

Abstract
Identifying binding hot spots in protein−protein interfaces is important for understanding the binding specificity and for the design of nonpeptide, small molecule inhibitors. Molecular dynamics simulation in the isopropanol/water cosolvent environment and in water was employed to investigate Bcl-xL protein, which has a highly flexible, large, and primarily hydrophobic binding site. Simulations of either the apo- or holocrystal structures of the Bcl-xL in pure water fail to generate conformations found in the cocrystal structures of Bcl-xL in complex with its binding partners due to hydrophobic collapse. In contrast, simulations in cosolvent starting either from the apo- or holocrystal structure of the Bcl-xL yield binding-site conformations similar to that found in the cocrystal structures of Bcl-xL. Hydrophobic binding hot spots identified using the conformations from the cosolvent simulations are in excellent agreement with experimental structural data of known inhibitors. Importantly, cosolvent simulations revealed the highly dynamic nature of the hydrophobic binding pockets in Bcl-xL and yielded new structural insights for the design of novel Bcl-xL small-molecule inhibitors.
Keywords: Protein−protein interfaces, Bcl-xL protein, cosolvent molecular dynamics simulation
Protein−protein interactions (PPIs) play a key role in regulating many cellular processes.1 Common features for many PPIs include their relatively large interface2 and the high plasticity observed at the protein−protein interface.3 Both features make the design of nonpeptide, small-molecule inhibitors to block PPIs a difficult task despite major progress made in recent years for several PPIs.4 Although a protein−protein interface can be large, an emerging concept is that the binding free-energy (binding affinity) between the binding partners is often dominated by a few key “hot spots”.5 An in-depth knowledge of the hot spots is useful not only for understanding the functions of proteins but also for the design of nonpeptide small-molecule inhibitors of protein−protein interactions.
In this study, we have investigated the use of molecular dynamic (MD) simulations in a cosolvent environment to identify and analyze the “hot spots” in the Bcl-xL protein. Bcl-xL is a key antiapoptotic regulator and a member of the Bcl-2 family of proteins.6 Bcl-xL has 233 amino acids and is a mainly α helix protein in which its C-terminal helix inserts into intracellular membranes including mitochondria. Bcl-xL inhibits apoptosis by heterodimerization with pro-apoptotic Bcl-2 proteins, such as Bad, Bim, and Bax proteins.6 The interactions between Bcl-xL and its binding partners are mediated by a large, hydrophobic groove in Bcl-xL and the BH3 (Bcl-2 Homology 3) domain of these proteins. The Bcl-xL protein is investigated for several reasons: (1) Bcl-xL interacts with its binding partners through a large, primarily hydrophobic binding groove.7 (2) Bcl-xL undergoes unfolding and refolding in its binding groove when bound to its binding partners with respect to its apo-form, and such large and extensive conformational changes expose a number of buried hydrophobic residues for effective interaction with its binding partners. (3) The interactions of Bcl-xL with its binding partners are dominated by a few key hydrophobic residues and one charged residue, hence a few “hot spots” based upon crystal structures and biochemical data.8 (4) There are intense research efforts9 toward the design of potent, nonpeptide, small-molecule inhibitors of Bcl-xL, and one such compound (ABT-263) has been advanced into clinical development.10 (5) A number of high-resolution crystal structures are available for Bcl-xL alone (the apo-form) and in complex with peptide-based and nonpeptide inhibitors.8,11−14
Cosolvents are frequently used to solubilize proteins. In the experimental multiple solvent crystal structures (MSCS) approach,15 they were used to locate potential protein binding sites. The MSCS method soaks the protein crystals in different cosolvent concentrations, allowing the cosolvent molecules to diffuse to different protein binding sites. Although highly elevated concentrations of organic solvents in water can induce unfolding of a protein, hen egg-white lysozyme remains stable in its native conformation and can be cocrystallized at 16−24% of cosolvent concentrations.16 Up to 60% of three different cosolvent concentrations have been used to stabilize the dynamic switch II in the H-Ras protein for crystal structure determination.17 Four different ratios of methanol, DMSO, and water cosolvent systems were used to study the dynamic transition of xylanase experimentally.18 Recently, Barril and co-workers19 investigated the maximum binding affinity of the binding sites in five different proteins by performing MD simulations of proteins in a 20% v/v isopropanol/water environment. We have recently shown20 that the binding free energy of the isopropanol in the binding site of thermolysin obtained using the cosolvent simulation is comparable to the values determined by the double decoupling method.21 In the present study, we used the same 20% v/v isopropanol/water condition for our cosolvent simulation of the Bcl-xL system.
In addition to the apo structure, crystal structures of Bcl-xL with several peptides derived from Bcl-2 homology domain 3 (BH3) of pro-apoptotic Bcl-2 proteins and with nonpeptide small-molecule inhibitors have been determined. These include complexes with the wild type BH3 domains of Bak,22 Bad,23 Bim,11,12 Beclin-1(Bec1),13 mutated BH3 domain peptides,14 foldamer peptides,12 and small molecule inhibitors.8 These crystal structures have revealed that when bound to BH3 peptides, Bcl-xL undergoes substantial backbone conformational changes at the α3 and α4 helices and significant changes at the α2 and α7 helices (cf. Figure 1C−E and S2). Unfolding of α3 in Bcl-xL is seen in the Bad- and Bec1-bound structures, and an additional helical turn at the end of α2 is observed in the Bec1-bound conformation. The backbone changes in the flexible α3 helix in Bcl-xL expose a large hydrophobic groove, which is used in binding to the amphipathic, helical BH3 domain peptides consisting of 20−25 amino acid residues.
Figure 1.
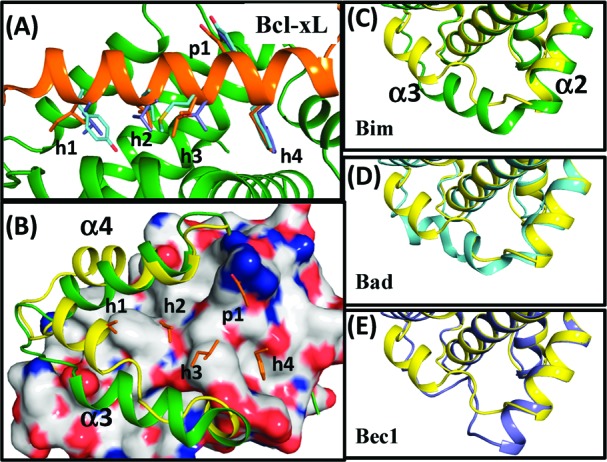
(A) Superimposition of Bcl-xL with the Bim (orange), Bad(cyan), and Bec1 (blue) BH3 peptides. (B) Structural alignment between the ligand-free Bcl-xL(yellow) and Bcl-xL with the Bim peptide(green). H1-4 and p1 residues of the Bim peptide are shown in orange. Conformational changes of the α3 helix in Bcl-xL when binding to (C) Bim (green), (D) Bad (cyan), and (E) Bec1 (blue) peptide. The reference structure of ligand-free Bcl-xL is shown in yellow. The PyMOL program (www.pymol.org) was used to view the protein models and prepare the graphics.
MD simulations were performed on the apo-Bcl-xL structure in water and in cosolvent to understand the effects of the cosolvent molecules on the energetics and conformations of the protein. The molecular mechanics Poisson−Boltzmann surface area (MM-PBSA) model24 was used to calculate and compare the conformational free energy (ΔGMM-PBSA) of Bcl-xL using conformations obtained from simulation trajectories in water and cosolvent. This comparison allows us to determine the energetic perturbation to the Bcl-xL conformations simulated in the cosolvent environment. Throughout the 32 ns simulations, the average ΔGMM-PBSA of the apo-Bcl-xL in cosolvent is similar to that in water (−1575 ± 29 in cosolvent versus −1581 ± 30 kcal/mol in water) (Figure S3D). However, conformations of the apo-Bcl-xL in cosolvent give higher surface accessible areas (ASAs) than those in water (8336 versus 7796 Å2). According to the PB solvation model, the differences of ASAs for the apo-Bcl-xL conformations in two different media contribute 3.89 kcal/mol in nonpolar solvation. Although the backbone rmsd of the apo-Bcl-xL (α2-α7) is maintained at 1.5 Å in both simulations, there are significant differences with respect to conformational changes between the two simulations. In cosolvent, the α3 and α4 helices of Bcl-xL undergo large conformational changes starting at 16 ns and become stabilized after 20 ns. In water, only minor backbone changes are found in the apo-Bcl-xL conformations. The conformations of the apo-Bcl-xL in cosolvent at 16 and 32 ns are shown in Figure S4C and D of the Supporting Information. Both figures indicate that interaction between the isopropanol and the apo-Bcl-xL drives the apo-Bcl-xL into a conformational state in which the buried h1 and h2 sites become exposed. In contrast, both sites remain buried in the conformations of apo-Bcl-xL throughout 32 ns of simulations in water (Figure S4A and B). Differences in the per residue ASA calculations of hydrophobic residues at the binding site identified greater surface exposure in Y101, A104, F105, L108, I114, and L130 (>10 Å2), involving primarily the α3 helix in the conformations obtained from cosolvent simulations (Figure S5B). Exposure of these residues, except Tyr101, is also seen when comparing three peptide-bound structures and the Apo-Bcl-xL structure (Figure S5A). Our simulations show that while the cosolvent molecules cause only small energetic perturbation, they induce significant backbone conformational changes in the α3 helix of the apo-Bcl-xL, exposing the h1 and h2 sites, similar to that observed in the crystal structures of Bcl-xL bound to different binding partners.
We next performed MD simulations of three different peptide-bound Bcl-xL crystal structures shown in Figure 1C but with the BH3 peptides removed from the structures (holo-Bcl-xL structures). In pure water simulations, the Bim-bound Bcl-xL structure remains in a conformational state with an intact α3 helix (Figure 2A); the Bad-bound Bcl-xL structure refolds its partially distorted α3 helix during the 32 ns simulation (Figure 2B); the Bec1-bound Bcl-xL structure partially loses its helicity for its α2 and α4 segments (Figure 2C). In all three simulations in water, a number of exposed hydrophobic residues in the binding groove become reburied and result in a conformational state, similar to the crystal structure of apo-Bcl-xL.
Figure 2.
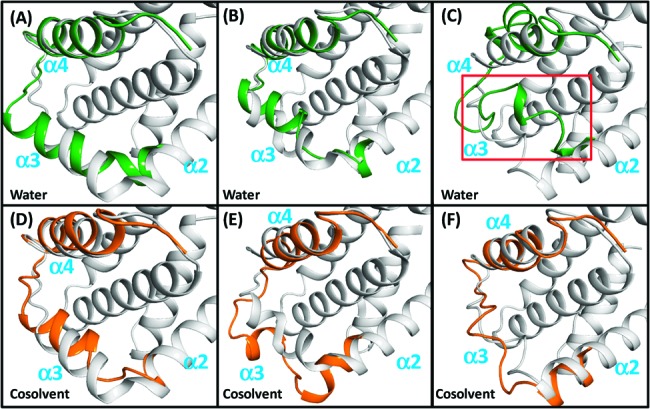
Comparison of the crystal structures of the Bim-bound (A, D), the Bad-bound (B, E), and the Bec1-bound (C, F) Bcl-xL with snapshots of conformations after 32 ns simulations. Conformations of crystal structures; snapshots in water and in cosolvent are colored in gray, green, and orange. Only residues F97−N136 in the snapshots are shown for clarity.
In the cosolvent simulations, small backbone changes in the Bim-bound holo Bcl-xL structure are observed (Figure 2D) while the Bec1-bound holo Bcl-xL structure remains stable (Figure 2F). In the case of the Bad-bound holo Bcl-xL structure, there is a large conformational change with respect to the region consisting of the tail of α2 and the whole α3 helix segments (F95−P116) (Figure 2E). However, in all cases, the three holo-Bcl-xL conformations maintain their exposed hydrophobic surface in the binding groove in the cosolvent simulations, in contrast to that observed in pure water simulations. Comparison of the ASAs of the hydrophobic residues around the binding site (A93−V155) between conformations obtained from both solvent media show that F97, F105, L108, L112, Y120, V126, and L130 are indeed more exposed to solvent in the cosolvent simulation than in water (Figure S5C). These residues are even more exposed when Bcl-xL binds to BH3 peptides (cf. Figure S5A and C).
Energetically, the ΔGMM-PBSA values of the holo-Bcl-xL conformations obtained in water and cosolvent are similar (−1586 versus −1605 kcal/mol for the Bim-bound, −1612 versus −1623 for the Bad-bound, and −1616 versus −1607 kcal/mol for the Bec1-bound). Although ASAs in the Bad-bound and Bec1-bound Bcl-xL conformations obtained in cosolvent are greater than those obtained in water, only small differences are found in the Bim-bound Bcl-xL conformations simulated in both media.
We next identified the hydrophobic hot spots using the cosolvent mapping method based on an ensemble of protein conformations obtained from the cosolvent simulations. In the cosolvent mapping method, the binding site of Bcl-xL is probed by atoms of the cosolvent molecules via their interaction with the protein. The observed frequency (Np) of the probe atom at a grid point around the protein binding site is compared with an expected frequency (N0) in a cosolvent mixture to give an estimate of the binding free energy of the probe atom at that grid point, i.e. ΔGCM = −kT log (Np/N0). Grid points with binding free energies higher than −0.83 kcal/mol were collected to form pseudoatoms (vertices with a radius = 1.4 Å), and a bonding distance of 2.5 Å between pseudoatoms (edge) was used to generate chemical graphs (details in Supporting Information). The derived chemical graphs represent the hot spots, whose specific physical properties depend on the types of probe atoms used. Here, the carbon atoms of the terminal methyl groups in isopropanol were used as probes to identify the hydrophobic hot spots in Bcl-xL.
Figure 3 shows the hydrophobic hot spots determined on the basis of conformations obtained from the 32 ns MD simulations in cosolvent for one apo-Bcl-xL and three holo-Bcl-xL structures. The relative rigidity of the α3 helix of the Bim-bound conformations yields a distribution of the hydrophobic hot spots covering the helical backbone of the Bim peptide and the h2 and h4 sites (Figure 3B). Differences in the conformational changes of the α3 helix in Bcl-xL in its binding with the Bad and Bec1 peptides are also reflected on the distribution of hot spots (Figure 3C and D). Hydrophobic hot spots determined by the Bad-bound Bcl-xL are skewed toward the h2 and h3 sites and less toward the h4 site. In contrast, the hot spots are distributed more toward the h3 and h4 sites and less deeply into the h2 site in the Bec1-bound Bcl-xL. Hot spots are mainly found at the h2, h3, and h4 sites from the apo-Bcl-xL simulation (Figure 3A), and additional hot spots situated more deeply in the protein are seen at the h2 and h3 sites. The locations of these hot spots are consistent with key components of either the Bcl-xL inhibitors ABT737 or W1101542, as seen in their crystal structures (Figure 4A). The conformation of apo-Bcl-xL obtained from the 32 ns cosolvent simulation gives an example that the binding site is better defined and suitable for small molecules than the conformation obtained from the 32 ns water simulation (cf. Figure 4B and C). A comparison of the hot spots distribution in the four different Bcl-xL conformations reveals a narrower consensus region covering predominantly the h2, h3, and h4 sites. Our hot spot analysis suggests that different scaffolds of small molecules of Bcl-xL may be designed to target different conformations of Bcl-xL.
Figure 3.
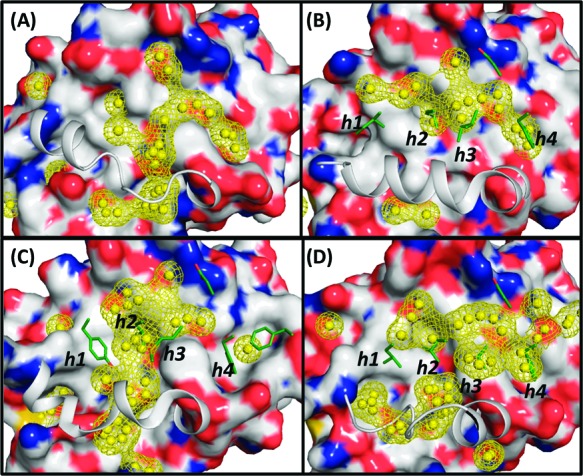
Hydrophobic hot spots (yellow balls) in the BH3 peptide binding groove detected from the cosolvent mapping method based on 32 ns of simulations. (A−D) Results using the apo-, Bim-bound, Bad-bound, and Bec1-bound Bcl-xL conformations, respectively. Four conserved hydrophobic residues and one acidic residue are shown in the stick model. Crystal structures of each conformation are used as the reference. The α3 helix is shown without the surface rendering for clarity.
Figure 4.
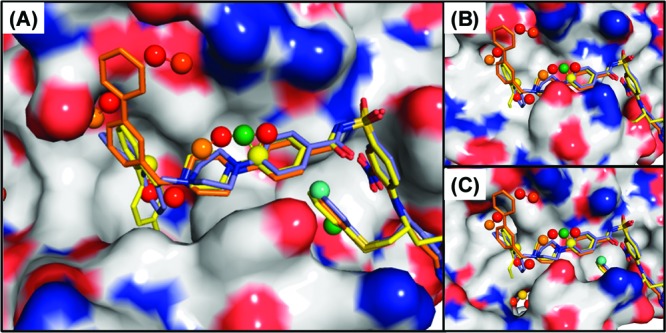
Alignment of the hydrophobic hot spots (pseudo-carbon atoms) determined from the conformations of the apo-Bcl-xL simulated in cosolvent with the crystal structures of Bcl-xL with ABT737 (yellow, PDB ID: 2YXJ) and conformation 1 (blue) and 2 (orange) of W1101542 (PDB ID: 3INQ). The red pseudoatoms yield lower binding affinity than the orange, yellow, green, and cyan ones. Bcl-xL is shown in surface representation. The reference protein structures are (A) the ABT737-bound Bcl-xL crystal structure and (B and C) the apo-Bcl-xL conformation (B) in water and (C) in cosolvent at 32 ns of MD simulation.
Our analysis also reveals the conformational dependence of the distribution of hot spots for Bcl-xL. Dynamical changes of the binding site conformations can be clearly seen with different hydrophobic hot spot patterns based on the conformations of the apo-Bcl-xL in every 4 ns window from the 32 ns cosolvent simulations in Figure 5. Initially, hot spots cluster at the h2, h3, and h4 sites in the first 20 ns of the simulation (Figure 5B−F). After 20 ns of simulation, additional hot spots spreading to the h1 site emerge as a result of the backbone conformational changes in the α3 helix (cf. Figure S3A).
Figure 5.

Evolution of hot spots (contour surface) every 4 ns determined from the cosolvent simulation started with the apo form of the Bcl-xL crystal structure. The Bim BH3 peptide is aligned, and key residues are green.
In summary, we have analyzed the Bcl-xL binding groove through MD simulations in pure water and in isopropanol/water cosolvent and hot spot mapping. When simulations are performed in water, the binding site of the apo-Bcl-xL has relatively minor conformational changes. Simulations of three holo-Bcl-xL structures in water show that the protein undergoes significant conformational changes and results in burying of several solvent-exposed hydrophobic residues in the binding groove, which are critical for effective interactions with the binding partners of Bcl-xL. In contrast, in the cosolvent, the apo-Bcl-xL structure has undergone large conformational changes, exposing large hydrophobic surfaces at the interface, which is similar to that observed in the crystal structures of Bcl-xL in complex with its peptide and nonpeptide ligands. Three holo-Bcl-xL structures retain their exposed hydrophobic surface throughout the 32 ns simulations, although there are significant conformational changes with respect to one segment in the Bad-bound holo Bcl-xL structure. Despite the substantial structural differences, the free energy differences of the Bcl-xL conformations obtained in both media are small, and they probably reflect the small perturbation by the cosolvent molecules and the plasticity of Bcl-xL. Hydrophobic hot spots determined using four different simulations in cosolvent reveal the high plasticity of the Bcl-xL binding sites. Importantly, hot spots determined based on the apo-Bcl-xL conformation are consistent with pharmacophore elements of known nonpeptide, small-molecule inhibitors. Furthermore, our simulations also reveal additional binding hot spots not found in the crystal cocomplex structures, which can be used for the design of novel and potent small-molecule inhibitors of Bcl-xL. Taken together, our present study suggests that cosolvent MD simulation is highly effective to investigate biologically relevant conformations for flexible and hydrophobic binding sites in proteins and is capable of yielding new structural insights not found in the crystal structures. We are currently employing this approach for investigation of other protein−protein interactions, which also possess highly flexible and hydrophobic binding sites.
Acknowledgments
We thank Dr. George W. A. Milne for his critical reading of the manuscript.
Supporting Information Available
Details of the computational methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Aloy P.; Russell R. B. Nat. Rev. Mol. Cell Biol. 2006, 7, 188. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Nature 2007, 450, 1001. [DOI] [PubMed] [Google Scholar]
- Winget J. M.; Mayor T. Mol. Cell 2010, 38, 627. [DOI] [PubMed] [Google Scholar]
- Fry D. C. Pept. Sci. 2006, 84, 535. [Google Scholar]
- Wells J. A. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle R. J.; Strasser A. Nat. Rev. Mol. Cell. Biol. 2008, 9, 47. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Olejniczak E. T.; Fesik S. W. Biochim. Biophys. Acta 2004, 1644, 83. [DOI] [PubMed] [Google Scholar]
- Lee E. F.; Czabotar P. E.; Smith B. J.; Deshayes K.; Zobel K.; Colman P. M.; Fairlie W. D. Cell Death Differ. 2007, 14, 1711. [DOI] [PubMed] [Google Scholar]
- Reed J. C.; Pellecchia M. Blood 2005, 106, 408. [DOI] [PubMed] [Google Scholar]
- Tomillero A.; Moral M. A. Methods Find. Exp. Clin. Pharmacol. 2008, 30, 761. [PubMed] [Google Scholar]
- Liu X.; Dai S.; Zhu Y.; Marrack P.; Kappler J. W. Immunity 2003, 19, 341. [DOI] [PubMed] [Google Scholar]
- Lee E. F.; Sadowsky J. D.; Smith B. J.; Czabotar P. E.; Peterson-Kaufman K. J.; Colman P. M.; Gellman S. H.; Fairlie W. D. Angew. Chem., Int. Ed. Engl. 2009, 48, 4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein A.; Jeffrey P. D.; Shi Y. J. Biol. Chem. 2007, 282, 13123. [DOI] [PubMed] [Google Scholar]
- Lee E. F.; Czabotar P. E.; Yang H.; Sleebs B. E.; Lessene G.; Colman P. M.; Smith B. J.; Fairlie W. D. J. Biol. Chem. 2009, 284, 30508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattos C.; Ringe D. Nat. Biotechnol. 1996, 14, 595. [DOI] [PubMed] [Google Scholar]
- Deshpande A.; Nimsadkar S.; Mande S. C. Acta Crystallogr., D: Biol. Crystallogr. 2005, 61, 1005. [DOI] [PubMed] [Google Scholar]
- Buhrman G.; de Serrano V.; Mattos C. Structure 2003, 11, 747. [DOI] [PubMed] [Google Scholar]
- Reat V.; Dunn R.; Ferrand M.; Finney J. L.; Daniel R. M.; Smith J. C. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seco J.; Luque F. J.; Barril X. J. Med. Chem. 2009, 52, 2363. [DOI] [PubMed] [Google Scholar]
- Yang C.-Y.; Wang S. ACS Med. Chem. Lett. 2010, 1, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Given J. A.; Bush B. L.; McCammon J. A. Biophys. J. 1997, 72, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M.; Liang H.; Nettesheim D.; Meadows R. P.; Harlan J. E.; Eberstadt M.; Yoon H. S.; Shuker S. B.; Chang B. S.; Minn A. J.; Thompson C. B.; Fesik S. W. Science 1997, 275, 983. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Nettesheim D. G.; Wang Y.; Olejniczak E. T.; Meadows R. P.; Mack J.; Swift K.; Matayoshi E. D.; Zhang H.; Thompson C. B.; Fesik S. W. Protein Sci. 2000, 9, 2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Acc. Chem. Res. 2000, 33, 889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.