

Abstract
Small insertions/deletions (INDELs) of ≤21 bp comprise 18% of all recorded mutations causing human inherited disease and are evident in 24% of documented Mendelian diseases. INDELs affect gene function in multiple ways: for example, by introducing premature stop codons that either lead to the production of truncated proteins or affect transcriptional efficiency. However, the means by which they impact post-transcriptional regulation, including alternative splicing, have not been fully evaluated. In this study, we collate disease-causing INDELs from the Human Gene Mutation Database (HGMD) and neutral INDELs from the 1000 Genomes Project. The potential of these two types of INDELs to affect binding-site affinity of RNA-binding proteins (RBPs) was then evaluated. We identified several sequence features that can distinguish disease-causing INDELs from neutral INDELs. Moreover, we built a machine-learning predictor called PinPor (predicting pathogenic small insertions and deletions affecting post-transcriptional regulation, http://watson.compbio.iupui.edu/pinpor/) to ascertain which newly observed INDELs are likely to be pathogenic. Our results show that disease-causing INDELs are more likely to ablate RBP-binding sites and tend to affect more RBP-binding sites than neutral INDELs. Additionally, disease-causing INDELs give rise to greater deviations in binding affinity than neutral INDELs. We also demonstrated that disease-causing INDELs may be distinguished from neutral INDELs by several sequence features, such as their proximity to splice sites and their potential effects on RNA secondary structure. This predictor showed satisfactory performance in identifying numerous pathogenic INDELs, with a Matthews correlation coefficient (MCC) value of 0.51 and an accuracy of 0.75.
INTRODUCTION
Micro-insertions and micro-deletions of ≤21 bp (INDELs) comprise the second largest category of pathogenic genetic variations in the human genome (after single-nucleotide substitutions), accounting for 18% of all documented genomic variants (1). Similar to single-nucleotide polymorphisms (SNPs) and large structural variations, INDELs are of significant clinical interest, owing to their potential to affect gene function and hence cause disease. Based on the Human Gene Mutation Database (HGMD) (2), single-nucleotide variations (SNVs) represent the largest class of genetic variant, responsible for>50% of known Mendelian diseases, followed by small INDELs, which are evident in 24% of known Mendelian diseases.
INDELs may affect gene function through multiple mechanisms. First, frameshifting INDELs insert/delete a number of nucleotides that are not divisible by three and therefore result in the shift of the entire reading frame and an altered protein sequence after the site of the INDEL; these INDELs often lead to premature termination of translation (3–10) and/or nonsense-mediated decay. Non-frameshifting INDELs, on the other hand, insert/delete a multiple of three nucleotides and lead to the addition or removal of amino acid residues at the INDEL locus, thereby also affecting protein function. In addition to altering protein amino acid sequences, INDELs within promoter regions have the potential to disrupt existing transcription factor-binding sites (or alternatively, generate new transcription factor-binding sites), thereby affecting gene expression (11,12). For example, an INDEL within the ACE gene promoter has been reported to be a causative factor for coronary heart disease (13). Overall, functions of INDELs have been studied in many types of disease, including inflammatory bowel disease (14), Alzheimer's disease (15,16), heart disease (17,18) and numerous cancers.
In addition to impacting protein function by altering amino acid sequence, INDELs in exonic and intronic regions can also interfere with binding sites of RNA-binding proteins (RBPs) and hence may influence RNA processing, including RNA editing (19), alternative splicing (20,21), microRNA binding and polyadenylation (22). Despite pathogenic small INDELs being a frequent cause of inherited disease, bioinformatics tools for prioritizing INDELs are not well established, with only a few tools available that utilize high-throughput sequencing-derived micro-insertion/micro-deletion data; such tools include PriVar (23), SIFT-INDEL (24) and DDIG-IN (25). However, all of these tools focus on the potential roles of INDELs in changing amino acid sequences (and hence protein structures) and do not attempt to assess their roles in RNA processing. Therefore, the prioritization of disease-causing INDELs that interrupt post-transcriptional regulation remains a little-studied topic and a formidable challenge.
To investigate the potential impact of INDELs on post-transcriptional regulation, we systematically evaluated several RNA-processing-related genomic features of disease-causing INDELs already documented in the HGMD. We found that pathogenic INDELs are preferentially located in alternatively spliced exons and often disrupt binding sites of RBPs. In the current work, we further performed a comparative study of disease-causing INDELs and neutral INDELs (documented in the 1000 Genomes Project database) to show how they may be discriminated in terms of sequence features, stabilization of RNA secondary structure and evolutionary conservation. Such characteristics, together with the effects of INDEL on RBP-binding affinity, were utilized to build a classifier (‘PinPor’, predicting pathogenic small insertions and deletions affecting post-transcriptional regulation) that can be used to predict the disease relevance/irrelevance of newly discovered INDELs, in relation to post-transcriptional regulation.
RESULTS
In this study, we investigated how INDELs affect post-transcriptional regulation by altering RNA processing, including the dysregulation of alternative-splicing patterns. In the RNA sequences flanking INDEL loci, we examined several nucleotide sequence-specific features that could affect splicing regulation. We systematically compared the differences between disease-causing (from the HGMD) and neutral (reported by the 1000 Genomes Project) INDELs. The selection of neutral INDELs was based on the fact that none of the individuals sequenced in the 1000 Genomes Project had any overt signs of disease. As most (96.4%) of the INDELs in the HGMD were located within gene-coding regions, we removed all examples of INDELs residing in intergenic, intronic and untranslated (UTR) regions from further analysis. In summary, our disease-causing and neutral datasets, respectively, included 27 422 and 1379 (non-UTR, exonic) INDELs.
Disease-causing INDELs tend to alter the binding affinities of RNA-binding proteins
To establish whether the presence of an INDEL would affect the binding affinity of an RBP, we evaluated RBP-binding score changes in the presence and absence of INDELs at the variant-containing loci; the scores were calculated based on the position-weight matrix (PWM) of the RBP and the RNA sequence of the putative RBP-binding site. Briefly, a posterior probability of the likelihood that an INDEL would change a given RBP-binding site and the magnitude of that change were calculated for each INDEL-RBP pair, using a strategy reported previously (see Materials and methods) (26). Positive and negative magnitude values indicate gain and loss of RBP binding, respectively. We focused our analysis on 53 RBPs whose PWMs were derived from experimental evidence and were documented in the RBPDB database (27). The PWMs of these RBPs were acquired using various technologies, including NMR (28,29), EMSA (30), SELEX (31) and CLIP-Seq (32).
We first examined whether disease-causing INDELs (documented in the HGMD) were more likely to affect the binding sites of the RBPs that play regulatory roles in RNA processing. We found that the disease-causing INDELs gave rise to significantly larger binding score deviations, when compared with the neutral INDELs (in the 1000 Genomes Project). Using the RBP ELAVL2 (embryonic lethal, abnormal vision and Drosophila-like 2) binding motif as an example, 811 (2.96%) and 12 (0.87%) of disease-causing and neutral INDELs, respectively, caused changes in the potentials of protein binding (odds ratio = 3.34, P-value < 0.01). Overall, among 53 RNA-binding motifs evaluated, 28 showed significantly higher rates (P-value < 0.05) of binding changes elicited by disease-causing versus neutral INDELs (Fig. 1). By contrast, only two RBPs showed significantly lower rates of binding changes (P-value < 0.05) caused by disease-causing INDELs than neutral ones. No significant differences were observed for the other 23 RBPs.
Figure 1.

Disease-causing INDELs tend to alter the binding sites of RBPs to a greater extent than neutral INDELs. The X-axis plots the proportion of disease-causing INDELs (derived from the HGMD) that change the RBP-binding affinity with a posterior probability of >0.5. The Y-axis plots the proportion of neutral INDELs (from 1000 Genomes Project data) that change the RBP-binding affinity with a posterior probability of >0.5. Each dot represents one RBP and is plotted against the proportion of disease-causing INDELs and neutral INDELs that have the potential to change RBP binding. Among the 53 RBPs, 28 were affected at a significantly (P-value < 0.05) higher rate by INDELs from HGMD than neutral ones (filled ellipses under the 45 degree line). Additionally, only two RBPs showed a significantly lower affection rate by INDELs from HGMD than neutral ones (P-value < 0.05). The open ellipses indicate RBPs with an insignificant ratio.
We further examined whether the potential for disease-causing INDELs to alter RBP binding was consistent among different disease states. We first identified all the diseases associated with >50 disease-causing INDELs documented in the HGMD database. For each RBP–disease pair, we calculated the proportion of disease-causing INDELs that could potentially change the binding of the specific RBP and further evaluated whether this proportion was statistically different from the neutral INDELs in the 1000 Genomes Project. For instance, of 68 INDELs in the HGMD documented as being associated with paraganglioma, 10 (14.7%) were predicted to change the binding of the RBP ELAVL2. This proportion was significantly higher than the proportion for neutral INDELs, which was only 0.87% (12 of 1379); the odds ratio for this difference was 19.6. Figure 2 is a heat map showing the odds ratios for all RBP–disease pairs, in which red and blue colors indicate higher and lower percentages of disease-causing INDELs (when compared with neutral ones) predicted to change the binding of the specific RBPs. The heat map clearly shows that, with a few exceptions, disease-causing INDELs tend to change RBP binding with a higher probability, when compared with neutral INDELs.
Figure 2.

Heat map of the relative proportion of INDELs that change RBP binding between disease-causing INDELs and neutral INDELs. Each dot, corresponding to one disease–RBP pair, represents the log2-transformed ratio of the proportion of disease-causing INDELs that change RBP-binding affinity, and the proportion of neutral INDELs. Only significant (P < 0.05) disease–RBP pairs are plotted. Red dots indicate significantly higher proportions of disease-causing INDELs potentially changing RBP binding than neutral INDELs, and blue dots indicate lower proportions.
Disease-causing INDELs are enriched in alternatively spliced exons
In both the disease-causing (HGMD) and neutral (from 1000 Genomes Project) datasets, we calculated the proportion of INDELs that were located within alternatively spliced exons (including upstream and downstream flanking exons, Table 1). We found disease-causing (HGMD) INDELs to be significantly enriched in those exons documented in the alternative-splicing database (33). Among 27 422 disease-causing INDELs listed in the HGMD, 6131 (22.4%) were found to reside within cassette exons (including flanking exons) derived from RefSeq, Ensembl, UCSC or other databases (34–36). By contrast, only 176 (12.8%) of the 1379 neutral INDELs in the 1000 Genomes Project dataset were located in these regions (P-value < 2.2 × 10−16). Similarly, disease-causing INDELs also displayed significant enrichment in gene regions, subject to alternative 5′ and 3′ splicing events (Fig. 3).
Table 1.
Proportion of INDELs in alternative-splicing events and non-alternative-splicing events
| Alt-event | HGMD (disease-causing) (%) | 1000 Genomes Project (neutral) (%) |
|---|---|---|
| 3′ SS | 2.21 | 2.47 |
| 5′ SS | 2.52 | 1.52 |
| Upper exon | 8.77 | 3.77 |
| Central exon | 4.80 | 2.76 |
| Down exon | 8.78 | 6.24 |
| Other(non-alternative) | 72.92 | 83.24 |
3′ SS, alternative 3′ splicing site; 5′ SS, alternative 5′ splicing site; upper exon, the exon which is located immediately upstream of a cassette exons; central exon, the cassette exon; down exon, the exon located immediately downstream of a cassette exon.
Figure 3.

Proportion of INDELs located in a portion of the gene that is involved in alternative processing events, comparing disease-causing INDELs and neutral INDELs. Each gray bar represents 1 of 15 diseases studied, whereas the dashed line represents neutral INDELs. The height of each bar and the dashed line represent the proportion of associated INDELs of a particular gene that is involved in one specific type of alternative processing event: (A) Cassette exon, including upstream and downstream flanking exons, (B) alternative 3′ splicing site (A3SS) and (C) alternative 5′ splicing site (A5SS).
INDELs in close proximity to splice sites tend to be disease-causing
The spatial relationship between RBP-binding-site positions and splice sites can provide important mechanistic insights to molecular function. It is reported that many RBPs, including SFRS1 and NOVA-2, tend to bind close to splice sites (37,38). In addition, we (38) and Pfarr et al. (39) have previously reported that disease-causing single-nucleotide substitutions tend to disrupt the RBP sites that are in close proximity to splicing junctions. Therefore, we examined whether the distances between splice sites and INDEL loci follow a different distribution between disease-causing and neutral INDELs. Based on a comparison between 27 422 and 1379 non-UTR, exonic INDELs in the HGMD and 1000 Genomes Project databases, we clearly observed that disease-causing INDELs tend to locate in closer proximity to splice sites than neutral INDELs, and this trend is consistent for both 5′ and 3′ splice sites (Fig. 4). The median distance (in nucleotides) between the variant loci and the 3′ end of the exons was 89 and 123 nt for HGMD and 1000 Genomes Project INDELs, respectively. Similarly, the median distance to the 5′ end of exons was 91 and 143 nt for HGMD and 1000 Genomes Project INDELs. This result is consistent with previous observations that disease-causing single-nucleotide variants (SNVs) locate closer to splice sites, when compared with non-disease-causing SNVs (39,40).
Figure 4.

Comparison of proximity to splice site between disease-causing INDELs and neutral INDELs. The solid line represents the proximity distribution of disease-causing INDELs, whereas the dashed line represents the proximity distribution of neutral INDELs. (A) Distributions of proximity to 5′ end boundary of exon (acceptor site). (B) Distributions of proximity to 3′ end boundary of exon (donor site).
GC content of flanking sequences
GC content influences pre-mRNA local structure and displays a positive correlation with structural stability, as measured by sequence minimum free energy (41). Consequently, the GC content difference of exons and adjacent introns may influence mRNA splicing, and INDELs located within the pre-mRNA coding regions with differing GC contents may elicit different splicing outcomes (42). For each INDEL, we calculated the GC content of ±50-bp sequence flanking the site of mutation. We observed clear differences in the GC content between the regions harboring HGMD INDELs, when compared with those harboring 1000 Genomes Project INDELs (Fig. 5), with disease-causing INDELs more prone to reside within regions with low GC content (0.35–0.5).
Figure 5.

Comparison of GC content of 100-bp fragments surrounding gene loci harboring disease-causing INDELs and neutral INDELs. The solid line represents the GC content for disease-causing INDELs, whereas the dashed line represents the GC content for neutral INDELs. (A) Distributions of GC content calculated from mutant form fragment. (B) Distributions of GC content calculated from reference form fragment.
Disease-causing INDELs tend to affect pre-mRNA secondary structure
Pre-mRNA structure conformational changes can influence the utilization of both splicing signals (5′ SS, 3′ SS, branch point) and cis-regulatory elements (exonic/intronic splice enhancers, and exonic/intronic splice silencers) (43–45). To evaluate the changes in pre-mRNA structure caused by INDELs, we compared the structural distance scores between the reference sequence and the mutated sequence using the RNADistance program in Vienna RNA package using default parameters (46). Structural distance is measured as the edit distance (the number of operations required to convert one structure into another) between two aligned RNA secondary structures. The RNA secondary structure was predicted using the RNAfold program (V2.0) in Vienna RNA package with default parameters (46). Similar to the evaluation of the GC content difference, 100-bp pre-mRNA sequences flanking the INDEL loci were extracted, with 50-bp on both the 5′ and 3′ sides. As shown in Figure 6, disease-causing INDELs tend to give rise to greater changes in RNA secondary structure.
Figure 6.

Comparison of INDEL effect to local RNA secondary structure between disease-causing INDELs and neutral INDELs. The solid line plots the distance distribution for disease-causing INDELs. The dashed line represents the distance distribution for neutral INDELs. When the edit distance between the RNA secondary structure of the mutation and reference forms is >20, the proportion of disease-causing INDELs is higher than that of neutral INDELs.
Sequence conservation
Nucleotide conservation is higher in constitutive exons than that in alternatively spliced exons (47–50). It is almost axiomatic that evolutionarily conserved DNA/RNA sequences are more likely to be of functional significance, because mutations in highly conserved regions are eliminated through natural selection. To examine this feature, phyloP (51) scores, which evaluate evolutionary conservation across 46 vertebrates, were downloaded from the UCSC Genome Browser conservation (phyloP46wayPrimates) track. For each INDEL, the average phyloP score for the nucleotides comprising the deletion site, or the average of the two nucleotides flanking the insertion site, was calculated. Positive or negative phyloP scores indicate that the site is evolutionarily conserved or fast-evolving, respectively. Our results indicate that HGMD INDELs tend to be disproportionately located at evolutionarily conserved sites, when compared with neutral INDELs from the 1000 Genomes Project (P-value < 2.2e–16, Fisher's Exact test, Fig. 7). This result strongly suggests that pathogenic INDELs tend to disrupt functional regulatory elements.
Figure 7.

Comparison of conservation score of deleted nucleotides (or two adjacent nucleotides) to inserted nucleotides. The solid line plots the phyloP score distribution for disease-causing INDELs. The dashed line plots the phyloP score distribution for neutral INDELs. Disease-causing INDELs exhibit a higher rate of occurrence at evolutionarily conserved regions than neutral INDELs.
Prioritizing INDELs with a role in inherited disease
In order to predict whether a newly discovered INDEL could be disease-causing, we constructed a machine-learning predictor, PinPor, based on the various genomic features that potentially affect RNA processing. The disease-causing and neutral INDEL datasets were, respectively, compiled from the HGMD and 1000 Genomes Project databases. When compiling these datasets, we aimed to remove all INDELs deemed likely to impact protein function through other known mechanisms. For instance, frameshifting INDELs may either change the protein sequence downstream of the INDEL or induce nonsense-mediated decay. Similarly, INDELs located in regions lacking stable tertiary structure (disordered region) are less likely to affect protein function, whereas INDELs within regions of very specific tertiary structure (structured regions) more likely affect protein function, by disrupting protein structures. Such INDELs were removed from the training dataset. Overall, of 27 422 exonic INDELs listed in the HGMD database, 3342 were non-frameshifting, and 624 of these were located in regions that were deemed unlikely to form structural proteins (with disorder score of >0.4). For the 1000 Genomes Project data, 685 of the 1379 exonic INDELs were non-frameshifting, whereas 531 located within disordered protein regions. These INDELs were further used as a ‘gold standard’ for training the predictors.
Features
The following genomic features that best discriminate between disease-causing and neutral INDELs were used for training the predictor: (1) distance to the splice donor sites (i.e. the 3′ end of an exon); (2) distance to the splice acceptor sites (the 5′ end of an exon); (3) GC content of the flanking sequence in the reference form (GCraw); (4) GC content of the flanking sequence in the mutated form (GCmut); (5) disruption of the RNA secondary structure, i.e. the edit distance of the flanking sequences in the reference and mutated forms (a higher edit distance indicates larger differences on RNA secondary structure in two forms); (6) conservation score, i.e. the phyloP scores (51) of the deleted nucleotides or the two nucleotides flanking the inserted sequences; (7) alt-event indicator, i.e. whether or not a given INDEL is located within an annotated alternative-splicing event; (8) maximal magnitude of RBP binding changes by an INDEL. This measurement represents the largest magnitude of potential change in binding [as defined in Eq. (5)] by an INDEL among 53 RBPs. (9) the number of RBPs whose binding could be potentially altered by the INDEL, as evaluated in Eq. (6).
To develop a computational model capable of predicting INDEL disease relevance, we evaluated five machine-learning and statistical classification algorithms available in the Weka software package (52), including Bayesian Network (BN) (53), Multilayer Perceptron (54), Naïve Bayes (55), Random Forest (56) and Logistic Regression (57). Each model was trained and tested based on the extracted genomic features using default parameters. To evaluate the performance of each classifier, 10-fold cross-validation was employed. Briefly, in each iteration, 9 of 10 of the gold standard (624 disease-causing and 531 neutral INDELs) dataset were used to train the model, and the remaining 1 of the 10 dataset was used to evaluate the model performance. The performance of each model was evaluated by several metrics, including Matthews correlation coefficient (MCC) (58); accuracy, the percentage of correctly classified samples; and area under curve (AUC) of the receiver operating characteristic (ROC) curve. Those measurements were averaged across all 10 iterations to determine the overall performance. Based on the testing results, as shown in Figure 8A, BN achieved the best performance of all five predictors, with MCC of 0.51, accuracy of 0.75 and AUC of 0.83.
Figure 8.
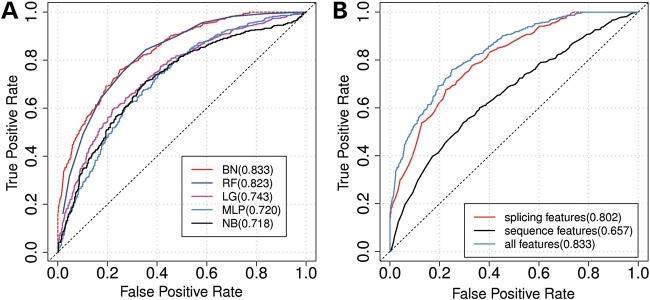
ROC curves of each individual classifiers and subcategories of features. (A) Performance comparison of different classification models evaluated using 10-fold cross-validation. BN, Bayesian Network; LG, Logistic Regression; MLP, Multilayer Perceptron; NB, Naïve Bayes; RF, Random Forest. Each ROC curve represents one model. The numbers in parentheses indicate the AUC of each ROC. The BN outperformed all other predictors. (B) ROC curves for using subcategory of features to classify disease-causing INDELs and neutral INDELs. Blue curve is the ROC curve generated by using all seven selected features. The red curve is generated by using only splicing-related features (i.e. change in RNA secondary structure, maximal magnitude of RBP binding changes by an INDEL and the number of RBPs whose binding is altered by the INDEL). The black curve is generated by using only sequence features (5′ end proximity, 3′ end proximity, GCmut and conservation score). The numbers in parentheses are the AUC. The dotted line is the 45 degree line.
To identify the most discriminative subset of features, we evaluated the performance of each subset of the nine features based on a BN classifier. In total, there were combinations of 511 (29 − 1) subset of features. For each of them, the same 10-fold cross-validation strategy was employed to evaluate its predicting power as an MCC value. The subset of features that achieved the best prediction performance (MCC as 0.51 and accuracy as 0.75) was composed of the following seven features: (1) distance to splice donor site, (2) distance to splice acceptor site, (3) GC content of mutated sequences, (4) disruption of the RNA secondary structure, (5) conservation score, (6) maximal magnitude of RBPs binding changes by an INDEL and (7) the number of RBPs whose binding could potentially be altered by the INDEL. We also divided the selected features into two subcategories: splicing-related features (i.e. disruption of RNA secondary structure, maximal magnitude of RBPs binding changes by an INDEL and number of RBPs whose binding can be potentially altered by the INDEL) and sequence composition features (i.e. 5′ end proximity, 3′ end proximity, GCmut and conservation score). The performance of each subcategory of features was tested separately using BN through 10-fold cross-validation. The subcategory of splicing-related features outperformed sequence composition features. The MCC values of using splicing-related features and sequence composition features alone were 0.448 and 0.246, respectively (Fig. 8B).
We further evaluated the contribution of each feature by the following steps: for each iteration, one feature was removed and the remaining features were used to train the BN predictor using the same 10-fold cross-validation strategy. Thus, larger decreases in MCC value caused by excluding that feature indicated a greater contribution. Among all the features tested, ‘disruption of RNA secondary structure’ was the most important feature, followed by ‘maximal magnitude of RBP binding changes by an INDEL’ and ‘3′ end proximity’ (Table 2).
Table 2.
Feature selection based on accuracy decrease, using BN
| Feature | MCC | FPR | TPR | ACC |
|---|---|---|---|---|
| RNA secondary structure | 0.375 | 0.281 | 0.659 | 0.687 |
| Maximal magnitude of RBPs binding changes by an INDEL | 0.463 | 0.435 | 0.873 | 0.732 |
| 3′ end proximity | 0.479 | 0.328 | 0.803 | 0.743 |
| Number of RBPs whose binding was altered by the INDEL | 0.486 | 0.412 | 0.875 | 0.743 |
| Conservation score | 0.493 | 0.299 | 0.791 | 0.750 |
| 5′ end proximity | 0.495 | 0.207 | 0.705 | 0.745 |
| GC_mut | 0.503 | 0.254 | 0.760 | 0.753 |
| Use all features | 0.506 | 0.250 | 0.760 | 0.755 |
MCC, Matthews correlation coefficient; FPR, false positive rate; TPR, true positive rate; ACC, accuracy.
DISCUSSION
In addition to their potential roles in disrupting protein structure and function, disease-causing genomic variants in exonic regions can also influence transcriptional and post-transcriptional regulation by changing the interaction between cis-acting RNA elements and trans-acting regulatory proteins. It is known, for example, that many diseases are caused by the dysregulation of splicing (59), with 15–50% of human disease mutations affecting splice site selection (60,61). It has been hypothesized that genetic variants can give rise to phenotypic differences by interfering with the splicing code (61), and we recently found that synonymous single-nucleotide variations (SNVs) residing in alternatively spliced exons have minor allele frequencies (MAFs) similar to non-synonymous SNVs, but lower than neutral SNVs (62). This finding suggests that dysfunctional RNA regulation is a major consequence of disease-causing SNVs, and it is reasonable to assume that INDELs can cause similar, if not greater, disruption of RNA regulation.
In this study, we systematically evaluated nucleotide sequence features that served to discriminate disease-causing from neutral INDELs, based on their potentials to disrupt interactions with RBPs. These features differ between disease-causing INDELs (cataloged in the HGMD database) and neutral INDELs (generated by the 1000 Genomes Project), indicating that these features have the potential to be used to predict disease-causing INDELs that disrupt post-transcriptional regulation.
Our analysis clearly showed significant differences between the neutral and disease-causing INDELs in terms of their potential to change the binding affinities of RBPs, based on the position–weight matrices of 53 RBPs documented in the RBPDB database (27). We found that disease-causing INDELs associated with significantly larger binding score deviations than neutral INDELs. Further analysis confirmed that this trend held true for most of the diseases studied, clearly suggesting that INDELs can give rise to new phenotypes by interacting with RBP-binding sites, consistent with previous findings on SNVs (26,62).
In addition to the potential for the direct disruption of RBP binding, we also found marked differences in other genomic features between disease-causing and neutral INDELs. For example, we noted that disease-causing INDELs tend to occur closer to splice sites (both 5′ donor and 3′ acceptor sites), when compared with neutral INDELs (Fig. 4A and B). The genomic loci harboring disease-causing INDELs tend to be more evolutionarily conserved (Fig. 7) and tend to be located within exons for which there is evidence for alternative splicing (Fig. 3). We also evaluated the GC content and potential disruption of RNA secondary structures of the nucleotide sequences adjacent to the INDEL sites (50 nt upstream and downstream); both features showed significant differences between disease-causing and neutral INDELs. Moreover, the DNA sequence surrounding disease-causing INDELs was generally found to be more prevalent in low-GC-content region than those DNA sequences flanking neutral INDELs (Fig. 5). We further observed that disease-causing INDELs tend to disrupt RNA secondary structure to a greater extent than neutral INDELs, as predicted by the RNADistance (46) program (Fig. 6). The ability of these features to clearly distinguish disease-causing from neutral INDELs confirms the importance of using RNA-based features for INDEL discrimination. All these measures indicate that INDELs significantly impact the regulation of RNA processing.
Of the five different machine-learning predictors tested, BN achieved the overall best performance. Using a greedy feature selection strategy, we identified the most informative subsets of features: RNA secondary structure, maximal magnitude of RBP binding changes by an INDEL, 3′ end proximity to splice junctions, number of RBPs whose binding could potentially be altered by the INDEL, conservation score, 5′ end proximity to splicing junction and GC content for the variant form.
We further ranked the seven most informative features based on their relative contribution to the overall performance measured by MCC value. Disruption of pre-mRNA secondary structure was shown to be the single most informative genomic feature. This is consistent with previous reports that RBPs recognize their target RNA not only by the sequence features of RBP-binding sites but also through target site accessibility, which is in part regulated by RNA secondary or tertiary structure conformation (43). In fact, several bioinformatics tools have used this as a major feature to study protein–RNA interactions (63–65). Our findings further confirm those observations and show that INDELs can disrupt RNA processing both by changing the structural conformation (rank no. 1) and by interrupting splicing factor assembly around the boundary of exons (donor/acceptor splice site) (rank nos. 3 and 6). Our model also demonstrated that the evolutionary conservation score of the locus harboring the INDEL is a major determinant for predicting INDEL pathological relevance (ranked no. 5 among all the features tested). Evolutionary conservation is widely regarded as one of major indicators of the biological functionality of a DNA sequence element. Indeed, many studies have reported that RBPs tend to bind to sites that are evolutionarily conserved (66–68). Thus, similar to RNA secondary structure, evolutionary conservation levels have been widely used as predictors of protein–RNA interactions (69,70).
The accuracy of the current model is limited by our current knowledge of RBP-binding motifs. Our current study was based on only 53 RBP-binding sites, and as this number only represents a small proportion of all RBPs, the current model is inevitably limited in terms of its general predictive potential. With rapid developments in high-throughput genomic technologies and supporting biological assays, our ability to identify RBP-binding sites should increase dramatically. Such information, once available, will help to increase the accuracy of model prediction and thus provide a better understanding of these very important elements (INDELs).
MATERIALS AND METHODS
INDEL lists from the 1000 genomes project and the human gene mutation database
The Human Gene Mutation Database (HGMD, http://www.hgmd.org/) contains 28,223 INDELs (micro-insertions/micro-deletions, HGMD professional release 2012.2) causing or associated with human inherited disease; 27,422 (97.15%) of these are located in non-UTR exons. The 1000 Genomes Project catalogs 1,443,514 small INDELs (version 3 of release 20101123, http://www.1000genomes.org/). We excluded those INDELs located in introns and UTR regions (3′ UTR and 5′ UTR), yielding a total of 1379 (0.096%) INDELs located in non-UTR exonic regions for further analysis.
Estimating the probability of an INDEL changing the binding affinity of an RNA-binding protein
Our analysis focused on 53 RBP-binding motifs cataloged in the RBPDB (27) database; these 53 RBP-binding domains represent the binding sites of 30 unique RBPs. For each of the 53 RBP-binding motifs, a PWM was derived from multiple sequence alignments of the experimentally determined RBP-binding sites. A PWM is a matrix of values that gives the count of each nucleotide at each locus of the binding site. The binding affinity between the n-nt DNA sequence and the PWM is described by a matching score S as follow:
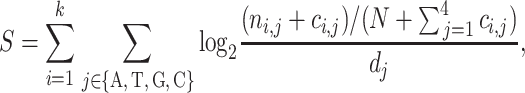 |
(1) |
where ni,j is the count of the jth nucleotide on the ith position in the PWM, k is the width of the binding site and ci,j is the pseudocount for the jth nucleotide on the ith position in the PWM. N is the total number of experimentally validated binding sites for each RBP. dj is the prior base frequency for the jth nucleotide (dj = 0.25 for j = A, T, G, C). Similar strategies have been used previously (71).
In Eq. (1), a high or low matching score indicates that the putative sequence has, respectively, a high or low likelihood to be a potential binding site. Each position of a binding site is assumed to be independent of the other. The matching score distributions for binding and non-binding events are both estimated based on the position-specific scoring matrix (PSSM) of an individual RBP. The PSSM is derived from the PWM, with each value at the ith column and the jth row defined as:
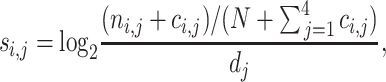 |
(2) |
where ni,j, ci,j, N and dj are the same as in the definition in Eq. (1).
The mean and variance of the binding scores for specific RBP-binding events are defined as:
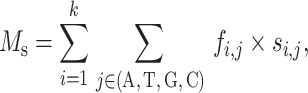 |
(3) |
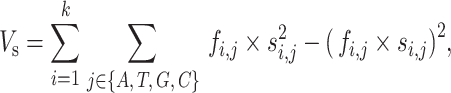 |
(4) |
where si,j is equivalent to the value of the ith column and the jth row in the PSSM and fi,j is the approximation of the true frequency of each nucleotide at each binding locus. For binding events,
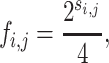 |
and for non-binding events, fi,j = 0.25.
Evaluating the magnitude of the change in RBP binding
As defined in our previous work (26), the magnitude M of an INDEL affecting the binding of an RBP is defined as a likelihood ratio of the INDEL loci being a binding event as opposed to it being a non-binding event in reference and alternative forms, respectively,
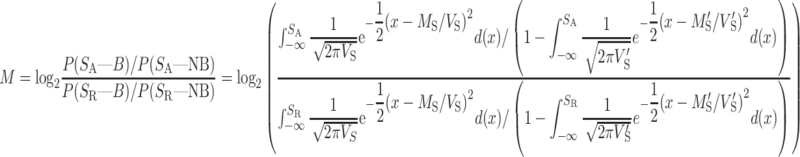 |
(5) |
where 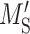 and
and 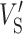 are, respectively, the mean and variance of the matching score for non-binding events. R and A indicate the reference and mutated sites, respectively, whereas B and NB denote binding and non-binding events, respectively. SR and SA each represent the matching scores of the reference and mutated sites. A positive score indicates a gain of an RBP-binding site, whereas a negative score indicates the loss of an RBP-binding site.
are, respectively, the mean and variance of the matching score for non-binding events. R and A indicate the reference and mutated sites, respectively, whereas B and NB denote binding and non-binding events, respectively. SR and SA each represent the matching scores of the reference and mutated sites. A positive score indicates a gain of an RBP-binding site, whereas a negative score indicates the loss of an RBP-binding site.
Bayesian posterior probability of RBP-binding-site gain/loss
We further calculate a Bayesian-based posterior probability for RBP-binding-site gain/loss, defined as the probability that a genetic locus could switch between binding and non-binding, with and without the INDEL variant:
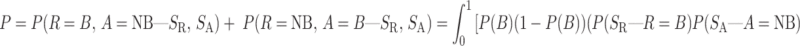 |
(6) |
where P(B) is the prior probability that a specific locus is a RBP-binding event. Here, we assign P(B) to be a beta distribution with a mode value as 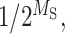 where MS is effectively equal to information content of specific motif. The terms P(R = B, A = NB|SR, SA) and P(R = NB, A = B|SR, SA) represent the probability density function (pdf) denoting loss or gain, respectively, of a RBP-binding site caused by an INDEL.
where MS is effectively equal to information content of specific motif. The terms P(R = B, A = NB|SR, SA) and P(R = NB, A = B|SR, SA) represent the probability density function (pdf) denoting loss or gain, respectively, of a RBP-binding site caused by an INDEL.
RNA secondary structure prediction
The effects on local RNA secondary structure with the presence of INDEL was evaluated using the programs RNAfold and RNADistance from the Vienna RNA package (46). The fragment flanking the locus of one INDEL with 50 bp on each side was extracted for both the reference form and the mutated form of the DNA sequence. First, the minimum free energy structure was calculated by the RNAfold program for each fragment. Then, the two structures were aligned and compared by means of the RNADistance program to calculate the edit distance between them.
Protein disorder score calculation
We used the program SPINE-D (72) to determine the disordered (or unstructured) region of each protein which overlapped an INDEL. Disordered regions are more flexible in three-dimensional structure than structured regions. Each amino acid was assigned a probability (disorder score) indicating whether it was located in a disordered region or not. The higher the disorder score, the more likely the amino acid was to be located in an unstructured region. One INDEL was taken as not affecting protein structure if it was located in a disordered region; these regions were defined as the average disorder scores of the corresponding amino acids of >0.4.
Evaluation of classifier performance
The performance of each classifier was evaluated by accuracy, MCC and the cumulative area under the ROC curve (AUC, which is often used for model comparison). The accuracy was defined as INDELs correctly classified out of all INDELs in the dataset. In other words, accuracy = (TP + TN)/(TP + TN + FP + FN), where TP denotes true positives (correctly classified non-frameshifting HGMD INDELs), TN denotes true negatives (correctly classified non-frameshifting neutral INDELs), FP denotes false positives (non-frameshifting neutral INDELs predicted to be disease-causing) and FN denotes false negatives (non-frameshifting disease-causing INDELs predicted to be neutral). Consequently,
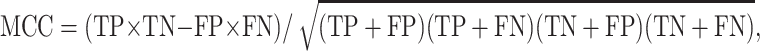 |
and MCC values range from −1 to +1, where −1 indicates all samples are incorrectly classified, +1 indicates all are correctly classified and 0 represents random prediction.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the grants from the U.S. National Institutes of Health (U54CA113001, R01AG041517 and R03NS083468) and Australia National Health and Medical Research project grant (1059775).
REFERENCES
- 1.Mullaney J.M., Mills R.E., Pittard W.S., Devine S.E. Small insertions and deletions (INDELs) in human genomes. Hum. Mol. Genet. 2010;19:R131–R136. doi: 10.1093/hmg/ddq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stenson P.D., Mort M., Ball E.V., Shaw K., Phillips A.D., Cooper D.N. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsater H., et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segditsas S., Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 5.Milward A., Metherell L., Maamra M., Barahona M.J., Wilkinson I.R., Camacho-Hubner C., Savage M.O., Bidlingmaier M., Clark A.J., Ross R.J., et al. Growth hormone (GH) insensitivity syndrome due to a GH receptor truncated after Box1, resulting in isolated failure of STAT 5 signal transduction. J. Clin. Endocrinol. Metab. 2004;89:1259–1266. doi: 10.1210/jc.2003-031418. [DOI] [PubMed] [Google Scholar]
- 6.Liao H., Zhao Y., Baty D.U., McGrath J.A., Mellerio J.E., McLean W.H. A heterozygous frameshift mutation in the V1 domain of keratin 5 in a family with Dowling-Degos disease. J. Invest. Dermatol. 2007;127:298–300. doi: 10.1038/sj.jid.5700523. [DOI] [PubMed] [Google Scholar]
- 7.Rosenstiel P., Till A., Schreiber S. NOD-like receptors and human diseases. Microbes Infect. 2007;9:648–657. doi: 10.1016/j.micinf.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Clark K.L., Yutzey K.E., Benson D.W. Transcription factors and congenital heart defects. Annu. Rev. Physiol. 2006;68:97–121. doi: 10.1146/annurev.physiol.68.040104.113828. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Gallardo E., Solano A., Herrero-Martin M.D., Martinez-Romero I., Castano-Perez M.D., Andreu A.L., Herrera A., Lopez-Perez M.J., Ruiz-Pesini E., Montoya J. NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet. 2009;46:64–67. doi: 10.1136/jmg.2008.060616. [DOI] [PubMed] [Google Scholar]
- 10.van Tintelen J.P., Entius M.M., Bhuiyan Z.A., Jongbloed R., Wiesfeld A.C., Wilde A.A., van der Smagt J., Boven L.G., Mannens M.M., van Langen I.M., et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 11.Sun T., Gao Y., Tan W., Ma S., Shi Y., Yao J., Guo Y., Yang M., Zhang X., Zhang Q., et al. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nat. Genet. 2007;39:605–613. doi: 10.1038/ng2030. [DOI] [PubMed] [Google Scholar]
- 12.Bhangale T.R., Rieder M.J., Livingston R.J., Nickerson D.A. Comprehensive identification and characterization of diallelic insertion-deletion polymorphisms in 330 human candidate genes. Hum. Mol. Genet. 2005;14:59–69. doi: 10.1093/hmg/ddi006. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz J., Blanche H., Cohen N., Velho G., Cambien F., Cohen D., Passa P., Froguel P. Insertion/deletion polymorphism of the angiotensin-converting enzyme gene is strongly associated with coronary heart disease in non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA. 1994;91:3662–3665. doi: 10.1073/pnas.91.9.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGovern D.P., Hysi P., Ahmad T., van Heel D.A., Moffatt M.F., Carey A., Cookson W.O., Jewell D.P. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 2005;14:1245–1250. doi: 10.1093/hmg/ddi135. [DOI] [PubMed] [Google Scholar]
- 15.Schutte D.L., Maas M., Buckwalter K.C. A LRPAP1 intronic insertion/deletion polymorphism and phenotypic variability in Alzheimer disease. Res. Theory Nurs. Pract. 2003;17:301–319. doi: 10.1891/rtnp.17.4.301.53188. discussion 335–308. [DOI] [PubMed] [Google Scholar]
- 16.Rogaeva E.A., Premkumar S., Grubber J., Serneels L., Scott W.K., Kawarai T., Song Y., Hill D.L., Abou-Donia S.M., Martin E.R., et al. An alpha-2-macroglobulin insertion-deletion polymorphism in Alzheimer disease. Nat. Genet. 1999;22:19–22. doi: 10.1038/8729. [DOI] [PubMed] [Google Scholar]
- 17.Cicoira M., Rossi A., Bonapace S., Zanolla L., Perrot A., Francis D.P., Golia G., Franceschini L., Osterziel K.J., Zardini P. Effects of ACE gene insertion/deletion polymorphism on response to spironolactone in patients with chronic heart failure. Am. J. Med. 2004;116:657–661. doi: 10.1016/j.amjmed.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 18.Catto A., Carter A.M., Barrett J.H., Stickland M., Bamford J., Davies J.A., Grant P.J. Angiotensin-converting enzyme insertion/deletion polymorphism and cerebrovascular disease. Stroke. 1996;27:435–440. [PubMed] [Google Scholar]
- 19.Simpson L., Aphasizhev R., Gao G., Kang X. Mitochondrial proteins and complexes in Leishmania and Trypanosoma involved in U-insertion/deletion RNA editing. RNA. 2004;10:159–170. doi: 10.1261/rna.5170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakhshi A., Guglielmi P., Siebenlist U., Ravetch J.V., Jensen J.P., Korsmeyer S.J. A DNA insertion/deletion necessitates an aberrant RNA splice accounting for a mu heavy chain disease protein. Proc. Natl. Acad. Sci. USA. 1986;83:2689–2693. doi: 10.1073/pnas.83.8.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong X., Liu J.R., Kyle J.W., Hanck D.A., Agnew W.S. A profile of alternative RNA splicing and transcript variation of CACNA1H, a human T-channel gene candidate for idiopathic generalized epilepsies. Hum. Mol. Genet. 2006;15:1497–1512. doi: 10.1093/hmg/ddl068. [DOI] [PubMed] [Google Scholar]
- 22.Patraquim P., Warnefors M., Alonso C.R. Evolution of Hox post-transcriptional regulation by alternative polyadenylation and microRNA modulation within 12 Drosophila genomes. Mol. Biol. Evol. 2011;28:2453–2460. doi: 10.1093/molbev/msr073. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L., Zhang J., Yang J., Ying D., Lau Y.L., Yang W. PriVar: a toolkit for prioritizing SNVs and indels from next-generation sequencing data. Bioinformatics. 2013;29:124–125. doi: 10.1093/bioinformatics/bts627. [DOI] [PubMed] [Google Scholar]
- 24.Hu J., Ng P.C. Predicting the effects of frameshifting indels. Genome Biol. 2012;13:R9. doi: 10.1186/gb-2012-13-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H., Yang Y., Lin H., Zhang X., Mort M., Copper D.N., Liu Y., Zhou Y. DDIG-in: discriminating between disease-associated and neutral non-frameshifting micro-indels. Genome Biol. 2013;14:R23. doi: 10.1186/gb-2013-14-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teng M., Ichikawa S., Padgett L.R., Wang Y., Mort M., Cooper D.N., Koller D.L., Foroud T., Edenberg H.J., Econs M.J., et al. regSNPs: a strategy for prioritizing regulatory single nucleotide substitutions. Bioinformatics. 2012;28:1879–1886. doi: 10.1093/bioinformatics/bts275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook K.B., Kazan H., Zuberi K., Morris Q., Hughes T.R. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oberstrass F.C., Auweter S.D., Erat M., Hargous Y., Henning A., Wenter P., Reymond L., Amir-Ahmady B., Pitsch S., Black D.L., et al. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science. 2005;309:2054–2057. doi: 10.1126/science.1114066. [DOI] [PubMed] [Google Scholar]
- 29.Yuan X., Davydova N., Conte M.R., Curry S., Matthews S. Chemical shift mapping of RNA interactions with the polypyrimidine tract binding protein. Nucleic Acids Res. 2002;30:456–462. doi: 10.1093/nar/30.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fushimi K., Ray P., Kar A., Wang L., Sutherland L.C., Wu J.Y. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc. Natl. Acad. Sci. USA. 2008;105:15708–15713. doi: 10.1073/pnas.0805569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Triqueneaux G., Velten M., Franzon P., Dautry F., Jacquemin-Sablon H. RNA binding specificity of Unr, a protein with five cold shock domains. Nucleic Acids Res. 1999;27:1926–1934. doi: 10.1093/nar/27.8.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue Y., Zhou Y., Wu T., Zhu T., Ji X., Kwon Y.S., Zhang C., Yeo G., Black D.L., Sun H., et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol. Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katz Y., Wang E.T., Airoldi E.M., Burge C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flicek P., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S., et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pruitt K.D., Tatusova T., Brown G.R., Maglott D.R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dreszer T.R., Karolchik D., Zweig A.S., Hinrichs A.S., Raney B.J., Kuhn R.M., Meyer L.R., Wong M., Sloan C.A., Rosenbloom K.R., et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 38.Sanford J.R., Wang X., Mort M., Vanduyn N., Cooper D.N., Mooney S.D., Edenberg H.J., Liu Y. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfarr N., Prawitt D., Kirschfink M., Schroff C., Knuf M., Habermehl P., Mannhardt W., Zepp F., Fairbrother W.G., Loos M., et al. Linking C5 deficiency to an exonic splicing enhancer mutation. J. Immunol. 2005;174:4172–4177. doi: 10.4049/jimmunol.174.7.4172. [DOI] [PubMed] [Google Scholar]
- 40.Cunninghame Graham D.S., Akil M., Vyse T.J. Association of polymorphisms across the tyrosine kinase gene, TYK2 in UK SLE families. Rheumatology. 2007;46:927–930. doi: 10.1093/rheumatology/kel449. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J., Kuo C.C., Chen L. GC content around splice sites affects splicing through pre-mRNA secondary structures. BMC Genomics. 2011;12:90. doi: 10.1186/1471-2164-12-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amit M., Donyo M., Hollander D., Goren A., Kim E., Gelfman S., Lev-Maor G., Burstein D., Schwartz S., Postolsky B., et al. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012;1:543–556. doi: 10.1016/j.celrep.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 43.Buratti E., Baralle F.E. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell. Biol. 2004;24:10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loeb D.D., Mack A.A., Tian R. A secondary structure that contains the 5′ and 3′ splice sites suppresses splicing of duck hepatitis B virus pregenomic RNA. J. Virol. 2002;76:10195–10202. doi: 10.1128/JVI.76.20.10195-10202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacquenet S., Ropers D., Bilodeau P.S., Damier L., Mougin A., Stoltzfus C.M., Branlant C. Conserved stem-loop structures in the HIV−1 RNA region containing the A3 3′ splice site and its cis-regulatory element: possible involvement in RNA splicing. Nucleic Acids Res. 2001;29:464–478. doi: 10.1093/nar/29.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L., Zheng S. Identify alternative splicing events based on position-specific evolutionary conservation. PLoS One. 2008;3:e2806. doi: 10.1371/journal.pone.0002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xing Y., Lee C. Alternative splicing and RNA selection pressure--evolutionary consequences for eukaryotic genomes. Nat. Rev. Genet. 2006;7:499–509. doi: 10.1038/nrg1896. [DOI] [PubMed] [Google Scholar]
- 49.Ermakova E.O., Nurtdinov R.N., Gelfand M.S. Fast rate of evolution in alternatively spliced coding regions of mammalian genes. BMC Genomics. 2006;7:84. doi: 10.1186/1471-2164-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Artamonova I.I., Gelfand M.S. Comparative genomics and evolution of alternative splicing: the pessimists' science. Chem. Rev. 2007;107:3407–3430. doi: 10.1021/cr068304c. [DOI] [PubMed] [Google Scholar]
- 51.Siepel A., Pollard K.S., Haussler D. New methods for detecting lineage-specific selection. Lect. Notes Comput. Sci. 2006;3909:190–205. [Google Scholar]
- 52.Hall M., Frank E., Holmes G., Pfahringer B., Reutemann P., Witten I.H. The WEKA Data Mining Software: An Update. SIGKDD Explorations. 2009;11 [Google Scholar]
- 53.Friedman N., Geiger D., Goldszmidt M. Bayesian network classifiers. Mach. Learn. 1997;29:131–163. [Google Scholar]
- 54.Hornik K., Stinchcombe M., White H. Multilayer feedforward networks are universal approximators. Neural Netw. 1989;2:359–366. [Google Scholar]
- 55.Good I.J. The Estimation of Probabilities; an Essay on Modern Bayesian Methods. Cambridge, MA: M.I.T. Press; 1965. [Google Scholar]
- 56.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 57.Chintagunta P.K. Logit modeling - practical applications - demaris,A. J. Mark. Res. 1993;30:391–392. [Google Scholar]
- 58.Matthews B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Blanco M.A., Baraniak A.P., Lasda E.L. Alternative splicing in disease and therapy. Nat. Biotechnol. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 60.Wang G.S., Cooper T.A. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 61.Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 62.Teng M., Wang Y., Wang G., Jung J., Edenberg H.J., Sanford J.R., Liu Y. Prioritizing single-nucleotide variations that potentially regulate alternative splicing. BMC Proc. 2011;5(Suppl 9):S40. doi: 10.1186/1753-6561-5-S9-S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X., Juan L., Lv J., Wang K., Sanford J.R., Liu Y. Predicting sequence and structural specificities of RNA binding regions recognized by splicing factor SRSF1. BMC Genomics. 2011;12(Suppl 5):S8. doi: 10.1186/1471-2164-12-S5-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kazan H., Ray D., Chan E.T., Hughes T.R., Morris Q. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput. Biol. 2010;6:e1000832. doi: 10.1371/journal.pcbi.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Foat B.C., Stormo G.D. Discovering structural cis-regulatory elements by modeling the behaviors of mRNAs. Mol. Syst. Biol. 2009;5:268. doi: 10.1038/msb.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim V.N. MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 67.Yan K.S., Yan S., Farooq A., Han A., Zeng L., Zhou M.M. Structure and conserved RNA binding of the PAZ domain. Nature. 2003;426:468–474. doi: 10.1038/nature02129. [DOI] [PubMed] [Google Scholar]
- 68.Lopez de Silanes I., Zhan M., Lal A., Yang X., Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bellucci M., Agostini F., Masin M., Tartaglia G.G. Predicting protein associations with long noncoding RNAs. Nat. Methods. 2011;8:444–445. doi: 10.1038/nmeth.1611. [DOI] [PubMed] [Google Scholar]
- 70.Brandman R., Brandman Y., Pande V.S. Sequence coevolution between RNA and protein characterized by mutual information between residue triplets. PLoS One. 2012;7:e30022. doi: 10.1371/journal.pone.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang G., Wang Y., Feng W., Wang X., Yang J.Y., Zhao Y., Wang Y., Liu Y. Transcription factor and microRNA regulation in androgen-dependent and -independent prostate cancer cells. BMC Genomics. 2008;9(Suppl. 2):S22. doi: 10.1186/1471-2164-9-S2-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang T., Faraggi E., Xue B., Dunker A.K., Uversky V.N., Zhou Y. SPINE-D: accurate prediction of short and long disordered regions by a single neural-network based method. J. Biomol. Struct. Dyn. 2012;29:799–813. doi: 10.1080/073911012010525022. [DOI] [PMC free article] [PubMed] [Google Scholar]