

Abstract
Purpose of review
Loss of cell growth control does not explain why tumors form as the immune system recognizes many malignant cells and keeps them in check. The local inflammatory microenvironment is a pivotal factor in tumor formation as tumor associated inflammation actively suppresses anti-tumor immunity. The purpose of this review is to evaluate emerging evidence that amino acid catabolism is a key feature of tumor-associated inflammation that supports tumor progression and immune resistance to therapy.
Recent findings
Enhanced amino acid catabolism in inflammatory tumor microenvironments correlates with carcinogen resistance and immune regulation mediated by tumor-associated immune cells that protect tumors from natural and vaccine-induced immunity. Interfering with metabolic pathways exploited by tumors is a promising anti-tumor strategy, especially when combined with other therapies. Moreover, molecular sensors that evolved to detect pathogens may enhance evasion of immune surveillance to permit tumor progression.
Summary
Innate immune sensing that induces amino acid catabolism in tumor microenvironments may be pivotal in initiating and sustaining local inflammation that promotes immune resistance and attenuates anti-tumor immunity. Targeting molecular sensors that mediate these metabolic changes may be an effective strategy to enhance anti-tumor immunity that prevents tumor progression, as well as improving the efficacy of cancer therapy.
Keywords: Amino acid catabolism, tumor tolerance, anti-tumor immunity, interferons, innate immune sensors
Introduction
Malignant cells with defective growth control create tumors but the immune system impedes tumor formation by eliminating many malignant cells (Fig. 1). Some malignant cells progress to form tumors by evading immune checkpoints to establish local microenvironments that protect malignant cells from immune-mediated destruction (1). Pre-established immune privilege explains tumor resistance to vaccines, which elicit weak clinical responses even if robust immune responses manifest. Tumor cells may suppress anti-tumor immunity themselves but tumor-associated inflammation, which extends to draining lymph nodes, is a critical factor regulating anti-tumor immunity.
Figure 1. Stages in tumor formation and therapy.
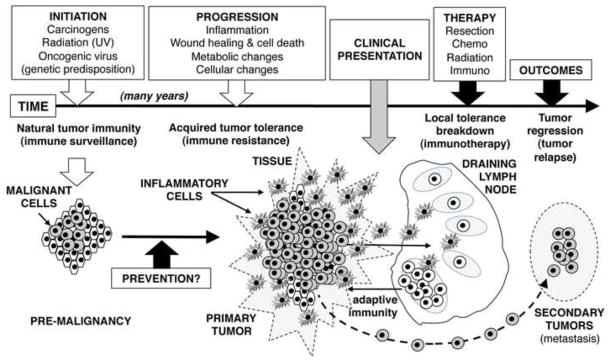
Once initiated, malignant lesions promote local inflammation that inhibits immune surveillance to facilitate tumor progression. Eventually, local lymph node involvement suppresses anti-tumor immunity. Tumor tolerance established prior to clinical presentation is a barrier to successful therapy and creates niches for metastasis and relapse.
The focus of this review is the emerging paradigm that increased amino acid catabolism is a frequent and important feature of inflammation that promotes tumor progression and inhibits anti-tumor immunity (Fig. 2). Recent reviews describe immune regulatory pathways involving dendritic cells (DCs), macrophages (MΦs), myeloid-derived suppressor cells (MDSCs), Natural Killer (NK), mast cells, and Foxp3-lineage regulatory CD4 T cells (Tregs) in chronic inflammatory syndromes (Table 1), including roles in tumor progression and immunotherapy (2–6). Biologic significance is still emerging but increased amino acid catabolism is commonly associated with inflammation that regulates rather than stimulates - immunity.
Figure 2. Amino acid catabolism during tumor progression, regression and relapse.
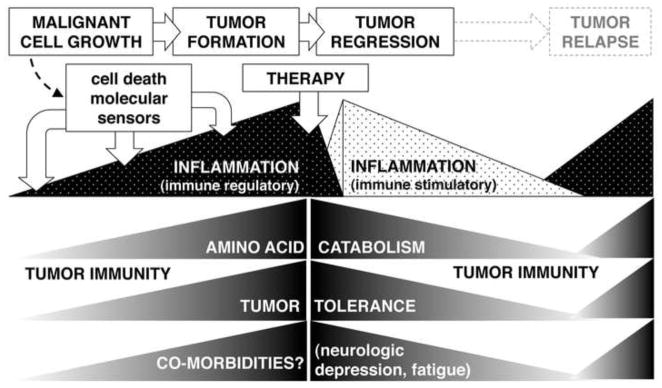
Amino acid catabolism increases during tumor formation, and correlates with increased local inflammation and cancer-associated morbidities. Therapeutic interventions modify local inflammation to break tumor tolerance and reduce amino acid catabolism and co-morbidities, while relapse correlates with the return of processes linked to cancer.
Table 1.
Immune regulatory pathways linked to tumor tolerance
| Pathway | Description |
|---|---|
| cell types: | |
| MDSCs | tumor infiltrating & circulating monocytic regulatory cells |
| DCs | specialized subsets suppress effector T cells & activate Tregs |
| MΦs | Specialized subsets are tumor infiltrating regulatory cells |
| Tregs | potent regulatory CD4 T cells (auto/tumor antigen-specific) |
| NK cells | non-antigen specific regulatory lymphocytes |
| Mast cells | innate immune cells active in wound healing, host defense & allergy |
|
| |
| molecules: | |
| CTLA4 | induces T cell anergy & induces IDO in DCs (blocked by Ipilimumab) |
| PD-1(PD-L) | inhibitory co-stimulator pathway in T cells & Tregs |
| GITR | mediates immune regulation to steroids |
| TGFβ, IL-10 | regulatory cytokines |
|
| |
| metabolic pathways: | |
| IFNs (I, II) | immune activating cytokines that can stimulate or suppress immunity |
| IDO1 (IDO2?) | catabolizes Trp to suppress effector T cells & activate Tregs |
| TDO | as for IDO |
| TPH-1 | consumes Trp to promote tolerance |
| ARG (I, II) | catabolizes Arg to suppress effector T cells and induce Tregs |
| mTOR | senses amino acid availability (nutrient check point control) |
| GCN2 kinase | amino acid depletion sensor (via tRNA); induces cell stress responses |
| AhR | senses Kyn (and other Trp catabolites) to regulate immunity |
Inflammation can stimulate or regulate immunity
The paradigm that inflammation stimulates immunity is a fundamental tenet of immunology but inflammation may also drive immune regulation. Thus interferons (IFNs) are known as ‘pro-inflammatory’ cytokines but they also induce immune regulation; moreover which functional response to IFNs is dominant is not always obvious (7). Local inflammation incited by malignancies that transition into tumors regulates tumor-specific immunity; hence, a therapeutic goal is to convert immune regulatory inflammation into stimulatory inflammation (Fig. 2).
Increased amino acid catabolism inhibits immunity in many chronic inflammatory syndromes (7–9). Immune tolerance to transplanted skin in mice correlated with enhanced amino acid catabolism in graft-associated DCs (10), and mast cells expressing tryptophan hydroxylase-1 (TPH-1) promoted tumor relapse after therapy and allograft tolerance (3). Genetically enhanced Trp catabolism mediated by indoleamine 2,3 dioxygenase (IDO) also suppressed rat lung allograft rejection (11, 12). Microbial infections often induce IDO (via IFNs), which may impede host immunity to promote pathogen persistence, a situation analogous to tumor persistence (8). Likewise, tumor tolerance is often linked to increased amino acid catabolism, though other regulatory pathways (Table 1) may be active simultaneously (13, 14). Thus tumors exploit natural immune regulatory pathways that evolved to protect healthy tissues from hyper-immunity. The paradigm that tumors are analogous to aseptic wounds that do not heal is useful since dying cells release cell contents such as DNA that are potentially immunostimulatory but natural regulation may reinforce ‘self’ tolerance under aseptic conditions.
Amino acid catabolism and tumor development
Genetic predisposition, carcinogens, radiation (UVB, ionizing) or oncogenic viruses synergize to generate malignant dividing cells (Fig. 1). Malignant cells depend on access to essential nutrients such as iron, and iron chelation impedes tumor growth (15). Correlations between tumor growth and increased local amino acid catabolism (i.e. elevated nutrient consumption) are not consistent with this paradigm. It is important to emphasize that amino acid catabolism triggers profound changes in immune cell functions via amino acid sensors and catabolite receptors, and it may not be necessary to actually ‘starve’ cells for immune regulatory effects to manifest. Merely reducing the pool of available amino acids may suffice to induce regulatory responses; if so, inhibiting amino acid catabolism may offer therapeutic opportunities. Trp catabolism is the most studied aspect of amino acid catabolism in tumor microenvironments and the recent literature reflects this bias, which was initiated by the finding that IDO activity protected fetal tissues from maternal immunity during pregnancy in mice (16, 17). However this discovery was perhaps anticipated by reports over 40 years ago describing increased Trp catabolism in breast and cervical cancer patients (18, 19).
Increased IDO activity often manifests in inflammatory lesions induced by tumor promoters such as oncogenic human papilloma (HPV) and murine leukemia virus (MuLV), phorbol myristate acetate (PMA or TPA) and UVB radiation (20–24), suggesting that Trp catabolism may suppress immune surveillance mechanisms. Indeed, IDO1-deficient (IDO1-KO) mice were more resistant to papilloma formation in the DMBA/TPA model of inflammation-driven carcinogenesis (24). IDO1-KO mice also exhibited more resistance to lung and breast tumors (25), and IDO1 loss correlated with reduced neo-vascularization, metastasis and functional maturation of myeloid derived suppressor cells (MDSCs) in the lung tumor model, suggesting that IDO facilitates these tumor supportive functions. However, it is unclear how developing tumors induce IDO. An emerging possibility is that dying tumor cells may trigger local IFN type I (IFNaβ) production via DNA sensors such as Toll-Like receptor-9 (TLR9) or the Stimulator of Interferon Genes (STING) adaptor, which then induces IDO (26, 27), though excessive STING activation also promotes autoimmunity (28, 29). Trp catabolites may also promote tumorigenesis as Trp catabolites activated β-catenin signaling to promote colon tumorigenesis via a T cell independent pathway in mice (30). Targeting amino acid catabolism may help prevent malignancies developing into tumors, though increased risk of autoimmunity is a potential undesirable consequence of such interventions.
Amino acid catabolism in tumor microenvironments
Elevated Trp and arginine (Arg) catabolism have been linked to regulation of anti-tumor immunity. Two different enzymes with oxygen binding iron-tetrapyrrole co-factors - IDO and tryptophan 2,3 dioxygenase (TDO) - catalyze oxidative Trp catabolism to generate kynurenine, and TPH-1 converts Trp into serotonin. IDO transcription is induced by IFNαβ and IFNγ in a range of cell types, including selected immune cells but measuring IDO enzyme activity is important as post-translational modifications may be required for IDO activity. Mice and humans possess two linked genes encoding IDO enzymes (IDO1, IDO2). IDO1 mediates regulatory responses to many inflammatory stimuli, including tumor growth (8). A role for IDO2 has not been defined, though recent studies on IDO2-deficient mice showed that IDO2 and IDO1 control cytokine expression differently and IDO2 did not phenocopy IDO1 in promoting inflammatory skin cancer (G. Prendergast, personal communication). IDO expression occurs in some cancer cells, stromal cells and certain immune cells such as some DCs and MΦs in tumor lesions and tumor-draining lymph nodes (TDLNs) in mice and cancer patients. Recent reports continue this trend with studies describing elevated IDO expression in endometrial carcinomas, brain (glioma), chronic lymphocytic leukemias, non-small cell lung cancers and laryngeal squamous cell carcinomas (31–36). These studies support the paradigm that IDO regulates anti-tumor immunity and is a potential prognostic marker for cancer, reinforcing the rationale for using IDO inhibitors to improve cancer therapy (37). Phase II oncology trials using IDO inhibitors are ongoing and it remains to be seen if this novel approach is effective in the clinic, though IDO was identified recently as a immune resistance factor following immunotherapy to block regulatory pathways involving CTLA4, PD-1/L and glucocorticoid-induced TNF receptor-related (GITR protein signaling (38).
Unlike IDO, TDO is expressed primarily in liver and is induced in the central nervous system (CNS) in response to stress-induced glucocorticoids. In liver, TDO regulates serum Trp levels by catabolizing Trp from dietary intake, and TDO expression in liver may account for the observed resistance of liver allografts to rejection (39). However, some tumor cells express TDO and potential roles for TDO in tumors were reviewed recently revealing striking parallels with the effects of IDO (40, 41). Thus, TDO transduced tumor cells were resistant to anti-tumor immunity and TDO-specific inhibitors restored the ability of mice to reject TDO-expressing tumors (42). Thus, tumors may evade innate immune tumor surveillance and regulate tumor-specific adaptive immunity by inducing IDO or TDO.
A critical role for TPH-1 in regulating anti-tumor immunity emerged from studies with TPH-1 deficient mice, which were more resistant to bladder carcinoma cell growth than TPH-1-suffiicent mice (3). Immune resistance was not serotonin dependent, implicating Trp depletion due to TPH-1 enzyme activity in mast cells in metabolic control of anti-tumor immunity. TPH-1 activity was also shown to be important in regulating skin allograft rejection and suppressing experimental autoimmune encephalitis (EAE). Thus, TPH-1 mediates immune regulation in several inflammatory settings of clinical significance.
Arg is catabolized by two iso-enzymes arginase (ARG) I and II. ARG-I is often co-expressed with nitric oxide synthase (NOS), which competes for Arg as a substrate. Retinoic acid induced ARG-I expression in DCs that promoted Treg differentiation (43), though MDSCs were reported to inhibit Treg differentiation (44). Several studies suggest that elevated Arg consumption regulates anti-tumor immunity. Increased ARG-I expression was detected in MDSCs from cancer patients with squamous cell carcinoma (45) and ARG-II expression was elevated in cancer-associated fibroblasts and correlated with worse outcomes in pancreatic cancer (46). MDSCs facilitate tumor immune resistance, though these enigmatic cells may use multiple immune regulatory mechanisms, including Trp (47) and Arg catabolism (48). Moreover MDSCs developed from fibrocyte precursors in some cancer patients and glutamine (Gln) catabolism promoted MDSC maturation (49, 50).
Mechanisms of immune regulation driven by amino acid metabolism
How does increased amino acid catabolism inhibit tumorigenesis and therapy? Though not fully resolved, studies in mice with chronic inflammatory syndromes such as tumors have provided some key insights. Amino acid catabolism depletes local pools and generates new catabolites, and amino acid sensors and catabolite receptors sense these changes in immune cells (Fig. 3).
Figure 3. Downstream effects of amino acid catabolism on tumor and immune cells.

Amino acid depletion and catabolite production have profound downstream effects on tumor and immune cells by triggering pathways that impact cell growth, proliferation, survival and cellular stress responses. Trp catabolites activate the AhR pathway but metabolites produced by other enzymes depicted may mediate critical effects via other pathways
Amino acid levels are sensed via the mammalian target of rapamycin (mTOR) and general control non-repressed-2 (GCN2). mTOR is a pivotal checkpoint governing cell cycle entry. Tumor cells may mutate this pathway to facilitate tumor growth and mTOR inhibitors such as everolimus may be effective therapies to control some cancers (51, 52). Amino acid depletion also restrains anti-tumor immunity since mTOR signals promote T cell responses. Hence local amino acid depletion and treatment with mTOR inhibitors may inhibit anti-tumor immunity despite the potential to inhibit tumor growth. Trp depletion via IDO to prevent mTOR activation and promote tumor autophagy has been reported (53), but effects on T cell mediated anti-tumor immunity were not evaluated.
GCN2 is a ribosome-associated kinase that senses binding of uncharged tRNAs. Activated GCN2 kinase incites the integrated stress response to shut down cellular protein synthesis and induce autophagy. GCN2 activation in T cells cultured with IDO-expressing DCs from TDLNs blocked T cell responses to antigens presented by DCs (54). GCN2 was also required for TDLN DCs to induce Tregs to acquire regulatory phenotypes via IDO (55). Thus GCN2 suppresses immunity and promotes immune regulation, though GCN2 activation in tumor cells may block proliferation and induce autophagy. Recent studies attest to the diverse effects of GCN2-mediated cell stress responses on tumor growth and anti-tumor immunity. Thus GCN2 promoted tumor angiogenesis (56), inhibited the anti-tumor effects of IFNαβ (57), modified mitochondrial functions in colon cancer cells (58), and supported tumor cell proliferation during restricted access to serine (59). GCN2 may also impact cancer therapy since GCN2 induced asparagine synthetase activity (a therapy resistance factor) in pediatric acute lymphoblastic leukemia (ALL) patients and GCN2 attenuated the efficacy of glucose competitors as anti-tumor drugs (60, 61), though GCN2 also promoted the anti-leukemic effects of pegylated ARG-I in ALL (62). Some anti-tumor reagents mimic the effects of amino acid depletion by inhibiting tRNA charging enzymes to activate GCN2. Thus the antibiotic borrelidin, which inhibits human threonine tRNA synthetase, induced apoptosis of ALL cells and GCN2 activation was elevated (63). Similarly, the anti-protozoal compound halofuginone, which inhibits prolyl-tRNA synthetase, may also inhibit cancers (64). However like mTOR, potential diametric effects on tumor cell growth and survival versus immune cell activation and differentiation need to be evaluated carefully as halofuginone inhibits effector TH17 T cell responses in autoimmune syndromes (65).
Trp catabolites also mediate potent effects on tumor and immune cells (Fig. 3). Kynurenine (Kyn), a Trp catabolite made by cells expressing IDO or TDO, is a natural ligand for the aryl hydrocarbon receptor (AhR), an orphan receptor expressed by many cells (66). Chemical toxins, known as dioxins, are artificial AhR ligands that have been studied extensively by toxicologists but relatively little is known about natural AhR ligands such as Kyn. Studies in mice have identified requirements for AhR signaling in T cells, Tregs and DCs to regulate immunity (7, 9). These findings provide a rationale for mechanistic links between IDO and TDO activity and regulation of anti-tumor immunity, justifying the use of AhR antagonists as potential therapies to block suppression of anti-tumor immunity. AhR signaling also enhanced NK cell anti-tumor activity in mice (67), indicating that AhR signaling can enhance or inhibit immunity, contingent on the physiologic setting (68). It is unclear how modulation of AhR signals will impact outcomes in cancer as AhR signals may mediate direct effects on tumor, stromal or immune cells to impede or promote tumor progression, survival, angiogenesis and anti-tumor immunity. Hence, the effects of ligands that modulate AhR signaling (in either direction) in settings of tumor growth and treatment requires more rigorous evaluation in physiologic settings of tumor growth to discern the impact of such interventions. For example, reports that Trp catabolites made by cells expressing IDO and TDO confer resistance to gliomas should be weighed against the known neurotoxic effects of some Trp catabolites (69, 70).
In summary, the downstream consequences of increased amino acid catabolism in tumor microenvironments are profound but complex, diametric and poorly defined. More studies to evaluate the role of specific pathways in physiologic settings of tumor development and therapy will be necessary to further elucidate specific contributions to tumor growth, survival and anti-tumor immunity.
Concluding remarks
Immune tolerance created by tumors is a major barrier to effective chemotherapy, radiotherapy and immunotherapy, and subsequent tumor relapse is associated with renewal of local tolerance. Increased amino acid catabolism may occur from the earliest stages of tumorigenesis due to inflammation associated with the formation malignant lesions that protects malignant cells from immune surveillance. Oncogenic viral infections, chemicals and radiation that promote carcinogenesis also stimulate increased amino acid catabolism that contributes to the local inflammatory responses to these insults and promotes resistance to natural and therapy-induced anti-tumor immunity.
Key Points.
Amino acid catabolism regulates immunity in settings of chronic inflammation, including cancer
Amino acid catabolism is enhanced prior to clinical presentation and may facilitate tumor progression
Amino acid depletion and production of catabolites both regulate immunity
Interventions targeting amino acid catabolism may be effective adjunct cancer therapies
Altered amino acid metabolism may yield useful prognostic markers as well as therapeutic targets.
Acknowledgments
A.L.M is a member of the Scientific Advisory Board of NewLink Genetics Corporation and receives remuneration from this source. The authors wish to acknowledge the advice and assistance of their colleagues in the Cancer immunology, Inflammation and Tolerance program at the GRU Cancer Center. Research in the authors laboratory is supported by grants from the NIH (AI083005, AI103347) and a generous donation from the Carlos and Marguerite Mason Trust in Atlanta (to A.L.M).
References
- 1.Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Annals of the New York Academy of Sciences. 2013;1284:1–5. doi: 10.1111/nyas.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunological Reviews. 2013;252:52–77. doi: 10.1111/imr.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3**.Nowak EC, de Vries VC, Wasiuk A, Ahonen C, Bennett KA, Le Mercier I, et al. Tryptophan hydroxylase-1 regulates immune tolerance and inflammation. The Journal of experimental medicine. 2012;209:2127–35. doi: 10.1084/jem.20120408. This study identifies tryptophan hydroxylase-1 (THP-1) expressed by mast cells as a regulatory pathway that promotes immune tolerance and facilitates tumor formation. THP-1 is the third Trp consuming enzyme to be linked to suppression of anti-tumor immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Frontiers in immunology. 2013;4:190. doi: 10.3389/fimmu.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montero AJ, Diaz-Montero CM, Kyriakopoulos CE, Bronte V, Mandruzzato S. Myeloid-derived suppressor cells in cancer patients: a clinical perspective. Journal of immunotherapy. 2012;35:107–15. doi: 10.1097/CJI.0b013e318242169f. [DOI] [PubMed] [Google Scholar]
- 7.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2012;34:137–43. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Huang L, Lemos H, Mautino M, Mellor AL. Altered tryptophan metabolism as a paradigm for good and bad aspects of immune privilege in chronic inflammatory diseases. In: Caspi Rachel R., editor. Frontiers in Immunological Tolerance. Article 109. 3 . 2012. pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGaha TL, Huang L, Lemos H, Metz R, Mautino M, Prendergast GC, et al. Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunological reviews. 2012;249:135–57. doi: 10.1111/j.1600-065X.2012.01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A. 2009;106:12055–60. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swanson KA, Zheng Y, Heidler KM, Mizobuchi T, Wilkes DS. CDllc+ cells modulate pulmonary immune responses by production of indoleamine 2,3-dioxygenase. Am J Respir Cell Mol Biol. 2004;30:311–8. doi: 10.1165/rcmb.2003-0268OC. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Liu L, Liu K, Bizargity P, Hancock WW, Visner GA. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. Journal of immunology. 2009;183:1022–31. doi: 10.4049/jimmunol.0900408. [DOI] [PubMed] [Google Scholar]
- 13.Monjazeb AM, Zamora AE, Grossenbacher SK, Mirsoian A, Sckisel GD, Murphy WJ. Immunoediting and antigen loss: overcoming the achilles heel of immunotherapy with antigen non-specific therapies. Frontiers in oncology. 2013;3:197. doi: 10.3389/fonc.2013.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson DJ, Ohashi PS. Molecular programming of steady-state dendritic cells: impact on autoimmunity and tumor immune surveillance. Annals of the New York Academy of Sciences. 2013;1284:46–51. doi: 10.1111/nyas.12114. [DOI] [PubMed] [Google Scholar]
- 15.Heath JL, Weiss JM, Lavau CP, Wechsler DS. Iron deprivation in cancer--potential therapeutic implications. Nutrients. 2013;5:2836–59. doi: 10.3390/nu5082836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 17.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, et al. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nature immunology. 2001;2:64–8. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 18.Rose DP. Tryptophan metabolism in carcinoma of the breast. Lancet. 1967;1:239–41. doi: 10.1016/s0140-6736(67)91301-3. [DOI] [PubMed] [Google Scholar]
- 19.Rose DP, Randall ZC. Tryptophan metabolism in early and advanced breast cancer and carcinoma of the cervix. Clinica chimica acta; international journal of clinical chemistry. 1972;40:276–80. doi: 10.1016/0009-8981(72)90284-7. [DOI] [PubMed] [Google Scholar]
- 20.Serbecic N, Beutelspacher SC. Indoleamine 2,3-dioxygenase protects corneal endothelial cells from UV mediated damage. Exp Eye Res. 2006;82:416–26. doi: 10.1016/j.exer.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 21.O’Connor M, Green WR. The role of indoleamine 2,3-dioxygenase in LP-BPM5 murine retroviral disease progression. Virology journal. 2013;10:154. doi: 10.1186/1743-422X-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshi M, Saito K, Hara A, Taguchi A, Ohtaki H, Tanaka R, et al. The absence of IDO upregulates type I IFN production, resulting in suppression of viral replication in the retrovirus-infected mouse. Journal of immunology. 2010;185:3305–12. doi: 10.4049/jimmunol.0901150. [DOI] [PubMed] [Google Scholar]
- 23.Mittal D, Kassianos AJ, Tran LS, Bergot AS, Gosmann C, Hofmann J, et al. Indoleamine 2,3-Dioxygenase Activity Contributes to Local Immune Suppression in the Skin Expressing Human Papillomavirus Oncoprotein E7. The Journal of investigative dermatology. 2013 doi: 10.1038/jid.2013.222. (EPub May 7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller AJ, Sharma MD, Chandler PR, DuHadaway JB, Everhart ME, Johnson BA, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A. 2008;105:17073–8. doi: 10.1073/pnas.0806173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discovery. 2012;2:722–35. doi: 10.1158/2159-8290.CD-12-0014. This study identifies critical roles for IDO in lung tumor progression, neovascularization, MDSC infiltration into the tumor microenvironment and tumor metastasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang L, Lemos HP, Li L, Li M, Chandler PR, Baban B, et al. Engineering DNA Nanoparticles as Immunomodulatory Reagents that Activate Regulatory T Cells. Journal of immunology. 2012;188:4913–20. doi: 10.4049/jimmunol.1103668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B, et al. DNA sensing via the Stimulator of Interferon Genes (STING) adaptor in myeloid dendritic cells induces potent tolerogenic responses. Journal of immunology. 2013 doi: 10.4049/jimmunol.1301419. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–91. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–31. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30*.Thaker AI, Rao MS, Bishnupuri KS, Kerr TA, Foster L, Marinshaw JM, et al. IDO1 Metabolites Activate beta-catenin Signaling to Promote Cancer Cell Proliferation and Colon Tumorigenesis in Mice. Gastroenterology. 2013;145:416–25. doi: 10.1053/j.gastro.2013.05.002. This study shows that kynurenines promote tumor growth by activating β-catenin, providing additional rationale for targeting Trp catabolism to treat cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Jong RA, Kema IP, Boerma A, Boezen HM, van der Want JJ, Gooden MJ, et al. Prognostic role of indoleamine 2,3-dioxygenase in endometrial carcinoma. Gynecologic oncology. 2012;126:474–80. doi: 10.1016/j.ygyno.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 32.Mei J, Li MQ, Ding D, Li DJ, Jin LP, Hu WG, et al. Indoleamine 2,3-dioxygenase-1 (IDO1) enhances survival and invasiveness of endometrial stromal cells via the activation of JNK signaling pathway. International journal of clinical and experimental pathology. 2013;6:431–44. [PMC free article] [PubMed] [Google Scholar]
- 33.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clinical cancer research. 2012;18:6110–21. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindstrom V, Aittoniemi J, Jylhava J, Eklund C, Hurme M, Paavonen T, et al. Indoleamine 2,3-dioxygenase activity and expression in patients with chronic lymphocytic leukemia. Clinical lymphoma, myeloma & leukemia. 2012;12:363–5. doi: 10.1016/j.clml.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Creelan BC, Antonia S, Bepler G, Garrett TJ, Simon GR, Soliman HH. Indoleamine 2,3-dioxygenase activity and clinical outcome following induction chemotherapy and concurrent chemoradiation in Stage III non-small cell lung cancer. Oncoimmunology. 2013;2:e23428. doi: 10.4161/onci.23428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye J, Liu H, Hu Y, Li P, Zhang G, Li Y. Tumoral indoleamine 2,3-dioxygenase expression predicts poor outcome in laryngeal squamous cell carcinoma. Virchows Archiv: an international journal of pathology. 2013;462:73–81. doi: 10.1007/s00428-012-1340-x. [DOI] [PubMed] [Google Scholar]
- 37.Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) 2012;4:734–45. doi: 10.2741/e414. [DOI] [PubMed] [Google Scholar]
- 38**.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. The Journal of experimental medicine. 2013;210:1389–402. doi: 10.1084/jem.20130066. This study identifies IDO as a key factor that inhibits effective responses to immunotherapy using novel reagents that block regulation mediated by CTLA4, PD1 and GITR pathways, justifying combinational therapies using IDO inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt SK, Muller A, Heseler K, Woite C, Spekker K, MacKenzie CR, et al. Antimicrobial and immunoregulatory properties of human tryptophan 2,3-dioxygenase. European journal of immunology. 2009;39:2755–64. doi: 10.1002/eji.200939535. [DOI] [PubMed] [Google Scholar]
- 40.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer research. 2012;72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 41.Munn DH. Indoleamine 2,3-dioxygenase, Tregs and Cancer. Curr Med Chem. 2011;18:2240–6. doi: 10.2174/092986711795656045. [DOI] [PubMed] [Google Scholar]
- 42*.Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109:2497–502. doi: 10.1073/pnas.1113873109. This study identifies TDO as a tumor immune resistance factor that, like IDO, catabolizes tryptophan in tumor microenvironments. TDO-specific inhibitors were effective in reversing tumor induced immune resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang J, Thangamani S, Kim MH, Ulrich B, Morris SM, Jr, Kim CH. Retinoic acid promotes the development of Arg1-expressing dendritic cells for the regulation of T-cell differentiation. European journal of immunology. 2013;43:967–78. doi: 10.1002/eji.201242772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Centuori SM, Trad M, LaCasse CJ, Alizadeh D, Larmonier CB, Hanke NT, et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-beta-induced differentiation of CD4+CD25+FoxP3+ Tregs from CD4+CD25-FoxP3- T cells. Journal of leukocyte biology. 2012;92:987–97. doi: 10.1189/jlb.0911465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. The Journal of clinical investigation. 2013;123:1580–9. doi: 10.1172/JCI60083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46*.Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, et al. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One. 2013;8:e55146. doi: 10.1371/journal.pone.0055146. This study on human subjects identifies ARG-II expressed in tumor-associated fibroblasts as a marker of poor prognosis in pancreatic ductal carcinoma patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47*.Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. Journal of immunology. 2013;190:3783–97. doi: 10.4049/jimmunol.1201449. This study on samples from breast cancer patients demonstrates that IDO is necessary for tumor infiltrating MDSCs to mediate immune suppression. [DOI] [PubMed] [Google Scholar]
- 48.Zanetti M. Cell-extrinsic effects of the tumor unfolded protein response on myeloid cells and T cells. Annals of the New York Academy of Sciences. 2013;1284:6–11. doi: 10.1111/nyas.12103. [DOI] [PubMed] [Google Scholar]
- 49.Hammami I, Chen J, Bronte V, DeCrescenzo G, Jolicoeur M. L-glutamine is a key parameter in the immunosuppression phenomenon. Biochemical and biophysical research communications. 2012;425:724–9. doi: 10.1016/j.bbrc.2012.07.139. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Maric I, Diprima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. 2013 doi: 10.1182/blood-2012-08-449413. (ePub Jun 11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paplomata E, O’Regan R. New and emerging treatments for estrogen receptor-positive breast cancer: focus on everolimus. Therapeutics and clinical risk management. 2013;9:27–36. doi: 10.2147/TCRM.S30349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilley R, Balmanno K, Cope CL, Cook SJ. Adaptation to chronic mTOR inhibition in cancer and in aging. Biochemical Society transactions. 2013;41:956–61. doi: 10.1042/BST20130080. [DOI] [PubMed] [Google Scholar]
- 53*.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1:1460–8. doi: 10.4161/onci.21716. This study identifies the mTOR pathway as a secondary target for the IDO inhibitor D-1MT, providing a new perspective on how D-1MT may enhance anti-tumor immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity. 2005;22:1–10. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. The Journal of clinical investigation. 2007;117:2570–82. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56*.Wang Y, Ning Y, Alam GN, Jankowski BM, Dong Z, Nor JE, et al. Amino Acid Deprivation Promotes Tumor Angiogenesis through the GCN2/ATF4 Pathway. Neoplasia. 2013;15:989–97. doi: 10.1593/neo.13262. This study provides evidence supporting a novel mechanism by which amino acid depletion in tumor microenvironments may stimulate tumor growth by enhancing neo-vascularization via GCN2 mediated stress responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhattacharya S, Huangfu WC, Dong G, Qian J, Baker DP, Karar J, et al. Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene. 2012 doi: 10.1038/onc.2012.439. (EPub Oct 8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez-Reyes I, Sanchez-Arago M, Cuezva JM. AMPK and GCN2-ATF4 signal the repression of mitochondria in colon cancer cells. Biochem J. 2012;444:249–59. doi: 10.1042/BJ20111829. [DOI] [PubMed] [Google Scholar]
- 59.Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A. 2012;109:6904–9. doi: 10.1073/pnas.1204176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balasubramanian MN, Butterworth EA, Kilberg MS. Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab. 2013;304:E789–99. doi: 10.1152/ajpendo.00015.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu H, Kurtoglu M, Cao Y, Xi H, Kumar R, Axten JM, et al. Conversion of 2-deoxyglucose-induced growth inhibition to cell death in normoxic tumor cells. Cancer Chemother Pharmacol. 2013;72:251–62. doi: 10.1007/s00280-013-2193-y. [DOI] [PubMed] [Google Scholar]
- 62.Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, et al. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia. 2013;27:569–77. doi: 10.1038/leu.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Habibi D, Ogloff N, Jalili RB, Yost A, Weng AP, Ghahary A, et al. Borrelidin, a small molecule nitrile-containing macrolide inhibitor of threonyl-tRNA synthetase, is a potent inducer of apoptosis in acute lymphoblastic leukemia. Invest New Drugs. 2012;30:1361–70. doi: 10.1007/s10637-011-9700-y. [DOI] [PubMed] [Google Scholar]
- 64.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol. 2012;8:311–7. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–8. doi: 10.1126/science.1172638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng S, Cao Z, Wang X. Role of aryl hydrocarbon receptor in cancer. Biochimica et biophysica acta. 2013;1836:197–210. doi: 10.1016/j.bbcan.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 67**.Shin JH, Zhang L, Murillo-Sauca O, Kim J, Kohrt HE, Bui JD, et al. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2013;110:12391–6. doi: 10.1073/pnas.1302856110. This study describes a novel NK cell anti-tumor mechanism dependent on AhR signaling, which stimulates NK cells to promote tumor killing activity, suggesting that tryptophan catabolites may have diverse and potentially diametric roles on immune cells in tumor microenvironments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hao N, Whitelaw ML. The emerging roles of AhR in physiology and immunity. Biochemical pharmacology. 2013 doi: 10.1016/j.bcp.2013.07.004. (EPub Jul 12) [DOI] [PubMed] [Google Scholar]
- 69.Sahm F, Oezen I, Opitz CA, Radlwimmer B, von Deimling A, Ahrendt T, et al. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer research. 2013;73:3225–34. doi: 10.1158/0008-5472.CAN-12-3831. [DOI] [PubMed] [Google Scholar]
- 70.Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, et al. The kynurenine pathway in brain tumor pathogenesis. Cancer research. 2012;72:5649–57. doi: 10.1158/0008-5472.CAN-12-0549. [DOI] [PubMed] [Google Scholar]