


Abstract
The free radical theory of aging proposes that reactive oxygen species (ROS)-induced accumulation of damage to cellular macromolecules is a primary driving force of aging and a major determinant of lifespan. Although this theory is one of the most popular explanations for the cause of aging, several experimental rodent models of antioxidant manipulation have failed to affect lifespan. Moreover, antioxidant supplementation clinical trials have been largely disappointing. The mitochondrial theory of aging specifies more particularly that mitochondria are both the primary sources of ROS and the primary targets of ROS damage. In addition to effects on lifespan and aging, mitochondrial ROS have been shown to play a central role in healthspan of many vital organ systems. In this article we review the evidence supporting the role of mitochondrial oxidative stress, mitochondrial damage and dysfunction in aging and healthspan, including cardiac aging, age-dependent cardiovascular diseases, skeletal muscle aging, neurodegenerative diseases, insulin resistance and diabetes as well as age-related cancers. The crosstalk of mitochondrial ROS, redox, and other cellular signaling is briefly presented. Potential therapeutic strategies to improve mitochondrial function in aging and healthspan are reviewed, with a focus on mitochondrial protective drugs, such as the mitochondrial antioxidants MitoQ, SkQ1, and the mitochondrial protective peptide SS-31.
Keywords: Mitochondria, Oxidative stress, Aging, Healthspan
Introduction
Denham Harman first proposed the free radical theory of aging in 1956, suggesting that free radical-induced accumulation of damage to cellular macromolecules is a primary driving force of aging and a major determinant of lifespan [1]. This theory, however, is a highly simplified view of the role of reactive oxygen species (ROS) in the biology of aging. There are a number of sources of intracellular ROS in mammals, including NADPH oxidases (NOX), mitochondria, xanthine oxidase, monoamine oxidase, and nitric oxide synthase. The term ROS itself, encompasses numerous species that range from highly reactive (OH.) to longer-lived and membrane permeant (H2O2). Under normal conditions, ROS are maintained at the physiological levels by several endogenous antioxidant systems, including superoxide dismutatase (SOD), catalase, glutathione peroxidases, and glutathione reductase (GR). Other antioxidant systems involving thiol-disulphide oxidoreductase systems include the cytosolic proteins thioredoxin (TRX) and glutaredoxin (GRX). These antioxidant systems are complex, located in different cellular compartments and are often redundant or complementary in various conditions. Physiological levels of ROS interact with redox state and play a role in mediating cell signaling, while pathological levels of ROS can result in oxidative damage to cellular components and activate several cell death pathways (Figure 1). The close interrelationship of redox balance to oxidative stress has in recent years become a more prominent aspect of the free radical theory of aging and has been the subject of several reviews [2-4].
Figure 1.

Illustration of the continuum of oxidative stress in health and pathology. The redox stress pathway emphasizes the signaling role of oxidative stress and focuses on reversible regulation and depends on the interaction between cellular components and the redox environment of the cell. In contrast, prolonged or high oxidative stress leads to structural changes in proteins, lipids, and DNA that are generally more irreversible. These represent two points along the continuum of how oxidative stress may contribute to aging phenotypes. Modified from Marcinek and Siegel [120].
Based on the free radical theory of aging, several scientists have attempted to increase lifespan by genetic manipulation of antioxidant system components, however, the results have generally been conflicting. In Caenorhabditis elegans, single or double SOD mutants have a normal lifespan, while mitochondrial SOD2 mutants (single or double with cytoplasmic SOD1) increased lifespan [5]. In Drosophila melanogaster early results were confounded by uncontrolled genetic background effects. Later analyses suggested that over-expression of catalase, SOD1, or SOD2 using the genes’ native promoters does not increase life span [6,7], but that when tissue-specific or conditional transgenic overexpression systems were used, elevated SOD2 did result in substantial life span increases [8]. It has also been suggested that the largest lifespan extensions were seen in backgrounds with shorter lifespan or under redox stress [3]. In transgenic mice, the overexpression of endogenous antioxidants, including CuZnSOD (SOD1, cytoplasmic), MnSOD (SOD2, mitochondrial), catalase, or combination of CuZnSOD/catalase and CuZnSOD/MnSOD failed to extend mouse lifespan [9-11]. While SOD1 knockout mice exhibit 30% shorter lifespan, the fact that their major cause of death is hepatocellular carcinoma and the absence of lifespan reduction in SOD1 heterozygous mice suggest the shorten lifespan in SOD1 knockout may not be due to accelerated aging. While complete deletion of SOD2 cause neonatal death, SOD2 heterozygous mice and SOD3 both shown normal lifespan. However, it is notable that in many disease models or when under environmental stress, the same transgenic overexpression mice may be healthier than their wild-type counterparts, and the converse for antioxidant under-expressing mice (reviewed by [12]).
Several clinical trials using antioxidant supplementation in various study populations have been performed during the last three decades and the results are often equivocal or conflicting. Meta-analyses of large numbers of individual reports are often required to reach conclusions, however these too vary. A large scale analysis of 68 randomized trials including 232,606 participants from general population or patients with heterogeneous diseases have reported no effect of antioxidant supplements on overall mortality, or even a significant increase in mortality in subjects receiving beta carotene, vitamin A, and vitamin E [13]. A recent widely cited meta-analysis including 50 randomized controlled trials with 294,478 participants showed no evidence to support the use of vitamin and antioxidant supplements for prevention of cardiovascular diseases [14]. In contrast, a recent meta-analysis of seven studies on the risk of Alzheimer’s disease showed that dietary intakes of vitamin E, vitamin C, and beta carotene can lower the risk of AD [15]. In spite of extensive study it remains clear that there is no consensus and/or those effects are disease-dependent.
Review
Mitochondrial free radical theory of aging
The lack of anti-aging effect with antioxidant supplements led Harman to modify his original theory to specify mitochondria as both the primary sources of ROS and the primary targets of ROS damage [16]. One of the features of the mitochondrial free radical theory is the central role that mitochondria play in generation of ROS from the electron transport chain, production of energy (ATP), and the numerous potential feedback loops in regulation of mitochondrial and cellular function, in which redox state and ROS might create ‘vicious cycles’ (Figure 2). These include mutations or deletions in mtDNA, which can result in damaged proteins, including important components of the electron transport chain that are encoded by mtDNA, as well as balances in mitochondrial redox state, including glutathione (GSH/GSSG) and nicotinamide dinucleotides. Even these are intertwined, as NADPH is used by glutathione reductase to regenerate glutathione (GSH) from oxidized glutathione (GSSG) (Figure 2). NADPH is also in equilibrium with NADH within mitochondrial through the activity of nicotinamide nucleotide transferase (NNT, also called mitochondrial NAD(P) transhydrogenase). The redox balance of NAD/NADH is the key regulator of the sirtuin histone deacetylases, including mitochondrial SIRT3. The latter has been shown to play a key role in the acetylation state of cyclophilin D, which in turn plays an important role in control of the mitochondrial permeability transition pore (mPTP) and apoptosis (Figure 2).
Figure 2.
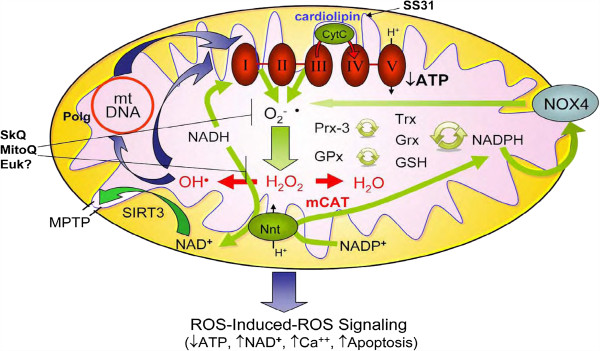
Interdependences of mtROS, nicotinamide nucleotides, and SIRT3: ROS-Induced ROS Signaling. Modified from Dai et al. [93].
The revised mitochondrial free radical theory suggests that failures of antioxidants to extend murine lifespan or failure of antioxidant supplements in clinical trials might be explained by poor distribution of antioxidants to mitochondria, and proposes that antioxidants targeted to mitochondria might be beneficial for lifespan extension. Several lines of evidence have supported the mitochondrial theory of aging. One of the most direct experimental evidence for the role of mitochondrial ROS in longevity was shown in mice overexpressing catalase targeted to mitochondria (mCAT), which resulted in a significant median and maximal lifespan extension in two independent lines of C57Bl6 mice [17]. Interestingly, similar overexpression of catalase targeted to peroxisome (pCAT), its normal location within the cell, or nuclear localization (nCAT) had modest and non-significant effects on murine lifespan. This indicates that mitochondrial localization of the catalase is key to lifespan extension in this model [17]. Consistently, mitochondria-targeted antioxidant SkQ1 has been shown to prolong the lifespan of inbred male mice in specific pathogen free (SPF) condition and outbred mice and dwarf hamster in conventional or outdoor cages [18].
Additional evidence for the involvement of mitochondrial ROS in aging comes from observations of mice with a targeted mutation of the p66Shc gene. These mice display reduced ROS generation and increased resistance to ROS-mediated apoptosis and thereby have a prolonged lifespan [19]. Further studies have shown that P66Shc is a mitochondrial redox enzyme located in the mitochondrial intermembrane space, which produces H2O2 from electron leakage during oxidative phosphorylation [20]. Phosphorylated p66Shc was later shown to accumulate within mitochondria to activate mitochondrial Ca2+ response, and subsequently induce apoptosis [21]. Further evidence supporting the role of mitochondria in aging was demonstrated using mice with homozygous mutation in exonuclease domain of the mitochondrial polymerase gamma (PolgaD257A/D257A , abbreviated Polgm/m). These mice were susceptible to accumulation of mtDNA point mutations and deletions with age [22,23]. They have shortened lifespan (maximal lifespan approximately 15 months) and display many phenotypes of ‘accelerated aging’, including kyphosis, graying and loss of hair, anemia, osteoporosis, sarcopenia (loss of muscle mass), and presbycusis (age-related hearing loss) [22]. Interestingly, the ‘premature’ aging-like phenotype in these mice showed good correlation with the accumulation of mtDNA deletions but not with the burden of mtDNA point mutations [24]. Moreover, the accumulation of mtDNA damage has been shown to increase apoptosis [23] and age-dependent cardiomyopathy and oxidative damage in the Polgm/m mouse heart was attenuated by mCAT [25].
In spite of these ‘attractive’ aspects of the mitochondrial free radical theory of aging, there remain many unsolved questions. Damage to mitochondrial DNA has been shown to increase with age, even more so than nuclear DNA [26]; however, it is necessary to distinguish point mutations from deletions; for example, it has been argued that the former do not [27] whereas the latter do correlate with lifespan of mice [24] This may be related to the fact that mitochondria have multiple copies of DNA, providing protection from heteroplasmic mutations. Recent improvements in DNA sequencing methods have also revealed the surprising result that age-related increases in mtDNA point mutations in human brains are primarily DNA transitions, whereas oxidative damage is expected to produce an excess of G ➔ T transversions [28]. Deletions, however, appear to accumulate and expand in the population of mtDNA during aging and some pathologies and it appears likely that mitochondrial respiratory failure only occurs with high loads of mtDNA deletion, such as has been observed in muscle fibers, intestinal crypts, and substantia nigra neurons (see review [29]). Finally, there is an increasing awareness that low levels of mitochondrial ROS may be ‘hormetic’ by inducing endogenous antioxidant defenses to prevent oxidative stress induced in pathological states and that mitochondrial ROS may be an important mediator of cell signaling (see section below).
The mitochondrial free radical theory has led to a focus on development and refinement of drugs to specifically target ROS specifically in the mitochondria of cells. The most common approach is based on delivery of known redox agents to the mitochondrial matrix by conjugation to delocalized cations (such as the triphenylphosphonium ion (TPP+)), including MitoQ and SkQ1 [30,31]. The Szeto-Schiller (SS) peptides represent a different chemical approach to reduce mitochondrial ROS. These aromatic-cationic tetrapeptides are targeted to cardiolipin on the inner mitochondrial membrane, and they have been shown to modulate electron flux in the electron transport chain and increases ATP production, while reducing electron leak and inhibiting excessive ROS production [32]. These mitochondria-targeted antioxidants are discussed in greater detail later (see section Mitochondrial protective strategies as potential therapeutics for aging-related diseases).
Mitochondrial signaling and the theory of mitohormesis
Apart from generating detrimental oxidative damage, ROS have numerous crucial biological roles in signaling and stress response (reviewed in [33-35]). Emerging evidence suggests that oxidative stress might promote longevity and metabolic health through the concept of mitochondrial hormesis (mitohormesis). The mitohormesis theory hypothesizes that low levels of oxidative stress induced by either caloric restriction, exercise [36], or other stimuli may trigger adaptive responses that improve overall stress resistance, probably through increased endogenous antioxidant defense, which may eventually reduce chronic oxidative damage [37] and subsequently achieve lifespan extension. This concept is supported by a study in C. elegans demonstrating that inhibition of respiration increases mitochondrial ROS production and significantly increases lifespan via mitochondrial ROS mediated activation of HIF-1 [38]. Low dose of oxidative stress induced by dietary restriction, especially glucose restriction, has been shown to preferentially induce mitochondrial metabolism and extend lifespan in various model organisms, including Drosophila melanogaster[39] and Caenorabditis elegans[40]. For instance, glucose restriction in C. elegans extends lifespan by inducing mitochondrial respiration and increasing oxidative stress, and this AMPK-dependent lifespan extension is abolished by pre-treatment of antioxidant N-acetyl cysteine, suggesting that oxidative stress is required for lifespan extension of dietary restriction [40]. Although the evidence of hormesis in lifespan regulation in mammalian models is still lacking, considerations should be taken when developing antioxidant therapy.
The theory of mitohormesis could have important translational implications as an ideal antioxidant therapy might be one that prevents oxidative damage induced under pathological conditions without interfering with ROS needed for hormesis and cellular signaling. We speculate that the targeted expression of catalase in mitochondria (mCAT) might be such an example, as there are beneficial effects of mCAT in aging and several disease models with negligible adverse effects (Table 1). Key to this may be that the Km of the catalytic activity of catalase is >10 mM, so that this enzyme is less likely to be effective at the lower intracellular H2O2 concentrations that may be involved in signaling or hormesis [41,42].
Table 1.
Mitochondrial targeted genetic and pharmacological manipulations on aging and healthspan
| Animal models | Description | Aging phenotypes | Healthspan phenotypes | |
|---|---|---|---|---|
|
Genotypes |
mCAT |
Overexpression of catalase targeted to mitochondria |
18% extension of lifespan [17]. Attenuated cardiac aging [43], aging-related sarcopenia [17], presbyacusis [44], and cancer incidence [45]. |
Protect against cardiac hypertophy and heart failure [46] |
| Reduce Aβ toxicity and oxidative injury, and extends the lifespan of Aβ PP overexpressing mice [47] | ||||
| Protective against mitochondrial ROS production and subsequent dopaminergic neuron degeneration in MPTP-induced Parkinson’s disease model [48] | ||||
| Attenuate lipid-induced insulin resistance in skeletal muscle [49] | ||||
| Polgm/m |
Homozygous mutation of mitochondrial polymerase gamma D257A |
‘Accelerated aging’: sarcopenia, graying and alopecia, kyphosis, presbyacusis, anemia [22,23], age-dependent cardiomyopathy [25] |
Aggravate heart failure in response to Angiotensin II [46] |
|
| p66shc |
Targeted mutation of the p66Shc gene |
Extension of lifespan. Reduction of ROS and apoptosis [19] |
Attenuate Angiotensin II induced LV hypertrophy and cardiomyocytes apoptosis; reduce oxidative damage in cardiac progenitor cells, cardiomyocytes and endothelial cells in diabetes [19,21,50,51] |
|
| SIRT3-/- |
SIRT3-deficient mice |
Accelerated cardiac aging, age-dependent increase in mitochondrial swelling due to increased mPTP opening [52] |
Early-age onset of hypertrophy associated with fibrosis |
|
| Abolish CR effect in reduction of oxidative damage, protection of cochlear neurons and prevention of presbycusis [53] |
Increased mortality after transverse aortic constriction [52] |
|||
| Pharmacological treatments | SS-31 |
Mitochondrial protective tetrapeptide |
Reverse age-related muscle weakness and muscle energy deficits [54] |
Attenuation of Angiotensin II induced cardiac hypertrophy and Gαq overexpression induced heart failure [55] |
| Ameliorate cardiac dysfunction after tranverse aortic constriction [56] | ||||
| Improve systolic function ischemic HF model [57,58] | ||||
| Attenuate cardiac I/R injury [59,60] | ||||
| Protect against renal I/R injury [61] | ||||
| Prevent high fat diet induced insulin resistance in skeletal muscle [62] | ||||
| Attenuation of diabetic retinopathy [63] | ||||
| Protective against ALS in SOD1 mutant mice [64] and Parkinson’s diseases in MPTP model [65] | ||||
| MitoQ |
Ubiquinone (antioxidant) conjugated with TPP+ |
|
Reduction of blood pressure and cardiac hypertrophy in spontaneous hypertensive rats [66] |
|
| SkQ | Plastoquinone conjugated with TPP+ | Prolonged lifespan. Attenuation of age-related decline in immunity. Protective against baldness and lordokyphosis in aged mice [18,67] | Attenuate heart arrhythmia, I/R injury, myocardial infarction, and kidney ischemia [68] |
|
| Delayed tumor development in p53-deficient mice [30] | ||||
| Protect against cataract and retinopathy in OXYS rats [69] |
Mitochondrial oxidative stress in healthspan
Cardiac aging
Increasing evidence suggests that abnormal mitochondrial ROS (mtROS) production and detoxification contributes to mitochondrial dysfunction and cardiomyopathy in old age (reviewed in [35,70,71]). An age-dependent reduction in cardiac mitochondrial oxidative phosphorylation function is related to the decline in mitochondrial state 3 respiration (maximal stimulated respiration) due to diminished activity of electron transport complexes I and IV (both have subunits encoded by mtDNA), while complexes II, III, and V are relatively unaffected (see review [72]). Impaired electron transport chain function is directly related to elevated electron leakage and generation of mtROS. Since the heart has a high metabolic demand and is rich in mitochondria, it produces ROS within mitochondria as a byproduct of oxidative phosphorylation and is, therefore, especially susceptible to oxidative damage. It has been shown that mitochondrial production of ROS significantly increases in the heart with advanced age [73].
The Framingham Heart Study and the Baltimore Longitudinal Study on Aging (BLSA) demonstrate that aging is associated with increased prevalence of left ventricular hypertrophy and decline in diastolic function (measured by the ratio of early to late ventricular filling (E/A) by Doppler echocardiography) in otherwise healthy individuals. Left ventricular (LV) wall thickness increases and maximal exercise capacity decreases with age in both sexes, indicative of LV hypertrophy, while systolic function is relatively preserved at rest (reviewed in [74,75]). Cardiac aging in murine models closely recapitulates those seen in humans [76], including cardiac hypertrophy (Figure 3A), a modest decline in systolic function (%FS, Figure 3B), a significant decline in diastolic function measured by Ea/Aa (Figure 3C), and worsening of the myocardial performance index (that is, an increased fraction of systole was spent during ineffective isovolumic contraction and relaxation, Figure 3D) [43]. The proportion of mice with diastolic dysfunction and left atrial dilatation also significantly increased with age [43].
Figure 3.

Echocardiography of cardiac aging in wild-type (WT) and mCAT mice (A-D) and Polgm/m mice in the presence or absence of mCAT (E-H). (A, E) Left ventricular mass index (LVMI), (B, F) % FS (fractional shortening), (C, G) Ea/Aa by tissue Doppler imaging (diastolic function), (D, H) the myocardial performance index (MPI). The increased linear trends across ages in WT mice were significant for all parameters (P <0.05 for all, left panels). The beneficial effect of mCAT versus WT was analyzed by the interaction between genotype and the linear age trend, and was significant in all cases (P <0.01 for all except fractional shortening, P = 0.03). *P <0.05 versus Polgm/m at age 4 to 6 months, #P <0.05 versus Polgm/m at age 13 to 14 months (right panels). LVMI, Left ventricular mass index; mCAT, catalase targeted to mitochondria. Modified from Dai et al. [25,43].
Data from our laboratory demonstrated that mCAT greatly attenuated many of these cardiac aging phenotypes (Figure 3A-D). The preserved cardiac aging phenotypes in mCAT mice were accompanied by reductions of age-dependent increases in mitochondrial protein carbonyls (Figure 4A) and mtDNA deletions (Figure 4B), suggesting prevention of mitochondrial oxidative damage as a mechanism of the cardiac aging protection. The success of mCAT protection in cardiac aging and the inability to confer similar protection by overexpression of peroxisomal catalase or the non-targeted antioxidant N-Acetyl Cysteine [55] underscores the importance of mitochondrial specificity in antioxidant intervention. Given the complexity of the systems involved, it is likely that mitochondrial dysfunction and aberrant ROS production may contribute to aging through both direct damage to cellular macromolecules and interference with normal signaling and energetics. There is an age-dependent increase in electron leakage and superoxide production. This makes a positive feedback between complex I inhibition and mitochondrial ROS production, as well as the more classical vicious cycle of mitochondrial DNA mutation and protein damage amplifying ROS (Figure 2). The effect of mitochondrial ROS in signaling and energetics may be a critical factor in cardiac (and other organ system) aging.
Figure 4.

Mitochondrial oxidative damage and mtDNA deletions in cardiac aging. (A). Mitochondrial protein carbonyl (nmol/mg) significantly increased in old wild-type (OWT, >24 months) and even more in middle-aged Polg (13.5 months) mouse hearts when compared with young WT mouse hearts. mCAT significantly reduced the age-dependent mitochondrial protein carbonylation. (B) Mitochondrial DNA deletion frequency significantly increased in OWT (>24 months) and young Polg (4 months) when compared with young WT, and this is dramatically increased in middle-aged Polg (13.5 months). mCAT overexpression significantly reduced the deletion frequency for both. *P <0.05 compared with YWT. Modified from Dai et al. [25,43].
As discussed above, mice with homozygous mutation of mitochondrial polymerase gamma (Polgm/m) have substantial increases in mtDNA mutations and deletions with age [22,23], shortened lifespan and exhibit several progeroid phenotypes, and developed cardiomyopathy in middle age (13 to 14 months) [22,25]. Middle age Polgm/m mice display cardiac hypertrophy (Figure 3E) and impaired systolic and diastolic function (Figure 3F-G) to an extent that is more severe than wild-type (WT) mice aged 24 to 30 months. Interestingly, mCAT partially rescues the mitochondrial damage and cardiomyopathy in Polgm/m mice (Figure 3E-H), supporting the role of mitochondrial ROS and mtDNA damage as part of a vicious cycle of ROS-induced ROS release (Figure 2) [25]. An interesting study shows that endurance exercise can prevent both skeletal muscle and cardiac progeroid phenotypes in Polgm/m mice [77]. The beneficial effect of exercise is thought to be mediated by the augmented level of mitochondrial biogenesis seen with exercise in these mice, which contributes to the preserved mitochondrial and, subsequently, organ function. Exercise induces ROS, and ROS stimulates upregulation of PGC1α [78], which is the master regulatory molecule in mitochondrial biogenesis and is known to improve endogenous antioxidant systems. The beneficial effect of exercise in this scenario is indeed a good example of mitochondrial hormesis mentioned above.
Cardiovascular diseases
Separate from the intrinsic decline in cardiac function during healthy aging mentioned above, old age is associated with an exponential increase in the prevalence of hypertension, stroke, coronary heart disease, and heart failure, especially in people aged over 65 years. Increased oxidative stress has been implicated in the pathogenesis of cardiovascular diseases, including hypertension, atherosclerosis, cardiac hypertrophy due to cardiac aging or pressure overload, cardiac ischemia-reperfusion injury, as well as cardiac failure. As in cardiac aging, a deficiency of mitochondrial energetics has been documented in human and experimental animals with heart failure [79]. Mechanisms may include mitochondrial biogenesis that does not keep up with the increasing demand (see review [80]), mitochondrial uncoupling and decreased substrate availability [81], and increased mitochondrial DNA deletions [46]. Mutations of genes encoding mitochondrial enzymes have been shown to be associated with various forms of idiopathic hypertrophic and dilated cardiomyopathies [82]. Mitochondrial DNA deletions have been found in experimental models of heart failure [83]. Studies on human hearts using 31P NMR spectroscopy indicated that the ATP content of failing hearts is generally 20% to 30% lower than that of normal hearts [84]. Furthermore, phosphocreatine, an important short-term reserve energy source that maintains a high phosphorylation potential to cope with acute increases in energy demand (for example, exercise), significantly declined by up to 60% in elderly heart failure patients [85]. The magnitude of this reduction is related to the severity of heart failure [86] and is shown to predict mortality in patients with dilated cardiomyopathy [87].
Hypertension is the most common cause of cardiac hypertrophy, which predisposes to chamber dilatation, heart failure, and sudden cardiac death [88]. Angiotensin II, a key molecule in the Renin-Angiotensin System which regulates hypertension, is well known to cause left ventricular hypertrophy and fibrosis [89]. At the molecular level, Angiotensin II binds to ATR1, a Gαq coupled-receptor, then activates NADPH oxidase through a PKC-dependent manner to produce ROS [90]. ROS from NADPH oxidase, particularly the NOX4 isoform, might increase mitochondrial ROS production, as previously shown in endothelial and vascular smooth muscle cells [91,92]. Mechanisms of ROS amplification in mitochondria might include ROS induced ROS release as well as a ROS-mtDNA damage vicious cycle (Figure 2) (see review [93]).
A study from our laboratory showed that Angiotensin II delivered for 4 weeks by an osmotic minipump induced increased blood pressure, cardiac hypertrophy, cardiac fibrosis, and diastolic dysfunction [55]. This experimental model of cardiac hypertrophy is associated with increased cardiac mitochondrial protein carbonyl content and the frequency of mitochondrial DNA deletions, indicating oxidative damage to mitochondria [46]. The accumulation of mitochondrial oxidative damage activated mitophagy, which in turn increased signaling for mitochondrial biogenesis through activation of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) and its target genes. Our observation is consistent with the report that PGC-1α is transcriptionally upregulated by ROS [78]. mCAT, but not pCAT, were resistant to cardiac hypertrophy, fibrosis, and diastolic dysfunction induced by Angiotensin II [46]. This strongly supports a central role of mitochondrial ROS in Angiotensin II-induced cardiomyopathy [46]. Additional evidence from other laboratories show that disruption of p66Shc prevents Angiotensin II-induced LV hypertrophy and cardiomyocyte apoptosis as well as reducing oxidative damage in cardiac progenitor cells, cardiomyocytes, and endothelial cells in a diabetic mouse model [50,51,94]. Moreover, mice deficient in mitochondrial deacetylase SIRT3 displayed early age onset of hypertrophy associated with fibrosis, age-dependent increase in mitochondrial swelling due to increased mPTP opening, increased mortality after transverse aortic constriction [52].
As noted above, Polgm/m mice have increased mitochondrial DNA mutations and develop heart failure at middle age or at young age when challenged with Angiotensin II, both of which are attenuated by mCAT [43,46]. This suggests that primary damage to mitochondrial DNA contributes directly to the phenotype of systolic heart failure, through increased mt ROS. Therefore, the protective effects of mCAT expression in Ang-induced cardiac hypertrophy and Gαq-induced heart failure provide direct evidence that amplification of ROS within mitochondria is a key mediator in these disease models [46]. Using the transverse-aortic constriction (TAC) mouse model, we further show that TAC-induced heart failure is associated with remodeling of the mitochondrial proteome, including decreased abundance of proteins involved in fatty acid metabolism and increased abundance of proteins in glycolysis, apoptosis, mitochondrial unfolded protein response, and proteolysis. Overexpression of mCAT mitigates the phenotype of heart failure, better preserves proteins involved in fatty acid metabolism, and attenuates the increases in apoptotic and proteolytic enzymes [95]. Thus, breaking the ROS vicious cycle within mitochondria by mCAT is effective in attenuating both cardiac hypertrophy and failure (Figure 2). In a highly parallel manner we also demonstrated that the mitochondrial protective peptide SS31 attenuates cardiac hypertrophy and diastolic dysfunction induced by chronic Angiotensin II, and the heart failure phenotypes induced by overexpression of Gαq or transverse aortic constriction (See section Mitochondrial protective strategies as potential therapeutics for aging-related diseases). Furthermore, SS-31 has also been shown to prevent hypoxia-reoxygenation induced apoptosis in renal tubular epithelial cell by downregulation of p66Shc[96].
Ischemic-reperfusion (I/R) injury often occurs during acute myocardial infarction, either due to spontaneous recanalization of the occluded artery or as a result of a reperfusion therapy. ROS are well known to be primary mediators in IR injury. ROS begin to accumulate during ischemia [97], causing mitochondrial respiratory complex dysfunction, which leads to a burst of ROS after reperfusion. Furthermore, post-ischemic reperfusion is associated with ROS accumulation, acidic pH, and a rise in [], conditions which have been shown to open the mPTP, which in turn triggers more mitochondrial ROS generation. This is one of the mechanism involved in mitochondrial ROS-induced ROS release [98] (Figure 2).
The aged myocardium has less tolerance to ischemia and hemodynamic stress than the young myocardium [99]. Aged cardiomyocytes have a lower threshold for ROS induced ROS release and increased susceptibility to mPTP opening [100]. Ischemic preconditioning is also impaired in the aged myocardium (reviewed by [100]). This loss of endogenous protective mechanisms of ischemic preconditioning in the aged heart might be due to a decrease in mitochondrial heat shock protein-70 [101], reduced nitric oxide bioavailability [102], damaged mitochondria that are vulnerable to stress, and diminished PKC translocation into mitochondria, all of which are required for the protective effect of ischemic preconditioning [103,104]. Cardiac aging and various models of cardiomyopathy in the context of mitochondrial ROS are summarized in Table 1.
Skeletal muscle aging
Sarcopenia is the loss of skeletal muscle mass and function with age. Sarcopenia is an important public health concern due to its role in exercise intolerance, increased morbidity, and loss of independence in the elderly [105-108]. This loss of independence is due to an inability to perform activities of daily living that require sustained muscle power, such as walking, dressing, and showering as well as an increased risk of falling [109]. The resulting increased rates of nursing home placement and hospitalization make the loss of skeletal muscle function with age a growing public health crisis in terms of both quality of life and economic costs to society. Janssen et al. [110] estimated these costs at $18 billion dollars in 2001 and predicted that a 10% reduction in sarcopenia prevalence would lead to a savings of $1.4 billion in healthcare costs (adjusted to 2010 dollars) [110].
Skeletal muscle, like heart, relies on mitochondria to meet the majority of the ATP demands for sustained muscle contraction. Mitochondrial function in skeletal muscle is very dynamic where the metabolic rate can vary by at least an order of magnitude during rest to work transitions, as well as varying with nutritional state. One consequence of this variation in mitochondrial function is that periods of increased mitochondrial ROS production are a normal part of the physiology of skeletal muscle [62,111]. Skeletal muscles also produce significant ROS from non-mitochondrial sources, primarily sarcolemmal NAD(P)H oxidases [112], that can also contribute to increased cellular and mitochondrial oxidative stress. These transient increases in oxidative stress modify muscle function and may play an important role in the beneficial adaptations to exercise training [36,113]. However, mitochondria in aged skeletal muscle have an increased capacity to produce H2O2 when measured under ex vivo conditions [54]. This increased mitochondrial oxidative stress can control mitochondrial function both in vivo[114,115] and ex vivo[116,117]. Inducing a mild oxidative stress in adult mice for 24 h using low doses of paraquat recapitulates the reduced mitochondrial coupling (P/O) and depression of skeletal muscle metabolism [114,115] observed in vivo in aged skeletal muscle in both mice [118] and humans [119]. This same paraquat treatment in old mice led to decreases in maximal mitochondrial ATP production (ATPmax), in addition to further decreases in P/O and resting metabolism [115]. This increased sensitivity is consistent with a decline in the ability of the aged skeletal muscle to buffer transient increases in oxidative stress.
Further support for a contribution of mitochondrial oxidative stress in age-related skeletal muscle dysfunction comes from experiments using the mitochondrial targeted peptide SS-31. SS-31 accumulates in the mitochondria by associating with the inner mitochondrial membrane [61] and reduces mitochondrial H2O2 production [54,62]. One hour after treatment with SS-31 age-related declines in mitochondrial P/O, ATPmax, and skeletal muscle metabolism were reversed (Figure 5) [54] and the skeletal muscle glutathione redox state was more reduced [120]. These metabolic changes were associated with improved fatigue resistance of the tibialis anterior muscle in situ and increased endurance capacity in the aged mice.
Figure 5.

Mitochondrial targeted SS-31 improves skeletal muscle function.In vivo mitochondrial coupling ratio (P/O) (A) and maximum mitochondrial ATP production (B) in the hindlimb muscles of aged mice were both increased 1 h after treatment with SS-31. In situ fatigue resistance in the aged mice was also increased 1 h after SS-31 treatment (C). Eight days of daily treatment with SS-31 led to increased endurance capacity in the aged mice (D) as well. Means ± SEM. n = 5-7 per group. **P <0.01 relative to age-matched control. ##P <0.01 relative to young control. Young - 5 months old; Old - 27 months old. Modified from Siegel et al. [54].
Genetic manipulation of mitochondrial antioxidants also supports a role for mitochondrial oxidative stress in controlling skeletal muscle function and metabolism. Deficiency of MnSOD (mitochondrial specific isoform of superoxide dismutase) in type IIB muscle fibers leads to an increase in mitochondrial oxidative stress and dysfunction in fast-twitch mouse muscles. Mitochondria from fast-twitch muscles in these mice had significantly reduced aconitase and succinate dehydrogenase (complex II of the electron transport chain) activities and increased capacity for superoxide production resulting in elevated F2-isoprostanes [121]. Both fatigue resistance in isolated muscles and whole body endurance performance were also decreased in the MnSOD deficient mice. Interestingly, MnSOD deficiency did not lead to an increase in muscle atrophy or change in maximal force production in aged mice. Conversely, mCAT preserved mitochondrial function and insulin sensitivity in skeletal muscle of aged mice [49], while vector delivery of mCAT into embryos led to an increase in exercise performance in 3-month-old mice [122]. However, the ectopic expression of mCAT had no effect on the contractility and fatigue resistance in isolated extensor digitorum longus muscle. The lack of effect in the isolated muscle may indicate that the increased exercise tolerance in this study was due to improved cardiac function as described above. Alternatively, the mosaic expression of mCAT in the skeletal muscles may have limited its effect on ex vivo skeletal muscle performance.
The current evidence strongly supports an important role of mitochondrial oxidative stress in the decline in skeletal muscle function with age, while its role in skeletal muscle atrophy with age is still controversial. The strongest evidence in support of a role for oxidative stress in age-related muscle atrophy comes from mice lacking CuZnSOD (SOD1). CuZnSOD is found in both the cytosol and the inner membrane space of the mitochondria. The absence of CuZnSOD leads to an accumulation of peroxinitrite and oxidative damage [123], increased mitochondrial ROS production, increased sensitivity to apoptotic loss of myonuclei, and mitochondrial dysfunction associated with a premature loss of skeletal muscle mass in aging mice [12]. Muscle fibers from CuZnSOD-/- knockout mice accumulated mitochondria around the neuromuscular junction, showed a loss of motor units and disruption of neuromuscular junctions. However, muscle specific knockout of CuZnSOD did not result in muscle atrophy, increased oxidative stress, nor mitochondrial dysfunction [124], but did have a loss of specific force throughout life. The lack of atrophy in the skeletal muscle specific deficiencies in CuZnSOD and MnSOD knockouts led these authors to suggest that increased oxidative stress in the myofibers are not primary cause of muscle atrophy with age. Instead they suggest that increased oxidative stress in the motorneurons leading to denervation may be the primary driving force behind loss of muscle mass. This conclusion is supported by the observation that direct stimulation of skeletal muscles in the CuZnSOD-/- mice leads to significant increases in force production over that achieved by nerve stimulation [124]. This result suggests that force is limited not by the muscle itself, but by the ability of the motorneuron to maximally stimulate the available myofibers. Thus, the data from the CuZnSOD-/- mice suggest an important role of increased oxidative stress in age-related muscle atrophy, although it remains unclear whether oxidative stress originating in the myofibers or the motorneurons are the primary drivers of this process. This conclusion is supported by recent work demonstrating that treatment with SS-31 during hind limb unloading ameliorated muscle atrophy and mitochondrial dysfunction [125].
There are multiple ways in which an increase in oxidative stress can affect skeletal mitochondrial and contractile function. Work to date has primarily focused on the accumulation of oxidative damage to proteins, lipids, and DNA. Most studies find a clear accumulation of oxidatively damaged macromolecules with age in skeletal muscle and most other tissues. However, oxidation of the mitochondrial and cellular redox environment can also exert control over cellular function through post-translational modification of proteins (Figure 1). Glutathionylation is a key redox dependent post-translational modification in the mitochondria. Increased oxidative stress has been found to lead to increased glutathionylation and inhibition of activity of electron transport chain proteins, F1F0 ATPase [126] and complex I [127], and of TCA cycle proteins, succinyl-CoA-transferase [126] and α-ketoglutarate dehydrogenase [128]. In addition recent evidence indicates that glutathionylation of UCP3 regulates proton leak under conditions of acute oxidative stress in skeletal muscle [129] resulting in increased proton leak and reduced P/O.
Redox modification of proteins can also affect contractile function. The skeletal muscle ryanodine receptor 1 (RyR1) in aged rats is oxidized by cysteine nitrosylation [130]. This leads to a loss of RyR1-calstabin1 interaction, destabilization of the channel and increased calcium leak from the SR, which causes a loss of specific force and reduced exercise tolerance with age. Stabilizing the channel and preventing calcium leak rescued force production and exercise tolerance. As pointed out by the authors, leaky RyR would lead to elevated cytosolic calcium and increased calcium loading by the mitochondria, and an elevation of mitochondrial ROS production. This could then lead to a feed-forward mechanism further exacerbating skeletal muscle dysfunction. Thus, oxidation of the cellular redox status in aged muscle [131] can contribute to energetic and contractile deficits through both reversible and post-translational modification of proteins.
Neurodegenerative disease
Old age is associated with progressive decline in the functional performance of the nervous system. This intrinsic nervous system aging includes slowed reaction times, degeneration of sensory and motor function, and a decline in cognitive performance. In addition to intrinsic nervous system aging, several neurodegenerative diseases demonstrate strong age-related onset including the highly prevalent Alzheimer’s disease (AD) and Parkinson’s disease (PD), among others.
Age-related sensorineural hearing loss
Age-related sensorineural hearing loss or presbycusis is gradual loss of hearing with aging. The prevalence in the elderly is estimated to be 30% to 35% of people aged 65 to 75 years and 40% to 50% of people aged older than 75 years [132]. The sensorineural hearing loss is usually more severe for high pitched sound, which eventually leads to difficulty in understanding speech. The pathology is characterized by age-dependent loss of sensory hair cells, spiral ganglion neurons, and stria vascularis cells in the inner ear cochlea. Someya et al. [44] reported that mice with the deletion of the mitochondrial pro-apoptotic gene Bak attenuated age-related apoptotic cell deaths and hence prevented presbycusis. While oxidative stress induced Bak expression in primary cochlear cells, mCAT suppressed Bak expression, reduced cell death and subsequently prevented presbycusis. These findings suggest a central role of mitochondrial ROS induced apoptotic pathway in presbycusis [44]. They further demonstrate that caloric restriction prevents presbycusis via reduction of oxidative damage by mitochondrial deacetylase SIRT3. In response to CR, SIRT3 directly deacetylates and activates mitochondrial isocitrate dehydrogenase 2, leading to increased NADPH levels and an increased ratio of reduced-to-oxidized glutathione in mitochondria and thereby enhancing the mitochondrial glutathione antioxidant defense system [53].
Alzheimer’s disease
AD is the most prevalent neurodegenerative disease, affecting approximately 5 million Americans. The clinical presentation of AD is primarily memory impairment and dementia. The early memory deficit in AD is often described as ‘recent memory impairment’ (for example, inability to recall a couple of words after a few minutes of distraction) [133]. Deficits in other cognitive functions may appear later after the development of memory impairment.
There are two principle pathologic lesions in AD: neurofibrillary tangle (NFT) and the amyloid plaque [134]. The NFTs consist of abnormal accumulations of abnormally phosphorylated tau within the cytoplasm of certain neurons. The amyloid plaques contain β-amyloid peptide (Aβ), which arises through proteolytic processing of amyloid precursor protein (APP) by β-secretase and γ-secretase (presenilin1/2). Each of these lesions has a characteristic distribution. The hierarchical pattern of NFTs among brain regions is so consistent that a staging scheme based on the topography of these lesions has been widely used [135]. The majority of AD cases are sporadic and occur very late in life, however, less than 1% of AD is familial AD cases, which have an early onset and are inherited in an autosomal dominant manner. Genes implicated in the early onset AD include β-amyloid precursor protein (APP), presenilin 1, and presenilin 2 [136]. Mutation of the APP gene affects the cleavage of APP by β-secretase or γ-secretase to generate various forms of Aβ. The Aβ peptides have a tendency to form oligomer aggregates and become toxic, especially the long form, Aβ1-42. Presenilins are integral membrane proteins that function as the proteolytic components of γ-secretase. Mutations of presenilins result in increased production of Aβ1-42.
Several lines of evidence have shown the central roles of mitochondria in AD (see reviews [137]). Both APP and presenilin have been isolated in mitochondrial fraction [138,139]. Moreover, Aβ is imported into the mitochondrial cristae through translocase of outer mitochondrial membrane complex (TOMM) [140]. Increased mitochondrial oxidative stress and damage to mitochondrial structural components and enzyme complexes are well documented in early AD [141-144]. One of the mechanisms involves Aβ, which inhibits mitochondrial function by inhibition of electron transport chain activity, especially complex III and IV, that further leads to increased ROS production, decreased ATP production, and facilitation of cytochrome c release [143,145,146]. Additional insults to mitochondria include altered Ca2+ homeostasis [147], increased mitochondrial DNA mutations, and deletions [141]. Furthermore, alterations of mitochondrial dynamics have been implicated in AD [148-150]. It has been shown that S-nitrosylation of Drp1 (a mitochondrial fission protein) mediates β-Amyloid-related mitochondrial fission and neuronal injury [148]. Increased production of Aβ interacts with Drp1, which is a critical factor in mitochondrial fragmentation, abnormal mitochondrial dynamics, and synaptic damage [150].
More direct evidence of the role of mitochondrial oxidative stress in AD is demonstrated by studies using mCAT mice or mitochondrial targeted antioxidants. Mao et al. [47] showed that mCAT decreases amyloid-beta (Aβ) toxicity and oxidative injury, and extends the lifespan of Aβ precursor protein (PP) overexpressing mice. This data provides direct evidence that mitochondrial oxidative stress plays a primary role in AD pathology, and supports the possibility that mitochondria-targeted antioxidants might be an effective therapeutic approach to treat patients with AD.
AD is associated with neuronal cell death, loss of synapses, as well as mitochondrial abnormalities. Incubation of N2a cells with Aβ led to reduced neurite outgrowth, lower cell viability, mitochondrial dysfunction, and fragmentation and loss of ATP, all of which were partially protected by simultaneous incubation with either SS-31 or MitoQ [125]. Primary neurons from the AβPP mouse model showed increased H2O2 production, reduced cytochrome oxidase activity, and decreased ATP levels [151]. There was also decreased anterograde mitochondrial movement, increased mitochondrial fission, and decreased fusion. Treatment with SS-31 restored mitochondrial transport and synaptic viability, and decreased the percentage of defective mitochondria [152].
Parkinson’s disease
PD, the second most common neurodegenerative disorder, is characterized by defects in motor functions, manifested as resting tremor, bradykinesia, rigidity, and postural instability. The hallmark pathology of PD is a gradual loss of pigmented dopaminergic neurons in the substantia nigra pars compacta, and accumulation of Lewy bodies in catecholaminergic neurons of the brainstem in the substantia nigra and locus ceruleus. Lewy bodies are abnormal aggregates of proteins composed predominantly of α-synuclein and ubiquitin.
There is substantial support for the central role of mitochondria in the pathogenesis of PD. A few genetic loci have been mapped in rare familial PD cases, and are sequentially named PARK1 to PARK11 (review in [153]). Several genes associated with familial PD have been identified in these loci, and the majority of them are related to mitochondria. PARK1 gene encodes α-synuclein, which has been implicated in the maintenance of mitochondrial membranes [154]. Increased amount of α-synuclein binding to mitochondria inhibits mitochondrial fusion and thereby triggers PD pathology, which can be rescued by PINK1, Parkin, and DJ-1 [154]. PARK8 encodes the leucine-rich repeat kinase 2 (LRRK2) and its mutations have been associated with mitochondrial oxidative phosphorylation dysfunction [155]. LRRK2 regulates mitochondrial dynamics by a direct interaction with DLP1, a mitochondrial fission protein [156]. PARK7 encodes DJ-1, the mutation of which is associated with complex I defects, increased mitochondrial ROS, reduced mitochondrial membrane potential, altered mitochondrial morphology, and dynamics [157-159]. PARK2 and PARK6 encode parkin and PTEN-induced kinase 1 (PINK1), which is involved in mitochondrial dynamics (fusion/fission) and turnover by mitophagy [160-162].
Several rodent models have been used to recapitulate pathology and pathophysiology of PD, including mice with genetic manipulation of many of the genes mentioned above and rodents treated with environmental toxins (see review [163]). Of note, the majority of the environmental toxins that recapitulate PD are mitochondrial complex I inhibitors, such as 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP), paraquat, or rotenone. Inhibition of complex I is associated with impaired mitochondrial respiration and leads to increased mitochondrial ROS production, increased oxidative damage to proteins, lipids, and DNA, which may further activate mitochondrial-dependent apoptotic pathways and cause dopaminergic neuronal cell death.
Direct evidence of the role of mitochondrial ROS in PD was shown by the protective effect of mCAT mouse brains against MPTP induced mitochondrial ROS production and subsequent dopaminergic neuron degeneration in substantia nigra pars compacta [48]. In contrast, harlequin mice with partial deficiency of apoptosis inducing factor, which is required for maintenance of complex I oxidative phosphorylation activity, are more susceptible to MPTP-induced dopaminergic neuronal cell death. The increased sensitivity of harlequin mice to MPTP is reversed by the antioxidant tempol (superoxide dismutase-mimetic) [48]. SS-31 was also shown to dose-dependently protect dopaminergic neurons and preserve striatal dopamine levels in mice treated with MPTP, with complete protection observed at 5 mg/kg [65]. Furthermore, SS-31 prevented MPP+-induced inhibition of oxygen consumption, ATP production, and mitochondrial swelling in isolated mitochondria. MitoQ was also reported to be protective against MPTP toxicity [164].
Insulin resistance, diabetes, and its complication
Growing evidence has implicated the involvement of oxidative stress in insulin resistance and the pathogenesis of diabetes. Hyperglycemia is associated with increased ROS production from glucose autoxidation, advanced glycosylation end-products (AGEs) formation, polyol pathway, and ROS-producing enzymes including NADPH oxidase [165]. Nishikawa et al. showed that, under hyperglycemia, increased glycolysis generates excess pyruvate, which overloads the mitochondria and leads to superoxide generation from the electron transport chain [166]. This mitochondrial superoxide production triggers the feed-forward cycle of mitochondrial ROS production in diabetes.
The involvement of mitochondrial oxidative stress in muscle insulin resistance has been demonstrated by the protection of mCAT mice in age-related reductions in mitochondrial function and lipid-induced insulin resistance in skeletal muscle [49]. This protection is associated with reduced mitochondrial oxidative damage and preserved mitochondrial respiration in muscle of old mCAT mice. In another study, Anderson and colleagues showed both mCAT mice and WT mice treated with SS-31 have reduced high-fat diet-induced mitochondrial H2O2 emission and showed preserved insulin sensitivity in skeletal muscle, further support of the mitochondrial ROS in muscle insulin resistance [62]. Thus mitochondrial oxidative stress plays an important role in the initiation of insulin resistance.
Diabetes is linked with multiple cardiovascular complications including accelerated atherosclerosis, augmented ischemic injury post-myocardial infarction, diabetic retinopathy, and nephropathy. In a mouse model of diabetes induced by streptozotocin injection, retina of diabetic mice had two-fold increase in superoxide levels, 40% reduction in GSH levels and 20% reduction in complex III activity, and increased mitochondrial membrane permeability. All these changes were attenuated in mice overexpressing MnSOD, which also experience reduced vascular histopathology, indicating the role of mitochondrial oxidative stress in retinopathy [167]. Cardiac mitochondria in diabetic mice also displayed increased mitochondrial membrane permeability, which has been shown to contribute to the increased propensity for I/R injury in diabetic hearts. Daily intraperitoneal injection of MTP-131 (analogous to SS-31) for 4 days partially reversed increased mPTP opening in diabetic heart mitochondrial. In the same study, Sloan et al. showed that the administration of MTP-131 peptide during reperfusion reduced I/R injury in diabetic hearts, supporting the role of mitochondrial ROS and mPTP opening in I/R injury in diabetic cardiomyopathy [168].
Despite the evidence supporting the role of mitochondrial oxidative stress in experimental models of diabetes, there are mixed results on the protective role of antioxidant treatments in diabetes or its complications from clinical trials [165]. For instance, the HOPE trial showed vitamin E treatment for 4.5 years fails to confer benefit in cardiovascular outcomes and nephropathy [169]. Results of SECURE trial and PPP trial also failed to demonstrate any protective effects with vitamin E treatment [165]. On the other hand, clinical trials on α-lipoic acid have shown more promising results than vitamin E trials. Multiple studies with α-lipoic acid, including ALADIN study, DEKAN study, and SYDNEY trial, have demonstrated its protective effect on diabetic neuropathy [165,170-174].
It is possible that increased mitochondrial ROS may play a larger role in the development of insulin resistance before the onset of chronic hyperglycemia. A recent study using the streptozotocin-induced mouse model of type 1 diabetes actually found reduced mitochondrial function and superoxide production in diabetic kidneys and suggested that this may be due to reduced mitochondrial biogenesis caused by lower PGC1α expression [175]. The investigators postulated that reduced mitochondrial biogenesis led to reduction in activity of AMPK, the master energy sensor. Activation of AMPK restored mitochondrial function and superoxide production, and this was associated with a beneficial reduction in renal pathology. Thus chronic mitochondrial oxidative stress may actually result in reduced mitochondrial function in the later stages of diabetes, and that restoration of mitochondrial structure and function may be necessary to prevent the decline in organ function.
It was recently reported that SS-31 significantly reduced diabetic retinopathy [63]. Daily treatment with SS-31 over 4 months in the rat streptozotocin model significantly prevented the loss of mitochondrial cristae and mitochondrial swelling in retinal epithelial cells. SS-31 also protected the inner blood-retinal barrier, and this was due to preservation of tight junctions in the retinal blood vessels, suggesting adequate ATP production is required to maintain the cytoskeleton of the endothelial cells. Oxidative markers such as 8-OHdG and nitrotyrosine were significantly reduced in the SS-31-treated diabetic animals. The upregulation of VEGFR2 was also significantly attenuated, and this suggests that SS-31 can reduce neovascularization. Interestingly, SS-31 had no effect on blood glucose, but clearly prevented the effects of hyperglycemia on retinal structure and function.
Age-related cancer
Mitochondrial ROS leads to oxidative damage in nucleic acids and proteins and has been implicated in carcinogenesis. A recent study demonstrates that loss of mitochondrial cytochrome oxidase is associated with the development of colonic dysplasia (precancerous state) in patients with ulcerative colitis [176]. Direct evidence for the role of mitochondrial ROS in age-related cancer is shown by the effect of mCAT to reduce the non-hematopoietic tumor burden in a mouse end-of-life pathology study [45]. The mCAT expression has also been shown to be protective in an experimental model of metastatic breast cancer (PyMT mice). The mCAT mice displayed reduced invasive grade of primary breast tumor and have 30% less pulmonary metastasis incidence. Both tumor cells and lung fibroblasts in mCAT expressing PyMT mice have reduced intracellular ROS and increased resistance to H2O2-induced cell death, which may confer the protective effects in mCAT mice [177].
Ataxia telangiectasia mutated (ATM) kinase plays a central role in the DNA-damage response and redox sensing by the phosphorylation of many key proteins that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair, or apoptosis. In addition to severe ataxia due to cerebellar degeneration, ataxia telangiectasia patients also have increased risk of lymphomas and leukemias, as well as immune defect [178]. ATM null mice (ATM-/-) develop thymic lymphomas, despite very mild neurodegenerative phenotypes. Reducing mitochondrial ROS by mCAT in ATM-/- mice reduced propensity to develop thymic lymphoma, improved bone marrow hematopoiesis, and macrophage differentiation in vitro, and partially rescued memory T-cell development [178].
Mitochondrial protective strategies as potential therapeutics for aging-related diseases
Meta-analyses of several clinical trials using antioxidant supplement have shown largely disappointing results [13]. With strong evidence of the central role of mitochondrial oxidative stress and damage in several age-related diseases as revealed by the mCAT model, there have been several attempts to develop mitochondria-targeted antioxidants. The most common approach used for delivering compounds into mitochondria have relied on the conjugation of known redox agents to triphenylphosphonium ion (TPP+) to take advantage of the potential gradient across the inner mitochondrial membrane. The second major category is aromatic-cationic tetrapeptides that selectively target the inner mitochondrial membrane without relying on mitochondrial potential.
TPP+ conjugated antioxidants
The mitochondrial inner membrane has a negative potential gradient (-150-180 mV) that is generated as a result of the release of protons from the mitochondrial matrix to the intermembrane space. The negative potential serves as a basis for the use of lipophilic cations to deliver redox agents into the mitochondrial matrix. This method can potentially result in 100- to 1,000-fold accumulation of drugs within the mitochondrial matrix [179]. TPP+ has been conjugated to coenzyme Q (MitoQ) and plastoquinone (SkQ1) [30,31]. By preferentially accumulating in the mitochondrial matrix, these TPP+-conjugated antioxidants are more potent than their lipophilic counterparts in reducing intracellular ROS, preserving reduced thiols, and reducing oxidative cell death [180,181], This lipophilic cation approach has also been used to generate other mitochondrial-targeted antioxidants to decrease superoxide (MitoSOD), hydrogen peroxide (MitoPeroxidase), ferrous iron (MitoTEMPO), and lipid peroxidation (MitoE2) (see review [182]).
MitoQ had been shown to improve pathology associated with antioxidant deficiency and prolong lifespan of SOD-deficient flies, however, it failed to show lifespan extension in normal WT flies [183]. Indeed, there was a dose-dependent increase in toxicity of MitoQ in flies [183]. MitoQ and SkQ1 have been shown to be effective in reducing ischemia-reperfusion injury [68,184,185]. Dikalova et al. reported that MitoQ treatment for 8 weeks reduced systolic blood pressure and cardiac hypertrophy in spontaneous hypertensive rats [66,186]. The plausible mechanism of blood pressure lowering effect is the improved bioavailability of endothelial nitric oxide. There is evidence that MitoQ can protect against endotoxin-induced cardiac dysfunction [187]. As mentioned earlier, MitoQ was also found to be protective in animal models of neurodegenerative diseases such as AD and PD [164,188]. A series of papers reported that SkQ1 prolonged lifespan, reduced ischemia-reperfusion injury, inhibited tumor development, and returned vision to blind animals [30,67-69].
However, recent reports suggest that MitoQ can actually increase superoxide production at Complex I [189,190] and both MitoQ and SkQ were reported to inhibit mitochondrial bioenergetics [191,192]. Thus while these TPP+-conjugated antioxidants can reduce mitochondrial ROS, they may also reduce oxidative phosphorylation and ATP production.
MitoQ has been evaluated in two clinical trials. A small trial of MitoQ in 30 patients with hepatitis C revealed a significant reduction in alanine aminotransferase after 28 days of treatment [193]. However, a double-blind, placebo-controlled trial in patients with PD showed that MitoQ treatment over 12 months did not slow the progression of PD [194]. It is unclear whether clinical development of MitoQ is being continued at this time. On the other hand, SkQ1 eye drops appear to have been approved for dry eye and are available in Russia.
SS peptides
The Szeto-Schiller (SS) compounds are tetrapeptides with an alternating aromatic-cationic amino acids motif, which was serendipitously found to preferentially concentrate in the inner mitochondrial membrane greater than 1,000-fold compared with the cytosolic concentration [68,190,195]. Although these peptides carry 3+ net charges, the mitochondrial uptake of these SS peptides is not dependent on mitochondrial potential, as they are also concentrated even in the depolarized mitochondria [190,195]. SS-31 (H-D-Arg-Dmt-Lys-Phe-NH2) was originally thought to exert its beneficial effect solely by the free radical scavenging activity of dimethyl tyrosine [66]. SS-31 is able to scavenge H2O2 hydroxyl radical and peroxynitrite in vitro in a dose-dependent manner [195,196].
A recent study revealed that in addition to this ROS scavenging capacity, SS-31 selectively binds to cardiolipin on the inner mitochondrial membrane via both electrostatic and hydrophobic interactions [61]. Cardiolipin is a phospholipid that is uniquely expressed on the inner mitochondrial membrane and plays an important role in the maintenance of cristae structure and formation of super complexes to facilitate electron transfer in the electron transport chain [197-199]. Cardiolipin also plays a role in anchoring cytochrome c to the inner mitochondrial membrane and facilitates electron transfer from complex III to complex IV [200,201]. Although electrostatic interaction with cardiolipin is important for cytochrome c to function as an electron carrier, hydrophobic interaction with cardiolipin tends to cause cytochrome c to unfold and dramatically enhances its peroxidase activity causing cardiolipin peroxidation [202-204]. The oxidation of cardiolipin disturbs cardiolipin microdomains on the inner mitochondrial membrane and causes the loss of cristae curvature and super complex formation. Disruption of super complex formation not only reduces oxidative phosphorylation but also increases ROS formation by complex I [205].
We recently showed that the binding of SS-31 to cardiolipin alters the interaction of cardiolipin with cytochrome c, and favors its electron carrier function while inhibiting peroxidase activity by protecting the Met80-heme ligand [32,206]. By promoting cytochrome c reduction, SS-31 increases electron flux in mitochondria and accelerates ATP production [206]. At the same time, SS-31 inhibits ROS generation and inhibits cytochrome c peroxidase activity, thereby preventing cardiolipin peroxidation and loss of cristae membranes [206]. Thus, SS-31 is a multifunctional mitoprotective compound that acts by promoting bioenergetics, reducing ROS production, scavenging excess ROS, inhibiting cardiolipin peroxidation, and preserving mitochondrial structure.
These unique properties of SS-31 are particularly effective in minimizing ischemia-reperfusion injury. After prolonged ischemia, the hydrophobic interaction between cardiolipin and cytochrome c is enhanced by low ATP concentration [207,208] and this would inhibit mitochondrial respiration at a time when ATP synthesis is necessary for survival. SS-31 is able to increase oxygen consumption and ATP synthesis under these conditions, thus accelerating ATP production upon return of blood flow to minimize cell death and promote organ recovery. By inhibiting cardiolipin peroxidation during reperfusion, SS-31 also preserves mitochondrial cristae and maintains ATP synthesis after ischemia. Numerous preclinical studies support these claims. Studies in models of renal ischemia reperfusion have demonstrated that SS-31 protects mitochondrial cristae architecture and prevents swelling during ischemia and reperfusion [61,209]. This results in more rapid ATP production upon reperfusion and preservation of the cytoskeletal integrity of the epithelial cells, and amelioration of acute kidney injury [61,209].
SS-31 has also been shown to reduce cardiac ischemia reperfusion injury and reperfusion arrhythmia and better preserve myocardial function in various infarct models [59,60,64,196]. SS-31 reduced infarct size in rabbits and sheep after coronary artery ligation, attenuated the extent of no-reflow in rabbits, and reduced infarct size in isolated perfused guinea pig hearts. SS-31 also reduced infarct size in a mouse model of cerebral ischemia and attenuated glutathione depletion when administered at the onset of reperfusion [210].
In addition to ischemia-reperfusion injury, SS-31 has shown impressive effects in preclinical models of heart failure. SS-31 ameliorated Angiotensin-II induced cardiac hypertrophy and diastolic dysfunction, as well as Gαq overexpression-induced heart failure, despite the absence of a blood pressure lowering effect [55]. SS-31 also reduced systolic heart failure in a pressure-overload model of transverse aortic constriction (TAC). Ultrastructural studies confirmed that SS-31 protected cardiac mitochondria in the TAC model and proteomic analyses showed that SS-31 attenuated the majority of the changes in mitochondrial and non-mitochondrial proteins [56]. By protecting mitochondrial function and bioenergetics in the heart, SS-31 prevented myocardial remodeling and fibrosis. The efficacy of SS-31 in combating heart failure has been confirmed in a post-myocardial infarction canine heart failure model, Sabbah et al. demonstrates that short-term administration of Bendavia for 2 h significantly increased ejection fraction, stroke volume, cardiac output, and LV contractility index (dP/dt) [57]. These findings suggest that the improvement of LV function is likely the result of improved cardiac energetics. Long-term administration for 3 months significantly improved ejection fraction and reduced LV end-diastolic pressure [58].
SS-31 has also been shown to be beneficial in many other models of age-associated diseases, including PD [65], AD [152], skeletal muscle aging, disuse skeletal muscle atrophy [210], [54] insulin resistance [62], and diabetic complications. Some of these studies were mentioned in the previous sections (see section Mitochondrial oxidative stress in healthspan above), and an extensive review of these studies was published recently [32].
Given the very promising preclinical efficacy data, SS-31 entered into clinical trials using a clinical formulation named Bendavia [32]. Several Phase I studies have assessed the safety, tolerability and pharmacokinetics of Bendavia in healthy male and female human subjects with intravenous and oral dosing. The highly predictable pharmacokinetics and safety profile of Bendavia have led to Phase II trials in patients. The first multinational phase II study is focused on cardiac ischemia-reperfusion injury for patients experiencing ST-elevation myocardial infarction [211]. A second ongoing phase II trial is for treatment of acute kidney injury in hypertension. A third phase II trial is planned for the treatment of congestive heart failure.
These clinical studies are generally designed to address the efficacy of SS-31 in the treatment of age-associated cardiorenal diseases. It will eventually be important to also establish whether these mitochondria-targeted antioxidants can delay aging and other age-related degenerative diseases.
Conclusion
Substantial evidence supports the central role of mitochondrial oxidative stress in aging and healthspan. Despite the disappointing outcomes of non-targeted antioxidants in clinical trials, there is growing evidence for the beneficial effects of mitochondrial-targeted antioxidants in aging and age-related diseases. Genetic and pharmacological approaches reducing mitochondrial oxidative stress (either by direct antioxidant or indirectly through preservation of mitochondrial structure and function) attenuate the phenotypes of cardiac aging, age-related cardiovascular diseases, skeletal muscle aging, neurodegenerative diseases, diabetes, and cancer various animal models (summarized in Table 1). Moreover, based on promising preliminary results in small and large mammals, mitochondrial-targeted antioxidants have moved into clinical trials. Further studies are necessary to investigate many of the remaining questions in this field, while examining the potential application of mitochondrial targeted therapeutics in the treatment or prevention of specific diseases as well as improved healthspan in general.
Competing interests
HH Szeto is the inventor of SS-31 and the Cornell Research Foundation (CRF) holds several patents covering the SS peptides and a patent application has been filed for the findings described in this article, with HH Szeto, PS Rabinovitch and DF Dai as inventors. CRF has licensed the SS peptide technology for further research and development to a commercial enterprise in which CRF and HH Szeto have financial interests.
Authors’ contributions
DFD and YAC drafted and revised the manuscript. DM helped to draft and made critical revision to the manuscript. HHS and PSR made critical revision to the manuscript. All authors read and approved the final manuscript.
Contributor Information
Dao-Fu Dai, Email: ddaofu@uw.edu.
Ying Ann Chiao, Email: annchiao@uw.edu.
David J Marcinek, Email: dmarc@uw.edu.
Hazel H Szeto, Email: hhszeto@med.cornell.edu.
Peter S Rabinovitch, Email: petersr@u.washington.edu.
Acknowledgements
This work was supported by grants from the American Federation for Aging Research, the Ellison Medical Foundation and the NIH (AT006526, AG038747, AG11370, HL101186, AG013280, AG042637, AG001751). YAC is supported by an Ellison Medical Foundation/AFAR Postdoctoral Fellowship.
References
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med. 2012;52:539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labunskyy VM, Gladyshev VN. Role of reactive oxygen species-mediated signaling in aging. Antioxid Redox Signal. 2013;19:1362–1372. doi: 10.1089/ars.2012.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett RJ, Orr WC, Rahmandar JJ, Benes JJ, Radyuk SN, Klichko VI, Sohal RS. Overexpression of Mn-containing superoxide dismutase in transgenic Drosophila melanogaster. Arch Biochem Biophys. 1999;371:260–269. doi: 10.1006/abbi.1999.1460. [DOI] [PubMed] [Google Scholar]
- Orr WC, Sohal RS. Does overexpression of Cu, Zn-SOD extend life span in Drosophila melanogaster? Exp Gerontol. 2003;38:227–230. doi: 10.1016/S0531-5565(02)00263-2. [DOI] [PubMed] [Google Scholar]
- Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 2005;126:365–379. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Huang TT, Carlson EJ, Gillespie AM, Shi Y, Epstein CJ. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J Gerontol A Biol Sci Med Sci. 2000;55:B5–B9. doi: 10.1093/gerona/55.1.b5. [DOI] [PubMed] [Google Scholar]
- Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, Qi W, Liu Y, Liang H, Chaudhuri A, Ikeno Y, Epstein CJ, Van Remmen H, Richardson A. Overexpression of Mn superoxide dismutase does not increase life span in mice. J Gerontol A Biol Sci Med Sci. 2009;64:1114–1125. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009;8:73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A, Van Remmen H. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;346:f10. doi: 10.1136/bmj.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FJ, Shen L, Ji HF. Dietary intakes of vitamin E, vitamin C, and beta-carotene and risk of Alzheimer’s disease: a meta-analysis. J Alzheimers Dis. 2012;31:253–258. doi: 10.3233/JAD-2012-120349. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Anisimov VN, Egorov MV, Krasilshchikova MS, Lyamzaev KG, Manskikh VN, Moshkin MP, Novikov EA, Popovich IG, Rogovin KA, Shabalina IG, Shekarova ON, Skulachev MV, Titova TV, Vygodin VA, Vyssokikh MY, Yurova MN, Zabezhinsky MA, Skulachev VP. Effects of the mitochondria-targeted antioxidant SkQ1 on lifespan of rodents. Aging (Albany NY) 2011;3:1110–1119. doi: 10.18632/aging.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, Rizzuto R. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. 2007;315:659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Chang CM, Chi CW. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res Rev. 2010;Suppl 1:S47–S58. doi: 10.1016/j.arr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9:e1003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet. 2007;3:e24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, Filenko OF, Kalinina NI, Kapelko VI, Kolosova NG, Kopnin BP, Korshunova GA, Lichinitser MR, Obukhova LA, Pasyukova EG, Pisarenko OI, Roginsky VA, Ruuge EK, Senin II, Severina II, Skulachev MV, Spivak IM, Tashlitsky VN, Tkachuk VA, Vyssokikh MY, Yaguzhinsky LS, Zorov DB. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. 2009;1787:437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Szeto HH. First-in-class cardiolipin therapeutic to restore mitochondrial bioenergetics. Br J Pharmacol. 2013. doi:10.1111/bph.12461. [DOI] [PMC free article] [PubMed]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21:894–899. doi: 10.1016/j.ceb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mech Ageing Dev. 2010;131:536–543. doi: 10.1016/j.mad.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Abe K, Makino N, Anan FK. pH dependency of kinetic parameters and reaction mechanism of beef liver catalase. J Biochem. 1979;85:473–479. doi: 10.1093/oxfordjournals.jbchem.a132353. [DOI] [PubMed] [Google Scholar]
- Agar NS, Sadrzadeh SM, Hallaway PE, Eaton JW. Erythrocyte catalase. A somatic oxidant defense? J Clin Invest. 1986;77:319–321. doi: 10.1172/JCI112294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Xu J, Kondo K, Ding D, Salvi RJ, Yamasoba T, Rabinovitch PS, Weindruch R, Leeuwenburgh C, Tanokura M, Prolla TA. Age-related hearing loss in C57BL/6 J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc Natl Acad Sci U S A. 2009;106:19432–19437. doi: 10.1073/pnas.0908786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF, Martin GM, Rabinovitch PS, Ladiges WC. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:813–822. doi: 10.1093/gerona/63.8.813. [DOI] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH, Rabinovitch PS, Reddy PH. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21:2973–2990. doi: 10.1093/hmg/dds128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, Bove J, Dehay B, Jackson-Lewis V, Rabinovitch PS, Przedborski S, Vila M. Apoptosis-inducing factor deficiency sensitizes dopaminergic neurons to parkinsonian neurotoxins. Ann Neurol. 2010;68:184–192. doi: 10.1002/ana.22034. [DOI] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota M, LeCapitaine N, Hosoda T, Boni A, De Angelis A, Padin-Iruegas ME, Esposito G, Vitale S, Urbanek K, Casarsa C, Giorgio M, Luscher TF, Pelicci PG, Anversa P, Leri A, Kajstura J. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ Res. 2006;99:42–52. doi: 10.1161/01.RES.0000231289.63468.08. [DOI] [PubMed] [Google Scholar]
- Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P, Luscher TF, Cosentino F. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci U S A. 2007;104:5217–5222. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS, Marcinek DJ. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12:763–771. doi: 10.1111/acel.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCross MJ, Rabinovitch PS. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013;6:1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Wang M, Zhang K, Gupta RC, Rastogi S. Acute intravenous infusion of Bendavia (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular systolic function in dogs with advanced heart failure. Circulation. 2012;126 doi: 10.1161/CIRCHEARTFAILURE.115.002206. A15385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Wang M, Zhang K, Gupta RC, Rastogi S. Long-term therapy with Bendavia (MTP-131), a novel mitochondria-targeting peptide, increases myocardial ATP synthesis and improves left ventricular systolic function in dogs with chronic heart failure. J Am Coll Cardiol. 2013;61:E709. doi: 10.1016/S0735-1097(13)60709-3. [DOI] [Google Scholar]
- Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, Gorman JH 3rd, Sloan RC, Frasier CR, Watson CA, Bostian PA, Kypson AP, Brown DA. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW III, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Li X, Li M, Li J, Xiao W, Ma W, Chen X, Liang X, Tang S, Luo Y. Mitochondria-targeted antioxidant peptide SS31 protects the retinas of diabetic rats. Curr Mol Med. 2013;13:935–945. doi: 10.2174/15665240113139990049. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, Calingasan NY, Szeto HH, Beal MF. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98:1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- Anisimov VN, Bakeeva LE, Egormin PA, Filenko OF, Isakova EF, Manskikh VN, Mikhelson VM, Panteleeva AA, Pasyukova EG, Pilipenko DI, Piskunova TS, Popovich IG, Roshchina NV, Rybina OY, Saprunova VB, Samoylova TA, Semenchenko AV, Skulachev MV, Spivak IM, Tsybul’ko EA, Tyndyk ML, Vyssokikh MY, Yurova MN, Zabezhinsky MA, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 5. SkQ1 prolongs lifespan and prevents development of traits of senescence. Biochemistry (Mosc) 2008;73:1329–1342. doi: 10.1134/S0006297908120055. [DOI] [PubMed] [Google Scholar]
- Bakeeva LE, Barskov IV, Egorov MV, Isaev NK, Kapelko VI, Kazachenko AV, Kirpatovsky VI, Kozlovsky SV, Lakomkin VL, Levina SB, Pisarenko OI, Plotnikov EY, Saprunova VB, Serebryakova LI, Skulachev MV, Stelmashook EV, Studneva IM, Tskitshvili OV, Vasilyeva AK, Victorov IV, Zorov DB, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke) Biochemistry (Mosc) 2008;73:1288–1299. doi: 10.1134/S000629790812002X. [DOI] [PubMed] [Google Scholar]
- Neroev VV, Archipova MM, Bakeeva LE, Fursova A, Grigorian EN, Grishanova AY, Iomdina EN, Ivashchenko Zh N, Katargina LA, Khoroshilova-Maslova IP, Kilina OV, Kolosova NG, Kopenkin EP, Korshunov SS, Kovaleva NA, Novikova YP, Philippov PP, Pilipenko DI, Robustova OV, Saprunova VB, Senin II, Skulachev MV, Sotnikova LF, Stefanova NA, Tikhomirova NK, Tsapenko IV, Shchipanova AI, Zinovkin RA, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 4. Age-related eye disease. SkQ1 returns vision to blind animals. Biochemistry (Mosc) 2008;73:1317–1328. doi: 10.1134/S0006297908120043. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Larsson NG. Mitochondrial dysfunction as a cause of ageing. J Intern Med. 2008;263:167–178. doi: 10.1111/j.1365-2796.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- Terzioglu M, Larsson NG. Mitochondrial dysfunction in mammalian ageing. Novartis Found Symp. 2007;287:197–208. doi: 10.1002/9780470725207.ch14. discussion 208-113. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.CIR.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.CIR.0000048893.62841.F7. [DOI] [PubMed] [Google Scholar]
- Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- Goffart S, von Kleist-Retzow J-C, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 2004;64:198–207. doi: 10.1016/j.cardiores.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004;364:1786–1788. doi: 10.1016/S0140-6736(04)17402-3. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Marin-Garcia J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc Res. 2001;52:103–110. doi: 10.1016/S0008-6363(01)00368-6. [DOI] [PubMed] [Google Scholar]
- Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274. doi: 10.1016/S0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae I, Mitsunami K, Omura T, Yabe T, Tsutamoto T, Matsuo S, Takahashi M, Morikawa S, Inubushi T, Nakamura Y, Kinoshita M, Horie M. Proton magnetic resonance spectroscopy can detect creatine depletion associated with the progression of heart failure in cardiomyopathy. J Am Coll Cardiol. 2003;42:1587–1593. doi: 10.1016/j.jacc.2003.05.005. [DOI] [PubMed] [Google Scholar]
- Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.CIR.96.7.2190. [DOI] [PubMed] [Google Scholar]
- Gardin JM, Lauer MS. Left ventricular hypertrophy: the next treatable, silent killer? JAMA. 2004;292:2396–2398. doi: 10.1001/jama.292.19.2396. [DOI] [PubMed] [Google Scholar]
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/S0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. doi: 10.1161/01.RES.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45:438–444. doi: 10.1161/01.HYP.0000157169.27818.ae. [DOI] [PubMed] [Google Scholar]
- Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia P, Cosentino F, Schiavoni M, Huang Y, Perna E, Camici GG, Luscher TF, Volpe M. p66(Shc) protein, oxidative stress, and cardiovascular complications of diabetes: the missing link. J Mol Med (Berl) 2009;87:885–891. doi: 10.1007/s00109-009-0499-3. [DOI] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WY, Han S, Zhang L, Zhu YH, Wang LM, Zeng L. Mitochondria-Targeted Antioxidant Peptide SS31 Prevents Hypoxia/Reoxygenation-Induced Apoptosis by Down-Regulating p66Shc in Renal Tubular Epithelial Cells. Cell Physiol Biochem. 2013;32:591–600. doi: 10.1159/000354463. [DOI] [PubMed] [Google Scholar]
- Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol. 1999;277:H2240–H2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Isoyama S, Nitta-Komatsubara Y. Acute and chronic adaptation to hemodynamic overload and ischemia in the aged heart. Heart Fail Rev. 2002;7:63–69. doi: 10.1023/A:1013701923065. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res. 2005;66:233–244. doi: 10.1016/j.cardiores.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Nitta Y, Abe K, Aoki M, Ohno I, Isoyama S. Diminished heat shock protein 70 mRNA induction in aged rat hearts after ischemia. Am J Physiol. 1994;267:H1795–H1803. doi: 10.1152/ajpheart.1994.267.5.H1795. [DOI] [PubMed] [Google Scholar]
- Chou TC, Yen MH, Li CY, Ding YA. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension. 1998;31:643–648. doi: 10.1161/01.HYP.31.2.643. [DOI] [PubMed] [Google Scholar]
- Korzick DH, Holiman DA, Boluyt MO, Laughlin MH, Lakatta EG. Diminished alpha1-adrenergic-mediated contraction and translocation of PKC in senescent rat heart. Am J Physiol Heart Circ Physiol. 2001;281:H581–H589. doi: 10.1152/ajpheart.2001.281.2.H581. [DOI] [PubMed] [Google Scholar]
- Tani M, Honma Y, Hasegawa H, Tamaki K. Direct activation of mitochondrial K(ATP) channels mimics preconditioning but protein kinase C activation is less effective in middle-aged rat hearts. Cardiovasc Res. 2001;49:56–68. doi: 10.1016/S0008-6363(00)00240-6. [DOI] [PubMed] [Google Scholar]
- Rondelli RR, Dal Corso S, Simoes A, Malaguti C. Methods for the assessment of peripheral muscle fatigue and its energy and metabolic determinants in COPD. J Bras Pneumol. 2009;35:1125–1135. doi: 10.1590/S1806-37132009001100011. [DOI] [PubMed] [Google Scholar]
- Smart N. Exercise training for heart failure patients with and without systolic dysfunction: an evidence-based analysis of how patients benefit. Cardiol Res Pract. 2011. doi:10.4061/2011/837238. [DOI] [PMC free article] [PubMed]
- Smart N, Haluska B, Jeffriess L, Marwick TH. Exercise training in systolic and diastolic dysfunction: effects on cardiac function, functional capacity, and quality of life. Am Heart J. 2007;153:530–536. doi: 10.1016/j.ahj.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Williams AD, Carey MF, Selig S, Hayes A, Krum H, Patterson J, Toia D, Hare DL. Circuit resistance training in chronic heart failure improves skeletal muscle mitochondrial ATP production rate–a randomized controlled trial. J Card Fail. 2007;13:79–85. doi: 10.1016/j.cardfail.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Bortz WM II. A conceptual framework of frailty: a review. J Gerontol A Biol Sci Med Sci. 2002;57:M283–M288. doi: 10.1093/gerona/57.5.M283. [DOI] [PubMed] [Google Scholar]
- Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52:80–85. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J Biol Chem. 2007;282:31257–31266. doi: 10.1074/jbc.M706129200. [DOI] [PubMed] [Google Scholar]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Nelson WB, Hudson MB. Exercise-induced oxidative stress in humans: cause and consequences. Free Radic Biol Med. 2011;51:942–950. doi: 10.1016/j.freeradbiomed.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Knowels G, Salmon A, Beyer R, Xie H, Van Remmen H, Smith SR, Marcinek DJ. Reduced coupling of oxidative phosphorylation in vivo precedes electron transport chain defects due to mild oxidative stress in mice. PLoS One. 2011;6:e26963. doi: 10.1371/journal.pone.0026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Wilbur T, Mathis M, Shankland EG, Trieu A, Harper ME, Marcinek DJ. Impaired adaptability of in vivo mitochondrial energetics to acute oxidative insult in aged skeletal muscle. Mech Ageing Dev. 2012;133:620–628. doi: 10.1016/j.mad.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, Adjeitey CN, Xuan JY, Harper ME. Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. FASEB J. 2012;26:363–375. doi: 10.1096/fj.11-189639. [DOI] [PubMed] [Google Scholar]
- Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol. 2005;569:467–473. doi: 10.1113/jphysiol.2005.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci U S A. 2007;104:1057–1062. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek DJ, Siegel MP. Targeting redox biology to reverse mitochondrial dysfunction. Aging (Albany NY) 2013;5:588–589. doi: 10.18632/aging.100590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustgarten MS, Jang YC, Liu Y, Muller FL, Qi W, Steinhelper M, Brooks SV, Larkin L, Shimizu T, Shirasawa T, McManus LM, Bhattacharya A, Richardson A, Van Remmen H. Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am J Physiol Cell Physiol. 2009;297:C1520–C1532. doi: 10.1152/ajpcell.00372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Lai Y, Yue Y, Rabinovitch PS, Hakim C, Duan D. Ectopic catalase expression in mitochondria by adeno-associated virus enhances exercise performance in mice. PLoS One. 2009;4:e6673. doi: 10.1371/journal.pone.0006673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Pye D, Vasilaki A, Zibrik L, Palomero J, Kabayo T, McArdle F, Van Remmen H, Richardson A, Tidball JG, McArdle A, Jackson MJ. Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu, Zn superoxide dismutase. Aging Cell. 2011;10:749–760. doi: 10.1111/j.1474-9726.2011.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV, Van Remmen H. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J. 2013;27:3536–3548. doi: 10.1096/fj.13-228130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate MA, Humphries KM, Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem. 2011;286:21865–21875. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, Forster MJ, Sohal RS. Association between life-span extension by caloric restriction and thiol redox state in two different strains of mice. Free Radic Biol Med. 2011;51:225–233. doi: 10.1016/j.freeradbiomed.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yueh B, Shapiro N, MacLean CH, Shekelle PG. Screening and management of adult hearing loss in primary care: scientific review. JAMA. 2003;289:1976–1985. doi: 10.1001/jama.289.15.1976. [DOI] [PubMed] [Google Scholar]
- Forstl H, Kurz A. Clinical features of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1999;249:288–290. doi: 10.1007/s004060050101. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012. doi:10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed]
- Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, Wallace DC. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–51660. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- Walls KC, Coskun P, Gallegos-Perez JL, Zadourian N, Freude K, Rasool S, Blurton-Jones M, Green KN, LaFerla FM. Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J Biol Chem. 2012;287:30317–30327. doi: 10.1074/jbc.M112.365890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, Amin SK, Hu L, Mei Y, Klann E, Pautler RG. Mitochondrial superoxide contributes to blood flow and axonal transport deficits in the Tg2576 mouse model of Alzheimer’s disease. PLoS One. 2010;5:e10561. doi: 10.1371/journal.pone.0010561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terni B, Boada J, Portero-Otin M, Pamplona R, Ferrer I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 2010;20:222–233. doi: 10.1111/j.1750-3639.2009.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H, Boyle K. Amyloid-beta peptide binds with heme to form a peroxidase: relationship to the cytopathologies of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:3381–3386. doi: 10.1073/pnas.0600134103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro E, Oliveira CR, Pereira CM. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008;30:331–342. doi: 10.1016/j.nbd.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Reddy PH. Mitochondria-Targeted Antioxidant SS31 Prevents Amyloid Beta-Induced Mitochondrial Abnormalities and Synaptic Degeneration in Alzheimer’s Disease. Pharmaceuticals (Basel) 2012;5:1103–1119. doi: 10.3390/ph5101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Spec No. 2):R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–2020. doi: 10.1212/WNL.0b013e3181ff9685. [DOI] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Cookson MR. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy. 2011;7:531–532. doi: 10.4161/auto.7.5.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem. 2012;121:830–839. doi: 10.1111/j.1471-4159.2012.07734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb Perspect Med. 2012;2:a009415. doi: 10.1101/cshperspect.a009415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Sun Y, Guo S, Lu B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet. 2011;20:3227–3240. doi: 10.1093/hmg/ddr235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose HL, Lincoln SJ, Tyndall GM, Farrer MJ. Parkinson’s disease: a rethink of rodent models. Exp Brain Res. 2006;173:196–204. doi: 10.1007/s00221-006-0461-3. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, Kalyanaraman B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic Biol Med. 2010;49:1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5. doi: 10.1186/1475-2840-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007;48:3805–3811. doi: 10.1167/iovs.06-1280. [DOI] [PubMed] [Google Scholar]
- Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, Brown DA. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52:1009–1018. doi: 10.1016/j.yjmcc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi Q, Zinman B, Bosch J, Dagenais G, Mann JF, Gerstein HC. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: results of the HOPE study and MICRO-HOPE substudy. Diabetes Care. 2002;25:1919–1927. doi: 10.2337/diacare.25.11.1919. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Hanefeld M, Ruhnau KJ, Hasche H, Lobisch M, Schutte K, Kerum G, Malessa R. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a 7-month multicenter randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-Lipoic Acid in Diabetic Neuropathy. Diabetes Care. 1999;22:1296–1301. doi: 10.2337/diacare.22.8.1296. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Hanefeld M, Ruhnau KJ, Meissner HP, Lobisch M, Schutte K, Gries FA. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study) Diabetologia. 1995;38:1425–1433. doi: 10.1007/BF00400603. [DOI] [PubMed] [Google Scholar]
- Reljanovic M, Reichel G, Rett K, Lobisch M, Schuette K, Moller W, Tritschler HJ, Mehnert H. Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): a two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy. Free Radic Res. 1999;31:171–179. doi: 10.1080/10715769900300721. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Schatz H, Conrad F, Gries FA, Ulrich H, Reichel G. Effects of treatment with the antioxidant alpha-lipoic acid on cardiac autonomic neuropathy in NIDDM patients. A 4-month randomized controlled multicenter trial (DEKAN Study). Deutsche Kardiale Autonome Neuropathie. Diabetes Care. 1997;20:369–373. doi: 10.2337/diacare.20.3.369. [DOI] [PubMed] [Google Scholar]
- Ametov AS, Barinov A, Dyck PJ, Hermann R, Kozlova N, Litchy WJ, Low PA, Nehrdich D, Novosadova M, O’Brien PC, Reljanovic M, Samigullin R, Schuette K, Strokov I, Tritschler HJ, Wessel K, Yakhno N. SYDNEY Trial Study Group. The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care. 2003;26:770–776. doi: 10.2337/diacare.26.3.770. [DOI] [PubMed] [Google Scholar]
- Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, Decleves AE, Andreyev A, Quach T, Ly S, Shekhtman G, Nguyen W, Chepetan A, Le TP, Wang L, Xu M, Paik KP, Fogo A, Viollet B, Murphy A, Brosius F, Naviaux RK, Sharma K. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussakli CH, Ebaee A, Binkley J, Brentnall TA, Emond MJ, Rabinovitch PS, Risques RA. Mitochondria and tumor progression in ulcerative colitis. J Natl Cancer Inst. 2013;105:1239–1248. doi: 10.1093/jnci/djt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh J, Enns L, Fatemie S, Hopkins H, Morton J, Pettan-Brewer C, Ladiges W. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer. 2011;11:191. doi: 10.1186/1471-2407-11-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza AD, Parish IA, Krause DS, Kaech SM, Shadel GS. Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol Ther. 2013;21:42–48. doi: 10.1038/mt.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KR, Brown VA, Jones DJ, Britton RG, Hemingway D, Miller AS, West KP, Booth TD, Perloff M, Crowell JA, Brenner DE, Steward WP, Gescher AJ, Brown K. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70:7392–7399. doi: 10.1158/0008-5472.CAN-10-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Antonenko YN, Avetisyan AV, Bakeeva LE, Chernyak BV, Chertkov VA, Domnina LV, Ivanova OY, Izyumov DS, Khailova LS, Klishin SS, Korshunova GA, Lyamzaev KG, Muntyan MS, Nepryakhina OK, Pashkovskaya AA, Pletjushkina OY, Pustovidko AV, Roginsky VA, Rokitskaya TI, Ruuge EK, Saprunova VB, Severina II, Simonyan RA, Skulachev IV, Skulachev MV, Sumbatyan NV, Sviryaeva IV, Tashlitsky VN, Vassiliev JM, Vyssokikh MY. et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry (Mosc) 2008;73:1273–1287. doi: 10.1134/S0006297908120018. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Magwere T, West M, Riyahi K, Murphy MP, Smith RA, Partridge L. The effects of exogenous antioxidants on lifespan and oxidative stress resistance in Drosophila melanogaster. Mech Ageing Dev. 2006;127:356–370. doi: 10.1016/j.mad.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horvath B, Zsengeller Z, Batkai S, Cao Z, Kechrid M, Holovac E, Erdelyi K, Tanchian G, Liaudet L, Stillman IE, Joseph J, Kalyanaraman B. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radic Biol Med. 2012;53:1123–1138. doi: 10.1016/j.freeradbiomed.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic Targeting of Mitochondrial Superoxide in Hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1095–R1102. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20(Suppl 2):S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley Y, Fink BD, Ross NC, Prisinzano TE, Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J Biol Chem. 2006;281:39766–39775. doi: 10.1074/jbc.M608268200. [DOI] [PubMed] [Google Scholar]
- Doughan AK, Dikalov SI. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid Redox Signal. 2007;9:1825–1836. doi: 10.1089/ars.2007.1693. [DOI] [PubMed] [Google Scholar]
- Fink BD, Herlein JA, Yorek MA, Fenner AM, Kerns RJ, Sivitz WI. Bioenergetic effects of mitochondrial-targeted coenzyme Q analogs in endothelial cells. J Pharmacol Exp Ther. 2012;342:709–719. doi: 10.1124/jpet.112.195586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87:40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- Rytomaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- Rytomaa M, Kinnunen PK. Reversibility of the binding of cytochrome c to liposomes. Implications for lipid-protein interactions. J Biol Chem. 1995;270:3197–3202. doi: 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, Pacheco AA, Winnica DE, Peterson J, Bayir H, Waldeck DH, Kagan VE. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiswedel I, Gardemann A, Storch A, Peter D, Schild L. Degradation of phospholipids by oxidative stress–exceptional significance of cardiolipin. Free Radic Res. 2010;44:135–145. doi: 10.3109/10715760903352841. [DOI] [PubMed] [Google Scholar]
- Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen xpecies from complex I. Antioxid Redox Signal. 2013;19:1469–1480. doi: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Bracken WC, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2013. doi:10.1111/bph.12468. [DOI] [PMC free article] [PubMed]
- Sinibaldi F, Howes BD, Piro MC, Polticelli F, Bombelli C, Ferri T, Coletta M, Smulevich G, Santucci R. Extended cardiolipin anchorage to cytochrome c: a model for protein-mitochondrial membrane binding. J Biol Inorg Chem. 2010;15:689–700. doi: 10.1007/s00775-010-0636-z. [DOI] [PubMed] [Google Scholar]
- Snider EJ, Muenzner J, Toffey JR, Hong Y, Pletneva EV. Multifaceted effects of ATP on cardiolipin-bound cytochrome c. Biochemistry. 2013;52:993–995. doi: 10.1021/bi301682c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- Chakrabarti AK, Feeney K, Abueg C, Brown DA, Czyz E, Tendera M, Janosi A, Giugliano RP, Kloner RA, Weaver WD, Bode C, Godlewski J, Merkely B, Gibson CM. Rationale and design of the EMBRACE STEMI study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am Heart J. 2013;165:509–514. doi: 10.1016/j.ahj.2012.12.008. [DOI] [PubMed] [Google Scholar]