B. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 407. [Google Scholar]
- Raymond J.; Siefert J. L.; Staples C. R.; Blankenship R. E. Mol. Biol. Evol. 2004, 21, 541. [DOI] [PubMed] [Google Scholar]
- Gruber N.; Galloway J. N. Nature 2008, 451, 293. [DOI] [PubMed] [Google Scholar]
- Burk D. Ergeb. Enzymforsch. 1934, 3, 23. [Google Scholar]
- Burk D.; Lineweaver H.; Horner C. K. J. Bacteriol. 1934, 27, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn S. E.; Boyd E. S.; Peters J. W.; Orphan V. J. Front. Microbiol. 2013, 3, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos P. C.; Fang Z.; Mason S. W.; Setubal J. C.; Dixon R. BMC Genomics 2012, 13, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber F. Naturwissenschaften 1922, 10, 1041. [Google Scholar]
- Haber F. Naturwissenschaften 1923, 11, 339. [Google Scholar]
- Boyd E. S.; Anbar A. D.; Miller S.; Hamilton T. L.; Lavin M.; Peters J. W. Geobiology 2011, 9, 221. [DOI] [PubMed] [Google Scholar]
- Burgess B. K.; Lowe D. J. Chem. Rev. 1996, 96, 2983. [DOI] [PubMed] [Google Scholar]
- Eady R. R. Chem. Rev. 1996, 96, 3013. [DOI] [PubMed] [Google Scholar]
- Beatty P. H.; Good A. G. Science 2011, 333, 416. [DOI] [PubMed] [Google Scholar]
- Godfray H. C. J.; Beddington J. R.; Crute I. R.; Haddad L.; Lawrence D.; Muir J. F.; Pretty J.; Robinson S.; Thomas S. M.; Toulmin C. Science 2010, 327, 812. [DOI] [PubMed] [Google Scholar]
- Rubio L. M.; Ludden P. W. Annu. Rev. Microbiol. 2008, 62, 93. [DOI] [PubMed] [Google Scholar]
- Macleod K. C.; Holland P. L. Nat. Chem. 2013, 5, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. C.; Mehn M. P. In Activation of Small Molecules: Organometallic and Bioinorganic Perspectives; Tolman W. B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp 81–119. [Google Scholar]
- Schrock R. R. Acc. Chem. Res. 2005, 38, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y.; Nishibayashi Y. Coord. Chem. Rev. 2013, 257, 2551. [Google Scholar]
- Jodin C. C. R. Acad. Paris 1862, 55, 612. [Google Scholar]
- Carnahan J. E.; Mortenson L. E.; Mower H. F.; Castle J. E. Biochim. Biophys. Acta 1960, 44, 520. [DOI] [PubMed] [Google Scholar]
- Bulen W. A.; Burns R. C.; LeComte J. R. Biochem. Biophys. Res. Commun. 1964, 17, 265. [DOI] [PubMed] [Google Scholar]
- Schneider K. C.; Bradbeer C.; Singh R. N.; Wang L. C.; Wilson P. W.; Burris R. H. Proc. Natl. Acad. Sci. U.S.A. 1960, 46, 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess B. K. Chem. Rev. 1990, 90, 1377. [Google Scholar]
- Hardy R. W.; Burns R. C.; Parshall G. W. In Bioinorganic Chemistry; Dessy R., Dillard J., Taylor L., Eds.; American Chemical Society: Washington, DC, 1971; Vol. 100, pp 219–247. [Google Scholar]
- Howard J. B.; Rees D. C. Chem. Rev. 1996, 96, 2965. [DOI] [PubMed] [Google Scholar]
- Thorneley R. N. F.; Lowe D. J. In Molybdenum Enzymes; Spiro T. G., Ed.; Wiley: New York, 1985; pp 221–284. [Google Scholar]
- Winter H. C.; Burris R. H. Annu. Rev. Biochem. 1976, 45, 409. [DOI] [PubMed] [Google Scholar]
- Bulen W. A.; LeComte J. R. Proc. Natl. Acad. Sci. U.S.A. 1966, 56, 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson L. E. Fed. Proc. 1965, 24, 233. [Google Scholar]
- Mortenson L. E. Biochim. Biophys. Acta 1966, 127, 18. [DOI] [PubMed] [Google Scholar]
- Hageman R. V.; Burris R. H. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean D. R.; Bolin J. T.; Zheng L. J. Bacteriol. 1993, 175, 6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. Annu. Rev. Biochem. 1994, 63, 235. [DOI] [PubMed] [Google Scholar]
- Kim J.; Rees D. C. Biochemistry 1994, 33, 389. [DOI] [PubMed] [Google Scholar]
- Bulen W. A.; Burns R. C.; LeComte J. R. Proc. Natl. Acad. Sci. U.S.A. 1965, 53, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns R. C.; Bulen W. A. Biochim. Biophys. Acta 1965, 105, 437. [DOI] [PubMed] [Google Scholar]
- Mortenson L. E. Biochim. Biophys. Acta 1964, 81, 473. [DOI] [PubMed] [Google Scholar]
- Mortenson L. E. Proc. Natl. Acad. Sci. U.S.A. 1964, 52, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah V. K.; Brill W. J. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. W.; Fisher K.; Newton W. E.; Dean D. R. J. Biol. Chem. 1995, 270, 27007. [DOI] [PubMed] [Google Scholar]
- Kim J.; Rees D. C. Science 1992, 257, 1677. [DOI] [PubMed] [Google Scholar]
- Ma L.; Brosius M. A.; Burgess B. K. J. Biol. Chem. 1996, 271, 10528. [DOI] [PubMed] [Google Scholar]
- Lowe D. J.; Fisher K.; Thorneley R. N. F. Biochem. J. 1993, 292, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadis M. M.; Komiya H.; Chakrabarti P.; Woo D.; Kornuc J. J.; Rees D. C. Science 1992, 257, 1653. [DOI] [PubMed] [Google Scholar]
- Kim J.; Rees D. C. Nature 1992, 360, 553. [DOI] [PubMed] [Google Scholar]
- Chan M. K.; Kim J.; Rees D. C. Science 1993, 260, 792. [DOI] [PubMed] [Google Scholar]
- Kim J.; Woo D.; Rees D. C. Biochemistry 1993, 32, 7104. [DOI] [PubMed] [Google Scholar]
- Jang S. B.; Seefeldt L. C.; Peters J. W. Biochemistry 2000, 39, 14745. [DOI] [PubMed] [Google Scholar]
- Jang S. B.; Seefeldt L. C.; Peters J. W. Biochemistry 2000, 39, 641. [DOI] [PubMed] [Google Scholar]
- Jang S. B.; Jeong M. S.; Seefeldt L. C.; Peters J. W. J. Biol. Inorg. Chem. 2004, 9, 1028. [DOI] [PubMed] [Google Scholar]
- Jeong M. S. Mol. Cells 2004, 18, 374. [PubMed] [Google Scholar]
- Strop P.; Takahara P. M.; Chiu H.-J.; Angove H. C.; Burgess B. K.; Rees D. C. Biochemistry 2001, 40, 651. [DOI] [PubMed] [Google Scholar]
- Sen S.; Igarashi R.; Smith A.; Johnson M. K.; Seefeldt L. C.; Peters J. W. Biochemistry 2004, 43, 1787. [DOI] [PubMed] [Google Scholar]
- Sen S.; Krishnakumar A.; McClead J.; Johnson M. K.; Seefeldt L. C.; Szilagyi R. K.; Peters J. W. J. Inorg. Biochem. 2006, 100, 1041. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Stowell M. H. B.; Soltis S. M.; Finnegan M. G.; Johnson M. K.; Rees D. C. Biochemistry 1997, 36, 1181. [DOI] [PubMed] [Google Scholar]
- Mayer S. M.; Lawson D. M.; Gormal C. A.; Roe S. M.; Smith B. E. J. Mol. Biol. 1999, 292, 871. [DOI] [PubMed] [Google Scholar]
- Sørlie M.; Christiansen J.; Lemon B. J.; Peters J. W.; Dean D. R.; Hales B. J. Biochemistry 2001, 40, 1540. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L. A.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. Science 2002, 297, 1696. [DOI] [PubMed] [Google Scholar]
- Schmid B.; Ribbe M. W.; Einsle O.; Yoshida M.; Thomas L. M.; Dean D. R.; Rees D. C.; Burgess B. K. Science 2002, 296, 352. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Tezcan F. A.; Haynes C. A.; Walton M. Y.; Andrade S.; Einsle O.; Howard J. B. Phil. Trans. R. Soc., A 2005, 363, 971. [DOI] [PubMed] [Google Scholar]
- Sarma R.; Mulder D. W.; Brecht E.; Szilagyi R. K.; Seefeldt L. C.; Tsuruta H.; Peters J. W. Biochemistry 2007, 46, 14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma R.; Barney B. M.; Keable S.; Dean D. R.; Seefeldt L. C.; Peters J. W. J. Inorg. Biochem. 2010, 104, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L. A.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Science 2011, 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Roemelt M.; Ettenhuber P.; Hu Y.; Ribbe M. W.; Neese F.; Bergmann U.; DeBeer S. Science 2011, 334, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Hu Y.; Bergmann U.; Ribbe M. W.; DeBeer S. J. Am. Chem. Soc. 2013, 135, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Lee C. C.; Ribbe M. W. Science 2012, 337, 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Lee C. C.; Hu Y.; Ribbe M. W. J. Am. Chem. Soc. 2013, 135, 4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-I.; Benton P. M. C.; Laryukhin M.; Igarashi R. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2003, 125, 5604. [DOI] [PubMed] [Google Scholar]
- Yang T.-C.; Maeser N. K.; Laryukhin M.; Lee H.-I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2005, 127, 12804. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Pelmenschikov V.; Maeser N.; Laryukhin M.; Yang T. C.; Noodleman L.; Dean D. R.; Case D. A.; Seefeldt L. C.; Hoffman B. M. Inorg. Chem. 2007, 46, 11437. [DOI] [PubMed] [Google Scholar]
- Chiu H.-J.; Peters J. W.; Lanzilotta W. N.; Ryle M. J.; Seefeldt L. C.; Howard J. B.; Rees D. C. Biochemistry 2001, 40, 641. [DOI] [PubMed] [Google Scholar]
- Schmid B.; Einsle O.; Chiu H.-J.; Willing A.; Yoshida M.; Howard J. B.; Rees D. C. Biochemistry 2002, 41, 15557. [DOI] [PubMed] [Google Scholar]
- Schindelin H.; Kisker C.; Schlessman J. L.; Howard J. B.; Rees D. C. Nature 1997, 387, 370. [DOI] [PubMed] [Google Scholar]
- Tezcan F. A.; Kaiser J. T.; Mustafi D.; Walton M. Y.; Howard J. B.; Rees D. C. Science 2005, 309, 1377. [DOI] [PubMed] [Google Scholar]
- Soboh B.; Boyd E. S.; Zhao D.; Peters J. W.; Rubio L. M. FEBS Lett. 2010, 584, 1487. [DOI] [PubMed] [Google Scholar]
- Dos Santos P. C.; Dean D. R.; Hu Y.; Ribbe M. W. Chem. Rev. 2004, 104, 1159. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Fay A. W.; Lee C. C.; Yoshizawa J.; Ribbe M. W. Biochemistry 2008, 47, 3973. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Acc. Chem. Res. 2010, 43, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Microbiol. Mol. Biol. Rev. 2011, 75, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Coord. Chem. Rev. 2011, 255, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. J. Biol. Chem. 2013, 288, 13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Biochim. Biophys. Acta 2013, 1827, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehder D. J. Inorg. Biochem. 2000, 80, 133. [DOI] [PubMed] [Google Scholar]
- Eady R. R. Coord. Chem. Rev. 2003, 237, 23. [Google Scholar]
- Crans D. C.; Smee J. J.; Gaidamauskas E.; Yang L. Chem. Rev. 2004, 104, 849. [DOI] [PubMed] [Google Scholar]
- Fisher K.; Dilworth M. J.; Newton W. E. Biochemistry 2006, 45, 4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W.; Holm R. H. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W. Science 2010, 329, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W. Angew. Chem., Int. Ed. 2011, 50, 5545. [DOI] [PubMed] [Google Scholar]
- Dance I. Dalton Trans. 2011, 40, 5516. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Lee C. C.; Ribbe M. W. Dalton Trans. 2012, 41, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval S.; Danyal K.; Shaw S.; Lytle A. K.; Dean D. R.; Hoffman B. M.; Antony E.; Seefeldt L. C. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeldt L. C.; Hoffman B. M.; Dean D. R. Curr. Opin. Chem. Biol. 2012, 16, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 17088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyvis M. G.; Wassink H.; Haaker H. Biochemistry 1998, 37, 17345. [DOI] [PubMed] [Google Scholar]
- Wilson P. E.; Nyborg A. C.; Watt G. D. Biophys. Chem. 2001, 91, 281. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Szilagyi R. K. Curr. Opin. Chem. Biol. 2006, 10, 101. [DOI] [PubMed] [Google Scholar]
- Danyal K.; Mayweather D.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2010, 132, 6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danyal K.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Biochemistry 2011, 50, 9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayweather D.; Danyal K.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Biochemistry 2012, 51, 8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J. M.; Christiansen J.; Dean D. R.; Seefeldt L. C. Biochemistry 1999, 38, 5779. [DOI] [PubMed] [Google Scholar]
- Nyborg A. C.; Johnson J. L.; Gunn A.; Watt G. D. J. Biol. Chem. 2000, 275, 39307. [DOI] [PubMed] [Google Scholar]
- Rupnik K.; Hu Y.; Lee C. C.; Wiig J. A.; Ribbe M. W.; Hales B. J. J. Am. Chem. Soc. 2012, 134, 13749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzilotta W. N.; Christiansen J.; Dean D. R.; Seefeldt L. C. Biochemistry 1998, 37, 11376. [DOI] [PubMed] [Google Scholar]
- Duyvis M. G.; Wassink H.; Haaker H. FEBS Lett. 1996, 380, 233. [DOI] [PubMed] [Google Scholar]
- Angove H. C.; Yoo S. J.; Münck E.; Burgess B. K. J. Biol. Chem. 1998, 273, 26330. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Dean D. R. Acc. Chem. Res. 1997, 30, 260. [Google Scholar]
- Chan J. M.; Ryle M. J.; Seefeldt L. C. J. Biol. Chem. 1999, 274, 17593. [DOI] [PubMed] [Google Scholar]
- Lanzilotta W. N.; Parker V. D.; Seefeldt L. C. Biochim. Biophys. Acta 1999, 1429, 411. [DOI] [PubMed] [Google Scholar]
- Ryle M. J.; Seefeldt L. C. J. Biol. Chem. 2000, 275, 6214. [DOI] [PubMed] [Google Scholar]
- Danyal K.; Inglet B. S.; Vincent K. A.; Barney B. M.; Hoffman B. M.; Armstrong F. A.; Dean D. R.; Seefeldt L. C. J. Am. Chem. Soc. 2010, 132, 13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth L. E.; Nguyen J. C.; Tezcan F. A. J. Am. Chem. Soc. 2010, 132, 13672. [DOI] [PubMed] [Google Scholar]
- Roth L. E.; Tezcan F. A. ChemCatChem 2011, 3, 1549. [Google Scholar]
- Roth L. E.; Tezcan F. A. J. Am. Chem. Soc. 2012, 134, 8416. [DOI] [PubMed] [Google Scholar]
- Durrant M. C. Biochem. J. 2001, 355, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant M. C. Biochemistry 2002, 41, 13934. [DOI] [PubMed] [Google Scholar]
- Durrant M. C. Biochemistry 2002, 41, 13946. [DOI] [PubMed] [Google Scholar]
- Cui Z.; Dunford A. J.; Durrant M. C.; Henderson R. A.; Smith B. E. Inorg. Chem. 2003, 42, 6252. [DOI] [PubMed] [Google Scholar]
- Thorneley R. N. F.; Angove H. C.; Durrant M. C.; Fairhurst S. A.; George S. J.; Sinclair A.; Tolland J. D.; Hallenbeck P. C. J. Inorg. Biochem. 2003, 96, 18. [Google Scholar]
- Durrant M. C.; Francis A.; Lowe D. J.; Newton W. E.; Fisher K. Biochem. J. 2006, 397, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnemann B.; Nørskov J. K. J. Am. Chem. Soc. 2003, 125, 1466. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Nørskov J. K. J. Am. Chem. Soc. 2004, 126, 3920. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Nørskov J. K. Phys. Chem. Chem. Phys. 2004, 6, 843. [Google Scholar]
- Hinnemann B.; Nørskov J. K. Top. Catal. 2006, 37, 55. [Google Scholar]
- Varley J. B.; Nørskov J. K. ChemCatChem 2013, 5, 732. [Google Scholar]
- Lovell T.; Li J.; Liu T.; Case D. A.; Noodleman L. J. Am. Chem. Soc. 2001, 123, 12392. [DOI] [PubMed] [Google Scholar]
- Lovell T.; Liu T.; Case D. A.; Noodleman L. J. Am. Chem. Soc. 2003, 125, 8377. [DOI] [PubMed] [Google Scholar]
- Torres R. A.; Lovell T.; Noodleman L.; Case D. A. J. Am. Chem. Soc. 2003, 125, 1923. [DOI] [PubMed] [Google Scholar]
- Pelmenschikov V.; Case D. A.; Noodleman L. Inorg. Chem. 2008, 47, 6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance I. J. Am. Chem. Soc. 2007, 129, 1076. [DOI] [PubMed] [Google Scholar]
- Harris T. V.; Szilagyi R. K. Inorg. Chem. 2011, 50, 4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neese F. Angew. Chem., Int. Ed. 2006, 45, 196. [DOI] [PubMed] [Google Scholar]
- Kästner J.; Blöchl P. E. J. Am. Chem. Soc. 2007, 129, 2998. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Rasche M. E.; Ensign S. A. Biochemistry 1995, 34, 5382. [DOI] [PubMed] [Google Scholar]
- Rasche M. E.; Seefeldt L. C. Biochemistry 1997, 36, 8574. [DOI] [PubMed] [Google Scholar]
- Mayer S. M.; Niehaus W. G.; Dean D. R. J. Chem. Soc., Dalton Trans. 2002, 802. [Google Scholar]
- Dos Santos P. C.; Mayer S. M.; Barney B. M.; Seefeldt L. C.; Dean D. R. J. Inorg. Biochem. 2007, 101, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi R. Y.; Dos Santos P. C.; Niehaus W. G.; Dance I. G.; Dean D. R.; Seefeldt L. C. J. Biol. Chem. 2004, 279, 34770. [DOI] [PubMed] [Google Scholar]
- Dos Santos P. C.; Igarashi R. Y.; Lee H.-I.; Hoffman B. M.; Seefeldt L. C.; Dean D. R. Acc. Chem. Res. 2005, 38, 208. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; McClead J.; Lukoyanov D.; Laryukhin M.; Yang T.-C.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Biochemistry 2007, 46, 6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeldt L. C.; Yang Z.-Y.; Duval S.; Dean D. R. Biochim. Biophys. Acta 2013, 1827, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K.; Dilworth M. J.; Kim C.-H.; Newton W. E. Biochemistry 2000, 39, 10855. [DOI] [PubMed] [Google Scholar]
- Yang Z.-Y.; Dean D. R.; Seefeldt L. C. J. Biol. Chem. 2011, 286, 19417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Lee C. C.; Ribbe M. W. Science 2011, 333, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y.; Moure V. R.; Dean D. R.; Seefeldt L. C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; Lee H.-I.; Dos Santos P. C.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Dalton Trans. 2006, 2277. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Hoffman B. M.; Dean D. R. Annu. Rev. Biochem. 2009, 78, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Dean D. R.; Seefeldt L. C. Acc. Chem. Res. 2009, 42, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. Acc. Chem. Res. 2013, 46, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y.; Khadka N.; Lukoyanov D.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson F. B.; Burris R. H. Science 1984, 224, 1095. [DOI] [PubMed] [Google Scholar]
- Davis L. C.; Henzl M. T.; Burris R. H.; Orme-Johnson W. H. Biochemistry 1979, 18, 4860. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; Laryukhin M.; Igarashi R. Y.; Lee H.-I.; Dos Santos P. C.; Yang T.-C.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Biochemistry 2005, 44, 8030. [DOI] [PubMed] [Google Scholar]
- Fisher K.; Dilworth M. J.; Newton W. E. Biochemistry 2000, 39, 15570. [DOI] [PubMed] [Google Scholar]
- Scott D. J.; May H. D.; Newton W. E.; Brigle K. E.; Dean D. R. Nature 1990, 343, 188. [DOI] [PubMed] [Google Scholar]
- Thomann H.; Bernardo M.; Newton W. E.; Dean D. R. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.-H.; Newton W. E.; Dean D. R. Biochemistry 1995, 34, 2798. [DOI] [PubMed] [Google Scholar]
- Dilworth M. J.; Fisher K.; Kim C.-H.; Newton W. E. Biochemistry 1998, 37, 17495. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; Lukoyanov D.; Igarashi R. Y.; Laryukhin M.; Yang T.-C.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Biochemistry 2009, 48, 9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton P. M. C.; Laryukhin M.; Mayer S. M.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Biochemistry 2003, 42, 9102. [DOI] [PubMed] [Google Scholar]
- Lee H.-I.; Igarashi R. Y.; Laryukhin M.; Doan P. E.; Dos Santos P. C.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2004, 126, 9563. [DOI] [PubMed] [Google Scholar]
- Igarashi R. Y.; Laryukhin M.; Dos Santos P. C.; Lee H.-I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2005, 127, 6231. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; Yang T.-C.; Igarashi R. Y.; Dos Santos P. C.; Laryukhin M.; Lee H.-I.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. J. Am. Chem. Soc. 2005, 127, 14960. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; Lukoyanov D.; Yang T.-C.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 17113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme-Johnson W. H.; Hamilton W. D.; Jones T. L.; Tso M. Y. W.; Burris R. H.; Shah V. K.; Brill W. J. Proc. Natl. Acad. Sci. U.S.A. 1972, 69, 3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M. J. Phys. Chem. 1994, 98, 11657. [Google Scholar]
- Hoffman B. M.; Sturgeon B. E.; Doan P. E.; DeRose V. J.; Liu K. E.; Lippard S. J. J. Am. Chem. Soc. 1994, 116, 6023. [Google Scholar]
- Hoffman B. M. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M. Acc. Chem. Res. 2003, 36, 522. [DOI] [PubMed] [Google Scholar]
- Schweiger A.; Jeschke G.. Principles of Pulse Electron Paramagnetic Resonance; Oxford University Press: Oxford, U.K., 2001. [Google Scholar]
- George S. J.; Barney B. M.; Mitra D.; Igarashi R. Y.; Guo Y.; Dean D. R.; Cramer S. P.; Seefeldt L. C. J. Inorg. Biochem. 2012, 112, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh B. H.; Henzl M. T.; Christner J. A.; Zimmermann R.; Orme-Johnson W. H.; Münck E. Biochim. Biophys. Acta 1980, 623, 124. [DOI] [PubMed] [Google Scholar]
- Yoo S. J.; Angove H. C.; Papaefthymiou V.; Burgess B. K.; Münck E. J. Am. Chem. Soc. 2000, 122, 4926. [Google Scholar]
- Lowe D. J.; Eady R. R.; Thorneley N. F. Biochem. J. 1978, 173, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K.; Newton W. E.; Lowe D. J. Biochemistry 2001, 40, 3333. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Barney B. M.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; Igarashi R. Y.; Dos Santos P. C.; Dean D. R.; Seefeldt L. C. J. Biol. Chem. 2004, 279, 53621. [DOI] [PubMed] [Google Scholar]
- Yates M. G.; Lowe D. J. FEBS Lett. 1976, 72, 121. [DOI] [PubMed] [Google Scholar]
- Kinney R. A.; Hetterscheid D. G. H.; Hanna B. S.; Schrock R. R.; Hoffman B. M. Inorg. Chem. 2010, 49, 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems J.-P.; Lee H.-I.; Burdi D.; Doan P. E.; Stubbe J.; Hoffman B. M. J. Am. Chem. Soc. 1997, 119, 9816. [Google Scholar]
- Kinney R. A.; Saouma C. T.; Peters J. C.; Hoffman B. M. J. Am. Chem. Soc. 2012, 134, 12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2010, 132, 2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. A. Chem. Rev. 2005, 105, 2365. [DOI] [PubMed] [Google Scholar]
- Henderson R. A. Coord. Chem. Rev. 2005, 249, 1841. [Google Scholar]
- Chiang K. P.; Scarborough C. C.; Horitani M.; Lees N. S.; Ding K.; Dugan T. R.; Brennessel W. W.; Bill E.; Hoffman B. M.; Holland P. L. Angew. Chem., Int. Ed. 2012, 51, 3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan P. E.; Telser J.; Barney B. M.; Igarashi R. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2011, 133, 17329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzini M.; Poli R.. Recent Advances in Hydride Chemistry; Elsevier Science B.V.: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Crabtree R. H.The Organometallic Chemistry of the Transition Metals, 5th ed.; Wiley: Hoboken, NJ, 2009. [Google Scholar]
- Oro L. A.; Sola E. In Recent Advances in Hydride Chemistry; Peruzzini M., Poli R., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2001; pp 299–328. [Google Scholar]
- Zimmermann R.; Münck E.; Brill W. J.; Shah V. K.; Henzl M. T.; Rawlings J.; Orme-Johnson W. H. Biochim. Biophys. Acta 1978, 537, 185. [DOI] [PubMed] [Google Scholar]
- Lee H.-I.; Sørlie M.; Christiansen J.; Yang T.-C.; Shao J.; Dean D. R.; Hales B. J.; Hoffman B. M. J. Am. Chem. Soc. 2005, 127, 15880. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M.; Tye J. W.; Hall M. B. Chem. Rev. 2007, 107, 4414. [DOI] [PubMed] [Google Scholar]
- Surawatanawong P.; Hall M. B. Inorg. Chem. 2010, 49, 5737. [DOI] [PubMed] [Google Scholar]
- Pickett C. J. J. Biol. Inorg. Chem. 1996, 1, 601. [Google Scholar]
- Chatt J.; Dilworth J. R.; Richards R. L. Chem. Rev. 1978, 78, 589. [Google Scholar]
- Chatt J.; Richards R. L. J. Organomet. Chem. 1982, 239, 65. [Google Scholar]
- Schrock R. R. Chem. Commun. 2003, 2389. [DOI] [PubMed] [Google Scholar]
- Yandulov D. V.; Schrock R. R. Science 2003, 301, 76. [DOI] [PubMed] [Google Scholar]
- Schrock R. R. Angew. Chem., Int. Ed. 2008, 47, 5512. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Dikanov S. A.; Yang Z.-Y.; Barney B. M.; Samoilova R. I.; Narasimhulu K. V.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2011, 133, 11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneley R. N.; Lowe D. J. Biochem. J. 1984, 224, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess B. K. In Metal Ions in Biology: Molybdenum Enzymes; Spiro T. G., Ed.; John Wiley and Sons: New York, 1985; pp 161–220. [Google Scholar]
- Thorneley R. N. F.; Eady R. R.; Lowe D. J. Nature 1978, 272, 557. [Google Scholar]
- Davis L. C. Arch. Biochem. Biophys. 1980, 204, 270. [DOI] [PubMed] [Google Scholar]
- Murakami J.; Yamaguchi W. Sci. Rep. 2012, 2, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. M.; Bill E.; Brennessel W. W.; Holland P. L. Science 2011, 334, 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. S.; Rittle J.; Peters J. C. Nature 2013, 501, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth M. J.; Eady R. R. Biochem. J. 1991, 277, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna C. E.; Simeonov A. M.; Eran H.; Bravo-Leerabhandh M. Biochemistry 1996, 35, 4502. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Dance I. G.; Dean D. R. Biochemistry 2004, 43, 1401. [DOI] [PubMed] [Google Scholar]
- Münck E.; Ksurerus K.; Hendrich M. P.. Metallobiochemistry Part D: Physical and Spectroscopic Methods for Probing Metal Ion Environment in Metalloproteins. In Methods in Enzymology; James F., Riordan B. L. V., Ed.; Academic Press: New York, 1993; Vol. 227, pp 463–479. [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Barney B. M.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D. J.; Fisher K.; Thorneley R. N. Biochem. J. 1990, 272, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- As summarized by Peters and Mehn, many of the attempts to understand nitrogen fixation theoretically treat a six-electron stoichiometry, and thus implicitly reject this central mechanistic feature of the LT scheme.
- Ballmann J.; Munhá R. F.; Fryzuk M. D. Chem. Commun. 2010, 46, 1013. [DOI] [PubMed] [Google Scholar]
- Kubas G. J. Chem. Rev. 2007, 107, 4152. [DOI] [PubMed] [Google Scholar]
- Tard C.; Pickett C. J. Chem. Rev. 2009, 109, 2245. [DOI] [PubMed] [Google Scholar]
- Crabtree R. H. Inorg. Chim. Acta 1986, 125, L7. [Google Scholar]
- Burgess B. K.; Wherland S.; Newton W. E.; Stiefel E. I. Biochemistry 1981, 20, 5140. [DOI] [PubMed] [Google Scholar]
- Li J.-L.; Burris R. H. Biochemistry 1983, 22, 4472. [DOI] [PubMed] [Google Scholar]
- Jensen B. B.; Burris R. H. Biochemistry 1985, 24, 1141. [DOI] [PubMed] [Google Scholar]
- Hoch G. E.; Schneider K. C.; Burris R. H. Biochim. Biophys. Acta 1960, 37, 273. [DOI] [PubMed] [Google Scholar]
- Jackson E. K.; Parshall G. W.; Hardy R. W. F. J. Biol. Chem. 1968, 243, 4952. [PubMed] [Google Scholar]
- Hartwig J.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Christiansen J.; Seefeldt L. C.; Dean D. R. J. Biol. Chem. 2000, 275, 36104. [DOI] [PubMed] [Google Scholar]
- Christiansen J.; Cash V. L.; Seefeldt L. C.; Dean D. R. J. Biol. Chem. 2000, 275, 11459. [DOI] [PubMed] [Google Scholar]
- Rivera-Ortiz J. M.; Burris R. H. J. Bacteriol. 1975, 123, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieber S. C. E.; Milsmann C.; Hoyt J. M.; Turner Z. R.; Finkelstein K. D.; Wieghardt K.; DeBeer S.; Chirik P. J. Inorg. Chem. 2012, 51, 3770. [DOI] [PubMed] [Google Scholar]
- Moret M.-E.; Peters J. C. J. Am. Chem. Soc. 2011, 133, 18118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret M.-E.; Peters J. C. Angew. Chem., Int. Ed. 2011, 50, 2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saouma C. T.; Moore C. E.; Rheingold A. L.; Peters J. C. Inorg. Chem. 2011, 50, 11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A.; Mankad N. P.; Peters J. C. J. Am. Chem. Soc. 2011, 133, 8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong H.; Moret M.-E.; Lee Y.; Peters J. C. Organometallics 2013, 32, 3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance I. Chem.—Asian J. 2007, 2, 936. [DOI] [PubMed] [Google Scholar]
- Dance I. Chem. Commun. 2013, 49, 10893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.