

Abstract
Various estrogen analogs were synthesized and tested for binding to human ERα using a fluorescence polarization displacement assay. Binding affinity and orientation were also predicted using docking calculations. Docking was able to accurately predict relative binding affinity and orientation for estradiol, but only if a tightly bound water molecule bridging Arg394/Glu353 is present. Di-hydroxyl compounds sometimes bind in two orientations, which are flipped in terms of relative positioning of their hydroxyl groups. Di-hydroxyl compounds were predicted to bind with their aliphatic hydroxyl group interacting with His524 in ERα. One nonsteroid-based dihdroxyl compound was 1000-fold specific for ERβ over ERα, and was also 25-fold specific for agonist ERβ versus antagonist activity. Docking predictions suggest this specificity may be due to interaction of the aliphatic hydroxyl with His475 in the agonist form of ERβ, versus with Thr299 in the antagonist form. But, the presence of this aliphatic hydroxyl is not required in all compounds, since mono-hydroxyl (phenolic) compounds bind ERα with high affinity, via hydroxyl hydrogen bonding interactions with the ERα Arg394/Glu353/water triad, and van der Waals interactions with the rest of the molecule.
Keywords: Estrogen receptor, Docking, Phenolic, Breast cancer, Endocrine disruptor
1. Introduction
Estrogen receptor-α (ERα) is a 595-residue, 66 kDa protein with a ligand binding domain of 245 residues (28 kDa). ERα, along with estrogen receptor-β (ERβ), belongs to the nuclear hormone family of intracellular receptors. It is one of the two principal receptors responsible for binding the endogenous estrogen, 17β-estradiol (E2), shown in Figure 1.1 In the nucleus, ER binds to DNA as a dimer, recruiting coactivators or corepressors that will result in activating or repressing the transcription of different genes.3 Binding of E2 activates the ER, regulating activity. Both ERα and ERβ forms are found in different tissue types. However, ERα is expressed more in breast tissue and is also known to be involved in the pathway that regulates breast cancer development.2,4 ERα antagonists such as raloxifene (Fig. 1) can bind to ER in the same ligand-binding domain as E2, and disrupt normal ER cellular function.4,5
Figure 1.
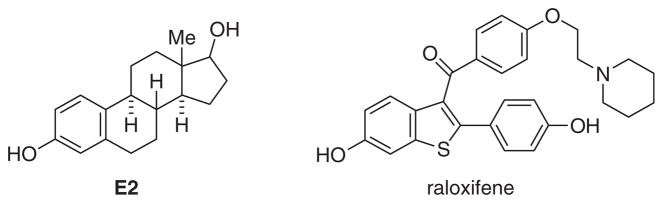
Structures of 17β-estradiol and raloxifene.
A key structural feature of E2 is the presence of two hydroxyl groups that are separated by 11 Å, which permits interaction with conserved binding site residues Arg394/Glu353 and His 524. But, the receptor is capable of binding many other compounds whose structures resemble that of the E2 hormone.6 Some of these compounds are endogeneous, such as estrone and other human estrogens; and, some are exogeneous, like the drugs raloxifene (Fig. 1) or tamoxifen that are used to treat breast cancer and osteoporosis.7 In addition to drugs, there exist other exogeneous compounds, some naturally occurring like phytoestrogens and some synthetic such as organochlorines, that have measurable estrogenic activity.5 Many of these latter compounds have been shown to be linked to breast cancer as well as birth defects.8,9 Through the National Institutes of Environmental Health Sciences, the BSB (Biomolecular Screening Branch), and other federal agencies, the government has developed a program to test many of the chemicals currently in our environment, to see if they have estrogenic activity.10
Because of the estrogen receptor’s prominent role as a breast cancer drug target, along with the threat posed by the potentially large number of estrogen agonists and antagonists in our environment (e.g., endocrine disruptors), it is essential to gain a better understanding of the binding requirements of the ERα ligand pocket. This understanding will allow for the design of better breast cancer drugs that interfere with the carcinogenic activity of estrogen agonists, and improve our ability to predict which pollutants might bind to ERα. Such predictions are strengthened by a better definition of the molecular features that trigger agonist or antagonist effects, as well as a validation of the docking methods used to predict binding.
One technique that can provide a quick and reliable experimental measurement of binding affinity is fluorescence polarization.11 A fluorescence polarization displacement assay can be used to screen non-fluorescent molecules, by displacing a fluorescent probe with the molecule of interest.12 Such fluorescence polarization displacement assays have been developed previously for ERα and ERβ, based on a fluorescein isothiocyanate (FITC)-tagged estradiol (F-E2).13,14 One such assay is available from Invitrogen.15 Subsequent studies in our lab improved the synthesis of F-E2 and examined the in vivo behavior of F-E2 in vivo, in fish. F-E2 was found to localize in cells that develop into reproductive organs, consistent with the proposed role of E2 in gender determination in fish.16 An analogous fluorescence polarization method was developed using an intrinsically fluorescent nonsteroid estrogen.17
Herein we present the synthesis of a series of phenolic mono-and di-hydroxyl estrogen analogs, which were tested for binding affinity for human ERα, using a fluorescence polarization displacement assay based on F-E2. Estrogen (E2) is a phenolic compound comprised of a steroid core and a second hydroxyl group that is 11 Å from the phenolic hydroxyl. Compounds synthesized herein have the phenolic core, but vary in terms of whether they: (a) are steroid-based, and (b) possess a second hydroxyl group, ~11 Å from the phenol. In addition to binding affinity measurements for compounds, docking calculations were performed. Docking is the process of positioning a ligand into the binding site of a protein and calculating a binding energy for each pose.18 It has become an important early-stage method for finding molecules likely to bind to a protein, allowing for many chemicals to be rapidly screened as potential drug leads.18–20 Docking has also proven useful for identifying compounds as targets for pollutant remediation.21 Besides predicting relative binding affinity, docking is used to predict the orientation or pose of a known ligand bound to a protein.22 Comparison of docking predictions with experimental affinity measurements allows one to rationalize binding site requirements, and also provides validation of the predictive ability of the docking calculations for a given target (e.g., ERα) and class of compounds (phenolic mono- and di-hydroxyl compounds). This is important because such experimental validation provides greater confidence in the docking calculations when they are done on larger sets of compounds, where experimental verification might not be feasible.
2. Results and discussion
2.1. Synthesis
Wittig olefination of estrone benzyl ether,23 followed by epoxidation with mCPBA gave the known24 epoxide 1 as a mixture of diastereomers (Scheme 1). Deprotonation of 1 with lithium diisopropylamine, followed by cleavage of the benzyl ether under dissolving metal conditions gave the allylic alcohol 2. Palladium catalyzed alkoxycarbonylation of the vinyl triflate derived from estrone benzyl ether, according to the literature procedure,25 gave n-propyl (20S)-3-(phenylmethoxy)-estra-1,3,5(10),16-tetraene- 17-carboxylate (3), which upon reduction in the presence of Raney-Ni gave the saturated ester 4. The skipped diene (20S)-3- (phenylmethoxy)-19,24-dinorchola-1,3,5(10),16,22-pentaene (5) was prepared by the literature procedure.25 Hydrogenation of the less substituted olefin in the presence of Wilkinson’s catalyst, followed by debenzylation gave 7. Hydroboration–oxidation of 5, by the literature procedure26 gave (20S)-3-(phenylmethoxy)-19,24- dinorchola-1,3,5(10),16-tetraen-23-ol (8). Subjecting 8 to acid resulted in the spirocyclic tetrahydrofuran 9 in quantitative yield, which upon catalytic hydrogenolysis gave 10. Alternatively, debenzylation of 8 afforded 11. Oxidation of 11 gave the aldehyde 12. Reaction of 12 with an excess of methyl Grignard, followed by work-up with saturated aqueous ammonium chloride proceeded by cyclization to afford the spirocyclic tetrahydrofuran 13 as a mixture of diastereomers.
Scheme 1.

Preparation of tetra- and pentacyclic ER analogs (ADD = 1,1′-(azodicarbonyl)dipiperidine).
A series of p-substituted phenols were also prepared (Scheme 2). Reduction of 4-(4′-hydroxyphenyl)cyclohexanone gave a separable mixture of trans-4-(4′-hydroxy-cyclohexyl)phenol 15 (86%) and its cis- diastereomer 14 (10%). The stereochemical assignments for each were made by comparison to their literature spectral data.27 Reaction of 4-(4′-hydroxyphenyl)cyclohexanone with hydroxylamine- hydrochloride gave the oxime 16. [4-((4′-Hydroxyphenyl) cyclohepta-2,6-dienyl)methanol 17 was prepared from p-acetoxystyrene according to the literature procedure.28 This involved cross metathesis with (1-methoxycarbonyl-2-vinyl-3-pentene- 1,5-diyl)Fe(CO)3 (21), followed by oxidatively induced reductive elimination. Reduction of the resultant cyclopropane-carboxylate and concomitant Cope [3,3]-rearrangement gave the cycloheptadiene 17. Catalytic reduction of 17 gave the saturated cycloheptane 18. Finally, Heck-type coupling of methyl 5-bromo- 2-furanoate with p-acetoxystyrene gave the trans-styrylfuranoate 19, which upon reduction with lithium aluminum hydride gave the furfuryl alcohol 20.
Scheme 2.

Preparation pf p-substituted phenols. Reagents and conditions: (a) 21,Grubbs 1st generation catalyst; (b) H2O2/NaOH; (c) LiAIH4, then 160 °C; (d) H2, 20% Pd/C, MeOH; (e) LiAIH4, Et2 O. (See above-mentioned reference for further information.)
2.2. Fluorescence polarization displacement and cell-based ERα and ERβ luminescence activity assays
Twelve compounds from Schemes 1 and 2 were screened using fluorescence polarization, for their ability to bind ERα (Table 1). Only six compounds showed any significant affinity for the receptor at concentrations as high as 1 μM. These compounds include five of the six steroid-core compounds—2, 4, 7, 11, and 13—and one bicyclic compound—18. Of the remaining six compounds which did not bind to ERα, one has the steroid core while the others contain the linked ring cores containing a flanking hydroxyl group—a structure whose hydrophobic interior and hydrophilic exterior resembles that of estrogen itself. The highest affinity ERα ligand was 2, with a Kd (32 nM) approaching that of E2 (3 nM). 18 is the only non-steroid core compound with measurable ERα binding affinity, but an accurate Kd could not be obtained (estimated to be >1 μM).
Table 1.
Dissociation constants (Kd) from the fluorescence polarization displacement assay and IC50 data from cell-based ERα and ERβ agonist assays and ERβ antagonist assays
Cell-based ERα and ERβ luminescence assays were performed to determine whether the ERα ligands were acting as agonists or antagonists, and whether they had specificity for the α isoform (Table 1, Fig. S1–6). Three compounds, 4, 13, and 2, showed agonist activity in the ERα assay; and, all six compounds showed ERβ agonist activity, with 4, 2, and 18 being the most potent; 18 is unique in its selectivity for ERβ over ERα, and is 25-fold more potent as an agonist, versus antagonist. 11, 7, and 18 displayed ERβ antagonist activity, with 7 being the most potent.
2.3. Docking
Compounds were computationally docked into human ERα and ERβ in agonist and antagonist conformations. Poses for ERα are shown in Fig. S7–8. Initial control docking studies were performed with E2, to validate the docking method by demonstrating an ability to reproduce the known binding mode from the crystal structure. Interestingly, E2 docked with similar predicted affinity in two distinct poses for the ERα agonist conformation (Fig. S9, Table S1), essentially flipping the positioning of the two hydroxyl groups with regard to interactions with Arg394/Glu353 and His524, located on opposite sides of the pocket. The predicted pose with the phenolic hydroxyl near Arg394/Glu353 is referred to as the ‘normal’ mode, and that with the phenolic hydroxyl near His524 as the ‘reversed’ mode. But, if docking is performed on receptor that has the tightly bound water present near Arg394/Glu353, then only the expected pose is obtained; and, E2 is the ligand with highest predicted affinity (Table 2), as expected. Thus, all docking was performed with the Arg394/Glu353 water present. This binding mode has been studied previously using molecular dynamics, and illustrates the important role of active site water molecules in ligand binding.30
Table 2.
Docking of compounds prepared in Schemes 1 and 2 into the agonist and antagonist conformations of ERα and ERβ
| Compound | Docking score for ERα agonist (kcal mol−1) | Docking score for ERα antagonist (kcal mol−1) | Docking score for ERβ agonist (kcal mol−1) | Docking score for ERβ antagonist (kcal mol−1) |
|---|---|---|---|---|
| E2 | −10.36 | −9.70 | −10.11 | −9.29 |
| 4 | −10.29 | −10.38 | −10.66 | −10.13 |
| 2 | −9.82 | −9.86 | −10.40 | −9.71 |
| 11 | −9.80 | −9.30 | −10.18 | −10.28 |
| 7 | −9.74 | −9.37 | −10.00 | −10.36 |
| 10 | −8.82 | −9.21 | −6.41 | −10.08 |
| 13 | −8.73 | −8.82 | −4.82 | −9.92 |
| 18 | −8.22 | −7.66 | −7.86 | −7.48 |
| 17 | −7.37 | −7.10 | −6.97 | −6.83 |
| 16 | −7.27 | −6.99 | −6.92 | −6.96 |
| 20 | −6.93 | −7.20 | −7.34 | −7.11 |
| 15 | −6.85 | −6.38 | −6.56 | −6.77 |
| 14 | −6.41 | −6.28 | −6.43 | −6.60 |
Compounds identified as having ERα affinity in the fluorescence polarization displacement assay are in bold.
Docking results were rank ordered according to the lowest energy pose for binding to the ERα agonist conformation, from the cluster with the highest population (Table 2). Identifying the compounds with measurable Kd values from the fluorescence polarization displacement assay (shown as bold in Table 2) indicates that the docking procedure using Autodock4 was able to separate the binding ligands from the non-binding ligands. ER is a unique docking target, since the binding site is comprised of a nearly closed hydrophobic pocket, flanked by hydrogen bonding groups that could provide specificity.31 Care in analyzing docking results is needed due to the large binding area in which ligands can potentially bind, and symmetry of the pocket. Three examples of reversed binding modes that are likely false are shown in Figure 2.
Figure 2.

Lowest energy docking poses from clusters where ligands were predicted to bind in two modes (A–B). The human ERα estrogen receptor that was used was in the agonist conformation (PDB code 1ere; chain A). Panel C shows the predicted binding orientation for 18 in ERβ, agonist conformation (PDB code 2jj3; chain A). Panel D shows the predicted binding orientation for 18 in ERβ, antagonist conformation (PDB code 1l2j; chain A).
Interestingly, while estradiol docked in only one orientation when the bound water is present, other compounds were still predicted to bind in two orientations (Table 2; Fig. 2), one normal (with the phenolic hydroxyl interacting with Arg394/Glu353/Water), and one ‘reversed,’ where the phenolic hydroxyl interacts with His524. This promiscuity in predicted binding mode may be due to symmetry in di-hydroxyl molecules like 2 (Fig. 2). Curiously, the mono-hydroxyl 4 also is predicted to bind in a reversed mode (Fig. 2), but with much lower affinity relative to the normal mode. This is likely due to the fact that 4 has only one hydroxyl group, the phenol, which provides significant binding energy via interaction with the Arg394/Glu353/water triad. It is also clear that the aliphatic hydroxyl interaction with His524 is not essential, since it is absent in 4 and 7, and yet both bind with reasonable affinity (IC50 = 160–320 nM). Indeed, this observation is consistent with the ability of phenolic endocrine disruptors, which contain only one hydroxyl group, to bind to ER.32
The docking of compounds 10 and 13 in the ERβ-agonist conformation displayed predicted binding energies that were weaker than expected in Table 2. Inspection of the binding site (Fig. S10) showed that these ligands experience steric clashes with binding site sidechains. Additionally, for structures 10 and 13, the oxygen atom in the tetrahydrofuran ring was not positioned near His475 for 10 or (for reversed mode binding) near Arg346, Glu305 for 13, to allow for hydrogen bond formation.
Compound 18 is in a unique class, in that it is not based on the steroid core, is selective for the β over the α ER isoform, and is 25- fold selective for ERβ agonist versus ERβ antagonist activity (Table 1). Docking pose predictions (Fig. 2C and D) show that 18 could form two hydrogen bonds (one with His475) in the ERβ agonist conformation, whereas in the ERβ antagonist conformation, hydrogen bonding is with Thr299, rather than His475. A molecular overlay of E2 and 18 (Fig. S11) shows the oxygen atoms of the two molecules are well-aligned.
3. Conclusions
Human ERα remains an important target for therapeutic interventions (cancer; osteoporosis). Estrogen has a key interaction between its phenolic hydroxyl and a binding site Arg394/Glu353/water triad, along with other important interactions including van der Waals interactions with the steroid core, and hydrogen bonding interactions between an aliphatic hydroxyl group and His524 (His475 in ERβ). The two estradiol hydroxyls are located 11 Å from each other. The studies presented herein probe the importance of interactions with the aliphatic hydroxyl and with the steroid core, using a series of novel mono- and di-hydroxyl compounds (Schemes 1 and 2).
The estrogen analog with highest measured affinity in the fluorescence polarization displacement assay(IC50 = 32 nM) and second highest predicted affinity is the di-hydroxyl steroid 2, which has a single point of unsaturation in the D-ring, and (relative to estradiol) has its aliphatic hydroxyl extended by one methylene group. Nonetheless, this gives an O–O distance essentially equivalent to that for estradiol. Di-hydroxyl steroid 2 behaves as an ERα agonist, and has no selectivity for α versus β ER isoforms. Indeed, 2 is a potent ERβ agonist and antagonist. In contrast, 18 binds weakly to ERα, yet has on O–O distance (11.1 Å) that is similar to 2. Of particular interest is the fact that 18 has the expected interaction with His475 in the ERβ agonist docking, whereas in the ERβ antagonist docking this aliphatic hydroxyl group is predicted to interact instead with Thr299 (Fig. 2). This could explain why 18 is so selective (25-fold) as an ERβ agonist, versus as an antagonist (Table 1). Most of the other compounds from Scheme 2 that lacked the steroid core did not bind to ERα, even though they possessed the phenolic hydroxyl. Compounds (4, 13, 2), which possessed ERα agonist activities, were also ERβ agonists; but, not ERβ antagonists. And, these compounds were more selective for ERβ over ERα.
In summary, several compounds have been identified that are potent ERα agonists, and also behave as ERβ agonists and antagonists (Table 1). The most potent is the dihydroxyl steroid 2. Also, the non-steroid dihydroxyl compound 18 is 1000-fold more selective for ERβ over ERα, and appears to adopt a different binding mode in these two targets (Fig. 2).
4. Experimental
4.1. General methods
The β-estradiol (min 98%) and fluorescein (FITC) were purchased from Sigma. The α-ER and α-ER screening buffer were from Invitrogen. The FITC-estradiol linked tracer used in the experiments was synthesized by as described previously. (1) DMSO-d6 was purchased from Cambridge Isotopes. The 96-well plates used were black, polystyrene, NBS (non-binding surface), flat-bottom plates obtained from Corning. A PolarStar Galaxy fluorescent plate reader was used and controlled with FLUOStar Galaxy software (version 4.30-0). Estrone benzyl ether23 and compounds 3,25 5,26 8,26 and 1728 were prepared by the literature procedures.
4.2. Estrogen analog synthesis
4.2.1. 3-Hydroxyestra-1,3,5(10),16-tetraene-17-methanol (2)
To a solution of methyl triphenylphosphonium bromide (589 mg, 1.65 mmol) in THF (10 mL) at −40 °C under N2, was added a solution of n-butyl lithium (0.66 mL, 2.5 M in hexanes, 1.7 mmol). The ylide solution was warmed to room temperature and a solution of estrone benzyl ether (200 mg, 0.556 mmol) in THF (7 mL) was added. The mixture was stirred for 12 h, and then heated at reflux for 5 h. The solution was cooled, and concentrated, and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 4:1) to afford the exocyclic methylene product (168 mg, 84%) as a colorless solid. This product was used in the next step without further characterization. To a solution of the olefin (100 mg, 0.279 mmol) in dichloromethane (6 mL) at 0 °C, was added solid m-chloroperoxybenzoic acid (57.5 mg, 0.333 mmol). The reaction mixture was 4 h, and then quenched with aqueous NaHCO3. The mixture was extracted several times with dichloromethane, dried and concentrated to afford the epoxide 1 (90 mg, 86%) as a colorless oil, which was used in the next step without further purification. To a solution of the epoxide (50 mg, 0.13 mmol) in hexanes (1 mL) and toluene (0.5 mL) was added HMPA (1 drop). The mixture was cooled to −78 °C, and then a solution of lithium diisopropylamine in hexanes (0.73 mmol) was added. The solution was warmed to room temperature and stirred for 10 h. The mixture was quenched with saturated aqueous NH4Cl, and the mixture extracted several times with ether. The combined extracts were dried (MgSO4) and concentrated, and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 3:2) to afford a colorless oil (29 mg, 58%) which was used without further characterization. To liquid ammonia (ca. 10 mL), at −78 °C was added lithium metal (24 mg, 3.5 mmol), followed by t-butyl alcohol (0.05 mL). To this solution was added a solution of the allylic alcohol (20 mg, 0.053 mmol) in THF (1 mL). The reaction mixture was stirred at −78 °C for 15 min, and then quenched with NH4Cl, and diluted with ether. The mixture was warmed to room temperature, and water (10 mL) was added. The mixture was extracted several times with ether followed by extraction with dichloromethane. The combined extracts were dried (MgSO4), concentrated and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 3:2) to afford 2 (9.0 mg, 60%) as a colorless solid. Mp 192–194 °C; 1H NMR (CDCl3, 400 MHz) δ 7.15 (d, J = 8.4 Hz, 1H), 6.64 (dd, J = 2.8, 8.4 Hz, 1H), 6.58 (d, J = 2.8 Hz, 1H), 5.65 (dd, J = 1.2, 2.8 Hz, 1H), 4.80 (br s, OH), 4.32–4.25 (m, 2H), 2.95–2.80 (m, 2H), 2.40–1.70 (m, 11 H), 0.87 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 155.2, 153.5, 138.5, 133.1, 126.4, 124.3, 126.4, 124.3, 115.5, 112.8, 60.4, 56.8, 46.4, 44.6, 37.4, 34.8, 31.1, 29.7, 27.9, 26.6, 16.5.
4.2.2. n-Propyl 3-hydroxyestra-1,3,5(10)-triene-17-carboxylate (4)
To a solution of 3 (177 mg, 0.411 mmol) in ethanol (10 mL) was added an aqueous slurry of Raney-Ni (60%, 0.6 mL). The reaction mixture was stirred under a H2 gas (balloon pressure) for 24 h, after which the mixture was filtered through a bed of filter-aid. The filter bed was washed several times with ethyl acetate, and the filtrate was concentrated under reduced pressure to afford 4 as a colorless solid (129 mg, 92%): mp 151.5–153 °C, +69.5 (c 0.388, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 7.17 (d, J = 8.4 Hz, 1H), 6.64 (dd, J = 2.8, 8.5 Hz, 1H), 6.57 (d, J = 2.7 Hz, 1H), 4.55 (br s, OH), 4.10 (dt, J = 10.8, 6.7 Hz, 1H), 4.02 (dt, J = 10.8, 6.7 Hz, 1H), 2.90–2.80 (m, 2H), 2.44 (t, J = 9.3 Hz, 1H), 2.35–2.15 (m, 3H), 1.90–1.75 (m, 3H), 1.68 (sextet, J = 7.2 Hz, 2H), 1.55–1.30 (m, 7H), 0.98 (t, J = 7.3 Hz, 3H), 0.71 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 174.5, 153.5, 138.4, 132.8, 126.7, 115.4, 112.8, 66.0, 55.6, 55.1, 44.3, 43.9, 39.0, 38.6, 29.8, 27.8, 26.7, 24.3, 23.7, 22.3, 13.7, 10.9. Anal. Calcd for C22H30O3·1/2H2O: C, 75.18; H 8.89. Found: C, 75.36; H, 8.28.
4.2.3. (20S) 3-(Phenylmethoxy)-19,24-dinorchola-1,3,5(10),16-tetraene (6)
To a solution of 5 (0.20 g, 0.50 mmol) in benzene (10 mL) in a Schlenk flask was added Rh(PPh3)3Cl (40 mg, 0.043 mmol). The reaction mixture was cooled with a dry ice–acetone bath, evacuated under high vacuum, and the system refilled to 1 atm with H2 gas. The mixture was stirred for 7 h at room temperature, and then the solvent was evaporated. The residue was extracted several times with ether, filtered, and concentrated. The residue was purified by column chromatography (SiO2, hexanes–CH2Cl2 = 10:1) to afford 6 (138 mg, 69%) as a colorless solid. Mp 82–83.5 °C, +67 (c 0.74, acetone); 1H NMR (CDCl3, 300 MHz) δ 7.46–7.30 (m, 5H), 7.20 (d, J = 8.4 Hz, 1H), 6.78 (br d, J = 8.4 Hz, 1H), 6.74 (br s, 1H), 5.35 (br s, 1H), 5.04 (s, 2H), 2.94–2.84 (m, 2H), 2.40–2.08 (m, 4H), 2.00–1.87 (m, 3H), 1.65–1.28 (m, 7H), 1.09 (d, J = 6.6 Hz, 3H), 0.89 (t, J = 7.3 Hz, 3H), 0.83 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 160.2, 155.9, 137.6, 136.7, 132.9, 128.0, 127.3, 127.0, 125.6, 120.4, 114.4, 111.8, 70.0, 56.4, 47.8, 44.7, 37.8, 35.4, 33.6, 31.3, 30.3, 30.2, 28.2, 27.0, 21.3, 17.1, 12.4. Anal. Calcd for C29H36O: C, 86.95; H, 9.06. Found: C, 86.99; H, 9.12.
4.2.4. (20S) 3-Hydroxy-19,24-dinorchola-1,3,5(10),16-tetraene (7)
Cleavage of the benzyl ether 6 (73 mg, 0.18 mmol) with sodium metal in n-butanol was carried out in a fashion similar to the cleavage of 8. Purification of the residue by column chromatography (SiO2, hexanes–ethyl acetate gradient = 5:1) gave unreacted starting material (17 mg) followed by 7 (46 mg, 81%) as a colorless solid. Mp 92–95 °C, +86.3 (c 0.32, acetone); 1H NMR (acetone-d6) δ 7.05 (d, J = 8.4 Hz, 1H), 6.56 (dd, J = 2.1, 8.4 Hz, 1H), 6.51 (d, J = 2.1 Hz, 1H), 5.35 (br s, 1H), 2.82–2.73 (m, 2H), 2.37–2.28 (m, 1H), 2.22–2.05 (m, 2H), 1.97–1.85 (m, 4H), 1.60–1.26 (m, 8H), 1.07 (d, J = 7.2 Hz, 3H), 0.87 (t, J = 7.5 Hz, 3H), 0.82 (s, 3H); 13C NMR (acetone-d6) δ 162.5, 156.7, 139.3, 133.2, 127.7, 122.7, 117.1, 114.7, 58.8, 50.0, 47.1, 40.4, 37.7, 35.8, 33.4, 32.5, 32.2, 30.6, 29.3, 23.2, 19.0, 14.1. Anal. Calcd for C22H30O·1/6H2O: C, 84.28; H, 9.75. Found: C, 84.28; H, 9.82.
4.2.5. (20S) 3-Hydroxy-19,24-Dinorchola-1,3,5(10),16-tetraen-23-ol (11)
To a solution of 8 (394 mg, 0.947 mmol) in n-butanol (20 mL), at 70 °C, was added sodium metal (0.87 g, 38 mmol) in small pieces. After all of the sodium had reacted, the reaction mixture was cooled to room temperature and quenched with water, followed by saturated aqueous NH4Cl. The reaction mixture was extracted several times with ether, the combined extracts were dried (MgSO4) and concentrated. The residue was purified by column chromatography (SiO2, hexanes–ethyl acetate gradient = 4:1 to 2:1) to afford unreacted starting material (91 mg) followed by 11 (150 mg, 49%) as a colorless solid. Mp 174.5–176 °C, +77.5 (c 1.50, acetone); 1H NMR (acetone-d6) δ 8.15 (s, phenol OH), 7.04 (d, J = 8.4 Hz, 1H), 6.56 (dd, J = 2.7, 8.4 Hz, 1H), 6.51 (d, J = 2.7 Hz, 1H), 5.38 (br s, 1H), 3.64–3.52 (m, 3H), 2.84–2.74 (m, 2H), 2.42–2.28 (m, 2H), 2.20–2.08 (m, 1H), 1.96–1.70 (m, 4H), 1.60–1.30 (m, 7H), 1.10 (d, J = 7.2 Hz, 3H), 0.82 (s, 3H); 13C NMR (acetone-d6) δ 162.8, 156.6, 139.2, 133.0, 127.6, 122.6, 117.0, 114.6, 61.4, 58.7, 49.9, 47.0, 43.0, 40.3, 37.5, 33.2, 32.0, 30.9, 30.5, 29.2, 23.7, 19.0. Anal. Calcd for C22H30O2: C, 80.94; H, 9.26. Found: C, 80.67; H, 9.32.
4.2.6. 17,23-Epoxy-3-(phenylmethoxy)-19,24-dinorchola-1,3,5(10)-triene (9)
To a solution of 8 (56 mg, 0.14 mmol) in CHCl3 (2 mL) was added a drop of concentrated HCl. The mixture was allowed to stand stirred for 24 h at room temperature, and then passed through a short column of silica gel using hexanes–ethyl acetate as eluent. Concentration of the eluent gave 9 (50 mg, 89%) as a colorless oil. +36 (c 1.0, CH2Cl2); 1H NMR (CDCl3, 300 MHz) δ 7.46–7.28 (m, 5H), 7.22 (d, J = 8.4 Hz, 1H), 6.87 (dd, J = 2.7, 8.4 Hz, 1H), 6.73 (d, J = 2.7 Hz, 1H), 5.04 (s, 2H), 3.87 (dt, J = 4.5, 7.8 Hz, 1), 3.62 (dt, J = 6.4, 7.8 Hz, 1H), 2.92–2.82 (m, 2H), 2.38– 1.20 (m, 16H), 1.10 (d, J = 6.9 Hz, 3H), 0.74 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 155.8, 137.6, 136.7, 132.8, 128.2, 127.3, 126.9, 125.8, 114.4, 111.8, 95.5, 70.0, 66.0, 50.0, 48.2, 44.0, 39.3, 36.9, 35.1, 31.3, 31.0, 30.3, 28.1, 26.6, 23.6, 19.0, 15.8. Anal. Calcd for C29H36O2: C, 83.61; H 8.71. Found: C, 83.35; H, 8.75.
4.2.7. 17,23-Epoxy-3-hydroxy-19,24-dinorchola-1,3,5(10)-triene (10)
To a solution of 9 (48.9 mg, 0.118 mmol) in methanol/CHCl3 (1:100, 6 mL) was added 10% Pd on carbon (5.6 mg). The mixture was stirred under H2 (ca. 46 psi) in a Paar hydrogenation apparatus for 3 h. The catalyst was removed by filtration through filter-aid and the filter bed was washed with copious CH2Cl2 and the combined filtrates were concentrated. The residue was purified by chromatography (SiO2, hexanes–ethyl acetate = 3:1) to afford 10 as a colorless solid (37.8 mg, 99%). Mp 172–174 °C; 1H NMR (CDCl3, 300 MHz) δ 7.15 (d, J = 8.4 Hz, 1H), 6.62 (dd, J = 2.7, 8.4 Hz, 1H), 6.55 (d, J = 2.7 Hz, 1H), 3.87 (dt, J = 4.5, 7.8 Hz, 1H), 3.60 (dt, J = 6.3, 8.1 Hz, 1H), 2.85–2.75 (m, 2H), 2.35–1.20 (m, 16H), 1.07 (d, J = 6.9 Hz, 3H), 0.70 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 153.3, 138.6, 133.2, 126.6, 115.4, 112.7, 96.0, 66.1, 50.0, 48.2, 43.9, 39.3, 36.8, 35.0, 31.2, 30.8, 30.0, 27.9, 26.4, 23.4, 18.8, 15.6. Anal. Calcd for C22H30O2·1/4H2O: C, 79.83; H 9.29. Found: C, 80.12; H, 9.33.
4.2.8. (20S) 3-Hydroxy-19,24-dinorchola-1,3,5(10),16-tetraen-23-al (12)
To a solution of 11 (100 mg, 0.296 mmol) in THF (4 mL) was added a solution of ethyl magnesium bromide in THF (0.67 mL, 1.0 M, 0.67 mmol). The solution was stirred at room temperature for 15 min, and then solid 1,1′-(azodicarbonyl)dipiperidine (0.17 g, 0.67 mmol) was added. The reaction mixture was stirred for 1 h, and then quenched with saturated aqueous NH4Cl and extracted several times with ether. The combined ethereal extracts were dried (MgSO4), concentrated and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 5:1) to afford 12 as a colorless solid (66 mg, 66%). Mp 168.5–171 °C, +78 (c 0.80, acetone); 1H NMR (acetone-d6, 300 MHz) δ 9.66 (t, J = 2.1 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.57 (dd, J = 2.5, 8.4 Hz, 1H), 6.51 (d, J = 2.5 Hz, 1H), 5.46 (br s, 1H), 2.90–2.75 (m, 4H), 2.62 (ddd, J = 1.8, 5.7, 16.2 Hz, 1H), 2.44–2.30 (m, 2H), 2.26–2.10 (m, 2H), 1.98–1.86 (m, 3H), 1.60–1.34 (m, 5H), 1.16 (d, J = 7.2 Hz, 3H), 0.88 (s, 3H); 13C NMR (acetone-d6, 75 MHz) δ 203.2, 161.4, 156.8, 139.5, 133.3, 127.9, 124.6, 117.2, 114.8, 59.2, 53.1, 50.2, 47.2, 40.5, 37.7, 33.6, 32.3, 30.7, 29.7, 29.4, 23.8, 19.3. Anal. Calcd for C22H28O2: C, 81.44; H, 8.70. Found: C, 81.21; H, 8.54.
4.2.9. 17,23-Epoxy-3-hydroxy-19-norchola-1,3,5(10)-triene (13)
To a solution of 12 (45.9 mg, 0.142 mmol) in THF (7 mL) at 0 °C was added a solution of methyl magnesium bromide in ether (0.10 mL, 3.0 M, 0.30 mmol). The reaction mixture was stirred for 3 h, and then quenched with saturated aqueous NH4Cl (15 mL). The mixture was extracted several times with CH2Cl2 and the combined extracts were dried (MgSO4) and concentrated. The residue was purified by chromatography (SiO2, hexanes–ethyl acetate = 5:1) to afford 13 as a colorless solid (44 mg, 92%). Analysis of the product by 1H NMR spectroscopy indicated this to be a 1:1 mixture of diastereomers. Mp 248–251 °C, 1H NMR (CDCl3, 300 MHz) δ 7.15 (d, J = 8.4 Hz, 1H), 6.62 (dd, J = 2.7, 8.4 Hz, 1H), 6.56 (d, J = 2.7 Hz, 1H), 4.18–4.07 (m, 1H), 3.85–3.74 (m, 1H), 2.85–2.75 (m, 2H), 2.35–1.20 (m, 15H), 1.23 & 1.20 (2 × d, J = 5.7 Hz, 3H total), 1.07 & 1.05 (2 × d, J = 6.9 Hz, 3H), 0.72 & 0.66 (2 × s, 3H total); 13C NMR (CDCl3, 75 MHz) δ 153.3, 138.6, 133.2, 126.6, 115.4, 112.7, 97.1 [95.8], 73.6 [71.3], 49.85 [49.80], 48.8, 47.1, 45.4, 43.9 [43.8], 43.5, 39.3 [39.2], 36.2, 34.5, 32.3, 31.2 [30.9], 30.6 [30.1], 27.8, 26.5 [26.4], 23.5 [23.4], 21.6, 19.2 [18.9], 16.3 [14.9]. Anal. Calcd for C23H32O2·1/2H 2O: C, 79.04; H, 9.52. Found: C, 79.34; H, 9.57.
4.2.10. cis- and trans-4-(4′-Hydroxycyclohexyl)phenol (14)
To a solution of 4-(4′-hydroxyphenyl)cyclohexanone (50 mg, 0.26 mmol) in methanol (1 mL) was added NaBH4 (15 mg, 4.0 mmol). The reaction mixture was stirred for 30 min, and then diluted with water. The mixture was extracted several times with ethyl acetate and the combined extracts were concentrated and purified by column chromatography (SiO2, hexanes–ethyl acetate = 2:1) to afford cis-14 (5.0 mg, 10%) followed by trans-15 (43 mg, 86%) both as colorless solids. Cis-14: 1H NMR (CD3OD, 400 MHz) δ 7.04–6.69 (AA′BB′, JAB = 8.8 Hz, 4H), 4.02 (narrow t, J = 2.8 Hz, 1H), 2.50–2.40 (m, 1H), 1.91–1.79 (m, 4H), 1.69–1.52 (m, 4H); 13C NMR (CD3OD, 75 MHz) δ 156.5, 140.1, 128.8, 116.1, 66.5, 44.5, 34.0, 29.4. Trans-15: 1H NMR (CD3OD, 400 MHz) δ 7.01–6.68 (AA′BB′, JAB = 8.4 Hz, 4H), 3.58 (tt, J = 4.4, 10.6 Hz, 1H), 2.39 (tt, J = 3.5, 11.8 Hz, 1H), 2.06–1.99 (m, 2H), 1.87–1.79 (m, 2H), 1.56–1.33 (m, 4H).
4.2.11. 4-(4-Hydroxyphenyl)-cyclohexanone oxime (16)
To a solution of 4-(4′-hydroxyphenyl)cyclohexanone (50 mg, 0.26 mmol), hydroxylamine hydrochloride (36.6 mg, 0.526 mmol) in ethanol (5 mL) was added Amberlyst (56 mg). After stirring for 2 h, the mixture was filtered, and the filtrate concentrated. The residue was partitioned between water and ethyl acetate, and the organic layer was concentrated and dried to give (±)-16 (44 mg, 82%) as a colorless solid. Mp 172–175 °C. 1H NMR (CD3OD, 400 MHz) δ 7.03–6.69 (AA′BB′, JAB = 8.8 Hz, 4H), 4.02 (narrow t, J = 2.8 Hz, 1H), 2.0–2.40 (m, 1H), 1.91–1.79 (m, 4H), 1.69–1.52 (m, 4H); 13C NMR (CD3OD, 75 MHz) δ 161.0, 156.8, 138.4, 128.7, 116.3, 44.3, 36.0, 34.7, 33.0, 25.2. HRMS (ESI): m/z calcd for C12H15NO2+Na+ [M+Na]+ 228.0995, found 228.0997.
4.2.12. cis-1-Hydroxymethyl-4-(4′-hydroxyphenyl)-cycloheptane (18)
To a solution of (±)-17 (75 mg, 0.35 mmol) in methanol (15 mL) in a heavy walled reaction vessel, was added a catalytic amount of 20% Pd/C. The mixture was stirred under H2 pressure (45 psi) for 75 min and then the reaction mixture was filtered through the pad of celite. The filtrate was concentrated and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 65:35) to afford (±)-18 (38 mg, 50%) as a colorless solid. Mp 60–61 °C; 1H NMR (CDCl3, 300 MHz) δ 7.06 and 6.75 (AA′BB′, JAB = 9.0 Hz, 4H), 3.48 (d, J = 6.3 Hz, 1H), 2.59–2.58 (m, 1H), 1.95– 1.08 (m, 13H); 13C NMR (CD3OD, 75 MHz) δ 127.9, 115.3, 68.6, 46.1, 41.4, 38.8, 33.1, 31.6, 28.5, 27.5. HRMS (ESI): m/z calcd for C14- H20O2+Na+ [M+Na]+ 243.1356, found 243.1356.
4.2.13. 5-[(1E)-2-(4-Hydroxyphenyl)ethenyl]-2-furanmethanol (20)
A solution of methyl 5-bromo-2-furanoate (1.03 g, 5.02 mmol), 4-acetoxystyrene (0.97 g, 6.0 mmol), palladium acetate (0.01 g, 0.05 mmol), tri-o-tolylphosphine (0.03 g, 0.2 mmol), and triethylamine (3 mL) was heated under nitrogen in a sealed heavy-walled Pyrex tube at 100 °C for 24 h. The reaction mixture was cooled, diluted with water and dichloromethane. The dichloromethane layer was separated, washed with water, and dried (MgSO4), and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 4:1) to afford 19 (350 mg, 24%), a pale yellow solid. Mp 110.5–112 °C; 1H NMR (CDCl3, 300 MHz) δ 7.51 (d, J = 8.1, 2H), 7.27 (d, J = 16.5 Hz, 1H), 7.20 (d, J = 3.6 Hz, 1H), 7.10 (d, J = 8.1 Hz, 2H), 6.86 (d, J = 16.5 Hz, 1H), 6.45 (d, J = 3.6 Hz, 1H), 3.92 (s, 3H, OMe), 2.32 (s, 3H, OAc). This product was used in the next step without further characterization. To a solution of diester (50 mg, 0.17 mmol) in anhydrous ether (1 mL) at 0 °C, was slowly added a solution of lithium aluminium hydride (0.52 mL, 1.0 M in THF, 0.52 mmol). Solution was stirred for 3 h at 0 °C and then saturated aqueous sodium bicarbonate (2 mL) was added follow by dilute sodium hydroxide. The mixture was warmed to room temperature, extracted several times with ethyl acetate. The combined extracts were dried (MgSO4), concentrated and the residue was purified by column chromatography (SiO2, hexanes–ethyl acetate = 1:1) gave 20 (28 mg, 74%) as a colorless solid. Mp 129–131 °C; 1H NMR (acetone- d6, 300 MHz) δ 8.59 (br s, 1H), 7.40 (d, J = 9.0 Hz, 2H), 6.97– 6.79 (m, 4H), 6.30 (s, 2H), 4.57 (br s, 2H), 3.05 (br s, 1H); 13C NMR (acetone-d6, 75 MHz) δ 158.2, 155.9, 154.1, 129.7, 128.6, 127.4, 116.5, 114.9, 109.9, 109.4, 57.4. HRMS (ESI): m/z calcd for C13H12O3+Na+ [M+Na]+ 239.0679, found 239.0681.
4.3. Fluorescence polarization
The assay was developed based on a commercially available kit from Invitrogen.15 Assays were run on a BMG POLARstar Galaxy reader with acquisition parameters as follows: 200 flashes, positioning delay 1.0 s, K factor ≤ 1.1 and ≥ 0.9, excitation filter of 485 ± 5 nm and emission filter of 520 ± 15 nm. For the IC50 determinations the [ER-α] was 30 nM and the [FITC-estradiol tracer] ([Tr]) was 10 nM. Sample volume was 150 μL. For each experiment the polarization was calibrated with a sample of FITC set at 20 mP. All proper blanks were used, including water for the FITC samples and blank samples containing only 30 nM ERα protein for the remaining data points. All protein samples contained 1% DMSO-d6, the maximum amount tolerated as stated by the supplier of the ERα protein, Invitrogen, to ensure the solubility of all hydrophobic compounds investigated. The Kd of the FITC-tagged estradiol for ER-α was determined by non-linear least squares fitting of the titration curve data to the following equation (where Tro is the F-E2 tracer):
4.4. Cell-based ERα and ERβ assays
ERα and ERβ assay kits for cell-based assays (Indigo Biosciences) allowed for investigation into the functional activity (i.e., agonist and/or antagonist) of the ligands identified to bind based on the initial fluorescence polarization displacement assay. Briefly, the cells contained a luciferase reporter gene that was functionally linked to either the ERα or ERβ-responsive promoter. By quantifying the luciferase expression via luminescence, the change in ER activity could be quantified. 1–2 mM stocks of the ligands were prepared in DMSO-d6 and diluted to final concentrations ranging from 3.2 nM to 2 μM, using the Compound Screening Medium provided in the kit. For the agonist assay, the cells were prepared by warming to 37 °C, plated, then the chemicals added. For the antagonist assay, the cells were prepared as above with the addition of E2 (for ERα 3.2 nM was added, approximating an IC75; and, for ERβ 160 pM was added, approximating an IC80). The cells were then plated, and the chemicals added. All plates were incubated in a cell culture incubator at 37 °C and 5% CO2 for 22 h. Each assay was performed in duplicate. Luminescence was characterized after removal of the incubating media and introduction of the Detection Substrate using a Molecular Devices SpectraMax M5 microplate reader. Data was fitted using GraphPad Prism and fit to the dose-response (four paramter) equation as follows.
4.5. Molecular docking
Ligand structures were drawn in PC Spartan Plus (Wavefunction) and three dimensional (3D) conformation was then optimized using semiempirical Austin Model 1 (AM1) calculations. Since compound 13 was afforded as a pair of diastereomers both were modeled and docked. The AM1 calculations provided geometries and bond distances for subsequent docking. AutoDock Tools (ADT) was used prepare the ligand files according to AutoDock requirements and assign Gasteiger charges.
The ERα receptor for agonist (pdb code 1ere)4 and antagonist (pdb code 1err)32 conformations were prepared for docking calculations using the ‘A’ chain. The ERβ receptor for agonist (pdb code 2jj3)33 and antagonist (pdb code 1l2j)34 conformations were prepared for docking calculations using the ‘A’ chain. ADT was used to further prepare the ER receptor files by adding hydrogen atoms and adding partial charges to each atom of the protein. The grid box was centered on the co-crystallized ligand, drawn to a box to incorporate amino acids Arg394, Glu353, and His524 for ERα and Arg346, Glu305, and His475 for ERβ, then the estradiol ligand was removed.35 AutoDock (v. 4.2) calculations were performed with default parameters, except with 100 genetic algorithmic runs and 2,500,000 evaluations per run.35–39
Supplementary Material
Acknowledgments
This work was supported by an NIH Instrumentation Grant (S10 RR019012). W.A.D. acknowledges financial support from the National Institutes of Health (GM-42641) and the National Science Foundation (CHE-0415771). D.S.S. acknowledges financial support from NIH Grants AI101975 and HL112639, and the use of resources at the Children’s Environmental Health Sciences Core Center (funded by National Institute of Environmental Health Sciences, P30 ES004184) at the University of Wisconsin Milwaukee. Dr. Phani Kumar Pullela is gratefully acknowledged for providing the synthesized F-E2 tracer. High-resolution mass spectra were obtained at the COSMIC lab at Old Dominion University.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.11.024.
References and notes
- 1.Manas ES, Xu ZB, Unwalla RJ, Somers WS. Structure. 2004;12:2197. doi: 10.1016/j.str.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Levin ER. Mol Endocrinol. 2005;19:1951. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M. Mol Cell Biol. 2004;24:7681. doi: 10.1128/MCB.24.17.7681-7694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 5.Payne J, Scholz M, Kortenhamp A. Environ Health Perspect. 2001;109:391. doi: 10.1289/ehp.01109391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, Tong W, Shi L, Perking R, Sheehan DM. Toxicol Sci. 2000;54:138. doi: 10.1093/toxsci/54.1.138. [DOI] [PubMed] [Google Scholar]
- 7.Deroo BJ, Korach KS. J Clin Invest. 2006;116:561. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colborn T, Saal FS, Soto AM. Environ Health Perspect. 1993;101:378. doi: 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brody JG, Rudel RA. Environ Health Perspect. 2003;111:1007. doi: 10.1289/ehp.6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tice R. National Toxicology Program. National Institute of Environmental Health Sciences. NIH, U.S. Dept. of Health and Human Services; 2001. Biomolecular Screening Branch. July 5, 2013 ( http://www.niehs.nih.gov/research/atniehs/labs/bmsb/index.cfm) [Google Scholar]
- 11.Nasir MS, Jolley ME. Comb Chem High Throughput Screening. 1999;2:177. [PubMed] [Google Scholar]
- 12.Burke TJ, Loniello KR, Beebe JA, Ervin KM. Comb Chem High Throughput Screening. 2003;6:183. doi: 10.2174/138620703106298365. [DOI] [PubMed] [Google Scholar]
- 13.Ohno K, Fukushima T, Santa T, Waizumi N, Tokuyama H, Maeda M, Imai K. Anal Chem. 2002;74:4391. doi: 10.1021/ac020088u. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki S, Ohno K, Santa T, Imai K. Anal Sci. 2003;19:1103. doi: 10.2116/analsci.19.1103. [DOI] [PubMed] [Google Scholar]
- 15.Parker GJ, Law TL, Lenoch FJ, Bolger RE. J Biomol Screen. 2000;5:77. doi: 10.1177/108705710000500204. [DOI] [PubMed] [Google Scholar]
- 16.Costache AD, Pullela PK, Kashi P, Tomasiewicz H, Sem DS. Mol Endocrinol. 2005;19:2979. doi: 10.1210/me.2004-0435. [DOI] [PubMed] [Google Scholar]
- 17.Bolger R, Wiese TE, Ervin K, Nestich S, Checovich W. Environ Health Perspect. 1998;106:551. doi: 10.1289/ehp.98106551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shoichet BK. Nature. 2004;432:862. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavasotto CN, Orry AJW. Curr Top Med Chem. 2007;7:1006. doi: 10.2174/156802607780906753. [DOI] [PubMed] [Google Scholar]
- 21.Suresh PS, Kumar A, Kumar R, Sihn VP. J Mol Graphics Modell. 2008;26:845. doi: 10.1016/j.jmgm.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Cross JB, Thompson DC, Rai BK, Baber JC, Fan KY, Hu Y, Humblet C. J Chem Inf Model. 2009;49:1455. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 23.De Riccardis F, Meo D, Izzo I, Di Filippo M, Casapullo A. Eur J Org Chem. 1998:1965. [Google Scholar]
- 24.Lam HYP, Begleiter A, Goldenberg GJ. J Med Chem. 1979;22:200. doi: 10.1021/jm00188a015. [DOI] [PubMed] [Google Scholar]
- 25.Li PK, Murakata C, Akinaga S. 6,288,050, 2001. US Patent.
- 26.He Z, Donaldson WA, Yi CS. Org Lett. 2003;5:1567. doi: 10.1021/ol030031+. [DOI] [PubMed] [Google Scholar]
- 27.Frigoli M, Mehl GH. Eur J Org Chem. 2004:636. [Google Scholar]; DeOrazio RJ, Nikam SS, Scott IL, Sherer BA, Wise LD. 01/81295 A1. PCT Int Appl WO. 2001
- 28.Indigo Biosciences. Human Estrogen Receptor Technical Manual [Google Scholar]
- 29.Pandey RK, Wang L, Wallock NJ, Lindeman S, Donaldson WA. J Org Chem. 2008;73:7236. doi: 10.1021/jo801446q. [DOI] [PubMed] [Google Scholar]
- 30.van Lipzig MMH, ter Laak AM, Jongegan A, Vermeulen NPE, Wamelink M, Geerke D, Meerman JHN. J Med Chem. 2004;47:1018. doi: 10.1021/jm0309607. [DOI] [PubMed] [Google Scholar]
- 31.Miteva MA, Lee WH, Montes MO, Villoutreix BO. J Med Chem. 2005;48:6012. doi: 10.1021/jm050262h. [DOI] [PubMed] [Google Scholar]
- 32.Norman BH, Richardson TI, Dodge JA, Pfeifer LA, Durst GL, Wang Y, Durbin JD, Krishnan V, Dinn SR, Liu S, Reilly JE, Ryter KT. Bioorg Med Chem Lett. 2007;17:5082. doi: 10.1016/j.bmcl.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 33.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzellenbogen JA, Agard DA, Greene GL. Nat Struct Biol. 2002;9:359. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 34.Tuccinardi T, Bertini S, Martinelli A, Minutolo F, Ortore G, Placanica G, Prota G, Rapposelli S, Carleson KE, Katzenellenbogen JA, Macchia M. J Med Chem. 2006;49:5001. doi: 10.1021/jm060560u. [DOI] [PubMed] [Google Scholar]
- 35.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639. [Google Scholar]
- 37.Goodsell DS, Morris GM, Olson AJ. J Mol Recognit. 1996;9:1. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Huey R, Morris BM, Olson AJ, Goodsell DS. J Comput Chem. 2007;28:1145. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Zhang H, Gibson M, Li J. Toxicol In Vitro. 2012;26:769. doi: 10.1016/j.tiv.2012.05.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.