

Abstract
Actinomycetes undergo a dramatic reorganization of metabolic and cellular machinery during a brief period of growth arrest (“metabolic switch”) preceding mycelia differentiation and the onset of secondary metabolite biosynthesis. This study explores the role of phosphorylation in coordinating the metabolic switch in the industrial actinomycete Saccharopolyspora erythraea. A total of 109 phosphopeptides from 88 proteins were detected across a 150-h fermentation using open-profile two-dimensional LC-MS proteomics and TiO2 enrichment. Quantitative analysis of the phosphopeptides and their unphosphorylated cognates was possible for 20 pairs that also displayed constant total protein expression. Enzymes from central carbon metabolism such as putative acetyl-coenzyme A carboxylase, isocitrate lyase, and 2-oxoglutarate dehydrogenase changed dramatically in the degree of phosphorylation during the stationary phase, suggesting metabolic rearrangement for the reutilization of substrates and the production of polyketide precursors. In addition, an enzyme involved in cellular response to environmental stress, trypsin-like serine protease (SACE_6340/NC_009142_6216), decreased in phosphorylation during the growth arrest stage. More important, enzymes related to the regulation of protein synthesis underwent rapid phosphorylation changes during this stage. Whereas the degree of phosphorylation of ribonuclease Rne/Rng (SACE_1406/NC_009142_1388) increased during the metabolic switch, that of two ribosomal proteins, S6 (SACE_7351/NC_009142_7233) and S32 (SACE_6101/NC_009142_5981), dramatically decreased during this stage of the fermentation, supporting the hypothesis that ribosome subpopulations differentially regulate translation before and after the metabolic switch. Overall, we show the great potential of phosphoproteomic studies to explain microbial physiology and specifically provide evidence of dynamic protein phosphorylation events across the developmental cycle of actinomycetes.
Microorganisms have evolved mechanisms that enable them to grow and rapidly adapt to changing environmental conditions. Regulation of protein activity can occur at transcriptional, translational, and/or post-translational levels. Transcriptional and translational control are slow and have high energy costs due to de novo synthesis of proteins (i.e. transcription, translation, and protein-folding processes). Conversely, protein post-translational modifications drive adaptive cellular responses more efficiently by adding or removing functional groups from specific protein residues (1). Among the post-translational modifications that regulate protein functionality, phosphorylation is by far the most studied in bacteria (2, 3).
Two-component systems involve the phosphorylation of histidine and aspartate residues and were the first studied bacterial signal transduction mechanisms (3). Pioneering studies in Escherichia coli and Bacillus subtilis demonstrated extensive serine, threonine, and tyrosine phosphorylation, predominantly at sites with eukaryotic-like phosphorylation signatures (4, 5). Since then, numerous studies have extended the repertoire of serine, threonine, and tyrosine kinases and eukaryotic-like phosphorylated proteins present in different bacteria (6–15). Apart from some studies on specific enzymes of interest in E. coli and other model organisms, these studies have focused on mapping phosphorylation sites rather than identifying the biological role of phosphorylation. In order for phosphorylation to play a role in adaptive responses, it must display quantitative and dynamic variation. Recently, in vivo phosphorylation controlling enzyme functionality was studied in E. coli and Streptomyces coelicolor developmental cycles (16, 17), showing the yet poorly explored effect of dynamic protein phosphorylation in microbial physiology.
Actinomycetes produce a large variety of secondary metabolites, including approximately half the antibiotics in current use (18). The production of secondary metabolites occurs after a critical culture transition known as the “metabolic switch” (19), believed to be triggered by nutrient limitation or oxidative stress. Understanding the regulatory mechanisms underpinning the reorganization of the metabolic and cellular machinery at the metabolic switch (20) is of both fundamental and practical importance, as efficient induction is essential for high-level production. Although some cellular regulatory mechanisms have been explained with regard to transcription (19, 20), in vivo protein phosphorylation has not been yet explored at the global level and promises to fill an important gap in the understanding of the biology of actinomycetes.
Saccharopolyspora erythraea is a soil-dwelling actinomycete from the Pseudonocaridaceae family. This soil bacterium contains within its 8.29-Mb genome the machinery required for the synthesis of more than 25 different secondary metabolites, including erythromycin, the first clinically used macrolide antibiotic (21). Although highly exploited in industry, the S. erythraea secondary metabolism remains mostly unexplored; in fact, more than 17 secondary metabolites produced by this bacterium have unknown function and chemical structure (22). In addition, even though the S. erythraea genome was completed more than half a decade ago, industrial titers of erythromycin are obtained mostly via classical methods of random mutagenesis and fermentation media optimization using complex carbon and nitrogen sources (23). Several genomic and transcriptomic studies have compared genome sequences and gene transcription between wild-type and industrial erythromycin overproducing strains (24–26). These investigations show that regulation of the erythromycin gene cluster is complex and may be regulated at the post-translational level.
Here, we present a dynamic phosphoproteomic study of the erythromycin-producing actinomycete S. erythraea. Using samples taken across a fermentation time course, a total of 109 phosphorylation sites were identified in discovery mode, before the degree of phosphorylation at each site was monitored using multiple reaction monitoring (MRM).1 Quantitatively significant changes in phosphorylation were observed for many proteins during the metabolic switch and the stationary phase, and several of these events can be directly linked to known metabolic effects. We thus present a time-resolved dynamic study of protein phosphorylation in S. erythraea that specifically provides new insights into the physiology of actinomycetes.
EXPERIMENTAL PROCEDURES
Strain and Culture Conditions
S. erythraea strain NRRL23338 was purchased from the American Type Culture Collection (ATCC number 11635TM). Unless otherwise specified, all chemicals were purchased from Sigma. Medium ISP 2 (yeast extract, 4 g/l; malt extract, 10 g/l; dextrose, 4 g/l; agar, 20 g/l) was used for spore germination and seed cultures. Medium MM-101 used in the bioreactors contained (per liter) 7 g of NH4Cl, 3 g of KH2PO4, 7 g of K2HPO4, 0.25 g of MgSO4·7 H2O, 0.0138 g of CaCl2·2 H2O, 40 g of glucose, and 4 ml of trace solution element. The trace solution composition (per liter) was 40 mg of ZnCl2, 200 mg of FeCl3·6 H2O, 10 mg of CuCl2·2 H2O, 10 mg of MnCl2·4 H2O, 10 mg of Na2B4O7·10 H2O, and 10 mg of (NH4)6Mo7O24·4 H2O. Samples were extracted from two different fermentations in 2-l Applikon reactors (Applikon Biosciences, Schiedam, The Netherlands) operated at working volumes of 1.4 l. The temperature and pH remained constant at 30 °C and 7, respectively, throughout the fermentation; the pH was controlled by the addition of 20% sodium hydroxide (NaOH) or 10.9% hydrochloric acid (HCl). Dissolved oxygen was maintained at 45% to 60% saturation by increasing the air flow and reactor mixing. Carbon dioxide production was measured using an HPR20 QIC mass spectrometer (Hiden Analytical Ltd., Warrington, UK) attached to the bioreactor's condensers.
Protein Extraction and Trypsin Digestion
To characterize and quantitate the phosphoproteome of S. erythraea, we used monitoring-initiated detection and sequencing with information-dependant acquisition. Samples were extracted at six time points across the fermentation: one time point during the exponential phase (34 h), three around the metabolic switch (48, 49, and 51 h), and finally two during the stationary phase (78 and 98 h). Proteins were extracted according to Ref. 4, incorporating some modifications. Cell pellets (100 ml of culture broth) were harvested from the bioreactor, pelleted (4 °C at 5000 rpm) using an Allegra X-15R centrifuge (Beckman Coulter), washed in 100 mm NaCl, 25 mm Tris-HCl (pH 7.5), and resuspended in lysis buffer (50 mm Tris-HCl, 5 mg/ml lysozyme, and 5 mm of each of the following phosphatase inhibitors: sodium fluoride, 2-glycerol phosphate, sodium vanadate, and sodium pyrophosphate). After 10 min of incubation at room temperature, N-octylglucoside was added at a final concentration of 1% for the solubilization of membrane proteins. Enhanced cell disruption was achieved by homogenizing cell lysates with glass beads for 5 min at 4800 rpm using a Mini-bead beater (Extech Equipment Pty Ltd, Wantirna South, Australia). Cellular debris was removed by centrifuging (10 min, 4 °C, 13,000 rpm) using a Microfuge (22R, Beckman Coulter). To remove nucleic acids, samples were incubated with DNase I (100 μg/ml; Fermentas, Vilnius, Lithuania) and RNase A (100 μg/ml; Fermentas) for 10 min at 37 °C. Finally, protein extracts were dialyzed for 16 h in deionized water using 3.5 molecular weight cutoff Side-A-Lyser dialysis cassettes (Thermo Scientific). The total protein content was quantified using a 2D Quant assay kit (GE Healthcare). Protein extract aliquots (10, 5, and 1 mg and 500, 200, 100, and 50 μg) were freeze-dried using Alpha 1–4 LSC (John Morris Scientific Pty Ltd, Willoughby, Australia) for 16 h and stored at −20 °C. Trypsin digestion was performed as described in Ref. 4. Briefly, 10 mg of dried protein was resuspended in 6 m urea, 2 m thiourea, and 2% CHAPS for protein denaturation and incubated for 45 min at room temperature with 1 mm DTT. Iodoacetamide (2.5 mm final concentration) was added, and samples were incubated for 45 min at room temperature in the dark. Samples were then diluted with 25 mm ammonium bicarbonate to obtain a final concentration of urea of <800 mm. Trypsin (Promega Gold, MS grade) was added at a ratio of 1/100 (trypsin/protein), and samples were incubated for 16 h at 37 °C. Reversed-phase chromatography was used to clean the digested peptide mixtures using C-18 cartridges (Sep-Pak tC18, Waters, Milford, MA) eluting with 60% acetonitrile (v/v). Residual acetonitrile was removed by vacuum centrifugation (Eppendorf, Hamburg, Germany) and resuspended in 0.1% formic acid prior to analysis. All samples were processed in parallel.
Phosphopeptide Enrichment
A total of three 10-mg samples (time points 34 and 48 h, time points 49 and 51 h, and time points 78 and 98 h) were fractionated (16 fractions) using strong cation exchange chromatography as described in Ref. 5. Briefly, samples were loaded onto a 1-ml Resource S column (GE Healthcare) in solvent A (5 mm KH2PO4, 30% acetonitrile, 0.1% trifluoroacetic acid, pH 2.7) at 1 ml/min. Sample elutions were collected in 16 2-ml fractions using a 0%–30% gradient of solvent B (5 mm KH2PO4, 30% acetonitrile, 350 mm KCl, 0.1% trifluoroacetic acid, pH 2.7) over 30 min. Each fraction was desalted using reversed-phase chromatography, concentrated by vacuum centrifugation, and pH adjusted (<2.7) with 0.1% formic acid. The resulting 48 fractions were enriched for phosphorylated peptides using titanium dioxide (TiO2) chromatography with Phos-TiO2 (GL Sciences Inc., Tokyo, Japan) according to the manufacturer's instructions.
Phosphopeptide Identification
Peptide identification was performed in an LC MS/MS QSTAR Elite (AB Sciex, Ontario, Canada). The LC system was equipped with a Vydac MS C18 300-Å, 150 mm × 0.3 mm column (Grace Davison Discovery Sciences, Deerfield, IL) operated at 30 °C with a 0%–80% acetonitrile gradient (in 0.1% formic acid) for 105 min at a flow rate of 3 μl/min. All MS/MS raw data are available online through the University of Queensland website (S. erythraea phosphoproteomic data (385 MB)). Proteins were identified via advanced information-dependent acquisition of the fragmentation spectra of one to five charged peptides with a precursor selection window of m/z 100–1800 using enhanced pulsed extraction of fragments (using Analyst 1.5.2; AB Sciex), employing specific features such as “Smart Collision” and “Smart Exit” (fragment intensity multiplier set to 2.0 and maximum accumulation time of 1.5 s) to obtain MS/MS spectra. Tandem mass spectra were acquired for 1 s, and fragmented peptides were selected for sequencing for 12 s in positive mode. The Paragon search algorithm (27) from Protein Pilot 4.0 software (Applied Biosystems, Foster City, CA) was used to identify all phosphoproteins. The mass tolerance values for precursor ions and fragment ions were set to the default values of the Paragon search algorithm. Trypsin was specified as the digesting protease, the iodoacetamide derivative of cysteine (carboxyamidomethylcysteine) was specified as the fixed modification, and urea denaturation and phosphorylation were specified as special factors. The sequence database used was taken from the S. erythraea Genome Project website (release version 15/03/2007). A false discovery rate analysis was performed for all searches. Hits were considered positive when at least two peptides with more than six residues and 90% confidence were detected. A monitoring-initiated detection and sequencing workflow was used for the relative quantification of peptide phosphorylation (28).
MRM Development
MRM assays for all the identified phosphopeptides and their unphosphorylated counterparts were developed and optimized from TiO2-enriched samples with MRMPilotTM 2.0 software (AB Sciex) according to the software manual. All quantitative experiments were performed from total protein extracts (without TiO2 enrichment or chromatographic fractionation) using a triple quadrupole mass spectrometer (QTRAPTM 4000, Applied Biosystems) with an electrospray ion source configured in positive mode. LC was performed using a 100 mm × 2.1 mm 2.6-μm, 100-Å Kinetix C18 column (Phenomenex, Torrance, CA) running a gradient of 2%–80% acetonitrile (in 0.1% formic acid) for 100 min. Mobile phase A was an aqueous solution of 2% acetonitrile and 0.1% formic acid, and solvent B was 2% MilliQ water and 0.1% formic acid in 100% acetonitrile. Every peptide and phosphorylated cognate was monitored by at least two transition ions overlapping in retention time in the LC chromatogram (supplemental Table S7). The MS scan was performed with the following parameters: ion source voltage, 5400 V; temperature, 350 °C; curtain gas, 20 psi; collisionally activated dissociation gas, high; TurboIonSpray nebulizer gas or Atmospheric-pressure chemical ionization nebulizer gas (Gas 1), 60 psi; TurboIonSpray heater gas (Gas 2), 60 psi; orifice (differential pressure), 80. To minimize technical variations, all injections were performed in triplicate.
Data Processing
NetPhos (29) and NetPhos-Bac (30) were used as primary sources for identifying phosphorylation sites. PhosCalc (31) was used to assign probabilities to the potential phosphorylation sites on the identified peptides. All possible phosphorylation positions, namely, tyrosine, threonine, serine, histidine, and aspartic acid, were evaluated. DAVID Bioinformatics Resources 6.7 (32) was used to perform gene-enrichment and functional annotation analysis. For all MRM experiments, automatic quantification methods were built in Analyst 1.5.2 (AB Sciex). All peaks were manually verified by visualizing their shape and correct elution time. This procedure was performed by two different people for two biological replicates in order to avoid bias in the analysis. The abundance for each peptide and its phosphorylated cognate was estimated as the average peak area of three technical replicates. The phosphorylation ratio was estimated from the area of the phosphorylated and unphosphorylated peptides. The phosphorylation ratios were log transformed to satisfy the linear model assumption of residual normality and to stabilize variances (phosphorylation ratio range of −3 to +3). The total protein amount was estimated from the logged sum of peak areas for the phosphorylated and nonphosphorylated peptide cognates. Cluster 3.0 was used for all hierarchical cluster analyses. Complete linkage clustering was used to compute distances between groups of phosphoproteins. Dendrograms were made using Java TreeView 1.1.5r2 (33). Similarities between biological replicates were evaluated according to the Spearman rank correlation coefficient using a coefficient cutoff of 0.7 and p values < 0.2. The statistical significance (p value) for the correlation coefficients was estimated using R.
Analytical Procedures
Cell growth was monitored by measuring A450 and by determining the cell dry weight during the fermentation. The cell dry weight was quantified by filtering 15 ml of broth using 22-μm nylon membranes (Millipore, Bedford, MA) and drying the samples at 60 °C for 8 h. The concentration of glucose was determined via high-performance liquid chromatography (Agilent 1200 HPLC system) (as described in Ref. 34) using a Phenomenex Rezex RHM Monosaccharide H+ column (300 × 7.8 mm; 8 μm) and detected by Refractive index. Erythromycin production was determined via LC-MS using a Dionex UltiMate 3000 liquid chromatography system (Dionex, Sunnyvale, CA) coupled with an AB Sciex 4000 QTRAP mass spectrometer (AB Sciex, Ontario, Canada) as described in Ref. 35.
RESULTS
S. erythraea Displays Biphasic Growth in a Bioreactor
S. erythraea fermentation displayed a two-stage growth curve common to secondary metabolite producers. For the first 48 h, biomass was produced exponentially at a constant rate of 0.11 h−1. A transient 30- to 60-min growth interruption (known as the metabolic switch) was observed around the 48th hour of culture. We monitored the metabolic switch online by connecting a mass spectrometer to the outlet of the bioreactor condenser. A drop of CO2 production was clearly observed in the middle of the exponential phase, which is an indicator of growth arrest (Fig. 1). After the metabolic switch, the culture recommenced growth at the initial rate for ∼10 h before entering the stationary phase. The main antibiotic produced by S. erythraea, erythromycin, was produced in a growth-independent manner for the first 90 h at a rate of 1.01 mg/g dry weight per hour. As the culture entered stationary phase, a significant increase in erythromycin production/release was observed (Fig. 1).
Fig. 1.
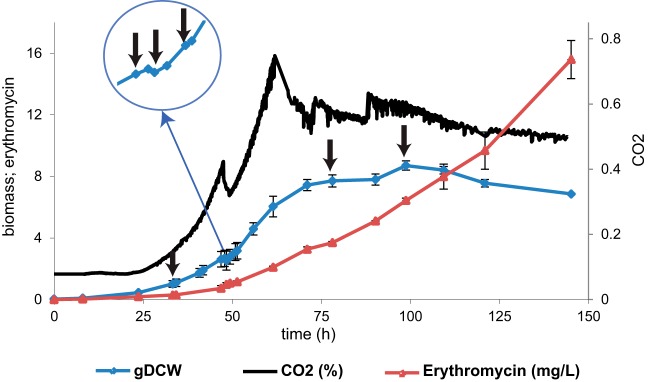
S. erythraea fermentation time course in chemically defined media (MM101). S. erythraea NRRL23338 displays two-stage behavior in MM101. Erythromycin production (red), dry cell weight (gDCW) (blue), and CO2 (black) were measured during the fermentation. The fermentation is characterized by a growth arrest stage known as the metabolic switch. To characterize post-transcriptional modifications involved in the metabolic switch, samples were harvested from the bioreactor at several time points, indicated in the graph with black arrows.
The S. erythraea Phosphoproteome: General Features
Open-profile two-dimensional LC-MS across six culture time points (see “Experimental Procedures”) identified a total of 109 phosphopeptides from 88 proteins (Fig. 2 and supplemental Table S1). Genes encoding the phosphorylated peptides were evenly distributed across the core region (53%), which encodes essential genes and primary metabolic functions, and the non-core region of the chromosome (47%), which codes for secondary metabolic functions (Fig. 2A). Enrichment scores (ESs) were estimated to systematically assess whether some subset of the phosphoproteins showed significant overrepresentation of certain cellular functions (the greater the ES, the more represented a certain cellular function is in the dataset). Functional annotation analysis revealed that the S. erythraea phosphoproteome is enriched for proteins related to ATP binding processes (ES = 1.23), acyl carrier activity (ES = 1.19), NAD(P) binding proteins (ES = 0.94), and FAD binding proteins (ES = 0.84). The least representative functional groups were glucose metabolism (ES = 0.68), phosphorous metabolism (ES = 0.51), zinc-binding proteins (ES = 0.46), oxidation/reduction-related proteins (ES = 0.4), proteolytic processes (ES = 0.31), and DNA-binding proteins (ES = 0.16) (Fig. 2A).
Fig. 2.

S. erythraea phosphoproteome topography. A, circular representation of the chromosome with the core (purple) and non-core (yellow) regions shown. The genes encoding the 88 identified phosphoproteins were evenly distributed in the chromosome. The bar chart indicates the S. erythraea phosphoproteome distribution according to gene enrichment functional analysis using DAVID Bioinformatic Resources 6.7 (32). The enrichment score reflects the degree to which a cellular function is overrepresented among a set of cellular functions. B, pie charts representing the phosphorylation residue distribution in S. erythraea. The upper panel shows the distribution according to the NetPhos (for eukaryotic-like phosphorylation signatures) (29) and NetPhos-Bac (for bacterium-like phosphorylation signatures) (30) algorithms. The lower panel shows the phosphosite distribution determined by PhosCalc (31).
Of all the identified proteins, 22 have been identified in other actinomycete phosphoproteomes (7, 13, 14, 16). For example, we found several common phosphorylated enzymes from central carbon metabolism, such as catalase, transketolase, acetyl/propionyl-CoA carboxylase, and 2-oxoglutarate dehydrogenase. In addition, enzymes involved in secondary metabolism were also found to be phosphorylated. Examples included polyketide synthase subunits, transposases, and TetR-family transcriptional regulators (7, 13, 14, 16). Regarding other bacterial phosphoproteomes (4–14, 16), a total of 54 phosphoproteins are shared with the dataset presented here. The most common phosphoproteins were those related to global cellular processes (e.g. cell division control proteins, elongation factors, and transcriptional regulators) and energy production proteins such as ATP synthase subunits. Finally, we found 12 phosphorylated proteins exclusive to the S. erythraea phosphoproteome, including trehalose-phosphatase, Rne/Rng family ribonuclease, isocitrate lyase, DNA recombination protein RecA, thiosulfate sulfurtransferase, porphobilinogen deaminase HemC, adenosine kinase, glycine dehydrogenase, Xaa-Pro dipeptidase, FMNH2 utilizing oxygenase, betaine-aldehyde dehydrogenase, and polyphosphate kinase (supplemental Table S2).
Only half of the phosphorylation sites identified were detected by NetPhos (29) or NetPhos-Bac (30) (Fig. 2B). This low identification rate is likely because these prediction tools were constructed based on model organisms such as E. coli, B. subtilis, and Saccharomyces cerevisiae. Using PhosCalc (31), 60% of the phosphosites were assigned to a single residue (supplemental Table S3). The most phosphorylated residue was serine (26.5%), followed by threonine (23%), histidine (5.3%), and tyrosine (4.4%) (Fig. 2B). Annotated spectra of all detected phosphopeptides and their corresponding NetPhos and NetPhos-Bac scores are presented in supplementary File S1.
The S. erythraea Phosphoproteome Displays Dynamic Behavior across the Fermentation Time Course
We estimated phosphorylation dynamics by developing MRM assays for all the identified phosphopeptides and their unphosphorylated cognates. To this end, data from discovery proteomics were used to build an MS/MS library of spectra and facilitate the design process. Discovery data were loaded into MRMPilot and used to design and select the best fragment ions. In order to rectify the identity of the analytes, all MRMs were verified by ensuring complete co-elution/overlap of at least four fragment ions for each analyte (supplementary File S2). Furthermore, we verified the quantifiable signal-to-noise ratio in total protein extracts sampled across six time points of the fermentation. Out of 109 phosphopeptides discovered from enriched samples, we restricted our analysis to 60 phosphopeptides (supplemental Table S4) that satisfied the following criteria: (i) at least two independent consistent MRMs each for the phosphorylated and unphosphorylated peptides, (ii) consistency in retention time, (iii) identified in both biological replicates, and (iv) signal intensity based on the limit of quantification and signal-to-noise ratio.
The degree of phosphorylation was approximated based on the ratio of the signal intensities of the phosphopeptide ion and its unmodified cognate, as previously done to assess protein functionality regulated by phosphorylation in yeast central carbon metabolism (36). Reproducible phosphorylation events among two independent experiments were selected by estimating the Spearman rank correlation coefficients (see “Experimental Procedures”). A total of 44 phosphopeptides with correlation coefficients of >0.5 were clustered and classified according to their degree of phosphorylation and functional annotation (Fig. 3).
Fig. 3.

Selection criteria employed for the identification of proteins potentially regulated by phosphorylation. The signal-to-noise ratio was first verified independently for two biological replicates. A total of 60 quantifiable phosphopeptides were obtained. The Spearman rank correlation coefficient was estimated using a cutoff threshold of 0.5 (horizontal axis). The resulting 44 phosphopeptides were clustered according to their phosphorylation ratio profiles and classified into three function categories (“Metabolism,” “Signaling and Cellular Processes,” and “Hypothetical Proteins”) according to the genome annotation. A total of 29 phosphopeptides showed dynamic behavior (table, second and third rows). The total amount of peptide quantified was estimated by adding the peak area of peptide + phosphopeptide across the time course, and a maximum average variation of 2% was used as a threshold (blue-shaded boxes). For simplicity, the recent genome annotation has been omitted from the figure.
Profile analysis identified seven distinct phosphorylation clusters (supplemental Fig. S1 and supplemental Table S5). Ignoring positional information, these clusters can be mapped into three general profiles (Fig. 3). Profile A includes 15 phosphopeptides with constant degrees of phosphorylation across the fermentation (see clusters III and V in the supplementary figures). Among this group we found proteins related to general cellular processes, such as elongation factor TufA (SACE_6838/NC_009142_6720) and cell division control protein Cdc48 (SACE_0519/NC_009142_05191), and enzymes related to metabolic functions, such as peptide ABC transporter (SACE_6324/NC_009142_6201) and type I polyketide synthase (SACE_4140/NC_009142_4094).
Proteins with an increased degree of phosphorylation during the metabolic switch and stationary phase were grouped in Profile B (see clusters I, IV, and VII in the supplementary figures). These proteins are presumably involved in the activation of cellular processes during the metabolic switch. Among this group, we found a ribonuclease of the Rne/Rng family (SACE_1406/NC_009142_1388) and ArsR-family transcriptional regulator (SACE_4355/NC_009142_4304). Central carbon metabolic enzymes found with this profile (isocitrate lyase (SACE_1449/NC_009142_1431), adenosine kinase (SACE_3899/NC_009142_3856), pyruvate carboxylase (SACE_6118/NC_009142_5998), 2-oxoglutarate dehydrogenase (SACE_6385/NC_009142_6262), and phosphoglycerate mutase (SACE_3448/NC_009142_3404)) suggest that repression of anabolic enzymes and activation of systems for nutrient recycling might occur during this stage of fermentation. Additionally, this profile also includes four hypothetical proteins located in the core region of the chromosome (SACE_3064/NC_009142_3026, SACE_3268/NC_009142_3226, SACE_3282/NC_009142_3240, and SACE_4206/NC_009142_4158) and three poorly characterized integral membrane proteins (SACE_6667/NC_009142_6544, SACE_6793/NC_009142_6674, and SACE_7119/NC_009142_7002).
Lastly, Profile C includes proteins with decreased phosphorylation during the metabolic switch and stationary phase (see clusters II and VI in the supplementary figures). Three enzymes with metabolic functions—acetyl/propionyl-CoA carboxylase (SACE_4237/NC_009142_4188), modular polyketide synthase (SACE_2875/NC_009142_2839), and FMNH2-dependent monooxygenase (SACE_4061/NC_009142_4018)—were found within this group. We also found a trypsin-like serine protease (SACE_6340/NC_009142_6216), a protein involved in cell dismantling and oxidative stress mechanisms in related actinomycetes (37, 38). Interestingly, ribosomal proteins S32 and S6 (SACE_6101/NC_009142_5981 and SACE_7023/NC_009142_6904, respectively) showed similar dephosphorylation behavior during the growth arrest stage. Lastly, SACE_6254/NC_009142_6132 and SACE_7023/NC_009142_6904, two hypothetical proteins located in the non-core region of the chromosome, were found in this group of proteins.
The changes observed in the phosphorylation ratios in Profiles B and C might reflect kinase- or phosphatase-mediated regulation events controlling metabolic and developmental processes. However, an altered ratio of protein phosphorylation may also result from dilution due to increased expression or preferential degradation of phospho- or nonphosphorylated proteins. The logged sum of peak areas for the phosphorylated and nonphosphorylated peptide cognates provides an indication of the total protein amount. For 20 out of 29 peptides in Profiles B and C, the difference across the profile was less than 2% (Fig. 3). Given that biological interpretation is more difficult when the total amount of protein changes across the developmental cycle, we limited our discussion to phosphoproteins that are differentially phosphorylated with less than 2% variation in the total amount of protein (Fig. 3, blue shaded boxes).
DISCUSSION
Actinomycetes undergo a dramatic reorganization of metabolic and cellular machinery during the metabolic switch leading to mycelia differentiation and the onset of secondary metabolite biosynthesis (38–40). We speculated that protein phosphorylation might play an important role in the regulation of this process. The S. erythraea phosphoproteome reported here consists of 109 phosphopeptides from 88 proteins (Fig. 2), which is similar in size to most phosphoproteomes reported for other bacteria (6–14). Sixty percent of phosphorylation events could be reliably assigned to a single site, and, as in other phosphoproteomes, serine and threonine accounted for the vast majority of events and were roughly equal in frequency (47% on serine and 45% on threonine) (41). Only half the phosphorylation events could have been predicted with NetPhos or NetPhos-Bac software.
Out of the 109 phosphopeptides, quantitative-dynamic MRM analysis was achieved for 60 phosphopeptides and their cognate non-phosphopeptides across six time points of the fermentation (Fig. 1). The failure to detect the remaining 49 peptides may be attributed to the absence of TiO2 enrichment for paired quantification. A total of 44 peptides showed reproducible phosphorylation profiles across the two experiments (Spearman rank correlation coefficient > 0.5).
Clustering analysis revealed three phosphorylation profiles (Fig. 3). Fifteen peptides showed constant phosphorylation throughout the fermentation (Profile A) and thus are unlikely to be involved in regulation. Of the 29 peptides displaying dynamic phosphorylation (Profiles B and C), 20 also had near-constant total protein contents, indicating kinase/phosphatase activity potentially regulating the developmental cycle. The phosphoproteins were functionally classified according to the genome annotation in three groups, namely, unknown and hypothetical proteins, metabolism, and cellular processes/signaling.
Unknown and Hypothetical Proteins
Seven hypothetical proteins (SACE_3064/NC_009142_3026, SACE_3268/NC_009142_3226, SACE_3282/NC_009142_3240, SACE_6667/NC_009142_6544, SACE_6254/NC_009142_6132, SACE_6793/NC_009142_6674, and SACE_7023/NC_009142_6904) were found with dynamic phosphorylation profiles across the fermentation (Fig. 4). A detailed sequence analysis identified SACE_6793/NC_009142_6674 as the only protein with a homolog among related actinomycetes. SACE_6793/NC_009142_6674 putatively encodes an integral membrane protein part of a secretion system highly conserved among actinomycetes (ESX-1) (42). In S. coelicolor, knock-out of ESX-1 generates deficient coordination of cell division with segregation of nucleoids (43). Although the functionality of the ESX-1 system depends on a seven-amino-acid peptide signal in Mycobacterium tuberculosis (44), it remains unknown how this system is targeted for secretion in S. coelicolor. It is unclear what role increased phosphorylation over the stationary phase may play, but its presence suggests that SACE_6793/NC_009142_6674 could be involved in the regulation of this important secretion system.
Fig. 4.

Two-dimensional plots of hypothetical proteins with dynamic phosphorylation profiles across the fermentation. The phosphorylation ratio (log P-ratio) and total peptide (log peak area (PA)) are plotted as solid red and dotted blue lines, respectively. Graphs represent SACE_3064/NC_009142_3026 (upper left panel); surface protein, SACE_6667/NC_009142_6544 (upper middle panel); SACE_3268/NC_009142_3226 (upper right panel); SACE_6793/NC_009142_6674 (middle left panel); selenium binding protein, SACE_3282/NC_009142_3240 (middle center panel); SACE_7023/NC_009142_6904 (middle right panel); and SACE_6254/NC_009142_6132 (lower left panel). Total peptide measurements were estimated as log (peak area of phosphopeptide + log peak area of peptide). For simplicity, the recent genome annotation has been omitted from the figure.
Metabolic Processes
Eight proteins related to either anabolic or catabolic pathways were detected with dynamic phosphorylation profiles. A putative acetyl-CoA carboxylase (SACE_4237/NC_009142_4188) was found to be phosphorylated only during the metabolic switch (Fig. 5). Acetyl-CoA carboxylases catalyze the conversion of acetyl-CoA to malonyl-CoA in the first step of the biosynthesis of fatty acids and the formation of polyketide precursors (45). Isocitrate lyase (ICL) and the E1 subunit of 2-oxoglutarate dehydrogenase (ODH-E1) had opposite phosphorylation profiles (Fig. 6). This result correlates with the natural competition for carbon flux between the glyoxylate shunt and the TCA cycle. Sequence analyses demonstrate that isocitrate lyase is highly conserved; however, their regulation seems to differ significantly. In E. coli, decreased isocitrate lyase activity is observed when the enzyme is dephosphorylated (46), whereas the opposite effect has been demonstrated in yeast (47). 2-Oxoglutarate dehydrogenase was dephosphorylated as cells entered the stationary phase, but the role of this phosphorylation event remains unknown. Phosphorylation of 2-oxoglutarate dehydrogenase has been identified in other bacterial phosphoproteomes (11). Beyond their well-known metabolic functions, both enzymes are involved in the biosynthesis of TCA intermediates, specifically succinate and succinyl-CoA. We consider this relevant, as succinyl-CoA is the precursor of the polyketide backbone molecule methyl malonyl-CoA (48). Several metabolic engineering studies have identified the availability of acyl-CoA derivatives as the main factor for improving polyketide production (49, 50). Correlating these phosphorylation events during the stationary phase (when secondary metabolism is active) suggests a regulatory mechanism that facilitates the biosynthesis of methyl malonyl-CoA. This result not only shows a link between enzyme phosphorylation and the transition between primary and secondary metabolism, but also provides new insights into the use of regulatory mechanisms as strategies to improve secondary metabolite producing strains.
Fig. 5.

Two-dimensional plots of proteins related to metabolic pathways with dynamic phosphorylation profiles across the fermentation. The phosphorylation ratio (log P-ratio) and total peptide (log peak area (PA)) are plotted as solid red and dotted blue lines, respectively. Graphs represent isocitrate lyase, SACE_1449/NC_009142_1431 (upper left panel); oxoglutarate dehydrogenase E1, SACE_6385/NC_009142_6262 (upper middle panel); acetyl-CoA carboxylase, SACE_4237/NC_009142_4188 (upper right panel); adenosine kinase, SACE_3899/NC_009142_3856 (middle left panel); adenylylsulfate kinase, SACE_1473/NC_009142_1456 (middle center panel); FAD-linked oxidoreductase, SACE_0195/NC_009142_0193 (middle right panel); FMNH2-dependent monooxygenase, SACE_4061/NC_009142_4018 (lower left panel); and cysteine desulfurase, SACE_2177/NC_009142_2150 (lower middle panel). Total peptide measurements were estimated as log (peak area of phosphopeptide + log peak area of peptide). For simplicity, the recent genome annotation has been omitted from the figure.
Fig. 6.

Two-dimensional plots of proteins related to cellular processes displaying dynamic phosphorylation profiles across the fermentation. The phosphorylation ratio (log P-ratio) and total peptide (log peak area (PA)) are plotted as solid red and dotted blue lines, respectively. Graphs represent Rne/Rng family ribonuclease, SACE_1406–2/NC_009142_1388–2 (upper left panel); trypsin-like serine protease, SACE_6340/NC_009142_6216 (upper middle panel); ribosomal protein S6, SACE_7351/NC_009142_7233 (upper right panel); ArsR family transcriptional regulator, SACE_4355/NC_009142_4304 (lower left panel); and ribosomal protein S32, SACE_6101/NC_009142_5981 (lower middle panel). Total peptide measurements were estimated as log (peak area of phosphopeptide + log peak area of peptide). For simplicity, the recent genome annotation has been omitted from the figure.
Another enzyme found with an increasing phosphorylation ratio during the late stage of the culture was adenosine kinase (SACE_3899/NC_009142_3856), which catalyzes the biosynthesis of adenosine monophosphate from adenosine. The phosphorylation site is not in the putative catalytic site, and increased phosphorylation may be linked to increased enzyme regulation. Adenosine kinase is essential for sporulation (51); thus the increase in phosphorylation as cells enter the stationary phase may reflect increased enzyme activity. Finally, several enzymes involved in sulfur and redox metabolism were identified: adenylylsulfate kinase (SACE_1473/NC_009142_1456), cysteine desulfurase (SACE_2177/NC_009142_2150), FAD-linked oxidoreductase (SACE_0195/NC_009142_0193), and FMNH2-dependent monooxygenase (SACE_4061/NC_009142_4018).
Signaling and Global Cellular Processes
Five proteins involved in global cellular processes with dynamic phosphorylation profiles were identified (Fig. 6). The first protein, SACE_4355/NC_009142_4304, encodes for a transcriptional regulator of the ArsR family. The phosphorylation degree of this regulator was constant at the early stages of the culture and increased during the metabolic switch and the stationary phase. Although this class of transcriptional regulators is conserved among actinomycetes, their function has not been deeply investigated. In several studies, this regulator has been related to the cellular response to hypoxic adaptation and the regulation of the phoP regulon (52, 53).
Ribonuclease E/G (SACE_1406/NC_009142_1388) phosphorylation dramatically increased during the metabolic switch (Fig. 6). Ribonucleases (RNases) of the Rne/Rng family are specific endonucleases involved in processing 16S and 5S RNA precursors (54). Targeted RNA degradation has been suggested as a global post-transcriptional mechanism for regulating the developmental cycle and antibiotic production in S. erythraea (20) and S. coelicolor (55). RNase catalysis regulated by phosphorylation has been reported in eukaryotic systems (56) and E. coli for the RNase catalytic domain of the polynucleotide phosphorylase (57). This study provides evidence that dynamic phosphorylation of the ribonuclease E/G is correlated with the metabolic switch, suggesting cross-talk between post-transcriptional and post-translational regulatory mechanisms.
Another protein found with dynamic phosphorylation across the developmental cycle was a trypsin-like serine protease (SACE_6340/NC_009142_6216). This enzyme, which was phosphorylated during early stages of the fermentation, rapidly decreased in phosphorylation degree at the metabolic switch (Fig. 6). Sequence alignment showed that this enzyme is homologous to high temperature requirement A (HtrA) protease, widely conserved in single-celled and multicellular organisms (58). HtrA protease is essentially involved in ATP-independent protein quality control. For example, bacterial HtrA proteases are essential for the stress response to protein denaturation (58). In addition, HtrA functions as a nuclear serine protease essential for apoptosis in S. cerevisiae (59). Despite being widely characterized, phosphorylation-mediated activation has been reported only for mitochondrial HtrA2 (60). In this study, the phosphorylation profile observed for HtrA suggested that this enzyme could be regulated at the post-translational level, and this provides new insights into the regulation of protein degradation processes during the metabolic switch in actinomycetes.
Finally, two ribosomal proteins, S32 and S6 (SACE_6101/NC_009142_5981 and SACE_7351/NC_009142_7233, respectively), were found to have notable phosphorylation dynamics. While constitutively transcribed, both proteins decreased dramatically in phosphorylation degree at the metabolic switch (Fig. 6). This result is consistent with the “ribosome filter hypothesis” (61), which establishes that the phosphorylation of some ribosomal proteins, particularly S6, generates variations in the stoichiometry of proteins associated with the ribosome. These ribosomal subpopulations selectively translate specific subsets of mRNAs, thus regulating translation (62). Evidence of ribosomal subpopulations in actinomycetes has been reported for S. erythraea (20) and S. coelicolor (63–65), in which rRNA and ribosomal protein operons are differentially expressed and undergo targeted degradation. The results presented here suggest that ribosomal subpopulations might exist in S. erythraea, regulating gene expression at the translational level.
In summary, this work represents the only in vivo dynamic phosphoproteomic study of S. erythraea to date and highlights the importance of studying phosphorylation dynamics to understand physiological processes in actinomycetes. In addition, the results presented provide new hypotheses for the regulation of central carbon metabolic enzymes and developmental programs during the metabolic switch and stationary phase.
Supplementary Material
Acknowledgments
We acknowledge Dr. Eduardo Rodriguez, Dr. Mark Hodson, and Dr. Robert Speight for their valuable comments on this paper. We thank Alun Jones and Amanda Nouwens from the IMB and SCMB mass spectrometry facilities for LC-MS injections.
Footnotes
Author contributions: C.L., E.M., and L.K.N. designed research; C.L., S.L., and E.M. performed research; C.L., S.L., and E.M. analyzed data; C.L., S.L., E.M., and L.K.N. wrote the paper.
* We gratefully acknowledge the financial support of the University of Queensland (Grant No. ECR 2010002194), the Mexican Council for Science and Technology (CONACyT), and the Australian Institute for Bioengineering and Nanotechnology (AIBN).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MRM
- multiple reaction monitoring
- CoA
- coenzyme A
- ES
- enrichment score
- HtrA
- high temperature requirement A
- TCA
- tricarboxylic acid.
REFERENCES
- 1. Deribe Y. L., Pawson T., Dikic I. (2010) Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 [DOI] [PubMed] [Google Scholar]
- 2. Mijakovic I. (2010) Protein phosphorylation in bacteria. Microbe 5, 21–25 [Google Scholar]
- 3. Kobir A., Shi L., Boskovic A., Grangeasse C., Franjevic D., Mijakovic I. (2011) Protein phosphorylation in bacterial signal transduction. Biochim. Biophys. Acta Gen. Subjects 1810, 989–994 [DOI] [PubMed] [Google Scholar]
- 4. Macek B., Gnad F., Soufi B., Kumar C., Olsen J. V., Mijakovic I., Mann M. (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 5. Macek B., Mijakovic I., Olsen J. V., Gnad F., Kumar C., Jensen P. R., Mann M. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 6. Voisin S., Watson D. C., Tessier L., Ding W., Foote S., Bhatia S., Kelly J. F., Young N. M. (2007) The cytoplasmic phosphoproteome of the Gram-negative bacterium Campylobacter jejuni: evidence for modification by unidentified protein kinases. Proteomics 7, 4338–4348 [DOI] [PubMed] [Google Scholar]
- 7. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C.-M., Bemis G., Church G. M., Steen H., Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidl S. R., Gronau K., Pietack N., Hecker M., Becher D., Stülke J. (2010) The phosphoproteome of the minimal bacterium Mycoplasma pneumoniae: analysis of the complete known ser/thr kinome suggests the existence of novel kinases. Mol. Cell. Proteomics 9, 1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun X., Ge F., Xiao C.-L., Yin X.-F., Ge R., Zhang L.-H., He Q.-Y. (2009) Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9, 275–282 [DOI] [PubMed] [Google Scholar]
- 10. Lin M.-H., Hsu T.-L., Lin S.-Y., Pan Y.-J., Jan J.-T., Wang J.-T., Khoo K.-H., Wu S.-H. (2009) Phosphoproteomics of Klebsiella pneumoniae NTUH-K2044 reveals a tight link between tyrosine phosphorylation and virulence. Mol. Cell. Proteomics 8, 2613–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ravichandran A., Sugiyama N., Tomita M., Swarup S., Ishihama Y. (2009) Ser/Thr/Tyr phosphoproteome analysis of pathogenic and non-pathogenic Pseudomonas species. Proteomics 9, 2764–2775 [DOI] [PubMed] [Google Scholar]
- 12. Soufi B., Gnad F., Jensen P. R., Petranovic D., Mann M., Mijakovic I., Macek B. (2008) The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493 [DOI] [PubMed] [Google Scholar]
- 13. Bendt A. K., Burkovski A., Schaffer S., Bott M., Farwick M., Hermann T. (2003) Towards a phosphoproteome map of Corynebacterium glutamicum. Proteomics 3, 1637–1646 [DOI] [PubMed] [Google Scholar]
- 14. Parker J. L., Jones A. M. E., Serazetdinova L., Saalbach G., Bibb M. J., Naldrett M. J. (2010) Analysis of the phosphoproteome of the multicellular bacterium Streptomyces coelicolor A3(2) by protein/peptide fractionation, phosphopeptide enrichment and high-accuracy mass spectrometry. Proteomics 10, 2486–2497 [DOI] [PubMed] [Google Scholar]
- 15. Soufi B., Kumar C., Gnad F., Mann M., Mijakovic I., Macek B. (2010) Stable isotope labeling by amino acids in cell culture (SILAC) applied to quantitative proteomics of Bacillus subtilis. J. Proteome Res. 9, 3638–3646 [DOI] [PubMed] [Google Scholar]
- 16. Manteca A., Ye J., Sanchez J., Jensen O. N. (2011) Phosphoproteome analysis of Streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J. Proteome Res. 10, 5481–5492 [DOI] [PubMed] [Google Scholar]
- 17. Soares N. C., Spät P., Krug K., Macek B. (2013) Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res. 12, 2611–2621 [DOI] [PubMed] [Google Scholar]
- 18. Berdy J. (2005) Bioactive microbial metabolites. J. Antibiotics 58, 1–26 [DOI] [PubMed] [Google Scholar]
- 19. Nieselt K., Battke F., Herbig A., Bruheim P., Wentzel A., Jakobsen O., Sletta H., Alam M., Merlo M., Moore J., Omara W., Morrissey E., Juarez-Hermosillo M., Rodriguez-Garcia A., Nentwich M., Thomas L., Iqbal M., Legaie R., Gaze W., Challis G., Jansen R., Dijkhuizen L., Rand D., Wild D., Bonin M., Reuther J., Wohlleben W., Smith M., Burroughs N., Martin J. (2010) The dynamic architecture of the metabolic switch in Streptomyces coelicolor. BMC Genomics 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marcellin E., Mercer T., Licona-Cassani C., Palfreyman R., Dinger M., Steen J., Mattick J., Nielsen L. (2013) Saccharopolyspora erythraea's genome is organised in high-order transcriptional regions mediated by targeted degradation at the metabolic switch. BMC Genomics 14, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oliynyk M., Samborskyy M., Lester J. B., Mironenko T., Scott N., Dickens S., Haydock S. F., Leadlay P. F. (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol. 25, 447–453 [DOI] [PubMed] [Google Scholar]
- 22. Nett M., Ikeda H., Moore B. S. (2009) Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26, 1362–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Enshasy H. A., Mohamed N. A., Farid M. A., El-Diwany A. I. (2008) Improvement of erythromycin production by Saccharopolyspora erythraea in molasses based medium through cultivation medium optimization. Bioresour. Technol. 99, 4263–4268 [DOI] [PubMed] [Google Scholar]
- 24. Li Y.-Y., Chang X., Yu W.-B., Li H., Ye Z.-Q., Yu H., Liu B.-H., Zhang Y., Zhang S.-L., Ye B.-C., Li Y.-X. (2013) Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S. erythraea E3 and NRRL23338 strains. BMC Genomics 14, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu W.-B., Yu W.-B., Gao S.-H., Ye B.-C. (2013) Genome sequence of Saccharopolyspora erythraea D, a hyperproducer of erythromycin. Genome Announc. 1, e00718–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peano C., Tala A., Corti G., Pasanisi D., Durante M., Mita G., Bicciato S., De Bellis G., Alifano P. (2012) Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL 2338 and a classically improved erythromycin over-producing strain. Microb. Cell Fact. 11, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 28. Unwin R. D., Griffiths J. R., Leverentz M. K., Grallert A., Hagan I. M., Whetton A. D. (2005) Multiple reaction monitoring to identify sites of protein phosphorylation with high sensitivity. Mol. Cell. Proteomics 4, 1134–1144 [DOI] [PubMed] [Google Scholar]
- 29. Blom N., Gammeltoft S., Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 30. Miller M. L., Soufi B., Jers C., Blom N., Macek B., Mijakovic I. (2009) NetPhosBac—a predictor for Ser/Thr phosphorylation sites in bacterial proteins. Proteomics 9, 116–125 [DOI] [PubMed] [Google Scholar]
- 31. MacLean D., Burrell M., Studholme D., Jones A. (2008) PhosCalc: a tool for evaluating the sites of peptide phosphorylation from mass spectrometer data. BMC Res. Notes 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang D. W., Sherman B. T., Lempicki R. A. (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 33. Saldanha A. J. (2004) Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248 [DOI] [PubMed] [Google Scholar]
- 34. Chen W. Y., Marcellin E., Hung J., Nielsen L. K. (2009) Hyaluronan molecular weight is controlled by UDP-N-acetylglucosamine concentration in Streptococcus zooepidemicus. J. Biol. Chem. 284, 18007–18014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Licona-Cassani C., Marcellin E., Quek L.-E., Jacob S., Nielsen L. (2012) Reconstruction of the Saccharopolyspora erythraea genome-scale model and its use for enhancing erythromycin production. Antonie Van Leeuwenhoek 102, 493–502 [DOI] [PubMed] [Google Scholar]
- 36. Oliveira A. P., Ludwig C., Picotti P., Kogadeeva M., Aebersold R., Sauer U. (2012) Regulation of yeast central metabolism by enzyme phosphorylation. Mol. Syst. Biol. 8, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Novotna J., Vohradsky J., Berndt P., Gramajo H., Langen H., Li X.-M., Minas W., Orsaria L., Roeder D., Thompson C. J. (2003) Proteomic studies of diauxic lag in the differentiating prokaryote Streptomyces coelicolor reveal a regulatory network of stress-induced proteins and central metabolic enzymes. Mol. Microbiol. 48, 1289–1303 [DOI] [PubMed] [Google Scholar]
- 38. Manteca A., Mäder U., Connolly B. A., Sanchez J. (2006) A proteomic analysis of Streptomyces coelicolor programmed cell death. Proteomics 6, 6008–6022 [DOI] [PubMed] [Google Scholar]
- 39. Holt T. G., Chang C., Laurent-Winter C., Murakami T., Garrels J. I., Davies J. E., Thompson C. J. (1992) Global changes in gene expression related to antibiotic synthesis in Streptomyces hygroscopicus. Mol. Microbiol. 6, 969–980 [DOI] [PubMed] [Google Scholar]
- 40. Manteca A., Jung H. R., Schwämmle V., Jensen O. N., Sanchez J. (2010) Quantitative proteome analysis of Streptomyces coelicolor nonsporulating liquid cultures demonstrates a complex differentiation process comparable to that occurring in sporulating solid cultures. J. Proteome Res. 9, 4801–4811 [DOI] [PubMed] [Google Scholar]
- 41. Mijakovic I., Macek B. (2012) Impact of phosphoproteomics on studies of bacterial physiology. FEMS Microbiol. Rev. 36, 877–892 [DOI] [PubMed] [Google Scholar]
- 42. Gey van Pittius N., Gamieldien J., Hide W., Brown G., Siezen R., Beyers A. (2001) The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. 2, research0044.1–0044.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akpe San Roman S., Facey P. D., Fernandez-Martinez L., Rodriguez C., Vallin C., Del Sol R., Dyson P. (2010) A heterodimer of EsxA and EsxB is involved in sporulation and is secreted by a type VII secretion system in Streptomyces coelicolor. Microbiology 156, 1719–1729 [DOI] [PubMed] [Google Scholar]
- 44. Champion P. A. D., Stanley S. A., Champion M. M., Brown E. J., Cox J. S. (2006) C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 313, 1632–1636 [DOI] [PubMed] [Google Scholar]
- 45. Gago G., Diacovich L., Arabolaza A., Tsai S.-C., Gramajo H. (2011) Fatty acid biosynthesis in actinomycetes. FEMS Microbiol. Rev. 35, 475–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoyt J. C., Reeves H. C. (1988) In vivo phosphorylation of isocitrate lyase from Escherichia coli D5H3G7. Biochem. Biophys. Res. Commun. 153, 875–880 [DOI] [PubMed] [Google Scholar]
- 47. da Silva Cruz A. H., Brock M., Zambuzzi-Carvalho P. F., Santos-Silva L. K., Troian R. F., Góes A. M., de Almeida Soares C. M., Pereira M. (2011) Phosphorylation is the major mechanism regulating isocitrate lyase activity in Paracoccidioides brasiliensis yeast cells. FEBS J. 278, 2318–2332 [DOI] [PubMed] [Google Scholar]
- 48. Reeves A. R., Brikun I. A., Cernota W. H., Leach B. I., Gonzalez M. C., Mark Weber J. (2007) Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab. Eng. 9, 293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mutka S. C., Bondi S. M., Carney J. R., Da Silva N. A., Kealey J. T. (2006) Metabolic pathway engineering for complex polyketide biosynthesis in Saccharomyces cerevisiae. FEMS Yeast Res. 6, 40–47 [DOI] [PubMed] [Google Scholar]
- 50. Cortés J., Velasco J., Foster G., Blackaby A. P., Rudd B. A. M., Wilkinson B. (2002) Identification and cloning of a type III polyketide synthase required for diffusible pigment biosynthesis in Saccharopolyspora erythraea. Mol. Microbiol. 44, 1213–1224 [DOI] [PubMed] [Google Scholar]
- 51. Rajkarnikar A., Kwon H.-J., Suh J.-W. (2007) Role of adenosine kinase in the control of Streptomyces differentiations: loss of adenosine kinase suppresses sporulation and actinorhodin biosynthesis while inducing hyperproduction of undecylprodigiosin in Streptomyces lividans. Biochem. Biophys. Res. Commun. 363, 322–328 [DOI] [PubMed] [Google Scholar]
- 52. Gao C.-H., Yang M., He Z.-G. (2011) An ArsR-like transcriptional factor recognizes a conserved sequence motif and positively regulates the expression of phoP in mycobacteria. Biochem. Biophys. Res. Commun. 411, 726–731 [DOI] [PubMed] [Google Scholar]
- 53. Gao C.-h., Yang M., He Z.-G. (2012) Characterization of a novel ArsR-like regulator encoded by Rv2034 in Mycobacterium tuberculosis. PLoS One 7, e36255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kime L., Jourdan S. S., McDowall K. J. (2008) Identifying and characterizing substrates of the RNase E/G family of enzymes. Methods in Enzymology 447, 215–241 [DOI] [PubMed] [Google Scholar]
- 55. Gatewood M. L., Bralley P., Weil M. R., Jones G. H. (2012) RNA-Seq and RNA immunoprecipitation analyses of the transcriptome of Streptomyces coelicolor identify substrates for RNase III. J. Bacteriol. 194, 2228–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang F., Peng Y., Schoenberg D. R. (2004) Endonuclease-mediated mRNA decay requires tyrosine phosphorylation of polysomal ribonuclease 1 (PMR1) for the targeting and degradation of polyribosome-bound substrate mRNA. J. Biol. Chem. 279, 48993–49002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu F., Cohen S. N. (1995) RNA degradation in Escherichia coli regulated by 3′ adenylation and 5′ phosphorylation. Nature 374, 180–183 [DOI] [PubMed] [Google Scholar]
- 58. Clausen T., Kaiser M., Huber R., Ehrmann M. (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 12, 152–162 [DOI] [PubMed] [Google Scholar]
- 59. Belanger K. D., Walter D., Henderson T. A., Yelton A. L., O'Brien T. G., Belanger K. G., Geier S. J., Fahrenkrog B. (2009) Nuclear localisation is crucial for the proapoptotic activity of the HtrA-like serine protease Nma111p. J. Cell Sci. 122, 3931–3941 [DOI] [PubMed] [Google Scholar]
- 60. Plun-Favreau H., Klupsch K., Moisoi N., Gandhi S., Kjaer S., Frith D., Harvey K., Deas E., Harvey R. J., McDonald N., Wood N. W., Martins L. M., Downward J. (2007) The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252 [DOI] [PubMed] [Google Scholar]
- 61. Mauro V. P., Edelman G. M. (2002) The ribosome filter hypothesis. Proc. Natl. Acad. Sci. U.S.A. 99, 12031–12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Volarević S., Thomas G. (2000) Role of S6 phosphorylation and S6 kinase in cell growth. Progress in Nucleic Acid Research and Molecular Biology 2000, 101–127 [DOI] [PubMed] [Google Scholar]
- 63. Mikulik K., Bobek J., Zikova A., Smetakova M., Bezouskova S. (2011) Phosphorylation of ribosomal proteins influences subunit association and translation of poly (U) in Streptomyces coelicolor. Mol. Biosyst. 7, 817–823 [DOI] [PubMed] [Google Scholar]
- 64. Kim H. L., Song W. S., Kim K., Lee K. (2008) Characterization of heterogeneous LSU rRNA profiles in Streptomyces coelicolor under different growth stages and conditions. Curr. Microbiol. 57, 537–541 [DOI] [PubMed] [Google Scholar]
- 65. Kim H. L., Shin E. K., Kim H. M., Ryou S. M., Kim S., Cha C. J., Bae J., Lee K. (2007) Heterogeneous rRNAs are differentially expressed during the morphological development of Streptomyces coelicolor. FEMS Microbiol. Lett. 275, 146–152 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.