

Abstract
Nitro-oleic acid (OA-NO2), acting as anti-inflammatory signaling mediators, are involved in multiple signaling pathways. Lipoprotein-associated phospholipase A2 (Lp-PLA2) is well known as a cardiovascular risk biomarker. Our results showed that OA-NO2 downregulated the expression of Lp-PLA2 in a time- and dose-dependent manner, whereas native OA had no such effect. Furthermore, OA-NO2 could repress Lp-PLA2 expression in the peripheral blood mononuclear cells of apo CIII-transgenic (apo CIII TG) pigs, which exhibited higher Lp-PLA2 expression and activity than did wild-type (WT) pigs. OA-NO2 inhibited Lp-PLA2 expression in macrophages, independent of nitric oxide formation and PPARγ-activation. However, OA-NO2 downregulates Lp-PLA2 by inhibiting the p42/p44 mitogen-activated protein kinase (MAPK) and the nuclear factor κB (NFκB) pathways. When used to mediate anti-inflammatory signaling, the regulation of inflammatory cytokines and SOD by OA-NO2 might be associated with the reduction of Lp-PLA2. These results suggested that OA-NO2 might exert a vascular-protective effect partially via Lp-PLA2 inhibition.
Nitrated fatty acids are derived from NO- and NO2−-dependent redox reactions with unsaturated fatty acids, including nitro-oleic acid (OA-NO2) and nitro-linoleic acid (LNO2)1. These fatty acids are involved in multiple signaling pathways, acting as anti-inflammatory signaling mediators. As a potent ligand of peroxisome proliferator-activated receptor-γ (PPARγ), they exert signaling effects via either PPARγ-dependent or PPARγ-independent mechanisms2. Nitrated fatty acids suppress proinflammatory reactions by inhibiting nuclear factor κB (NFκB)-dependent gene expression and suppressing the proinflammatory signal transducer and activator of transcription-1 (STAT-1)3,4. For example, nitrated fatty acids inhibit lipopolysaccharide (LPS)-induced inflammatory cytokines (IL-6, TNFα and MCP-1) in RAW264.7 macrophages4. Additionally, nitrated fatty acids upregulate pulmonary epithelial heme oxygenase-1 (HO-1) expression, mediated by the mitogen-activated protein kinase (MAPK) signaling pathway, and induce heat shock factor- and nuclear factor-erythroid 2-related factor 2/Kelch-like ECH-associating protein 1 (Keap1/Nrf2)-dependent phase II gene expression5,6. In addition, nitrated fatty acids regulate enzyme function, cell signaling and protein trafficking via reversible post-translational protein modification7,8,9.
Nitrated fatty acids are found in the plasma, red cells and urine of healthy humans, and they can inhibit the neointimal hyperplasia induced by arterial injury and angiotensin II–induced hypertension10,11,12. In addition, it has been demonstrated that the nitro–fatty acids reduce atherosclerosis in apolipoprotein E–deficient mice13.
Lipoprotein-associated phospholipase A2 (Lp-PLA2), also known as platelet-activating factor acetylhydrolase (PAF-AH), is a unique member of the phospholipase A2 superfamily. Previous clinical and epidemiological studies have shown that Lp-PLA2 acts as a biomarker of cardiovascular risk due to its correlation with coronary disease and stroke14,15. Lp-PLA2 hydrolyzes oxidized phospholipids in oxLDL as a result of the generation of two key types of proinflammatory mediators: lysophosphatidylcholines (lysoPCs) and oxidized non-esterified fatty acids (oxNEFAs), both of which are thought to play roles in atherogenesis. For example, lysoPCs can generate reactive oxygen species (ROS) and upregulate the expression of proinflammatory cytokines and adhesion molecules16,17,18. The regulation of Lp-PLA2 is important in the development of atherosclerosis. However, no previous study has examined whether the nitrated fatty acid OA-NO2 has an effect on the expression of Lp-PLA2.
Herein, we investigated the effect of OA-NO2 on the expression of Lp-PLA2 in THP-1 cells. As a coronary heart disease (CHD) biomarker, a considerable amount of evidence supports the idea that Lp-PLA2 plays an important role in atherogenesis and development. Inhibition of Lp-PLA2 will thus help to ameliorate atherosclerosis. Therefore, the roles of Lp-PLA2 in the inflammatory response and in SOD activity regulated by OA-NO2 were also assessed.
Results
OA-NO2 downregulates Lp-PLA2 expression
To determine whether OA-NO2 regulates Lp-PLA2 expression, THP-1 cells were stimulated with 0.05 μM PMA following OA-NO2 treatment8, through which monocytes were transformed into macrophages. Lp-PLA2 mRNA expression was examined at 4 h, 12 h and 24 h after cell activation via quantitative real-time PCR. As shown in Figures 1A and 1C, Lp-PLA2 mRNA expression was increased at 12 h and 24 h after induction by PMA. OA-NO2 suppressed Lp-PLA2 transcription compared with PMA induction, and the suppression of Lp-PLA2 by OA-NO2 was both dose- and time-dependent. However, no change in the basal level of Lp-PLA2 mRNA was observed in the absence of cell differentiation induced by PMA. However, native OA had no effect on Lp-PLA2 expression (Supplementary figure S1). The regulation of Lp-PLA2 by OA-NO2 in protein showed the same effect as above in mRNA (Figure 1B). These results suggested that OA-NO2 inhibited the expression of Lp-PLA2 in THP-1 upon stimulation by PMA. The regulation of Lp-PLA2 by OA-NO2 was also evaluated in HUVECs, and it showed the same effect as in THP-1 (Supplementary figure S2).
Figure 1. OA-NO2 downregulates Lp-PLA2 expression.

THP-1 cells were incubated with OA-NO2 and OA at the indicated concentrations for 1 h and then stimulated with PMA (0.05 μM). (A), The relative Lp-PLA2 mRNA expression levels were determined after stimulation with PMA for 12 h. (B), The Lp-PLA2 protein expression levels were determined after stimulation with PMA for 24 h. (C), The relative Lp-PLA2 mRNA expression levels were determined after stimulation with PMA for 4 h, 12 h and 24 h. (D), Peripheral blood mononuclear cells isolated from ApoC III TG pigs were incubated with OA-NO2 at the indicated concentrations for 12 h, and the relative Lp-PLA2 mRNA expression levels were then determined. All data are presented as the mean ± SEM (n = 3); *P < 0.05, **P < 0.005 compared with PMA treatment based on analysis of variance.
Hypertriglyceridemia has recently come to be considered an independent risk factor for CHD, and human apo CIII-transgenic (apo CIII TG) pigs have been generated by our group as an animal model for studying hypertriglyceridemia19. Significantly higher Lp-PLA2 expression was observed in the apo CIII TG pigs than in wild-type (WT) pigs (unpublished). To identify the effect of OA-NO2 with respect to Lp-PLA2 downregulation, peripheral blood mononuclear cells isolated from the apo CIII TG pigs and the WT pigs were treated with OA-NO2 at different concentrations, showing that Lp-PLA2 was markedly suppressed in a dose-dependent manner in apo CIII TG pigs (Figure 1D), but it had no effect on the WT pigs (Supplementary figure S3). It might be concluded that OA-NO2 could inhibit the expression of endogenous PLA2G7 activated by exogenous Apo CIII, but have no significant effect on the basic expression of endogenous PLA2G7 without activation by Apo CIII. These results suggested that OA-NO2 downregulated the expression of Lp-PLA2 both in THP-1-derived macrophages and in apo CIII TG pigs' peripheral blood mononuclear cells.
The cell viability affected by the addition of OA-NO2 and OA have been detected by a Cell Counting Kit-8 (CCK-8, Dojindo, Japan) assay. OA-NO2 inhibits cell proliferation in dose-dependent manner, while OA have no effect on the cell proliferation (Supplementary figure S4).
OA-NO2 downregulates Lp-PLA2 via an NO-independent mechanism
To investigate whether OA-NO2, as an NO donor, regulates Lp-PLA2 in a manner that is dependent on NO release, THP-1 cells were pretreated with carboxy-PTIO (an NO scavenger) and sodium nitroprusside (SNP, a nitric oxide donor) for 1 h prior to PMA stimulation. As shown in Figures 2A and B, carboxy-PTIO had no effect on Lp-PLA2 expression under either basal or downregulated levels of OA-NO2 treatment. However, SNP upregulated Lp-PLA2 transcription, contrary to the results observed under OA-NO2 treatment, and carboxy-PTIO decreased the effect of SNP on Lp-PLA2 transcription. OA-NO2 also could inhibit the upregulation of PLA2G7 mediated by SNP (Figure 2C). These results demonstrated that the regulation of Lp-PLA2 expression by OA-NO2 was independent of nitric oxide release.
Figure 2. The OA-NO2-induced downregulation of lp-PLA2 occurs via an NO-independent mechanism.

THP-1 cells were incubated with OA-NO2, SNP and PTIO at the indicated concentrations for 1 h and then stimulated with PMA (0.05 μM). (A and C), The relative Lp-PLA2 mRNA expression levels were determined after stimulation with PMA for 12 h. (B), The Lp-PLA2 protein expression levels were determined after stimulation with PMA for 24 h.
OA-NO2 downregulates Lp-PLA2 via a PPARγ-independent mechanism
As a potent ligand of PPARγ, OA-NO2 exerts signaling effects via both PPARγ-dependent and PPARγ-independent mechanisms2. To address whether the regulation of Lp-PLA2 expression by OA-NO2 is PPARγ-dependent, THP-1 cells were treated with Rosiglitazone and GW9662, which are a known PPARγ ligand and PPARγ inhibitor, respectively, as previously described. With an increase in the concentration of Rosiglitazone, Lp-PLA2 transcription showed the opposite tendency of that induced by OA-NO2 treatment. In contrast, GW9662 showed no effect on the regulation of Lp-PLA2 expression by OA-NO2 in terms of either mRNA or protein levels (Figure 3B and 3C), which suggested that the inhibition of PPARγ was not involved in the regulation of Lp-PLA2. These results suggested that downregulation of Lp-PLA2 expression by OA-NO2 occurred via a PPARγ-independent mechanism.
Figure 3. The OA-NO2-induced downregulation of lp-PLA2 occurs via a PPARγ-independent mechanism.

THP-1 cells were incubated with OA-NO2, Rosiglitazone and GW9662 at the indicated concentrations for 1 h and then stimulated with PMA (0.05 μM). (A and C), The relative Lp-PLA2 mRNA expression levels were determined after stimulation with PMA for 12 h. (B), The activation of PPARγ and Lp-PLA2 protein expression levels were determined after stimulation with PMA for 24 h.
OA-NO2 downregulates Lp-PLA2 via the p42/p44 MAPK pathway
The MAPK signaling pathway was examined as possible mechanism of the regulation of Lp-PLA2 expression by OA-NO2. Inhibition of the phosphorylation of p42/p44 MAPK using PD98059 suppressed the expression of Lp-PLA2 in a manner similar to that of OA-NO2, and OA-NO2 also inhibited the phosphorylation of p42/p44 MAPK (Figure 4A and 4B). These results demonstrated that the downregulation of Lp-PLA2 expression by OA-NO2 occurred via the p42/p44 MAPK pathway.
Figure 4. OA-NO2 downregulates Lp-PLA2 expression via the p42/p44 MAPK pathway.
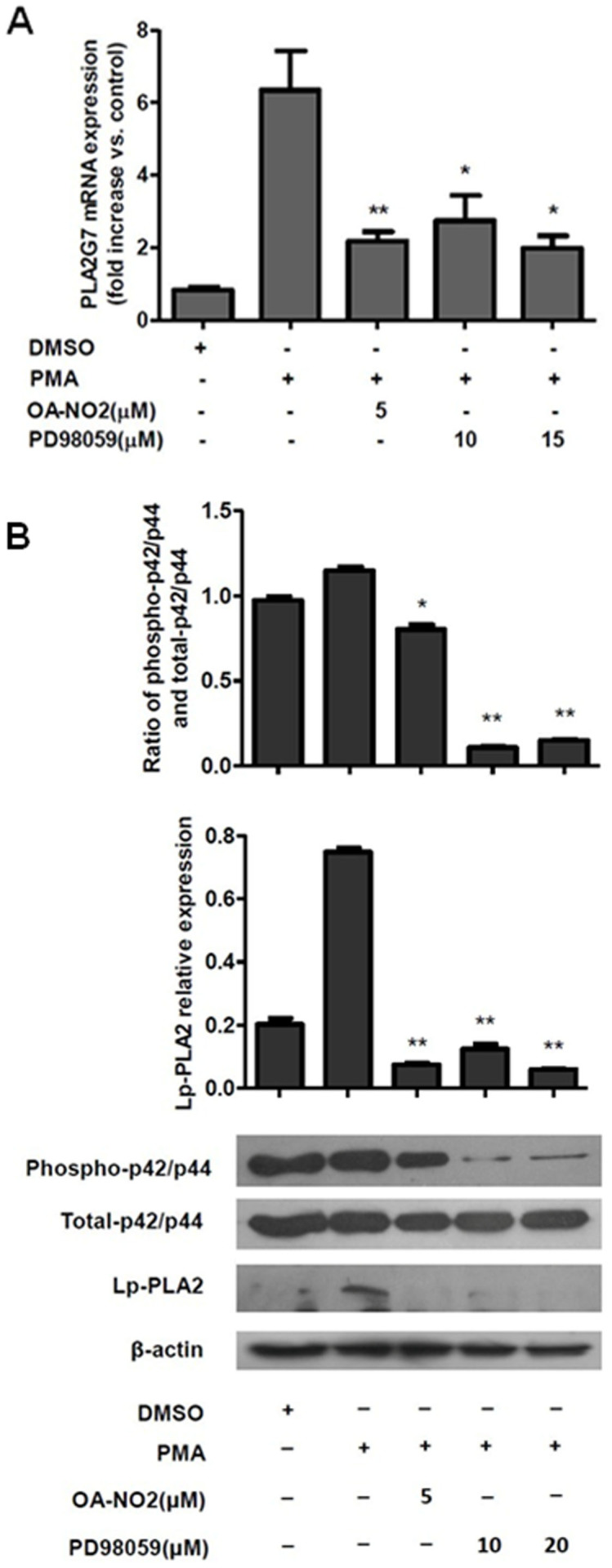
THP-1 cells were incubated with OA-NO2 and PD98059 at the indicated concentrations for 1 h and then stimulated with PMA (0.05 μM). (A), The relative Lp-PLA2 mRNA expression levels were determined after stimulation with PMA for 12 h. (B), The phosphorylation of p42/p44 MAPK and the Lp-PLA2 protein expression levels were determined after stimulation with PMA for 24 h.
OA-NO2 downregulates Lp-PLA2 via the NFκB pathway
The NFκB signaling pathway was also examined as possible mechanism of the regulation of Lp-PLA2 expression by OA-NO2. Inhibition of the phosphorylation of p65 NFκB using PDTC suppressed the expression of Lp-PLA2 in a manner similar to that of OA-NO2, and OA-NO2 also inhibited the phosphorylation of p65 NFκB (Figure 5A and 5B). Furthermore, lipopolysaccharide (LPS), which has been shown to induce NFκB-dependent gene expression in macrophages20, upregulated the expression of Lp-PLA2 via phosphorylation of NFκB, and this effect was abolished by OA-NO2 treatment (Figure 5A and 5B). These results demonstrated that the downregulation of Lp-PLA2 expression by OA-NO2 occurred via the NFκB pathway. As is shown in Figure 5B, OA-NO2 didn't inhibit the phosphorylation of p65 NFκB significantly in the presence of LPS, but it strongly reduced the expression of Lp-PLA2 protein. These results may suggested that there were other pathways involving in the regulation of Lp-PLA2 except for the p65 NFκB. OA-NO2 may downregulate PLA2G7 expression partially through reducing the phosphor-p65 NFκB.
Figure 5. OA-NO2 downregulates Lp-PLA2 expression via the p65 NFκB pathway.

THP-1 cells were incubated with OA-NO2, PDTC and LPS at the indicated concentrations for 1 h and then stimulated with PMA (0.05 μM). (A), The relative Lp-PLA2 mRNA expression levels were determined after stimulation with PMA for 12 h. (B), The phosphorylation of p65 NFκB and the Lp-PLA2 protein expression levels were determined after stimulation with PMA for 24 h. (C), the NFκB/DNA binding activity was measured by EMSA.
To determine whether OANO2 inhibit the activity of NFκB, the NFκB/DNA binding activity was measured by electrophoretic mobility shift assay (EMSA). As shown in Figure 5C, OA-NO2 could reduce the NFκB/DNA binding activity significantly. It could be concluded that OA-NO2 downregulated the expression of Lp-PLA2 through inhibiting the p65 NFκB pathway.
The regulation of the classical NFκB pathway by OA-NO2 has also been investigated by assessing the degradation of IκBα. PDTC increased the IκBα expression and LPS decreased the IκBα expression compare with PMA treatment, but OA-NO2 inhibited the expression of IκBα significantly (Supplementary figure S5). These results suggested that PDTC and LPS might regulate the expression of PLA2G7 through the IκB-NFκB pathway and the inhibition of phosphor-p65 by OA-NO2 might not through the classical NFκB pathway. As it is reported before, the electrophilic nature of OA-NO2 could result in nitroalkylation of p65 protein in macrophages, and represse NFκB-dependent target gene expression3. The reduction of phosphor-p65 NFκB by OA-NO2 may have a relationship with the nitroalkylation of p65.
The regulation of inflammatory cytokines and SOD by OA-NO2 are associated with the reduction of Lp-PLA2
To evaluate whether the anti-inflammatory effect of nitrated fatty acids is related to the regulation of Lp-PLA2, we pretreated THP-1 cells with either 5 μM OA-NO2 or 75 μM 1-linoleoyl glycerol (1-LG, Lp-PLA2 inhibitor) for 1 h prior to stimulation with 0.05 μM PMA and 1 μg/ml LPS. Following stimulation with PMA and LPS for 24 h, the levels of 3 different inflammatory cytokines in the media were measured. As shown in Figure 6A, 6B and 6C, LPS stimulated the release of inflammatory cytokines, and OA-NO2 suppressed the LPS-induced production of the cytokines IL-6, MCP-1 and TNFα. The Lp-PLA2 inhibitor 1-LG also suppressed the LPS-induced production of inflammatory cytokines, indicating that downregulation of Lp-PLA2 reduced the release of proinflammatory cytokines. Therefore, these results suggested that OA-NO2 might suppress the release of inflammatory cytokines partially through downregulating the expression of Lp-PLA2.
Figure 6. OA-NO2 downregulates Lp-PLA2 expression and is associated with the reduction of proinflammatory cytokines and ROS.

(A–C), THP-1 cells were incubated with OA-NO2 and 1-LG at the indicated concentrations for 1 h and then stimulated with PMA (0.05 μM) and LPS (1 μg/ml) for 24 h. The release of proinflammatory cytokines (IL6, MCP-1 and TNFα) into the medium was determined by ELISA. (D), Both shRNA-Lp-PLA2 and a negative shRNA control were transfected into THP-1 cells for 24 h prior to stimulation with 0.05 μM PMA for another 24 h. (E and F), The down-regulation of the expression of Lp-PLA2 by shRNA. (G), An Lp-PLA2 overexpression plasmid and pcDNA3.1 plasmid were transfected into 293T cells for 24 h, and the medium were used to treat THP-1 cells together with PMA. Then, SOD activity was determined. (H), The PAF-AH activity in the medium of the transfected 293T cells.
Both inflammation and ROS play important roles in the development of atherosclerosis. As an inflammatory regulator, OA-NO2 can suppress the generation of ROS21. To characterize the relationship between Lp-PLA2 and SOD, we treated macrophages induced from THP-1 cells with shRNA-Lp-PLA2 and an Lp-PLA2 overexpression plasmid and then quantified the SOD activity in those cells. The mRNA and protein expression of Lp-PLA2 was downregulated by shRNA-Lp-PLA2, and the activity of Lp-PLA2 in the medium of cells overexpressing Lp-PLA2 was much higher than in the control medium (Figure 6E, 6F and 6H). Furthermore, SOD activity was increased in shRNA-Lp-PLA2-transfected cells and decreased in Lp-PLA2-treated cells (Figure 6D and 6G). We concluded that Lp-PLA2 repressed the activity of SOD in macrophages, whereas OA-NO2 increased the SOD activity in a dose-dependent manner (Figure 6D and 6G). OA-NO2 could upregulate the activity of SOD through downregulating the expression of Lp-PLA2. In summary, the regulation of inflammatory cytokines and SOD by OA-NO2 was associated with the reduction of Lp-PLA2.
Discussion
Due to its correlation with coronary disease and stroke, Lp-PLA2 is considered a cardiovascular risk marker14,15. OA-NO2, a nitrated fatty acid, has been reported to have relevant biological effects on the vasculature and can be used as a therapeutic tool to investigate vascular and cardiac tissue damage in cardiovascular models of disease10,21,22. In the present study, we examined the potential regulatory relationship between OA-NO2 and the expression of Lp-PLA2 and explored possible pathways underlying this relationship. We found the first evidence that OA-NO2 could downregulate Lp-PLA2 expression in macrophages. Our results may provide a new perspective on the treatment of coronary disease and stroke.
Nitrated fatty acids are formed via NO-dependent oxidative reactions23. These fatty acids act not only as NO reservoirs but also as endogenous donors of NO that can upregulate the expression and activity of endothelial NO synthase to increase NO levels22,24,25,26. NO has been applied as a medicine for the treatment of cardiovascular conditions based on its modulation of angiogenesis and its protective role in the vasculature27. Peroxisome proliferator-activated receptors (PPARs) are a series of nuclear receptors involved in a multitude of physiological processes, such as anti-inflammatory effects in several cell types, including smooth muscle cells28,29. Moreover, PPARγ-regulated gene products play critical modulatory roles in cardiovascular disease30. As a potent ligand of PPARγ, OA-NO2 exerts signaling effects via both PPARγ-dependent and PPARγ-independent mechanisms2. It seems likely that OA-NO2 might regulate Lp-PLA2 expression via PPARγ activation and NO formation. However, our results suggested that OA-NO2 regulated Lp-PLA2 expression via PPARγ- and NO-independent mechanisms. The mechanism of the regulation of Lp-PLA2 was observed to be similar to that of the PPARγ-independent inhibitory effects of OA-NO2 on cytokine production and inflammation2. This result is consistent with a recent report that OA-NO2 inhibits the LPS-induced secretion of proinflammatory cytokines in macrophages, independent of nitric oxide formation and PPARγ activation3.
In addition to PPARγ activation, OA-NO2 is involved in multiple other signaling pathways, such as the NFκB, STAT-1, MAPK and Keap1/Nrf2 pathways2,3,4,5,6. The NFκB pathway is an essential signaling pathway involved in conveying inflammatory signals. Nitrated fatty acids can suppress the expression of NFκB-dependent target genes, including inflammatory cytokine-related genes, by inhibiting the alkylation of the p65 NFκB protein to inhibit its DNA-binding activity3. The MAPK family is an important mediator of signal transduction and can be activated by many different stimuli. Therefore, both the NFκB and MAPK pathways are potentially related to the regulation of Lp-PLA2 by OA-NO2. This possibility led us to investigate whether the NFκB and MAPK pathways were involved in the downregulation of Lp-PLA2. Our findings suggested that the NFκB and p42/p44 MAPK pathways did indeed play significant roles in the regulation of Lp-PLA2 expression.
Atherosclerosis is characterized by a chronic state of vascular oxidative stress and inflammation31. Oxidative stress is associated with the formation of lipid oxidation products, which increase vascular inflammation, and it is thought to play a key role in the development of atherosclerosis32. Inhibition of oxidative stress and inflammation is important for the prevention and treatment of atherosclerosis. Lp-PLA2, as a biomarker of CHD, might exert regulatory effects related to anti-atherogenesis that are induced by OA-NO2, and our results indeed showed that the downregulation of Lp-PLA2 induced by OA-NO2 suppressed the release of inflammatory cytokines and increased the activity of SOD, confirming the potential for a protective role of OA-NO2 in cardiovascular contexts.
In humans, Lp-PLA2 is expressed mainly by macrophages and is being evaluated as a potential therapeutic target in coronary heart disease (CHD)33, for which it is currently in phase III clinical trials. Research addressing the regulatory mechanism between OA-NO2 and Lp-PLA2 in human macrophages might provide insight related to the treatment of atherosclerosis. However, the correlation between the endogenous OA-NO2 and Lp-PLA2 in coronary disease and stroke remains unknown and requires further clinical investigation.
In summary, our data demonstrate that OA-NO2 inhibits Lp-PLA2 expression in macrophages, and this inhibition is associated with the reduction of inflammatory cytokines and the increase of SOD activity. Therefore, OA-NO2 might exert a vascular-protective effect partially mediated by Lp-PLA2 inhibition.
Methods
OA-NO2 (9-Nitrooleate and 10-Nitrooleate), oleic acid and 1-linoleoyl glycerol were purchased from the Cayman Chemical Company Inc., USA. Antibodies against p42/p44, phosphorylated-p42/p44, p65 and phosphorylated-p65 were obtained from Cell Signaling Technology Inc., USA. PPARγ and β-actin antibodies were purchased from Proteintech Group Inc., China. An Lp-PLA2 antibody was purchased from Bioss Inc., China. GW9662 and Rosiglitazone were obtained from Sigma-Aldrich Inc., USA. All other reagents, unless otherwise specified, were purchased from Beyotime Inc., China.
Cell culture
THP-1 cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum (Hyclone) at 37°C in a humidified atmosphere with 5% CO2. Prior to the experiments, the medium was changed to medium containing 0.5% serum for 16 h. Then, the cells were treated with the specified drugs diluted in 0.5% serum-containing RPMI1640 medium. Every treatment was performed in triplicate.
Peripheral blood mononuclear cells isolation
Blood was collected from pigs using anticoagulant EDTA tubes, and macrophages were isolated from the whole blood with Histopaque (SIGMA Inc., USA) according to the manufacturer's instructions.
Quantitative real-time PCR
THP-1 cells (85% confluent) were pretreated with the specified drugs for 1 h prior to stimulation with 0.05 μM PMA for 12 h. Then, the cells were harvested for RNA extraction8. Total RNA was isolated from cultured cells using TRIzol-A+ reagent (Tiangen Inc., China) according to the manufacturer's instructions. First-strand cDNAs were synthesized from 1 μg of total RNA using a BioRT cDNA first stand synthesis kit (Bioer Inc., China), and the samples were analyzed with a Bioeasy SYBR green I real-time PCR kit (Bioer Inc., China) with gene expression primers for Lp-PLA2 and GAPDH (a housekeeping gene). The sequences of the primers were 5′-TAATGATCGCCTTGACACCCT -3′ (forward) and 5′-TACAGCAGCAACTATAAACCC -3′ (reverse) for Lp-PLA2 and 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse) for GAPDH.
Western blot analysis
Cells treated under different experimental conditions were washed twice with PBS and lysed in lysis buffer for Western and IP analyses (Beyotime Inc., China) with 1 mM PMSF and a phosphatase inhibitor for 30 min on ice, followed by centrifugation. The obtained protein concentrations were quantified using the Enhanced BCA protein assay kit (Beyotime Inc., China). Equal amounts of protein were fractionated via 12% SDS–PAGE and then transferred to a nitrocellulose membrane, blocked with 5% (wt/vol) nonfat milk in TBST (50 mM Tris, PH 7.5, 250 mM NaCl, 0.2% Tween 20) and probed with antibodies overnight at 4°C. Next, the membranes were washed in TBST three times and exposed to the appropriate secondary antibodies for 1 h at room temperature. Immunoreactive bands were visualized using BeyoECL Plus (Beyotime Inc., China) according to the manufacturer's instructions.
Detection of PAF-AH and SOD activity
The PAF-AH levels in the culture media were measured with a PAF Acetylhydrolase Kit (Cayman Inc., USA) following the manufacturer's instructions. The SOD levels in the cells were quantified with a Total Superoxide Dismutase Assay Kit with WST-1 (Beyotime Inc., China) according to the manufacturer's instructions.
Electrophoretic mobility shift assay
Nuclear proteins were prepared from THP-1 cells using Nuclear and cytoplasmic protein extraction kit (Beyotime Inc., China) as the manufacturer's instructions described. Nuclear proteins (3 μg) were incubated with biotin-labeled and un-labeled NFκB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGG-3′) which were purchased from Beyotime Institute of Biotechnology. The reaction mixture was separated in 6.5% nondenaturing polyacrylamide gel, and then exposed to the streptavidin-HRP conjugate. Immunoreactive bands were visualized using BeyoECL Plus (Beyotime Inc., China) according to the manufacturer's instructions.
Quantification of proinflammatory cytokines
The levels of proinflammatory cytokines in the culture media were measured with a Human IL-6 ELISA Kit, a Human TNFα ELISA Kit and a Human MCP-1 ELISA Kit (Boster Inc., China), following the manufacturer's instructions.
Statistical analysis
The results are expressed as the mean ± SEM throughout the paper. The data were analyzed using an unpaired two-tailed Student's t-test unless otherwise specified. P < 0.05 was considered to be statistically significant.
Author Contributions
X.T. conceived and directed the project. G.W. and Y.J. designed the experiments. G.W., Y.J., N.G., Q.S., Z.L., X.H., L.Q., T.W., W.H., H.O. and D.P. carried out the experiments, conducted the data analysis and interpreted the results. G.W. wrote and edited the paper. All authors reviewed the manuscript.
Supplementary Material
supplementary figures
Acknowledgments
This work was supported by grants from the China Postdoctoral Science Foundation (No. 2012M510886), the China National Key Basic Research Program (973 Program, No. 2011CBA01000), and the National Natural Science Foundation (No. 31201874).
References
- Lundberg J. O. et al. Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol 5, 865–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopfer F. J. et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci U S A 102, 2340–5 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui T. et al. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. J Biol Chem 281, 35686–98 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa T. et al. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: a critical role of mitogen-activated protein kinase phosphatase 1. Endocrinology 149, 4086–94 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles K. E. et al. Fatty acid transduction of nitric oxide signaling: nitrolinoleic acid mediates protective effects through regulation of the ERK pathway. Free Radic Biol Med 46, 866–75 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen E. et al. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J Biol Chem 284, 33233–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batthyany C. et al. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem 281, 20450–63 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacci G. et al. Electrophilic fatty acids regulate matrix metalloproteinase activity and expression. J Biol Chem 286, 16074–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen E. et al. Electrophilic nitro-fatty acids activate NRF2 by a KEAP1 cysteine 151-independent mechanism. J Biol Chem 286, 14019–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole M. P. et al. Nitro-fatty acid inhibition of neointima formation after endoluminal vessel injury. Circ Res 105, 965–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. et al. Nitro-oleic acid inhibits angiotensin II-induced hypertension. Circ Res 107, 540–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker P. R. et al. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem 280, 42464–75 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph T. K. et al. Nitro-fatty acids reduce atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 30, 938–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatine M. S. et al. Prognostic utility of lipoprotein-associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler Thromb Vasc Biol 27, 2463–9 (2007). [DOI] [PubMed] [Google Scholar]
- Tsimikas S. et al. Lipoprotein-associated phospholipase A2 activity, ferritin levels, metabolic syndrome, and 10-year cardiovascular and non-cardiovascular mortality: results from the Bruneck study. Eur Heart J 30, 107–15 (2009). [DOI] [PubMed] [Google Scholar]
- Davis B. et al. Electrospray ionization mass spectrometry identifies substrates and products of lipoprotein-associated phospholipase A2 in oxidized human low density lipoprotein. J Biol Chem 283, 6428–37 (2008). [DOI] [PubMed] [Google Scholar]
- MacPhee C. H. et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem J 338 (Pt 2), 479–87 (1999). [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T., Kobayashi T. & Kamata K. Role of lysophosphatidylcholine (LPC) in atherosclerosis. Curr Med Chem 14, 3209–20 (2007). [DOI] [PubMed] [Google Scholar]
- Wei J. et al. Characterization of a hypertriglyceridemic transgenic miniature pig model expressing human apolipoprotein CIII. FEBS J 279, 91–9 (2012). [DOI] [PubMed] [Google Scholar]
- Sharif O., Bolshakov V. N., Raines S., Newham P. & Perkins N. D. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol 8, 1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles B. et al. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circ Res 91, 375–81 (2002). [DOI] [PubMed] [Google Scholar]
- Rudolph V. et al. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc Res 85, 155–66 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell V. B. et al. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem Res Toxicol 12, 83–92 (1999). [DOI] [PubMed] [Google Scholar]
- Faine L. A. et al. Bioactivity of nitrolinoleate: effects on adhesion molecules and CD40-CD40L system. J Nutr Biochem 21, 125–32 (2010). [DOI] [PubMed] [Google Scholar]
- Khoo N. K. et al. Activation of vascular endothelial nitric oxide synthase and heme oxygenase-1 expression by electrophilic nitro-fatty acids. Free Radic Biol Med 48, 230–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopfer F. J. et al. Fatty acid transduction of nitric oxide signaling. Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J Biol Chem 280, 19289–97 (2005). [DOI] [PubMed] [Google Scholar]
- Fukumura D., Kashiwagi S. & Jain R. K. The role of nitric oxide in tumour progression. Nat Rev Cancer 6, 521–34 (2006). [DOI] [PubMed] [Google Scholar]
- Francis G. A., Fayard E., Picard F. & Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol 65, 261–311 (2003). [DOI] [PubMed] [Google Scholar]
- Staels B. et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 393, 790–3 (1998). [DOI] [PubMed] [Google Scholar]
- Chawla A. et al. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 7, 48–52 (2001). [DOI] [PubMed] [Google Scholar]
- Libby P., Ridker P. M. & Maseri A. Inflammation and atherosclerosis. Circulation 105, 1135–43 (2002). [DOI] [PubMed] [Google Scholar]
- Witztum J. L. & Berliner J. A. Oxidized phospholipids and isoprostanes in atherosclerosis. Curr Opin Lipidol 9, 441–8 (1998). [DOI] [PubMed] [Google Scholar]
- White H. et al. Study design and rationale for the clinical outcomes of the STABILITY Trial (STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY) comparing darapladib versus placebo in patients with coronary heart disease. Am Heart J 160, 655–61 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary figures