

Abstract
In Saccharomyces cerevisiae, the essential mismatch repair (MMR) endonuclease Mlh1-Pms1 forms foci promoted by Msh2-Msh6 or Msh2-Msh3 in response to mispaired bases. Here we analyzed the Mlh1-Mlh2 complex, whose role in MMR has been unclear. Mlh1-Mlh2 formed foci that often colocalized with and had a longer lifetime than Mlh1-Pms1 foci. Mlh1-Mlh2 foci were similar to Mlh1-Pms1 foci: they required mispair recognition by Msh2-Msh6, increased in response to increased mispairs or downstream defects in MMR, and formed after induction of DNA damage by phleomycin but not double-stranded breaks by I-SceI. Mlh1-Mlh2 could be recruited to mispair-containing DNA in vitro by either Msh2-Msh6 or Msh2-Msh3. Deletion of MLH2 caused a synergistic increase in mutation rate in combination with deletion of MSH6 or reduced expression of Pms1. Phylogenetic analysis demonstrated that the S. cerevisiae Mlh2 protein and the mammalian PMS1 protein are homologs. These results support a hypothesis that Mlh1-Mlh2 is a non-essential accessory factor that acts to enhance the activity of Mlh1-Pms1.
Author Summary
Lynch syndrome (hereditary nonpolyposis colorectal cancer or HNPCC) is a common cancer predisposition syndrome. In this syndrome, predisposition to cancer results from increased accumulation of mutations due to defective mismatch repair (MMR) caused by a mutation in one of the human mismatch repair genes MLH1, MSH2, MSH6 or PMS2. In addition to these genes, various DNA replication factors and the excision factor EXO1 function in the repair of damaged DNA by the MMR pathway. In Saccharomyces cerevisiae, the MLH2 gene encodes a MutL homolog protein whose role in DNA mismatch repair has been unclear. Here, we used phylogenetic analysis to demonstrate that the S. cerevisiae Mlh2 protein and the mammalian Pms1 protein are homologs. A combination of genetics, biochemistry and imaging studies were used to demonstrate that the Mlh1-Mlh2 complex is recruited to mispair-containing DNA by the Msh2-Msh6 and Msh2-Msh3 mispair recognition complexes where it forms foci that colocalize with Mlh1-Pms1 foci (note that scPms1 is the homolog of hPms2) and augments the function of the Mlh1-Pms1 complex. Thus, this work establishes the Mlh1-Mlh2 complex as a non-essential accessory factor that functions in MMR.
Introduction
DNA mismatch repair (MMR) recognizes single base and insertion/deletion mispairs generated by errors in DNA replication and some forms of chemically damaged bases [1]–[5]. Both types of errors can lead to mutations if uncorrected. Arguably, the best understood MMR system is the Escherichia coli methyl-directed MMR system where mispair excision is targeted to the transiently unmethylated newly synthesized DNA strand before Dam methylase acts on the newly synthesized strand. MMR is initiated by the MutS homodimer, which directly recognizes mispaired bases in DNA. After mispair recognition, MutS recruits the MutL homodimer, which promotes the MutH-mediated cleavage of the unmethylated strand at hemi-methylated GATC sites. The MutH-generated strand discontinuity (nick) functions as the initiation site for an excision reaction that results in the degradation of a stretch of the newly synthesized strand followed by its resynthesis. However, there are other bacteria that lack MutH and do not use DNA methylation for strand discrimination [6]–[8]. In eukaryotes, the early steps of MMR are conserved with those in E. coli [1], [3]–[5] with the partially redundant MutS-related complexes, the Msh2-Msh6 and Msh2-Msh3 heterodimers, recognizing mispairs followed by recruitment of MutL-related complexes. Genetic evidence in the yeast Saccharomyces cerevisiae indicates that the Mlh1-Pms1 heterodimer (called MLH1-PMS2 in humans) is the primary MutL homolog complex that functions in promoting post-replication MMR [5]. In contrast to E. coli, the steps during MMR following the recruitment of the MutL homologs have remained poorly understood in eukaryotes and other organisms that lack methyl-directed MMR. Recent experiments in S. cerevisiae have indicated that MMR proteins are coupled to DNA replication and have demonstrated the existence of a short window of time after DNA is replicated during which MMR has to initiate [2]. These and other results suggest that some aspect of the DNA replication intermediates themselves may play a role in mediating strand discrimination in organisms that lack methyl-directed MMR [2], [9].
Most eukaryotes encode multiple MutL homologs that function as heterodimers. Mlh1-Pms1 (called MLH1-PMS2 in humans) possesses an endonuclease activity that can introduce single-stranded nicks into double-stranded DNA, suggesting that Mlh1-Pms1 functions as the equivalent of a combination of both the bacterial MutL and MutH proteins [10]–[12]. This endonuclease activity is required for MMR in vivo as well as for suppression of homeologous recombination and responses to DNA methylating agents [10], [13]–[15]. This endonuclease activity is also present in MutL homologs from bacteria lacking methyl-directed MMR [16]–[20].
S. cerevisiae encodes two additional MutL complexes, Mlh1-Mlh3 and Mlh1-Mlh2. Mlh1-Mlh3 plays only a minor role during MMR [21]–[23] but plays a major role in the resolution of recombination intermediates during meiosis [24]–[26]. Mlh3 contains the conserved metal-binding motif that is required for the endonuclease activity of Pms1, and mutations affecting this motif in MLH3 cause defects in MLH3-dependent MMR and meiotic crossing over [27]; consistent with this, the Mlh1-Mlh3 complex has recently been directly shown to have endonuclease activity [28], [29]. Mlh1-Mlh2 is more poorly understood than either Mlh1-Pms1 or Mlh1-Mlh3 [22], [25], [30]–[32]. Mlh2 lacks the metal binding motif present in Pms1 and Mlh3, and in most studies reported, deletion of MLH2 causes a weak or no mutator phenotype, and the results of double mutant analysis have been taken to suggest a partial redundancy between MLH2 and MLH3 in MSH3-dependent MMR [22]. It has also been reported that deletion of MLH2 increases the frequency of reversion of the lys2ΔA746 frameshift mutation reporter due to the formation of large deletions [22]. Deletion of MLH2 does not affect meiotic crossing over or meiotic MMR, unlike deletion of MLH3 or PMS1 [25], [31]. The mlh2Δ mutation does increase the frequency of gene conversion events, suggesting a partial role for Mlh2 in preventing heteroduplex formation, but not in subsequent mismatch correction; this property is unique among the S. cerevisiae MMR genes [25], [31]. Consistent with the idea that Mlh2 plays a role in recombination, simultaneous deletion of PMS1, MLH2 and MLH3 was required to cause defects in a mitotic heteroduplex rejection assay equivalent to that caused by an mlh1Δ mutation [32]. An mlh2Δ mutation as well as deletions of MSH2, MSH3, MSH6 and MLH1 but not PMS1 have also been reported to cause a modest increase in resistance to some DNA damaging agents like cisplatin, reminiscent of that seen in human and mouse cells [30]. However, it is unclear how MLH2 contributes to either recombination or MMR and why loss of Mlh2 only results in weak phenotypes.
Here, we employ live cell imaging in S. cerevisiae, complemented by genetic and biochemical assays, to analyze Mlh2 function in MMR. A similar approach applied to Pms1 previously revealed that the accumulation of Pms1 foci can be used to distinguish between genetic defects that affect events prior to Mlh1-Pms1 loading and those affecting downstream steps [33]. We found that Mlh2 formed nuclear foci similar to Pms1, whereas Mlh3 did not. Mlh2 foci partially colocalized with Pms1 foci and were dependent on MSH2, MSH6, and MLH1 but not MSH3. Mlh2 foci increased in abundance in strains with increased mispair formation and in strains containing mutations that disrupt downstream steps in MMR similarly to what was previously observed for Pms1 foci [33]. In vitro, Mlh1-Mlh2 was recruited to mispair-containing DNA by both Msh2-Msh6 and Msh2-Msh3. Deletion of MLH2 caused a synergistic increase in mutation rates when combined with a deletion of MSH6 or a promoter substitution that reduced the expression of Pms1; by contrast, no synergy was observed when deletion of MLH2 was combined with deletions of MSH3 or EXO1. Together, these data support the hypothesis that MLH2 encodes a homolog of MutL that functions in conjunction with Mlh1 as a MMR accessory factor whose roles become increasingly important under conditions when other MMR components are limiting.
Results
Pms1 and Mlh2, but not Mlh3, form nuclear foci
We recently visualized Pms1 in live S. cerevisiae cells and demonstrated that Mlh1-Pms1 foci are intermediates in MMR [33]. To gain insight into the roles of Mlh1-Mlh2 and Mlh1-Mlh3, we integrated a cassette encoding a 4×GFP tag at the 3′ end of the chromosomal MLH2 or MLH3 genes. Live cell imaging of these strains revealed that Mlh2-4×GFP formed nuclear foci similar to those observed for Pms1-4×GFP in ∼8% of unsynchronized cells (Figure 1A and B). Mlh2-4×GFP foci were almost exclusively observed in small budded cells, characteristic of cells in S-phase (Figure 1C). The observation that the Mlh2 foci visualized using different tags (4×GFP or a monomer tdTomato tag) were similar in number and appearance (Figure 1; see below) indicate that the fluorescent tags do not contribute to focus formation. In contrast to Mlh2-4×GFP, few if any cells had Mlh3-4×GFP foci (Figure 1B). The reason for the lack of Mlh3 foci is unclear, but this could indicate a limited role of Mlh3 in MMR or that the levels of Mlh3 at repair sites were too low to visualize.
Figure 1. Mlh2 forms foci that partially colocalize with Pms1.

(A) Images of cells expressing Pms1-4×GFP or Mlh2-4×GFP and Nic96-mCherry as a marker of the nuclear pore complex reveal the presence of nuclear Pms1 and Mlh2 foci. (B) Quantitation of Mlh2-4×GFP, Pms1-4×GFP, and Mlh3-4×GFP foci. Error bars indicate the standard error of the mean (SEM), and “n” indicates the number of cells examined. (C) Distribution of Mlh2-4×GFP foci according to bud size: no bud or small (<1.5 µm), medium (1.5–3 µm), or large (>3 µm) budded cells. “n” indicates the number of cells examined. (D) Images of cells expressing Mlh2-tdTomato and Pms1-4×GFP reveal foci that contain both proteins as well as foci containing only one of them. (E) Quantitation of Pms1 foci and Pms1 foci that colocalize with Mlh2. (F) Time-lapse images of cells with colocalized Mlh2-tdTomato and Pms1-4×GFP foci at one-minute intervals. White arrowheads indicate the start of colocalization and yellow arrowheads indicate Mlh2-tdTomato foci that persist after loss of the Pms1-4×GFP signal.
To test if Mlh2 localizes to the same MMR intermediates as Pms1, we examined colocalization of Mlh2-tdTomato with Pms1-4×GFP by live cell imaging. Interestingly, the Pms1 and Mlh2 foci partially colocalized, with ∼50% of Pms1 foci showing colocalization with Mlh2 foci at the limit of resolution of deconvolution microscopy (Figure 1D and E). To further examine the relationship between the Pms1 and Mlh2 foci, image stacks were taken at one-minute intervals to observe the localization of Pms1 and Mlh2 foci over time. We analyzed 50 cases where we could follow the formation, retention for at least two images, and disappearance of an Mlh2 or Pms1 focus. In agreement with the single images, approximately half (21/50, 42%) of the foci displayed colocalization between Pms1 and Mlh2 at some point during their lifetimes (Figure 1F), 36% (18/50) were Pms1 foci with no colocalization of Mlh2, and 22% (11/50) were Mlh2 foci with no colocalization of Pms1.
For the 21 cases of colocalization observed by time-lapse imaging, Pms1 and Mlh2 first appeared within the same frame for the majority of events (13/21), indicating that both proteins were recruited to the same site within the one minute temporal resolution of the imaging (Figure 1F, bottom). In 3/21 events, a Pms1 focus preceded the colocalized Mlh2 focus, and in the remaining 5/21 events, the Mlh2 focus preceded the colocalized Pms1 focus. Interestingly, Mlh2 foci frequently persisted after the colocalized Pms1 was no longer detectable (11/21 cases); the Pms1 foci were usually visible for 1 to 4 min, whereas the Mlh2 foci were visible for up to 7 min (average = 4 min) after the associated Pms1 focus was no longer detected (11/21 cases, Figure 1F). In the remaining 10 cases, the colocalized Pms1 and Mlh2 foci disappeared at the same time. Strikingly, Pms1 foci were never present after the disappearance of Mlh2, suggesting that Mlh2 frequently marks the site of Pms1 foci and likely the site of MMR after Pms1 has been removed.
Mlh2 foci behave similarly to Pms1 foci that are MMR intermediates
In our previous study [33], we identified Pms1 foci as an MMR repair intermediate based on the following observations: (i) the formation of the Pms1 foci depended on mispair recognition by Msh2-Msh6 or Msh2-Msh3; (ii) the abundance of Pms1 foci increased with increasing levels of mispaired bases; and (iii) Pms1 foci increased in abundance in cells defective in MMR at steps that were downstream of recruitment of Mlh1-Pms1 [33]. Given the partial colocalization of Pms1 and Mlh2, we investigated Mlh2 foci by performing the same set of perturbations used to analyze Pms1 foci. Deletion of MSH2, which eliminates the Msh2-Msh3 and Msh2-Msh6 mispair recognition complexes, completely abolished Mlh2 foci (Figure 2A). Similarly, other mutations that disrupted mispair recognition also eliminated Mlh2 foci (Figure 2A), including the msh3Δ msh6Δ double mutation that eliminates both the Msh2-Msh3 and Msh2-Msh6 complexes and the msh3Δ msh6-F337A double mutation that eliminates Msh2-Msh3 and eliminates mispair binding by Msh2-Msh6 [34]. Deletion of MSH6 alone also greatly reduced the number of Mlh2 foci, whereas deletion of MSH3 had no effect, suggesting that the majority of Mlh2 foci were dependent upon Msh2-Msh6 but not Msh2-Msh3 (Figure 2A). The DNA polymerase epsilon and delta active site mutations (pol2-M644G and pol3-L612M, respectively) [35], [36] or a mutation causing a defect in the 3′-5′-exonuclease activity of DNA polymerase delta (pol3-01) [37], all of which increase the level of misincorporated bases, greatly increased the abundance of Mlh2 foci (Figure 2B). Deletion of EXO1, which encodes the 5′-3′ exonuclease that participates in the mispair excision reaction, increased the percentage of nuclei with Mlh2 foci to ∼50% (Figure 2A). Together, these results mirror what was previously observed for Pms1 foci, with the one notable exception that Pms1 foci were substantially increased and not decreased in an msh6Δ mutant suggesting that Pms1 and Mlh2 differ in their ability to be recruited by Msh2-Msh6 and Msh2-Msh3 in vivo [33].
Figure 2. Mutations that perturb the abundance of Pms1 foci also perturb the abundance of Mlh2 foci.

(A) The percentages of nuclei containing Mlh2 foci were quantified in strains containing mutations in MMR genes or (B) genes encoding the catalytic subunits of the DNA polymerases Pol2 and Pol3. (C) Quantification of the percentages of nuclei containing Pms1 foci in an mlh2Δ strain. (D) Quantification of the percentages of nuclei containing Mlh2 foci in strains containing the pms1Δ mutation, the pms1Δ mutation in combination with an msh2Δ mutation or the endonuclease active site pms1 mutation pms1-E707K. Error bars indicate the SEM, and “n” indicates the numbers of cells examined.
We next examined the interdependencies of Pms1 and Mlh2 on their ability to form foci. Mlh2 has been shown to interact with Mlh1, but not with Pms1 or Mlh3, by yeast two-hybrid and affinity-capture mass spectrometry [25], [38]–[40], indicating the existence of an Mlh1-Mlh2 heterodimer that is distinct from Mlh1-Pms1 and Mlh1-Mlh3 heterodimers. Consistent with this, deletion of MLH1 eliminated the vast majority of Mlh2 foci (Figure 2A). Deletion of MLH2 had no effect on the number of Pms1 foci (Figure 2C). In contrast, deletion of PMS1 drastically increased the percentage of cells containing Mlh2 foci to ∼95% (Figure 2D). This increase in Mlh2 foci could either be due to increased formation of the Mlh1-Mlh2 complex because of loss of competition for the Mlh1 partner protein by Pms1 or due to loss of Mlh1-Pms1 endonuclease activity and the consequent inhibition of downstream steps in MMR. To differentiate between these two possibilities, we measured the frequency of Mlh2 foci in cells containing the endonuclease active site pms1-E707K mutation that results in expression of an endonuclease-defective Mlh1-Pms1 complex. We observed a similar increase in the number of Mlh2 foci in pms1Δ and pms1-E707K mutant cells (Figure 2D), suggesting that the increase in Mlh2 foci in pms1Δ cells is likely due to the inhibition of downstream steps in MMR. The high levels of Mlh2 foci seen in the pms1Δ mutant were completely abolished by an msh2Δ mutation (Figure 2D), confirming that the increased recruitment of Mlh1-Mlh2 into foci in cells lacking Pms1 was dependent on Msh2.
Mlh2 and Pms1 foci are increased by phleomycin treatment but are not present at a double-strand break generated by the I-SceI endonuclease
In human cells, Mlh1 and other MMR components are recruited to sites where DNA damage has been induced by UV-laser micro-irradiation [41]–[43]. This has been interpreted as the recruitment of MutL homolog complexes to double-strand breaks (DSBs). Consistent with these observations, treatment of S. cerevisiae cells with the radiomimetic drug phleomycin greatly increased the percentage of cells containing Pms1 and Mlh2 foci (∼5-fold and ∼3-fold, respectively) (Figure 3A). This was unlikely to be the result of simply activating the DNA damage response (DDR) because treatment with hydroxyurea, which also activates the DDR by causing replication fork stalling by depleting dNTP pools, did not cause an increase in the abundance of Pms1 or Mlh2 foci (Figure 3A). As with foci formed in untreated cells, foci induced by phleomycin treatment were not observed in msh2Δ strains.
Figure 3. Pms1 and Mlh2 foci are induced upon treatment with phleomycin but not hydroxyurea and do not colocalize with DNA double-strand breaks.
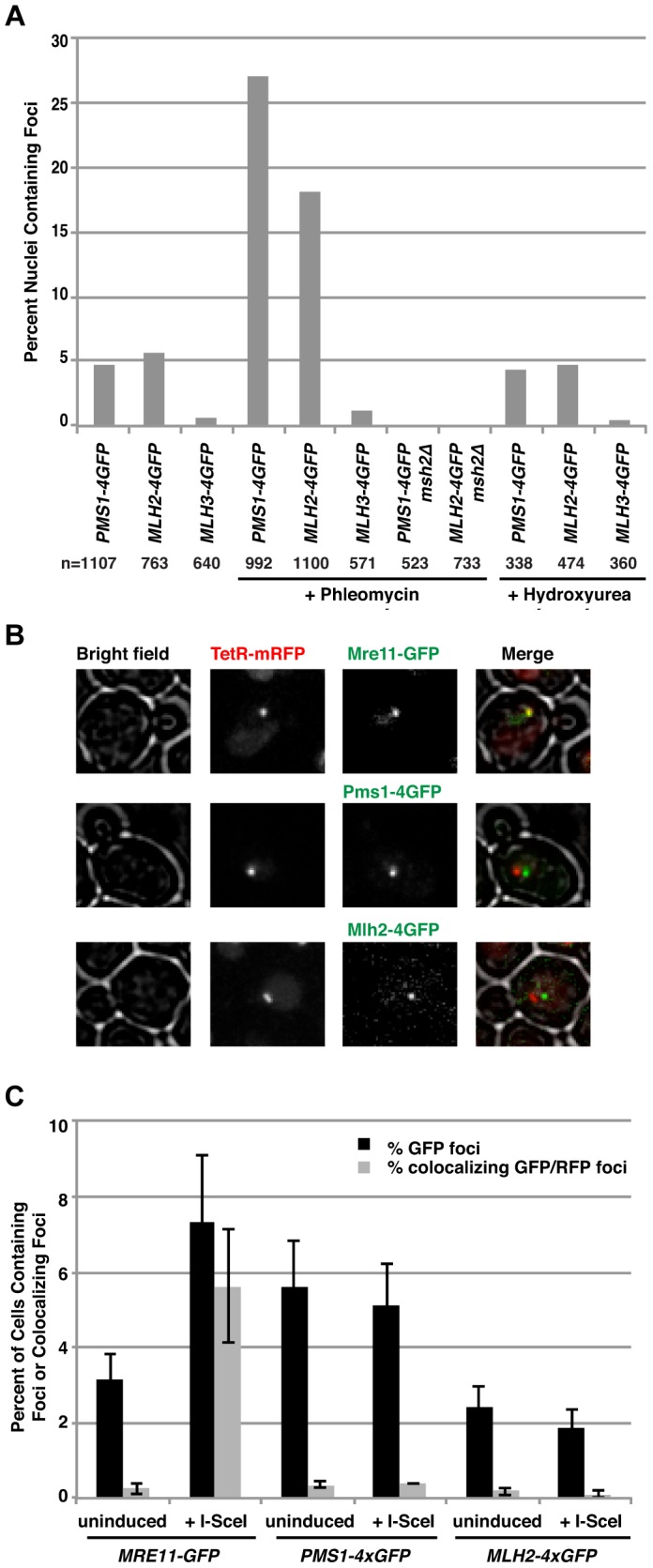
(A) Phleomycin, but not hydroxyurea, increases the number of Pms1-4×GFP and Mlh2-4×GFP foci in an MSH2-dependent manner. “n” indicates the numbers of live cells examined. (B) Images of cells expressing Mre11-GFP, Pms1-4×GFP, or Mlh2-4×GFP in a strain expressing TetR-mRFP and containing a tandem array of tetO sequences adjacent to an I-SceI restriction site after induction of I-SceI reveal that Mre11, but not Pms1 or Mlh2, colocalizes with the double-strand break. (C) Quantitation of cells containing Mre11, Pms1, and Mlh2 foci and their colocalization with the tetO array with and without I-SceI induction.
To determine if the Msh2-dependent Pms1 and Mlh2 foci were formed at DSBs and not other types of DNA lesions generated during phleomycin treatment, we investigated the recruitment of Pms1 and Mlh2 to a site-specific DSB generated by a galactose-inducible I-SceI endonuclease. The DSB was generated adjacent to a tandem array of tetO sequences on chromosome V that was marked by expression of TetR-mRFP1 [44]. Cells expressing Pms1-4×GFP, Mlh2-4×GFP, or Mre11-GFP (as a positive control) were monitored before and after the addition of galactose to induce the DSB. Consistent with published results [45], Mre11 rapidly formed foci that colocalized with the tetO array (Figure 3B and C). In contrast, neither Pms1 nor Mlh2 formed foci that colocalized with the tetO array. These results suggest that the recruitment of Pms1 and Mlh2 to lesions induced by phleomycin (and the similar recruitment of mammalian MMR proteins to the sites of laser micro-irradiation) may not be due to recognition of DSBs but rather due to recognition of the other types of DNA damage induced by these treatments that might mimic mispaired bases.
MLH2 suppresses frameshift mutations when other MMR components are limiting
We next investigated the effects of deleting MLH2 on MMR using the hom3-10 and lys2-10A frameshift reversion and CAN1 forward mutation rate assays (Table 1A). Deletion of MLH2 alone did not cause a significant increase in mutation rate in either the frameshift reversion or forward mutation assays, in agreement with previous work [22]. We next tested if the mlh2Δ mutation exacerbated the defects caused by mutations in other MMR genes. The mlh2Δ msh3Δ or mlh2Δ exo1Δ double mutant strains did not exhibit mutation rates that were higher than the mutation rates of the single mutants. In contrast, the mlh2Δ msh6Δ double mutant strain exhibited a synergistic increase in the hom3-10 and lys2-10A frameshift reversion assays but not in the CAN1 forward mutation assay that, in addition to frameshift mutations, detects base substitution mutations and other kinds of mutations [46]. Although the frameshift reversion rates of the mlh2Δ msh6Δ double mutant were higher than that of the respective single mutants, the rates were still substantially lower than caused by the msh3Δ msh6Δ double mutation that eliminates mispair recognition and causes a complete MMR defect. The combination of the specificity of MLH2 for suppressing frameshift mutations and synergy of the mlh2Δ mutation with the msh6Δ mutation but not with the msh3Δ mutation suggests that MLH2 contributes preferentially to MSH3-dependent MMR.
Table 1. Mutation rate analysis of mlh2Δ in combination with mismatch repair mutations.
| Mutation rate (fold increase)a | |||||
| Relevant genotype | RDKY | Thr+ | Lys+ | CanR | |
| A | wild-type | 5964 | 2.5 [1.8–3.8]×10−9 (1) | 1.5 [1.0–5.0]×10−8 (1) | 6.8 [3.9–8.7]×10−8 (1) |
| mlh2Δ | 7926 | 5.5 [3.0–8.4]×10−9 (2) | 3.1 [1.3–4.0]×10−8 (2) | 6.1 [4.2–11.7]×10−8 (1) | |
| msh6Δ | 7965 | 9.9 [8.7–18.9]×10−9 (4) | 4.1 [2.9–7.2]×10−7 (27) | 5.1 [4.3–8.6]×10−7 (8) | |
| msh3Δ | 6051 | 3.1 [2.0–4.2]×10−8 (12) | 1.2 [1.0–3.1]×10−7 (8) | 1.2 [0.7–4.5]×10−7 (2) | |
| exo1Δ | 7884 | 1.2 [0.5–3.0]×10−8 (5) | 1.0 [0.5–1.7]×10−8 (7) | 7.7 [5.9–13]×10−7 (11) | |
| mlh2Δ msh3Δ | 7923 | 9.0 [7.3–15.1]×10−9 (4) | 8.3 [3.9–12.8]×10−8 (6) | 7.5 [3.9–9.6]×10−8 (1) | |
| mlh2Δ msh6Δ | 7924 | 8.5 [3.5–12.4]×10−8 (34) | 2.7 [1.2–3.9]×10−6 (178) | 7.0 [2.7–12.0]×10−7 (10) | |
| mlh2Δ exo1Δ | 7925 | 6.7 [4.0–16.1]×10−9 (3) | 8.2 [3.4–22.2]×10−8 (5) | 3.8 [2.0–4.7]×10−7 (6) | |
| msh3Δ msh6Δ | 6098 | 5.1 [2.9–13]×10−6 (2040) | 3.4 [2.0–5.0]×10−5 (2267) | 2.0 [0.8–5.6]×10−6 (29) | |
| B | tetO2-PMS1 (YPD) | 8160 | 3.0 [1.5–5.9]×10−9 (1) | 3.2 [1.9–4.6]×10−8 (2) | 4.4 [3.4–8.4]×10−8 (1) |
| tetO2-PMS1 (+Dox) | 8160 | 4.0 [2.5–5.8]×10−8 (16) | 1.3 [0.9–1.5]×10−6 (89) | 1.0 [0.9–1.4]×10−7 (2) | |
| mlh2Δ (YPD) | 8159 | 1.0 [0.7–2.4]×10−8 (4) | 7.9 [3.8–10.1]×10−8 (5) | 1.0 [0.3–1.5]×10−7 (2) | |
| mlh2Δ (+Dox) | 8159 | 6.0 [5.3–14.1]×10−9 (2) | 6.9 [3.8–11.3]×10−8 (5) | 1.1 [0.7–1.6]×10−7 (2) | |
| tetO2-PMS1 (YPD) mlh2Δ | 8161 | 5.2 [4.0–8.4]×10−9 (2) | 1.4 [0.7–1.7]×10−7 (9) | 4.3 [2.7–5.6]×10−8 (1) | |
| tetO2-PMS1 (+Dox) mlh2Δ | 8161 | 1.2 [1.0–1.7]×10−7 (48) | 3.1 [2.5–4.0]×10−6 (207) | 1.7 [1.4–2.0]×10−7 (3) | |
Median rates of hom3-10 and lys2-10A reversion and inactivation of the CAN1 gene with the 95% C.I. in square brackets and fold increase relative to the wild-type in parentheses.
tetO2-PMS1, PMS1 driven by the tetO2 promoter; Dox, doxycycline.
Given the results of the Mlh2 localization studies, we hypothesized that Mlh2 becomes more important for MMR under conditions where the major MutL-related complex Mlh1-Pms1 is limiting. To test this idea, we took advantage of the previously reported tetracycline repressible system [47] to regulate Pms1 expression (tetO2 promoter) in a doxycycline-dependent manner. After titrating doxycycline, we found that 10 µg/ml of doxycycline resulted in partial downregulation of Pms1 protein expression (Figure S1) and a weak MMR defect in the frameshift reversion assays but not in the CAN1 forward mutation assay (Table 1B). Consistent with the hypothesis, we observed a synergistic increase in the mutation rate in the frameshift reversion assays when the mlh2Δ mutation was combined with reduced expression of Pms1. Thus, Mlh2 becomes more important for MMR when the level of Pms1 is reduced, suggesting that Mlh2 normally plays an accessory role in MMR.
Mlh1-Mlh2 is recruited to mispair-containing DNA by Msh2-Msh6 and by Msh2-Msh3 in vitro
The genetics of Mlh2 foci formation suggested that Mlh1-Mlh2 is primarily recruited to mispair-containing DNA by Msh2-Msh6 (Figure 2A), whereas the genetics of frameshift mutation reversion suggested that MLH2 functions primarily in an MSH3-dependent pathway (Table 1A), suggesting that Mlh2 can function in conjunction with either Msh2-Msh6 or Msh2-Msh3 depending on the assay tested. To address this possibility, we purified the S. cerevisiae Mlh1-Mlh2 complex and tested its ability to be recruited to mispair-bound Msh2-Msh6 or Msh2-Msh3 using a previously developed surface plasmon resonance assay [48]. As previously demonstrated [48], Msh2-Msh6 has low affinity for a substrate lacking a mispair (the ‘GC’ substrate) and a higher affinity for substrates with a central T:G mispair (the ‘TG’ substrate) or a +T insertion (the ‘+1’ substrate) (Figure 4A–C). As expected, Mlh1-Pms1 readily bound Msh2-Msh6 on all of these substrates and the increase in resonance units correlated with the amount of pre-bound Msh2-Msh6. Msh2-Msh6 also recruited Mlh1-Mlh2 and, similar to Mlh1-Pms1, the increase in resonance units correlated with the amount of pre-bound Msh2-Msh6 (Figure 4A–C). However, the kinetics of Mlh1-Mlh2 recruitment differed from Mlh1-Pms1 in that initial binding was slower and the binding failed to saturate. Msh2-Msh3 had a low affinity for both the GC and TG substrates, but bound well to the +1 substrate (Figure 4D–F), consistent with the function of MSH3 in the repair of insertion/deletion mispairs and an inability to function in the repair of many kinds of base-base mispairs [46], [49], [50]. As seen with Msh2-Msh6, both Mlh1-Pms1 and Mlh1-Mlh2 were recruited to substrates bound by Msh2-Msh3 with the level of recruitment correlating with the amount of Msh2-Msh3 bound (Figure 4D–F). The ability of Msh2-Msh3 to recruit Mlh1-Pms1 was consistent with the fact that Pms1 foci form in msh6Δ and msh3Δ strains but not in msh2Δ and msh3Δ msh6Δ strains and with our previous study demonstrating the recruitment of Mlh1-Pms1 to mispair-containing DNA by Msh2-Msh3 in vitro [33], [51]. Overall, these results support the idea that Mlh1-Mlh2 can function in conjunction with both Msh2-Msh3 and Msh2-Msh6, although the extent of involvement of Mlh2 may depend on the assay used and hence the exact MMR substrate being acted on.
Figure 4. Msh2-Msh6 and Msh2-Msh3 can recruit Mlh1-Pms1 and Mlh1-Mlh2 to mispaired bases.

(A–C) Recruitment of Mlh1-Pms1 or Mlh1-Mlh2 by Msh2-Msh6 to (A) homoduplex DNA, (B) DNA containing a central TG mispair, and (C) DNA containing a central +T insertion. (D–F) Recruitment of Mlh1-Pms1 or Mlh1-Mlh2 by Msh2-Msh3 to (D) homoduplex DNA, (E) DNA containing a central TG mispair, and (F) DNA containing a central +T insertion. Binding of Msh2-Msh6 or Msh2-Msh3 was monitored by surface plasmon resonance for 100 seconds (black lines), following which Mlh1-Pms1 (orange lines) or Mlh1-Mlh2 (blue lines) or no MutL homolog (dashed black line) was included in the binding reaction. Increases in resonance units (RU) indicate the binding of the proteins to the DNA on the chip. The curves shown were obtained after subtraction of the signals from the reference flow cell as well as the signals obtained by Mlh1-Pms1 or Mlh1-Mlh2 binding to the DNA alone, which did not exceed 10 to 20% of the signal attributable to recruitment of Mlh1-Pms1 or Mlh1-Mlh2 by Msh2-Msh6 or Msh2-Msh3.
Overexpression of Mlh2 and Mlh3, but not Pms1, causes MMR defects
Because MLH2 lacks conserved endonuclease motifs and mutations abolishing pms1 endonuclease function cause a weakly dominant MMR defect that is enhanced by overexpression [13], we tested if overexpression of MLH2 would cause an MMR defect. We therefore engineered S. cerevisiae strains in which the endogenous promoters of the MLH2, MLH3 and PMS1 genes were replaced by the strong promoter of the glyceraldehyde 3-phosphate dehydrogenase gene (pGPD) and monitored these strains for mutator phenotypes using the hom3-10 and lys2-10A frameshift reversion assays and the CAN1 forward mutation assay. Overexpression of PMS1 did not cause increased mutation rates; however, overexpression of MLH2 or MLH3 drastically increased the mutation rates up to levels that were almost indistinguishable from an MMR defective strain (msh3Δ msh6Δ) (Table 2). Similar results were obtained upon the expression of these genes driven by their native promoters on high copy number plasmids in wild-type cells (data not shown). The endogenous expression level of Pms1 was roughly 5–10-fold higher than that of either Mlh2 or Mlh3 (Figure 5A), and the pGPD promoter increased the expression of each MutL homolog by >50-fold relative to the endogenous level of Pms1 (Figure 5B). The mismatch repair defect caused by the overexpression of MLH2 was largely suppressed by the simultaneous overexpression of PMS1 (Table 2). These data suggest that increasing the level of Mlh2 or Mlh3 by overexpression allows Mlh2 or Mlh3 to outcompete Pms1 for binding to the Mlh1 present in the cell, thereby preventing the formation of sufficient levels of Mlh1-Pms1 complex to support MMR, and that neither Mlh2 nor Mlh3 is sufficient to replace Pms1 function in MMR. In the case of Mlh1-Mlh2, this is most likely because Mlh1-Mlh2 lacks endonuclease activity. In the case of Mlh1-Mlh3, it is possible that Mlh1-Mlh3 lacks sufficient endonuclease activity to substitute for Mlh1-Pms1 or it does not function sufficiently in the Msh2-Msh6 pathway to promote MMR [21]. It is also possible that overexpression leads to much higher levels of Mlh1-Mlh2 or Mlh1-Mlh3 complexes, which then outcompete the Mlh1-Pms1 complex for a key substrate.
Table 2. Mutation rate analysis of strains overexpressing Pms1, Mlh2 or Mlh3.
| Relevant genotype | RDKY | Mutation rate (fold increase)a | ||
| Thr+ | Lys+ | CanR | ||
| wild-type | 5964 | 2.5 [1.8–3.8]×10−9 (1) | 1.5 [1.0–5.0]×10−8 (1) | 6.8 [3.9–8.7]×10−8 (1) |
| pGPD-PMS1 | 7897 | 1.0 [5.3–17.0]×10−8 (4) | 8.7 [7.1–14.9]×10−8 (6) | 8.4 [6.1–13.9]×10−8 (1) |
| pGPD-MLH2 | 7895 | 2.4 [1.6–5.4]×10−6 (968) | 5.8 [4.5–6.9]×10−5 (3848) | 1.6 [1.3–2.1]×10−6 (23) |
| pGPD-MLH3 | 7896 | 1.7 [1.1–2.6]×10−6 (667) | 5.7 [3.4–8.3]×10−5 (3821) | 1.3 [1.1–2.4]×10−6 (19) |
| pGPD-(MLH2 + PMS1) | 7904 | 2.8 [2.0–5.7]×10−8 (11) | 7.4 [5.3–15.9]×10−7 (49) | 9.9 [7.9–12.2]×10−8 (1) |
| msh3Δmsh6Δ | 6098 | 5.1 [2.9–13]×10−6 (2040) | 3.4 [2.0–5.0]×10−5 (2267) | 2.0 [0.8–5.6]×10−6 (29) |
Median rates of hom3-10 and lys2-10A reversion and inactivation of the CAN1 gene with the 95% C.I. in square brackets and fold increase relative to the wild-type in parentheses.
Figure 5. Expression of Pms1, Mlh2 and Mlh3 under control of the endogenous and pGPD promoters.
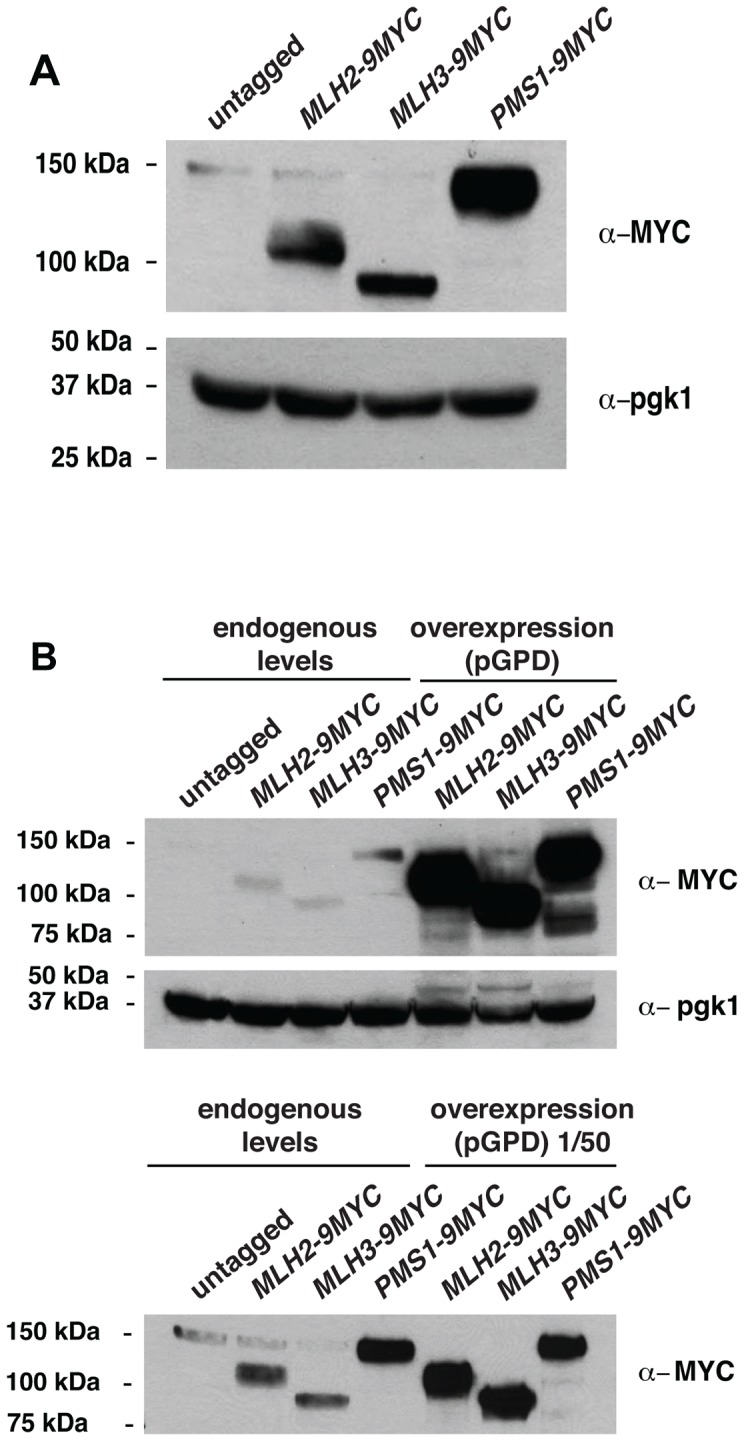
(A) Expression level of Mlh2-9MYC, Mlh3-9MYC, and Pms1-9MYC driven from their endogenous promoters monitored by immunoblotting with an anti-MYC antibody. Immunoblotting with an anti-Pgk1 antibody was used a loading control. (B) Comparison of the levels of Mlh2-9MYC, Mlh3-9MYC, and Pms1-9MYC by immunoblotting with an anti-MYC antibody when expressed from the endogenous promoter or the pGPD promoter. Bottom panel, comparison after 50-fold dilution of the pGPD samples.
Mlh2 is a widely conserved MutL homolog lacking endonuclease motifs
Because S. cerevisiae MLH2 plays only an accessory role in MMR, we examined the conservation of MLH2. We first identified MutL homologs using BLAST [52] in the Saccharomycotina subphylum of Ascomycota, which includes S. cerevisiae. Obvious homologs of MLH1, PMS1, MLH3, and MLH2 were identified (Figure 6 and S2; Table S1) by reciprocal BLAST, by analysis of conserved synteny [53], and/or by the characteristic C-terminal sequence motifs of MLH1, PMS1, and MLH3 involved in endonuclease activity. The origin of MLH2 predated the whole-genome duplication that occurred ∼100 million years ago and led to S. cerevisiae and related yeasts [54], because MLH2 homologs were present in species that diverged from S. cerevisiae prior to the genome duplication and two MLH2 ohnologs (paralogs produced by the whole-genome duplication [55], [56]) were present in Vanderwaltozyma polyspora. A few clades in Saccharomycotina have lost MLH2, including the ‘CTG’ yeast that encode serine instead of leucine with the codon CTG [57] and species in the Lachancea genus (Figure S2, Table S1).
Figure 6. Evolutionary conservation of MutL homologs.

The presence of a conserved MLH1, S. cerevisiae PMS1/human PMS2, MLH3, and S. cerevisiae MLH2/human PMS1 homolog in sequenced unikont genomes is indicated by a ‘Y’. ‘N’ indicates the lack of an identifiable homolog. MLH1 and ScPMS1/hPMS2 are extensively conserved, whereas MLH3 and ScMLH2/hPMS1 are less well conserved. Alternating light and dark grey backgrounds indicate separations between major unikont groups (Fungi, Nucleariidae and Fonticula, Ichthyosporea, Choanoflagellata, Metazoa, Apusozoa, and Amoebozoa).
We also identified MutL homologs in other sequenced fungi (Figure 6, Table S1). MLH1, PMS1, MLH3, and MLH2 genes were found in the Pezizomycotina subphylum of Ascomycota, but the early diverging Taphrinomycotina subphylum, which includes Schizosaccharomyces pombe, only contained MLH1 and PMS1. In a small number of species, the MLH2 homologs contained stop codons and frameshifts, which could reflect errors in the genome sequences, loss of non-essential portions of MLH2 or inactivation of the MLH2 homologs (Table S2). MLH2 homologs were also not identified in Basidiomycetes but were observed in the basal fungi Mucor circinelloides (Mucoromycotina) and Allomyces macrogynus (Blastocladiomycota), which was consistent with the loss of MLH2 in Basidiomycetes rather than the gain of MLH2 in Ascomycetes. Phylogenetic analysis of the full-length Mlh2 protein sequences was consistent with the major divisions within fungi (Figure S3).
S. cerevisiae MLH2 has a number of similarities to metazoan PMS1 (note that PMS2 in metazoans is the name for the homolog of S. cerevisiae PMS1). Like S. cerevisiae MLH2, metazoan PMS1 lacks endonuclease motifs and does not support MMR reactions in vitro [58], [59], and deletion of metazoan PMS1 causes an extremely weak mutator phenotype [60]. To examine the relationship between metazoan Pms1 and fungal Mlh2, we performed phylogenetic analysis on the sequences of the N-terminal domains of MutL homologs from select unikont species (Figure S4). This analysis identified distinct clades with strong support (clade credibility scores of 100%) for the homologous human PMS2 and fungal Pms1 proteins, human MLH3 and fungal Mlh3 proteins, as well as the human PMS1 and fungal Mlh2 proteins. Thus, this analysis suggests that human PMS1 is evolutionarily related to fungal Mlh2 and that the accessory MMR function is conserved across diverse eukaryotes (Figure 6).
Discussion
In this study, we demonstrated that both Pms1 and Mlh2 form foci that often colocalize and that the formation of these foci depends upon Mlh1, Msh2-Msh6 and, in the case of Pms1, can also be promoted by Msh2-Msh3 [33]. The frequency of both types of foci increase in strains with increased mispair formation or defects in the downstream steps of MMR. In contrast, no Mlh3 foci were detected despite the fact that Mlh3 was expressed at levels similar to Mlh2. Deletion of MLH2 did not cause a significant increase in the mutation rate in frameshift reversion assays unless Pms1 levels were reduced, and an MLH2 deletion synergized with a deletion of MSH6, but not with a deletion of MSH3. In vitro, both Msh2-Msh6 and Msh2-Msh3 could recruit Mlh1-Mlh2 to mispair-containing DNA. These results are consistent with a role for Mlh2 in MMR. However, the lack of endonuclease motifs in Mlh2 suggests that its ability to promote MMR must involve mechanisms other than Mlh1-Mlh2-mediated cleavage of DNA. Together, these data suggest that Mlh1-Mlh2 acts as an MMR accessory or stimulatory factor that functions in conjunction with Mlh1-Pms1.
The studies performed here could be taken to present an apparent discrepancy in the placement of MLH2 in known MMR sub-pathways. The genetics of foci formation suggest that Mlh2 recruitment is primarily mediated by Msh2-Msh6 but not Msh2-Msh3, whereas the frameshift reversion assays indicate the involvement of MLH2 in MSH3-dependent but not MSH6-dependent MMR. In contrast, the ability of Mlh1-Mlh2 to be recruited to mispair-containing DNA in vitro by both Msh2-Msh6 and Msh2-Msh3 is consistent with a role for Mlh2 in both pathways. One potential explanation for this apparent discrepancy is that Mlh2 plays a role in both pathways, but the recruitment and function of Mlh1-Mlh2 in MMR are highly dependent on the type of mispair that is recognized. The relative contribution of Mlh1-Mlh2 to repair may be influenced by the sequence context of the mispair and whether repair occurs by substitution, deletion or insertion. Thus, the different assay-dependent activities of Mlh1-Mlh2 observed could reflect the fact that the three different assays used in our studies all by necessity use different mispaired substrates. Additionally, the in vitro mispair-dependent Mlh1-Mlh2 recruitment assays use different ratios of Msh2-Msh6 and Msh2-Msh3 than present in cells, resulting in different apparent efficiencies of recruitment of Mlh1-Mlh2 by Msh2-Msh6 and Msh2-Msh3 in vitro and in vivo. These types of differential activities have been clearly documented in the case of the Mlh1-Mlh3 complex [21]–[23], [25], [28].
We propose the following hypothesis for the role of Mlh2 in MMR. The Mlh1-Mlh2 complex may have some functional overlap with the non-endonuclease functions of the Mlh1-Pms1 complex in MMR such as recruitment of downstream MMR components and discrimination of the newly synthesized DNA strand, allowing MMR to occur at lower levels of Mlh1-Pms1 at the sites of repair. This role of Mlh1-Mlh2 in reducing the requirement for Mlh1-Pms1 while not being able to replace the activity of Mlh1-Pms1 suggests that Mlh1-Mlh2 acts as a non-essential accessory or stimulatory factor in MMR. It is also possible that Mlh1-Mlh2 in some way regulates the availability or activity of Mlh1-Pms1. Because the Mlh1-Pms1 endonuclease activity is essential for MMR in vivo, this hypothesis suggests that it might be possible to reveal these Mlh2 functions through the isolation of separation-of-function mutations in Pms1 that eliminate MMR in the absence of Mlh2 but leave the endonuclease activity of Mlh1-Pms1 intact. In addition, this hypothesis predicts that Mlh1-Pms1 and Mlh1-Mlh2 might be loaded onto the same DNA substrate, as suggested by the colocalization observed between Pms1 and Mlh2 foci as well as the accumulation of Mlh2 foci in the absence of Mlh1-Pms1 endonuclease activity; future studies will be required to determine if both complexes are recruited to the same mispaired substrate and if this is functionally significant. Our results are reminiscent of bacteriophage Lambda site-specific recombination where the biochemical requirement for the FIS protein during excision in vitro, which acts as an accessory factor, could only be revealed at sub-optimal levels of XIS protein [61].
Our studies have provided strong evidence that S. cerevisiae Mlh2 is the homolog of mammalian PMS1. Mammalian PMS1 is known to form a complex with MLH1, although the possible role of mammalian PMS1 in MMR is unclear [58], [60]. Consequently, the results described here may also provide new insights into a possible role of the mammalian MLH1-PMS1 complex in MMR. In addition, the ability of S. cerevisiae Mlh1-Mlh2 to be recruited by mispairs and the mutator phenotype caused by overexpression of MLH2 (and MLH3) suggests that increased expression of human PMS1 (or human MLH3) might represent a mechanism that could lead to MMR inactivation and promote tumorigenesis, analogous to MMR defects in Lynch Syndrome and other types of sporadic cancer characterized by microsatellite instability [1], [3]–[5].
Materials and Methods
Media and strains
S. cerevisiae strains were grown at 30°C in yeast extract-peptone-dextrose media (YPD) or appropriate dextrose-containing synthetic dropout media for selection of Lys+ or Thr+ revertants or canavanine-resistant (CanR) mutants. All strains used for mutation analyses in this study (Table S3) were derivatives of the S288c strain RDKY3686 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A) [62]. Strains used for the colocalization experiments with the inducible I-SceI-generated double strand break contained the pGAL-I-SceI construct, the I-SceIcs restriction site adjacent to the 3xURA3-tetOx112 array, and TetR-mRFP derived from W9561-17A [63] (generously provided by R. Rothstein, Columbia Medical School).
Gene deletion, tagging and promoter replacements for gene overexpression (pGPD) were performed using standard PCR-based recombination-mediated gene targeting methods [64] followed by confirmation with PCR. The correct insertion of tags/promoters and the absence of additional mutations were confirmed by sequencing.
Strains expressing Pms1 under the tetracycline repressible promoter (tetO2) were generated as previously described [47]. The parental strain (RDKY8158) containing the chimeric repressor (tetR′-SSN6), the transactivator (tTA) and the MMR reporters (lys2-10A and hom3-10) was obtained after mating RDKY3686 with the CML476 strain (MATa ura3-52 leu2Δ1 his3Δ200 GAL2 CMVp(tetR′-SSN6)::LEU2 trp1::tTA (Euroscarf). Plasmid pCM324 (Euroscarf) was used to introduce the tetO2 promoter immediately upstream of the Pms1 start codon.
Specific point mutations were introduced in chromosomal genes using standard integration/excision methods and the following integrating plasmids: msh6-F337A was introduced with pRDK1602 [33]; pms1-E707K was introduced with pRDK1583 [33]; and the DNA polymerase alleles pol2-M644G, pol3-01, and pol3-L612M were integrated as previously described [35], [37], [65], [66]. The presence of the desired mutation and absence of additional mutations was confirmed by DNA sequencing.
For visualization of the low abundance proteins Pms1 and Mlh2, we tagged them at the C-terminus at the endogenous gene locus with four tandem copies of GFP (4×GFP) using the pSM1023 plasmid (gift of E. Schiebel, University of Heidelberg). Testing of the tagged strain RDKY7893 (MLH2-4GFP.KanMX6) for sensitivity to cisplatin as described previously [30] showed that it had the same sensitivity as the wild-type parental strain RDKY5964 and was more sensitive than the mlh2Δ control strain RDKY7926 (mlh2::KanMX4), indicating that the tag on the C-terminus of Mlh2 was unlikely to have an effect on Mlh2 function [30]. The nuclear pore protein Nic96 was tagged at the C-terminus with mCherry using the plasmid pBS35 as a template (Yeast Resource Center YRC). The C-terminus of Mlh2 was tagged with tdTomato using a PCR-based strategy and the plasmid pRDK1663. This plasmid was derived from pYM25 yeGFP.hphNT1 (Ref. [64]; obtained from Euroscarf) by excising the HindIII-BglII fragment containing the yeGFP coding sequence, replacing it with a HindIII-BglII fragment (generated by gene synthesis at Integrated DNA Technologies, IDT) encoding an S. cerevisiae codon optimized tdTomato gene and a linker (Gly Ala)5 immediately upstream of the Met start codon of tdTomato.
Immunoblotting
S. cerevisiae whole-cell extracts were prepared using the Yeast Extract Buffer (1.85 M NaOH, 7.5% beta-mercaptoethanol), and the soluble proteins were precipitated by addition of an equal volume of 50% trichloroacetic acid. The protein extracts were analyzed by SDS-PAGE using 4%–15% gradient gels (BioRad) and immunoblotting. Detection of GFP-tagged proteins was performed using the anti-GFP antibody, clone B34 (Covance). Using the anti-Pgk1 antibody (clone 22C5D8; Invitrogen), Pgk1 expression was monitored as a loading control. MYC-tagged proteins were detected with the monoclonal anti-MYC antibody (clone 4A6; Millipore).
Genetic assays
Mutation rates were determined using the hom3-10 and lys2-10A frameshift reversion assays and CAN1 inactivation assay by fluctuation analysis [67] as previously described [46], [62]. The mutation rates presented in Table 1B were determined in the absence or presence of 10 µg/ml of doxycycline (present in liquid cultures as well as in agar plates).
Protein purification
All the proteins were expressed from plasmid expression vectors in either E. coli or S. cerevisiae as indicated below. Typical yields ranged from 100 µg to 500 µg per liter of expressing cells. All the protein preparations were confirmed to be greater that 95% pure as judged by SDS-PAGE followed by staining of the resulting gels with Coomassie Blue.
Purification of Mlh1-Pms1
S. cerevisiae Mlh1-Pms1 was overexpressed in S. cerevisiae and purified according to a previously published procedure using an overexpression strain derived by the transformation of RDKY1293 (MATα, ura3-52, leu2Δ1, trp1, his3Δ200, pep4::HIS3, prb1Δ1.6R, can1) with the plasmids pRDK573 (pGAL1-10-MLH1 TRP1) and pRDK1099 (pGAL1-10-PMS1-FLAG LEU2) [13], [68].
Purification of Mlh1-Mlh2
S. cerevisiae Mlh1-Mlh2 was overexpressed in the S. cerevisiae strain RDKY8153 that was generated by transformation of RDKY1293 (MATα, ura3-52, leu2Δ1, trp1, his3Δ200, pep4::HIS3, prb1Δ1.6R, can1) with the plasmids pRDK573 (pGAL1-10-MLH1 TRP1) and pRDK1664 (pGAL1-10-Mlh2-FLAG LEU2). pRDK1664 was generated by replacement of the PMS1 ORF with MLH2 using gap-repair. Briefly, pRDK1099 was linearized with SphI and used to co-transform wild-type S. cerevisiae with a PCR product containing the MLH2 ORF and flanking sequences homologous to the pGAL1-10 promoter and FLAG at the 5′ and 3′ end, respectively. Gap-repaired plasmids were recovered from Leu+ transformants and verified by sequencing.
The overexpressing strain RDKY8153 was grown as previously described for the purification of Mlh1-Pms1 [69]. A 100 ml culture was harvested by centrifugation for 10 min at 3,000 rpm in a swinging bucket rotor in a Sorvall Legend RT centrifuge at 20°C and then the cells were resuspended in 25 ml of Buffer A200 [50 mM Tris-Cl (pH 8.0), 10% glycerol, 200 mM NaCl, 2 mM β-mercaptoethanol, protease inhibitor mixture PIC D (final concentrations of 1 mM phenylmethanesulfonyl fluoride (PMSF), 1 µg/L chymostatin, and 1 µg/L pepstatin A) and protease inhibitor mixture PIC W (final concentrations of 1 mM benzamidine, 0.5 µg/L bestatin, 1 µg/L aprotinin, and 1 µg/L leupeptin)] at 4°C. Then, 4 ml of Cell Lytic Y Cell Lysis Reagent (Sigma) was added to the resuspended cells, and the cells were distributed into 1 ml aliquots in microcentrifuge tubes. The tubes were rocked for 45 min at 4°C, and then the cells were sonicated for 25 s with a 5-s pulse on and a 1-s pulse off for three cycles. The tubes were then centrifuged at 14,000 rpm in a tabletop Eppendorf centrifuge for 30 min at 4°C, and the supernatants were pooled. Then, 4 ml of FLAG antibody resin equilibrated in Buffer A200 was added to the pooled supernatant, and the resulting suspension was rocked for 1 hr at 4°C. The resin was then poured into a column that was washed 5 times with 1 ml of buffer A200, and then the protein was washed 7 times with 1 ml of buffer A200 containing 200 µg/ml of FLAG peptide. Fractions containing the protein were pooled and concentrated as previously described [13], and then aliquots were frozen in liquid nitrogen and stored at −80°C.
Purification of Msh2-Msh6
S. cerevisiae wild type Msh2-Msh6 was overexpressed in E. coli BL21 (DE) RIL using the pET11 MSH2-MSH6 plasmid and purified according to a previously published procedure [70], [71].
Purification of Msh2-Msh3
S. cerevisiae Msh2-Msh3 was expressed in S. cerevisiae RDKY2418 MATα, ura3-52, leu2Δ1, trp1, his3Δ200, pep4::HIS3, prb1Δ1.6R, can1, msh2::hisG, msh6::hisG [72] transformed with the expression plasmids pRDK354 (pGAL1-10-MSH2 URA3) and pRDK1596 (pGAL1-10 MSH3-FLAG LEU2) and purified as described previously [51].
Surface plasmon resonance analysis
The recruitment of Mlh1-Pms1 or Mlh1-Mlh2 by Msh2-Msh6 or Msh2-Msh3 bound to DNA was analyzed using a Biacore T100 instrument essentially as described previously [13], [68]. Biotinylated 236 bp-long double-stranded DNAs containing the terminal lacO sequence and a central TG mispair, +T insertion or GC base pair were conjugated to 3 flow cells of a streptavidin-coated Biacore SA chip (GE Healthcare) to obtain a signal of ∼100 Resonance Units (RU). The signal from the unmodified flow cell was used for reference subtraction in all experiments. A constant flow rate of 20 µl/min was maintained and experiments were performed at 25°C. First, 30 nM purified LacI tetramer (a gift from Kathleen Matthews, Rice University) in reaction buffer (25 mM Tris-Cl (pH 8.0), 4 mM MgCl2, 110 mM NaCl, 0.01% Igepal, 2 mM DTT and 2% glycerol) was injected over the chip for 60 s. Next, at time t = 0 a sample containing 30 nM LacI, 20 nM Msh2-Msh6 or 20 nM Msh2-Msh3 and 250 µM ATP in reaction buffer was injected for 100 s, followed by the immediate injection of a sample containing the same mixture and in addition 40 nM Mlh1-Pms1 or 40 nM Mlh1-Mlh2, or no MutL homolog for 150 s. The chip surface was regenerated using a 60 s pulse of 2 M NaCl. Control experiments were performed in which the first injection consisted of 30 nM LacI, the sond injection consisted of 30 nM LacI and 250 µM ATP, and the third injection consisted of 30 nM LacI, 250 µM ATP and either 40 nM Mlh1-Pms1 or 40 nM Mlh1-Mlh2. These data were subtracted from the data obtained using Msh2-Msh6/Msh2-Msh3 in the second and third injections, and the subtracted curves are presented. Data were analyzed using the BiaEvaluation 2.0.3 (GE Healthcare) and Prism 6.0 software (GraphPad).
Live-cell imaging and image analysis
Exponentially growing cultures were washed and resuspended in water and placed on minimal media agar pads, covered with a coverslip, and sealed with valap (a 1∶1∶1 mixture of Vaseline, lanolin, and paraffin by weight). Cells were imaged on a Deltavision (Applied Precision) microscope with an Olympus 100×1.35NA objective. Fourteen 0.5 µm z sections were acquired and deconvolved with softWoRx software. For time-lapse imaging of Pms1 foci, images were collected every min with fewer z sections to minimize photobleaching. Further image processing, including maximum intensity projections and intensity measurements, was performed using ImageJ.
For drug treatments, cells that were growing logarithmically in YPD medium were treated with either 200 mM hydroxyurea or 5 µM phleomycin for 3 hr and prepared for microscopy as described above. Cell cycle arrest was confirmed by examining cell morphology using a microscope. For DSB induction by I-SceI, strains RDKY7906, RDKY7907, and RDKY7908 were grown overnight in medium containing 2% raffinose. The cultures were diluted into the same medium and after 3 hr, pelleted and resuspended in medium containing either 2% raffinose or 2% galactose, incubated at 30°C for 2.5 hr, and prepared for microscopy.
Foci were considered to be colocalized if over half of their diameters overlapped. Colocalization was scored if at least one focus per nucleus displayed colocalization in the same z section. Images with the same fluorescent fusion protein in the same figure have identical contrast adjustment. The data presented here contain representative images and quantitative data from at least two independent experiments, each performed using two independent strain isolates, which gave similar results. The total number of cells/nuclei (n) analyzed for each strain is indicated.
Phylogenetic analysis
MutL homologs were identified using Protein BLAST [52] against the non-redundant protein sequences (nr) database. For some MutL homologs, alignments of the protein sequences revealed that strongly conserved regions were missing. We analyzed the genomic sequences encoding these genes and discovered that these were often due to incorrect assignment of exons, as the missing regions tended to either be at exon boundaries, suggesting the inappropriate identification of a predicted splice site, or in introns, suggesting the failure to identify an exon. For these genes, we re-annotated the exons, typically merging in-frame introns with the surrounding exons or identifying missing exon sequences, and retranslated the protein sequences (Table S2). The criteria for re-annotation of the exons were to improve homology to the conserved portions of the protein sequence alignment and maintain conservation of the intron/exon structure with closely related species. In some cases, a frameshift or stop codon was present in an exon. In these cases, the surrounding protein sequence was translated to prevent truncations from having an inordinately large effect on the phylogenetic analysis; however, this analysis could not distinguish between sequencing errors or species having mutations that inactivated the gene. The cases in which a protein was translated in spite of frameshifts or stop codons in the reference genomic sequence are explicitly labeled in Table S2. To decipher relationships between MutL homologs, amino acid sequences of the N-terminal domains were aligned with MAFFT [73] to avoid misalignments due to effects of the rapidly evolving and likely unstructured linker between the MutL N- and C-terminal domains. Phylogenetic analyses were performed with MrBayes [74] using the mixed amino acid rate matrices model and 1,000,000 generations. Clade credibility values above 75 were considered significant.
Supporting Information
Downregulation of S. cerevisiae Pms1 expression upon addition of doxycycline. Expression levels of Pms1 (tagged with 9MYC) of strains containing the repressible tetO2-Pms1 promoter (lanes 1+2) or the endogenous Pms1 promoter (lanes 3+4). Cells were grown for 3 hours in YPD in the presence (or absence) of 10 µg/ml of doxycycline, as indicated. Whole-cell extracts were prepared and analyzed by SDS-PAGE and immunoblotting. The anti-Myc antibody was used to detect Pms1 expression and Pgk-1 was used as a loading control. Relates to Table 1.
(EPS)
Conservation of MLH1, MLH2, MLH3, and PMS1 in species of Saccharomycotina with sequenced genomes. ‘WGD clade’ indicates the group of species that have undergone whole-genome duplication. ‘CTG clade’ indicates the group of species that encode serine instead of leucine with the CTG codon. Phylogenetic relationships between species were derived from previously published analyses [75]–[77].
(EPS)
Clustering of the full-length Mlh2 sequences from fungi. This phylogenetic analysis reconstructed the Saccharomycotina and Pezizomycotina subphyla within Fungi as well as class and family relationships, consistent with homology between fungal MLH2 genes. Clade support values of 75 or higher are indicated on the phylogenetic tree.
(EPS)
Clustering of sequences of the N-terminal domains of MutL homologs from sequenced unikont species provided clear support for the homology of S. cerevisiae MLH2 and human PMS1, S. cerevisiae MLH3 and human MLH3, as well as S. cerevisiae PMS1 and human PMS2. Clade support values of 75 or higher are indicated on the phylogenetic tree.
(EPS)
MutL homologs in sequenced unikont genomes.
(XLS)
Re-annotation of MutL genes in fully sequenced genomes.
(DOCX)
S. cerevisiae strains used in this study.
(DOCX)
Acknowledgments
The authors would like to thank the Kolodner and Desai Lab members for helpful discussions.
Funding Statement
This work was supported by NIH grants GM50006 (RDK), GM074215 (AD) and CA23100 (AD, RDK), a Damon Runyon Cancer Research Foundation Fellowship (CSC) and to the Harald zur Hausen Fellowship from the German Cancer Research Center, DKFZ (HH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Harfe BD, Jinks-Robertson S (2000) DNA mismatch repair and genetic instability. Annu Rev Genet 34: 359–399. [DOI] [PubMed] [Google Scholar]
- 2. Hombauer H, Srivatsan A, Putnam CD, Kolodner RD (2011) Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334: 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iyer RR, Pluciennik A, Burdett V, Modrich PL (2006) DNA mismatch repair: functions and mechanisms. Chem Rev 106: 302–323. [DOI] [PubMed] [Google Scholar]
- 4. Modrich P, Lahue R (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 65: 101–133. [DOI] [PubMed] [Google Scholar]
- 5. Kolodner RD, Marsischky GT (1999) Eukaryotic DNA mismatch repair. Curr Opin Genet Dev 9: 89–96. [DOI] [PubMed] [Google Scholar]
- 6. Lobner-Olesen A, Skovgaard O, Marinus MG (2005) Dam methylation: coordinating cellular processes. Curr Opin Microbiol 8: 154–160. [DOI] [PubMed] [Google Scholar]
- 7. Brezellec P, Hoebeke M, Hiet MS, Pasek S, Ferat JL (2006) DomainSieve: a protein domain-based screen that led to the identification of dam-associated genes with potential link to DNA maintenance. Bioinformatics 22: 1935–1941. [DOI] [PubMed] [Google Scholar]
- 8. Barbeyron T, Kean K, Forterre P (1984) DNA adenine methylation of GATC sequences appeared recently in the Escherichia coli lineage. J Bacteriol 160: 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, et al. (2010) PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci U S A 107: 16066–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O'Donnell M, et al. (2007) Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem 282: 37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kadyrov FA, Dzantiev L, Constantin N, Modrich P (2006) Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126: 297–308. [DOI] [PubMed] [Google Scholar]
- 12. Kosinski J, Plotz G, Guarne A, Bujnicki JM, Friedhoff P (2008) The PMS2 subunit of human MutLalpha contains a metal ion binding domain of the iron-dependent repressor protein family. J Mol Biol 382: 610–627. [DOI] [PubMed] [Google Scholar]
- 13. Smith CE, Mendillo ML, Bowen N, Hombauer H, Campbell CS, et al. (2013) Dominant mutations in Saccharomyces cerevisiae PMS1 identify the Mlh1-Pms1 endonuclease active site and an Exonuclease 1-independent mismatch repair pathway. PLoS 9: e1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Erdeniz N, Nguyen M, Deschenes SM, Liskay RM (2007) Mutations affecting a putative MutLalpha endonuclease motif impact multiple mismatch repair functions. DNA Repair (Amst) 6: 1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deschenes SM, Tomer G, Nguyen M, Erdeniz N, Juba NC, et al. (2007) The E705K mutation in hPMS2 exerts recessive, not dominant, effects on mismatch repair. Cancer Lett 249: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, et al. (2010) Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell 39: 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mauris J, Evans TC (2009) Adenosine triphosphate stimulates Aquifex aeolicus MutL endonuclease activity. PLoS One 4: e7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Correa EM, Martina MA, De Tullio L, Argarana CE, Barra JL (2011) Some amino acids of the Pseudomonas aeruginosa MutL D(Q/M)HA(X)(2)E(X)(4)E conserved motif are essential for the in vivo function of the protein but not for the in vitro endonuclease activity. DNA Repair (Amst) 10: 1106–1113. [DOI] [PubMed] [Google Scholar]
- 19. Namadurai S, Jain D, Kulkarni DS, Tabib CR, Friedhoff P, et al. (2010) The C-terminal domain of the MutL homolog from Neisseria gonorrhoeae forms an inverted homodimer. PLoS One 5: e13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duppatla V, Bodda C, Urbanke C, Friedhoff P, Rao DN (2009) The C-terminal domain is sufficient for endonuclease activity of Neisseria gonorrhoeae MutL. Biochem J 423: 265–277. [DOI] [PubMed] [Google Scholar]
- 21. Flores-Rozas H, Kolodner RD (1998) The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc Natl Acad Sci U S A 95: 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harfe BD, Minesinger BK, Jinks-Robertson S (2000) Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr Biol 10: 145–148. [DOI] [PubMed] [Google Scholar]
- 23. Romanova NV, Crouse GF (2013) Different roles of eukaryotic MutS and MutL complexes in repair of small insertion and deletion loops in yeast. PLoS Genet 9: e1003920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Muyt A, Jessop L, Kolar E, Sourirajan A, Chen J, et al. (2012) BLM helicase ortholog Sgs1 is a central regulator of meiotic recombination intermediate metabolism. Mol Cell 46: 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang TF, Kleckner N, Hunter N (1999) Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci U S A 96: 13914–13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zakharyevich K, Tang S, Ma Y, Hunter N (2012) Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 149: 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nishant KT, Plys AJ, Alani E (2008) A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics 179: 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rogacheva MV, Manhart CM, Chen C, Guarne A, Surtees J, et al. (2014) Mlh1-Mlh3, A Meiotic Crossover and DNA Mismatch Repair Factor, is a Msh2-Msh3-Stimulated Endonuclease. J Biol Chem 289: 5664–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ranjha L, Anand R, Cejka P (2014) The Saccaromyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem 289: 5674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Durant ST, Morris MM, Illand M, McKay HJ, McCormick C, et al. (1999) Dependence on RAD52 and RAD1 for anticancer drug resistance mediated by inactivation of mismatch repair genes. Curr Biol 9: 51–54. [DOI] [PubMed] [Google Scholar]
- 31. Abdullah MF, Hoffmann ER, Cotton VE, Borts RH (2004) A role for the MutL homologue MLH2 in controlling heteroduplex formation and in regulating between two different crossover pathways in budding yeast. Cytogenet Genome Res 107: 180–190. [DOI] [PubMed] [Google Scholar]
- 32. Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE (2004) Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci U S A 101: 9315–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD (2011) Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147: 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowers J, Tran PT, Liskay RM, Alani E (2000) Analysis of yeast MSH2-MSH6 suggests that the initiation of mismatch repair can be separated into discrete steps. J Mol Biol 302: 327–338. [DOI] [PubMed] [Google Scholar]
- 35. Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA (2007) Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 317: 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nick McElhinny SA, Stith CM, Burgers PM, Kunkel TA (2007) Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem 282: 2324–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morrison A, Johnson AL, Johnston LH, Sugino A (1993) Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J 12: 1467–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- 39. Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, et al. (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636. [DOI] [PubMed] [Google Scholar]
- 40. Collins SR, Kemmeren P, Zhao XC, Greenblatt JF, Spencer F, et al. (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics 6: 439–450. [DOI] [PubMed] [Google Scholar]
- 41. Hong Z, Jiang J, Hashiguchi K, Hoshi M, Lan L, et al. (2008) Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J Cell Sci 121: 3146–3154. [DOI] [PubMed] [Google Scholar]
- 42. Liberti SE, Andersen SD, Wang J, May A, Miron S, et al. (2011) Bi-directional routing of DNA mismatch repair protein human exonuclease 1 to replication foci and DNA double strand breaks. DNA Repair (Amst) 10: 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roesner LM, Mielke C, Fahnrich S, Merkhoffer Y, Dittmar KE, et al. (2013) Stable expression of MutLgamma in human cells reveals no specific response to mismatched DNA, but distinct recruitment to damage sites. J Cell Biochem 114: 2405–2414. [DOI] [PubMed] [Google Scholar]
- 44. Lisby M, Mortensen UH, Rothstein R (2003) Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577. [DOI] [PubMed] [Google Scholar]
- 45. Lisby M, Barlow JH, Burgess RC, Rothstein R (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713. [DOI] [PubMed] [Google Scholar]
- 46. Marsischky GT, Filosi N, Kane MF, Kolodner R (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev 10: 407–420. [DOI] [PubMed] [Google Scholar]
- 47. Yen K, Gitsham P, Wishart J, Oliver SG, Zhang N (2003) An improved tetO promoter replacement system for regulating the expression of yeast genes. Yeast 20: 1255–1262. [DOI] [PubMed] [Google Scholar]
- 48. Mendillo ML, Mazur DJ, Kolodner RD (2005) Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem 280: 22245–22257. [DOI] [PubMed] [Google Scholar]
- 49. Habraken Y, Sung P, Prakash L, Prakash S (1996) Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr Biol 6: 1185–1187. [DOI] [PubMed] [Google Scholar]
- 50. Dowen JM, Putnam CD, Kolodner RD (2010) Functional studies and homology modeling of Msh2-Msh3 predict that mispair recognition involves DNA bending and strand separation. Mol Cell Biol 30: 3321–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Srivatsan A, Bowen N, Kolodner RD (2014) Mispair-specific recruitment of the Mlh1-Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2-Msh3 complex. J Biol Chem 289: 9352–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 53. Byrne KP, Wolfe KH (2006) Visualizing syntenic relationships among the hemiascomycetes with the Yeast Gene Order Browser. Nucleic Acids Res 34: D452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wolfe KH, Shields DC (1997) Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708–713. [DOI] [PubMed] [Google Scholar]
- 55. Byrne KP, Wolfe KH (2005) The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res 15: 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wolfe K (2000) Robustness–it's not where you think it is. Nat Genet 25: 3–4. [DOI] [PubMed] [Google Scholar]
- 57. Massey SE, Moura G, Beltrao P, Almeida R, Garey JR, et al. (2003) Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res 13: 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raschle M, Marra G, Nystrom-Lahti M, Schar P, Jiricny J (1999) Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J Biol Chem 274: 32368–32375. [DOI] [PubMed] [Google Scholar]
- 59. Siehler SY, Schrauder M, Gerischer U, Cantor S, Marra G, et al. (2009) Human MutL-complexes monitor homologous recombination independently of mismatch repair. DNA Repair (Amst) 8: 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, et al. (1998) Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet 18: 276–279. [DOI] [PubMed] [Google Scholar]
- 61. Thompson JF, Moitoso de Vargas L, Koch C, Kahmann R, Landy A (1987) Cellular factors couple recombination with growth phase: characterization of a new component in the lambda site-specific recombination pathway. Cell 50: 901–908. [DOI] [PubMed] [Google Scholar]
- 62. Amin NS, Nguyen MN, Oh S, Kolodner RD (2001) exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol Cell Biol 21: 5142–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mine-Hattab J, Rothstein R (2012) Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol 14: 510–517. [DOI] [PubMed] [Google Scholar]
- 64. Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, et al. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962. [DOI] [PubMed] [Google Scholar]
- 65. Li L, Murphy KM, Kanevets U, Reha-Krantz LJ (2005) Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase delta in Saccharomyces cerevisiae. Genetics 170: 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morrison A, Bell JB, Kunkel TA, Sugino A (1991) Eukaryotic DNA polymerase amino acid sequence required for 3′----5′ exonuclease activity. Proc Natl Acad Sci U S A 88: 9473–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lea DE, Coulson CA (1948) The distribution of numbers of mutants in bacterial populations. J Genet 49: 264–285. [DOI] [PubMed] [Google Scholar]
- 68. Hargreaves VV, Shell SS, Mazur DJ, Hess MT, Kolodner RD (2010) Interaction between the Msh2 and Msh6 nucleotide-binding sites in the Saccharomyces cerevisiae Msh2-Msh6 complex. J Biol Chem 285: 9301–9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bowen N, Smith CE, Srivatsan A, Willcox S, Griffith JD, et al. (2013) Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. Proc Natl Acad Sci U S A 110: 18472–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shell SS, Putnam CD, Kolodner RD (2007) Chimeric Saccharomyces cerevisiae Msh6 protein with an Msh3 mispair-binding domain combines properties of both proteins. Proc Natl Acad Sci U S A 104: 10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Antony E, Hingorani MM (2003) Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry 42: 7682–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Harrington JM, Kolodner RD (2007) Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol Cell Biol 27: 6546–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kurtzman CP, Robnett CJ (2013) Relationships among genera of the Saccharomycotina (Ascomycota) from multigene phylogenetic analysis of type species. FEMS Yeast Res 13: 23–33. [DOI] [PubMed] [Google Scholar]
- 76. Fitzpatrick DA, Logue ME, Stajich JE, Butler G (2006) A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang H, Xu Z, Gao L, Hao B (2009) A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol Biol 9: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Downregulation of S. cerevisiae Pms1 expression upon addition of doxycycline. Expression levels of Pms1 (tagged with 9MYC) of strains containing the repressible tetO2-Pms1 promoter (lanes 1+2) or the endogenous Pms1 promoter (lanes 3+4). Cells were grown for 3 hours in YPD in the presence (or absence) of 10 µg/ml of doxycycline, as indicated. Whole-cell extracts were prepared and analyzed by SDS-PAGE and immunoblotting. The anti-Myc antibody was used to detect Pms1 expression and Pgk-1 was used as a loading control. Relates to Table 1.
(EPS)
Conservation of MLH1, MLH2, MLH3, and PMS1 in species of Saccharomycotina with sequenced genomes. ‘WGD clade’ indicates the group of species that have undergone whole-genome duplication. ‘CTG clade’ indicates the group of species that encode serine instead of leucine with the CTG codon. Phylogenetic relationships between species were derived from previously published analyses [75]–[77].
(EPS)
Clustering of the full-length Mlh2 sequences from fungi. This phylogenetic analysis reconstructed the Saccharomycotina and Pezizomycotina subphyla within Fungi as well as class and family relationships, consistent with homology between fungal MLH2 genes. Clade support values of 75 or higher are indicated on the phylogenetic tree.
(EPS)
Clustering of sequences of the N-terminal domains of MutL homologs from sequenced unikont species provided clear support for the homology of S. cerevisiae MLH2 and human PMS1, S. cerevisiae MLH3 and human MLH3, as well as S. cerevisiae PMS1 and human PMS2. Clade support values of 75 or higher are indicated on the phylogenetic tree.
(EPS)
MutL homologs in sequenced unikont genomes.
(XLS)
Re-annotation of MutL genes in fully sequenced genomes.
(DOCX)
S. cerevisiae strains used in this study.
(DOCX)