

Abstract
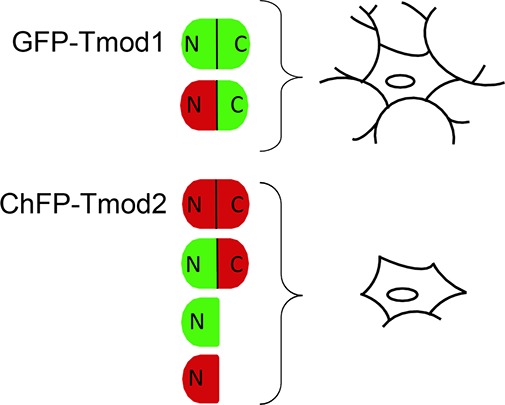
Actin dynamics is fundamental for neurite development; monomer depolymerization from pointed ends is rate-limiting in actin treadmilling. Tropomodulins (Tmod) make up a family of actin pointed end-capping proteins. Of the four known isoforms, Tmod1–Tmod3 are expressed in brain cells. We investigated the role of Tmod’s C-terminal (LRR) domain in the formation of neurite-like processes by overexpressing Tmod1 and Tmod2 with deleted or mutated LRR domains in PC12 cells, a model system used to study neuritogenesis. Tmod1 overexpression results in a normal quantity and a normal length of processes, while Tmod2 overexpression reduces both measures. The Tmod2 overexpression phenotype is mimicked by overexpression of Tmod1 with the LRR domain removed or with three point mutations in the LRR domain that disrupt exposed clusters of conserved residues. Removal of Tmod2’s LRR domain does not significantly alter the outgrowth of neurite-like processes compared to that of Tmod2. Overexpression of chimeras with the N-terminal and C-terminal domains switched between Tmod1 and Tmod2 reinforces the idea that Tmod1’s LRR domain counteracts the reductive effect of the Tmod N-terminal domain upon formation of processes while Tmod2’s LRR domain does not. We suggest that the TM-dependent actin capping ability of both Tmods inhibits the formation of processes, but in Tmod1, this inhibition can be controlled via its LRR domain. Circular dichroism, limited proteolysis, and molecular dynamics demonstrate structural differences in the C-terminal region of the LRR domains of Tmod1, Tmod2, and the Tmod1 mutant.
Neuritogenesis is the process by which a developing neuron extends protrusions that later become dendrites and the axon. The process is complex, involving the entirety of the cytoskeleton with significant cross talk among the microfilaments, intermediate filaments, and microtubules. Many details of this process have been reviewed elsewhere.1−4 The careful regulation of these interacting systems is important for the development of the adult nervous system. Neurite formation commences during migration when postmitotic neurons utilize internal stores of actin to develop growth cones, composed of filament actin-based filopodia and lamellipodia. These structures sense the extracellular environment and regulate actin dynamics to provide the force necessary for neurite elongation. Actin is regulated by a host of proteins and has been shown to influence the direction of neurite outgrowth.
There are two characteristic ends of actin filaments (F-actin): the fast-growing barbed end and the slow-growing pointed end. Addition of monomers to the distally oriented barbed end provides protrusive forces on the membrane that contribute to neurite extension, while depolymerization from the pointed end provides a constant supply of monomers for continued extension. Depolymerization from the pointed end is often the rate-determining step of actin dynamics.5 Tmod is a 40 kDa tropomyosin-dependent actin-capping protein that binds at the pointed end and inhibits depolymerization, thereby stabilizing F-actin.6 Although four isoforms of Tmod are known in vertebrates,7−10 research on the structure of Tmods has primarily been conducted on Tmod1, and the crystal structure of this isoform has been determined.11 Tmod1 is found in many tissues, including the brain, but has been mainly studied in erythrocytes, heart, and slow skeletal muscle. Tmod2 is solely expressed in neuronal tissue. Tmod3 is ubiquitously expressed. Tmod4 is primarily expressed in fast skeletal muscle. Isoform concentrations vary by the type of tissue, cellular location, and stage of development. Tmod1–Tmod3 have been shown to be expressed in the brain, but only Tmod1 and Tmod2 have been shown to have a role in neural development.12
Tmods have two structurally distinct functional halves (Figure 1): an unstructured N-terminal half and a tightly folded C-terminal half, a so-called LRR domain.13 The N-terminal half of Tmod1 acquires tertiary structure upon binding to tropomyosin (TM) and actin at the pointed end.14,15 This domain holds Tmod’s two TM binding sites and a TM-dependent actin-capping site.14,16,17 In contrast to the N-terminal half, the LRR domain is stable and compact and forms a cooperatively melting domain.11,15 The LRR domain is highly conserved across species and contains five leucine rich repeats (LRRs) and the C-terminal sixth α-helix. LRR folds are a repeated pattern of α-helices and β-strands, which are important for protein–protein recognition.18 Though removal of the LRR domain did not change the capping activity of Tmod1 in vitro,17 it has been shown to impair Tmod1 localization to the pointed end of thin filaments in cardiomyocytes;19 this indicates that the domain is crucial for targeting Tmod1 to thin filaments during sarcomere assembly in muscle cells. This domain also holds a TM-independent actin-capping site that is believed to be close to the C-terminus.20 Both actin-capping sites are important for Tmod’s nucleating ability.21
Figure 1.

Schematic of structural and functional sites of Tmod1 and Tmod2. The TM-binding sites are colored yellow, and the actin-capping sites are colored green. Indicated by an asterisk is a region that interacts with thymosin β-10.52 The C-terminal half is a leucine rich repeat domain that has a repeated pattern of α-helices and β-strands. The highlighted sequences contain the V8 protease (red) and trypsin (blue) cut sites as determined by N-terminal sequencing and mass spectrometry of the proteolytic fragments.
Tmod’s affinity for actin is greatly enhanced in the presence of TM, a coiled-coil protein that binds along F-actin.6 There are more than 40 isoforms of TMs, and they have been shown to have isoform specific roles in recruiting other actin-associated proteins; a thorough discussion of TM can be found in refs (22) and (23). With regard to neural systems, several TM isoforms are expressed in neurons and are important in regulating neuron size and shape; for example, TM5NM1 is believed to impact shape by recruiting myosin IIb.24 Recently, TMs have been implicated in neuritogenesis and neurite branching patterns in B35 neuroblastoma cells.25 Previously, we have shown that mutations in the TM binding sites in Tmod1 resulted in the altered localization of Tmod and a decreased length of neurite-like processes in PC12 cells.26
While Tmod1–Tmod3 are found in brain tissue, only Tmod2 is restricted to the nervous system. Mice lacking Tmod2 demonstrated reduced sensorimotor gating, hyperactivity, and impaired learning and memory.27 The authors suggested that control of actin filament length through pointed end regulation could be important in these processes. Proteomic studies have indicated that Tmod2 expression is significantly altered in fetal Down syndrome,28 mesial temporal lobe epilepsy,29 and post-seizure,30 post-stroke,31 and post-methamphetamine exposure conditions.32 Both Tmod1 and Tmod2 are localized within growth cones during development, but their location, level of expression, and role in neurite extension differ significantly. Tmod1 levels increase during neurite extension and proximally localize along actin filament bundles in the lamellipodia and to growth cones later in development.12,26 Conversely, Tmod2 levels remain fairly constant within the cytoplasm throughout neuritogenesis with some enrichment in the central region of growth cones. Knockdown of Tmod2 in N2a cells doubled the number of neuronal cells that developed neurites and the mean length of primary neurites, while Tmod1 knockdown led to an increased number of neurites per neuron.12 These results suggest an isoform specific inhibition of different components of neurite extension. Structural differences between the two isoforms may account for their unique actin pointed-end capping activity, though these differences remain unknown.
To further characterize the functional and structural differences between Tmod1 and Tmod2, we first investigated the role of Tmod’s LRR domain in neurite formation by overexpressing Tmod1 and Tmod2 with deleted LRR domains in PC12 cells, a model system used to study early stages of neuritogenesis. Additionally, a Tmod1 mutant with three point mutations in the LRR domain that resulted in disrupted localization in cardiac cells,19 was overexpressed. We further characterized the structural differences between the LRR domain of Tmod1, a Tmod1 mutant, and Tmod2 by circular dichroism (CD), limited proteolysis, and molecular dynamics simulation (MDS). We demonstrated the lower stability of the Tmod1 mutant and Tmod2 compared to that of Tmod1 and localized structural differences to the region of the C-terminal (sixth) α-helix. Two chimeric Tmods were created with the LRR domains exchanged between Tmod1 and Tmod2; this allowed us to investigate the role of Tmod’s LRR domain while maintaining the TM binding properties of the N-terminal domain. From these data, we suggest that the TM-dependent actin capping ability of both Tmods inhibits neurite formation, but in Tmod1, this inhibition can be controlled via its LRR domain. Altogether, our data unravel the structural and functional differences between the LRR domains of Tmod1 and Tmod2 and relate these changes to neurite outgrowth.
Experimental Procedures
Constructs for Tmod Expression
For transfection experiments and expression in Escherichia coli, mouse Tmod2 (accession number NM_016711) was subcloned into pReceiver-M55 with an mCherry tag (mChFP) and into pReceiver-B01 with a His tag (GeneCopoeia, Rockville, MD), respectively. Mouse Tmod1 (accession number NM_21883), Tmod1[1–159], and Tmod1[V232D/F263D/L313D] (Tmod1[DDD])19 were subcloned into pEGFP-C1 for transfection experiments. The construct with His-tagged Tmod1[DDD] was used for expression in E. coli. Chimera constructs GFP-Tmod1[1–159]/Tmod2[162–351] in mammalian expression vector pEZ-M29 and mChFP-Tmod2[1–161]/Tmod1[160–359] in mammalian expression vector pReceiver-M55 were ordered from GeneCopoeia. Our attempts to change the codon for Pro162 in mChFP-Tmod2 to a stop codon were not successful. Therefore, to create truncated Tmod2, the codon for Val163 was changed to a stop codon by Mutagenex Inc. (Hillsborough, NJ).
Tmod Purification
(His)Tmod1[DDD] and (His)Tmod2 were overexpressed in E. coli BL21 plysE and BL21 cells, respectively. Proteins were purified as described in refs (19) and (26), respectively. The Tmod concentration was determined using a PerkinElmer Lambda 2 UV–vis spectrometer by dissolving 60 μL of protein in cuvettes containing 500 μL of 6 M guanidine base (pH 12.5) or 6 M guanidine-HCl (pH 7.1). A baseline was taken from 320 to 270 nm with the pH 7.1 solution in the reference compartment and the pH 12.5 solution in the sample compartment. The concentration in moles per liter of Tmods in the cuvette was determined through the equation A/(2357Y + 830W), where A is the absorbance at 294 nm, Y is the number of tyrosines, and W is the number of tryptophans as in ref (33).
Limited Proteolysis, N-Terminal Sequencing, and Mass Spectrometry
Limited proteolysis was performed using trypsin and Staphylococcus aureus V8 protease at room temperature for wild-type Tmod2 or Tmod1[DDD] containing three point mutations in its C-terminal domain (V232D, F263D, and L313D). At 1, 5, 15, and 60 min, aliquots were removed and proteolysis was terminated with 10 mM Pefabloc or by mixing with sample buffer for sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The enzyme:protein ratio was 1:200 (w/w) for trypsinolysis and 1:50 for V8 proteolysis. Samples of the reaction mixture were inhibited with Pefabloc, dialyzed, and sent for mass spectrometry. The bands of corresponding fragments were cut from the gel and sent for N-terminal sequencing. N-Terminal sequencing and mass spectrometry of fragments were conducted at The Keck Biotechnology Resource Laboratory at Yale University (New Haven, CT). Positions of fragments were determined using the N-terminal sequence, the full-length protein’s sequences, and Expasy’s molecular mass/pI calculator.34−36
Circular Dichroism (CD) and Fluorescence Measurements
The CD spectra in the wavelength range of 190–260 nm for Tmod1 and Tmod2 were measured at 0.5 nm intervals on a spectropolarimeter (Aviv model 400). A cylindrical fused quartz cell with a path length of 0.1 cm was used. For titration experiments, two solutions containing proteins dissolved in 0.1 M sodium phosphate buffer with or without 10 M urea were combined to make protein solutions in 0–10 M urea. Each urea solution was measured in the wavelength range of 221–223 nm, and the signal at 222 nm was recorded. Differential scanning fluorimetry was conducted using an Agilent Technologies Stratagene Mx 3005 P system with 0.5–1.0 mg/mL samples.
Molecular Dynamics Simulation (MDS) of Tmod1, Tmod1[DDD], and Tmod2 Structure Prediction
X-ray and simulated structures were visualized using CHIMERA. Mutated Tmod1 structures were generated by editing Protein Data Bank (PDB) entry 1IO0 for the protein.11 Incomplete side chains in the mutated protein were generated using the Leap function in AMBER11.37 The amino acid sequence of Tmod2 has no insertions or deletions relative to Tmod1. Over the length of the fragment in 1IO0, the level of sequence identity between 1IO0 and mouse Tmod2 is 72 and 78%. Given this level of sequence homology, a structure for Tmod2 was generated by editing the PDB file of Tmod1 with the amino acids appropriate for Tmod2. As necessary, incomplete side chains were added to the generated Tmod2 structure using the Leap function in AMBER11.
MDS are conducted using the AMBER11 suite of codes. Hydrogen atoms are added to the X-ray structure on the basis of bond angles. Counterions (Na+ or Cl−) are added using a Coulombic potential on a grid to ensure charge neutrality. The protein is then placed in a box of TIP3P water molecules38 with a minimal distance of 10 Å from the protein to the edge of the box of waters. Periodic boundary conditions are used to ensure the maintenance of the ensemble and statistical mechanical measures. The protein/water system is then energy minimized using 1500 steps of steepest descent and 1500 steps of conjugant gradient minimization. The time evolution of the system is followed using the particle mesh Ewald method39−41 for calculating the electrostatic part of the potential energy term at constant pressure, with gradual heating to physiological temperature. Temperature is maintained via coupling to an external bath using the Berendsen algorithm.42 The SHAKE algorithm is employed43 with a time step of 2 fs. Simulations are run for 40 ns and then restarted for an additional 40 ns with the iwrap function active.
Cell Culture and Imaging
Undifferentiated PC12 cells were grown in DMEM (Invitrogen) supplemented with 5% fetal bovine serum (FBS, Invitrogen) and 10% horse serum (HS, Invitrogen) and subcultured every 3 days. For nerve growth factor (NGF)-induced differentiation, PC12 cells were grown on 100 μg/mL poly-d-lysine-coated eight-well plates or 35 mm culture dishes (ibidi LLC) in DMEM supplemented with 0.5% FBS, 1% HS, and 100 ng/mL NGF (Invitrogen). PC12 cells were transfected with GFP-Tmod1, GFP-Tmod1[1–159], GFP-Tmod1[DDD], mChFP-Tmod2, mChFP-Tmod2[1–162], or GFP and mChFP constructs using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. GFP-Tmod1/Tmod2 and mChFP-Tmod2/Tmod1 chimeras were transfected separately in PC12 cells before being plated and cocultured together before NGF induction. Twenty-four hours after transfection, NGF was used to induce differentiation outgrowth of neurite-like processes (neurites) monitored on days 3 and 6 on a laser scanning confocal microscope (LSM710, Zeiss) with a 63× oil immersion lens (Zeiss). Images were acquired with Zen software (Zeiss), and the number and length of neurites were analyzed with Imaris (Bitplane). Statistical analysis was performed by one-way analysis of variance (ANOVA).
Results
Tmod’s LRR Domain Is Influential in Differentiating PC12 Cells
The C-terminal domain, or the LRR domain, of Tmod is highly conserved between Tmod isoforms. This domain is important for localization in myocytes and contains a TM-independent actin-capping site. We used PC12 cells, a cell line derived from pheochromocytoma of rat adrenal gland and can be reversibly induced to produce neurite-like processes by growth in media with NGF. For the sake of simplicity, PC12 neurite-like processes will be called neurites hereafter. These cells are a model for early stages of neurite outgrowth.44−46
It has been reported that there is a compensatory expression of Tmod isoforms after knockdown of a single isoform.12 To prevent complications with compensatory expression, Tmod knockdown was not utilized in these experiments. As all of our constructs maintain the TM binding sites in the N-terminal domain, we therefore expect the overexpressed proteins to replace endogenous protein for pointed ends of actin filaments, which allows us to see the effect of the altered LRR domain.
Constructs for expression of GFP-Tmod1, the GFP-Tmod1[V232D/F263D/L313D] triple mutant (GFP-Tmod1[DDD]), and a 159-amino acid N-terminal fragment with the LRR domain deleted (GFP-Tmod1[1–159]) were generated with an N-terminal GFP. Tmod1[DDD] was developed to disrupt exposed hydrophobic clusters and resulted in poor localization to pointed ends in cardiac cells.19 The various constructs were transfected into PC12 cells to assess their effect on neurite outgrowth. After NGF-induced differentiation for 3 days, the number and length of neurites of transfected cells were analyzed for each Tmod construct and compared to those of PC12 cells overexpressing GFP only. Representative confocal images of cells overexpressing GFP-Tmod1, GFP-Tmod1[1–159], and GFP-Tmod1[DDD] are shown in Figure 2a, and the number of neurites per transfected cell and the average length of neurites corresponding to each construct are shown in Figure 2c. In agreement with our previous observations,26 overexpression of GFP-Tmod1 did not significantly alter the neurite formation (3.2 ± 0.91 neurites/cell) or length (64.4 ± 15.81 μm) compared to the values seen with GFP-overexpressing cells, 3.4 ± 0.81 neurites/cell and 74.2 ± 14.32 μm, respectively. In addition, no difference in the length of the longest neurite can be observed (data not shown). In contrast, overexpression of GFP-Tmod1[DDD] resulted in an 80% reduction in the number of neurites (0.7 ± 0.61 neurites/cell), an 80% reduction in the average length of the neurites (14.2 ± 12.69 μm), and a 65% reduction in the longest neurite (data not shown) relative to the GFP-Tmod1 control. Surprisingly, the reduction in the level of neurite formation and growth in cells overexpressing GFP-Tmod1[DDD] is greater than the reduction observed in cells overexpressing GFP-Tmod1[1–159] with the deletion of the entire LRR domain. GFP-Tmod1[1–159] resulted in reductions of 60, 45, and 30% in the number of neurites (1.4 ± 0.58 neurites/cell), average length (34.7 ± 14.1 μm), and longest neurite length (data not shown), respectively. The differences between GFP-Tmod1[DDD] and GFP-Tmod1[1–159] are statistically significant for all three measures (p < 0.005; by ANOVA). We also observed that a significant proportion of GFP-Tmod1[DDD] is targeted to the nucleus compared to the proportion of GFP-Tmod1[1–159]-overexpressing cells (data not shown).
Figure 2.

Effect of deletion or mutations of the LRR domain in Tmod1 and Tmod2 on neurite formation. Confocal imaging of PC12 cells overexpressing GFP, GFP-Tmod1, GFP-Tmod1[1–159], and GFP-Tmod1[DDD] (a) and mChFP, mChFP-Tmod2, and mChFP-Tmod2[1–162] (b) after NGF-induced differentiation for 3 days. The bar is 30 μm. Bar graphs representing the quantification of the number of neurites per cell and the average length of neurites in PC12 cells overexpressing the various Tmod1 (c) and Tmod2 (d) constructs: GFP (n = 29), Tmod1 (n = 51), Tmod1[1–159] (n = 32), Tmod1[DDD] (n = 40), mChFP (n = 30), Tmod2 (n = 30), and Tmod2[1–162] (n = 30). Error bars represent standard deviations (ns, not specific; ***p < 0.0005; one-way ANOVA).
Similarly, we next tested the effect of Tmod2 with a deleted LRR domain. Constructs for the expression of full-length mChFP-Tmod2 and a 162-amino acid N-terminal fragment (mChFP-Tmod2[1–162]) were generated with an N-terminal mChFP. Representative confocal images of PC12 cells transfected with mChFP-Tmod2 and mChFP-Tmod2[1–162] are shown in Figure 2b, and the number of neurites per transfected cell and the average length of neurites corresponding to each constructs are shown in Figure 2d. Overexpression of mChFP-Tmod2[1–162] resulted in a 70% reduction in the number of neurites per cell (0.9 ± 0.64 neurites/cell) and a 75% reduction in the average neurite length (22.9 ± 11.42 μm) relative to the values of control cells overexpressing mChFP only, 3.1 ± 0.63 neurites/cell and 72.1 ± 17.38 μm, respectively. After NGF-induced differentiation for 3 days, there was no statistical difference between the cells overexpressing mChFP-Tmod2 and mChFP-Tmod2[1–162] (p > 0.18), which shows that with or without its LRR domain, Tmod2 significantly impairs neurite formation. Interestingly, a similar degree of reduction is observed between GFP-Tmod1[DDD] and mChFP-Tmod2[1–162] with no statistical difference between either the number of neurites per cell or the longest neurite length.
These data demonstrated that the difference in the effects of Tmod1 and Tmod2 on the initiation and extension of neurites is due to their LRR domains. The N-terminal domains from both isoforms inhibit neurite formation. Tmod1’s LRR domain counteracts the inhibition by its N-terminal half, while Tmod2’s LRR domain lacks the ability to counteract the inhibition by its N-terminal half. The V232D, F263D, and L313D mutations in Tmod1’s LRR domain reduced the rate of neurite outgrowth to an extent similar to that of Tmod2 overexpression. It was shown using CD that these mutations caused a decrease in the stability of the LRR domain.19 To determine if there is a similar correlation between the stability of the LRR domain and its function in the formation of neurite-like processes in PC12 cells, we looked to uncover structural differences among Tmod1, Tmod1[DDD] and Tmod2 using three different approaches: CD, limited proteolysis, and MDS.
Tmod2 and Tmod1[DDD] Are Less Stable Than Tmod1
CD Experiments
CD is a common technique for determining a protein’s stability and secondary structure. In our previous studies, we showed that the V232D, F263D, and L313D mutations in the LRR domain of Tmod1 did not change its overall secondary structure but did decrease its stability.19 The stability of Tmod2 was compared with that of Tmod1 using urea denaturation (Figure 3b) and thermal shift, or differential scanning fluorimetry (data not shown). As the N-terminal half of Tmod2 is disordered, we actually measured the stability of the C-terminal half, or LRR domain, in these experiments. Titration experiments indicated that the midpoint of the two-state transition corresponds to ∼3.5 M urea (Figure 3b) for Tmod2, while it was 5 M for Tmod1 and 4 M for Tmod1[DDD].19 Melting temperatures determined from the thermal shift experiment are 48 °C for Tmod2 and 59 °C for Tmod1. These results demonstrate that the LRR domain of Tmod2 is less stable than that of Tmod1. The CD spectrum of Tmod2 was measured (Figure 3a). The α-helical content calculated from the spectrum was found to be 30.8% (predicted value of 37.6%), which is lower than Tmod1’s α-helical content (44%).13
Figure 3.

Structural properties of Tmod2. (a) CD spectrum of Tmod2. (b) Comparison of titration of Tmod2 (left y-axis) and Tmod1 (right y-axis) with urea. The data for Tmod1 were taken from ref (19).
Limited Proteolysis and Fragment Localization
Limited proteolysis is a simple technique for checking the structural stability of a protein. Proteins and protein domains that are tightly folded are more resistant to proteases than those that are flexible and extended. Limited proteolysis has already been conducted on Tmod1 and is used as a reference.13 Tmod1 trypsinolysis yielded stable fragments with molecular masses of 20 and 17 kDa. Digestion by V8 protease yielded a stable 20 kDa fragment. The 17 kDa tryptic fragment and the 20 kDa V8 proteolytic fragment were resistant to further proteolysis and could be stored in the reaction mixture for several days.13 In contrast, Tmod1[DDD] produced only a 17 kDa fragment by trypsinolysis and two fragments, 20 and 16 kDa, by V8 protease digestion. These results indicate the different accessibilities of Arg/Lys and Glu, respectively, in mutated Tmod1. The 16 kDa tryptic and 20 kDa V8 proteolytic fragments (boxed in Figure 4a) were cut from the gel and sent for N-terminal sequencing. These fragments have N-terminal sequences of YKPVP and GLNSV for trypsin and V8 proteases, respectively. The N-termini of these fragments correspond to those obtained by limited proteolysis of wild-type Tmod1.13 The maintained N-termini with lower molecular masses indicate that the C-terminus of the mutant is more susceptible to proteases. We suggest that this could be a result of structural changes in the region of the sixth α-helix.
Figure 4.
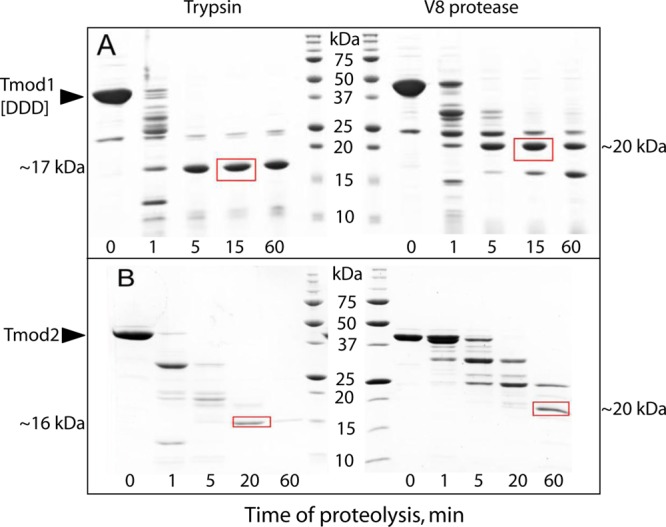
Time course proteolysis of (A) Tmod1[DDD] and (B) Tmod2. Trypsin and S. aureus V8 protease were added to Tmods [1:200 and 1:50 (w/w) ratios, respectively]. The arrowheads indicate the positions of Tmods. The molecular masses shown on the sides of the images correspond to boxed bands that were used for N-terminal sequencing. Molecular mass standards are 250, 150, 100, 75, 50, 37, 25, 20, 15, and 10 kDa.
Tmod2 was found to be even more susceptible to proteases than the triple mutant (Figure 4b). The 16 and 20 kDa fragments were produced by trypsin and V8 proteolysis, respectively. While the corresponding fragments from Tmod1 were stable for several days, the Tmod2 fragments degraded within a few hours. These fragments were cut from the gel and sent for N-terminal sequencing; these bands are boxed in Figure 4. The tryptic fragment’s N-terminal sequence is AKPVF. The V8 proteolytic fragment’s N-terminal sequence is KAKPV. The tryptic fragment had a mass of 15734 Da, and the V8 proteolytic fragment had a mass of 19790 Da. The positions of the fragments were then approximated using the determined molecular masses and N-terminal sequences; the tryptic fragment localized to A172–R312 and the V8 proteolytic fragment to K171–E347 (Figure 1). The mass of the fragments was found to be 1 and 13 Da different from that predicted by protein sequences for trypsinolysis and V8 proteolysis, respectively. The localization of the fragments is shown in Figure 1. The N-terminal sequence points of these fragments are very similar to those of Tmod1’s fragments; however, rapid digestion of Tmod2’s fragments indicates a greater degree of flexibility in the LRR domain.
One confounding factor of this conclusion is the presence of several more proteolytic sites in Tmod2 than in Tmod1. Tmod1 has 12 V8 proteolytic sites and 20 trypsin proteolytic sites in its C-terminal half, while the Tmod2 sequence has 18 V8 proteolytic sites and 25 trypsin proteolytic sites. Of the additional proteolytic sites in Tmod2, only one of the trypsin cut sites is in the loop immediately before the sixth α-helix. Although a structural difference is a conclusion that fits the data, additional data are necessary to rule out the impact of these additional cut sites.
Tmod1[DDD] and Tmod2 Have Altered Structure at Their C-Terminal Ends
Using MDS, we can look for possible changes to protein structure by using a known structure. The known structure can be manipulated to include the sequence of an unknown protein and then, through simulation of forces on individual atoms of the unknown protein, to produce an approximation of the structure quite rapidly. The Protein Data Bank structure of Tmod1 (entry 1IO0(11,47)) was used as the starting point for the triple mutant simulation, and the mutations were substituted into the protein’s structure. Simulations of the native (control) and mutated protein were conducted for 40 ns and then restarted and run for an additional 40 ns. Plots of the root-mean-square deviation (rmsd) of the simulated protein from the original structure suggest that the structures had reached a new quasi-steady state; these are shown in Figure 5. Shown in Figure 6 is an overlay of the final frame of the simulations: Tmod1 colored blue, Tmod1[DDD] purple, and Tmod2 yellow. There are modest differences between Tmod1 and Tmod1[DDD], with an increase in curvature between the two and a small angle and offset of the sixth α-helix. A closer look yields the observation that a hydrogen bond is added between D313 and N326 because of the mutations of Tmod1[DDD].
Figure 5.

Plots of rmsd calculations for the MD simulations.
Figure 6.
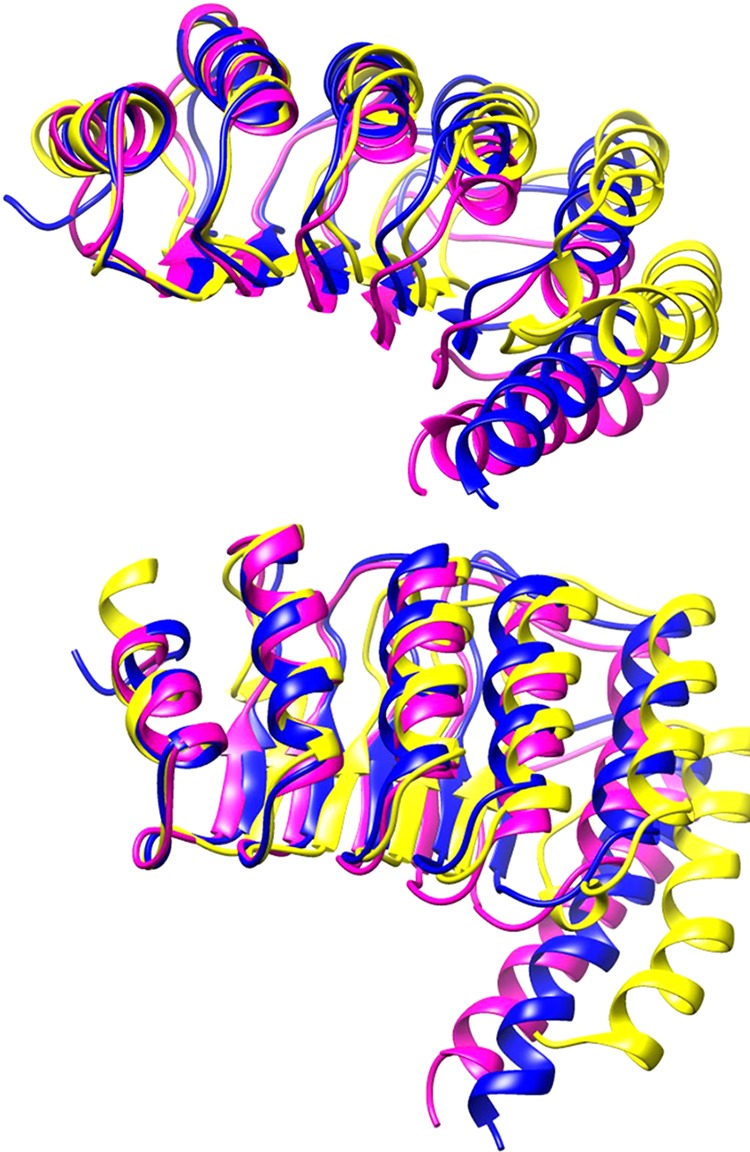
Molecular dynamics simulations of Tmod structures. Time averages of the last 10 ns of the simulations for Tmod1 (blue), Tmod1[DDD] (purple), and Tmod2 (yellow). The figure shows two different views of the results of the simulations. Structures have the N-termini on the left side; the top panel has the C-terminal α-helix directed out of the page, and the bottom panel has the C-terminal α-helix pointing down the page. The structures are matched over the α-carbons of the 44 N-terminal amino acids.
In a manner similar to that used for Tmod1[DDD], a file was developed with all of the substitutions necessary to mutate Tmod1 into Tmod2. This file was used for a 40 ns simulation and restarted for an additional 40 ns. The rmsd from the initial structure as a function of time in Figure 5 shows that the simulation had reached a new pseudosteady state. The final frame of the simulation is shown in Figure 6. From these structures, we see significant deviation from the Tmod1 control, with a reduction in curvature along the α-helices of the LRR domain and an offset and angled sixth helix. Additionally, curvature is observed in the final helix. A hydrophobic surface plot (Figure 7) of Tmod2 shows that there is a hydrophobic pocket formed in the area analogous to the added hydrogen bond in Tmod1[DDD].
Figure 7.
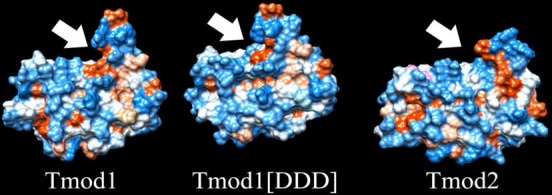
Hydrophobic surface plots of Tmod structures. Hydrophobic surface plots of Tmod1 (right) and Tmod2 (left) oriented such that the last α-helix is positioned at the top of the frame with the β-strands facing toward the reader. Indicated by arrows are hydrophobic (red) pockets.
Together with the data obtained by CD, thermal shift, and limited proteolysis, we can conclude that both Tmod1[DDD] and Tmod2 are less stable than Tmod1 and the difference in stability is caused by changes in the very C-terminal region of the LRR domain close to the sixth α-helix.
Phenotypic Difference between Overexpression of Tmod Chimeras
In the presence of TMs, Tmod’s actin capping behavior is determined by the N-terminal half, which holds two TM binding sites that flank the TM-dependent actin-capping site (Figure 1). Tmod’s actin nucleating and sequestering abilities require the actin-capping sites in both domains. Tmod’s targeting is attributed to only the LRR domain.19 To investigate the impact of Tmod’s LRR domains on neurite formation, we produced a set of chimeras with the C-terminal and N-terminal halves of Tmod1 and Tmod2 switched between the two isoforms. These chimeras allow us to maintain TM binding of a single isoform while switching the LRR domains. Two chimeric proteins were constructed; mChFP-Tmod2/Tmod1 contained the N-terminal half of Tmod2 (residues 1–161) and the C-terminal half of Tmod1 (residues 160–359), and GFP-Tmod1/Tmod2 contained the N-terminal half of Tmod1 (residues 1–159) and the C-terminal half of Tmod2 (residues 162–351). Representative confocal images of PC12 cells transfected with mChFP-Tmod2/Tmod1 and GFP-Tmod1/Tmod2 and cocultured in the presence of NGF for 3 days are shown in Figure 8a. The distributions of the number of neurites per cell and the length of neurites are shown in Figure 8b, and the corresponding average values are shown in Figure 8c. After NGF-induced differentiation for 3 days, PC12 cells overexpressing the GFP-Tmod1/Tmod2 chimera had significantly fewer (1.57 ± 0.51 neurites/cell) and shorter (29.2 ± 14.7 μm) neurites than cells overexpressing the mChFP-Tmod2/Tmod1 chimera, 3.17 ± 0.63 neurites/cell and 57.5 ± 16.8 μm, respectively. The number and length of neurites in the mChFP-Tmod2/Tmod1 chimera were similar to the number and length of neurites observed in control cells overexpressing mChFP only (Table 1). These data strongly argue in favor of a differential role of Tmod1 and Tmod2’s LRR domains. Tmod1’s LRR domain may act as a positive regulator of neurite formation.
Figure 8.

Comparison of the effect of Tmod1/Tmod2 and Tmod2/Tmod1 chimeras on PC12 cell differentiation. (a) Confocal imaging of PC12 cells overexpressing GFP-Tmod1/Tmod2 cocultured with PC12 cells overexpressing mChFP-Tmod2/Tmod1 after NGF-induced differentiation for 3 days. The bar is 50 μm. (b) Distribution of the number of neurites per cell and distribution of the length of the neurites in PC12 cells expressing the GFP-Tmod1/Tmod2 chimera (green traces and bars) and PC12 cells expressing the mChFP-Tmod2/Tmod1 chimera (red traces and bars). (c) Average numbers of neurites and average lengths of neurites in PC12 cells expressing the GFP-Tmod1/Tmod2 chimera (n = 35; light gray) and the mChFP-Tmod2/Tmod1 chimera (n = 42; dark gray). Error bars represent the standard deviation (***p < 0.0005; one-way ANOVA).
Table 1. Summary of Results from PC12 Cell Experiments with Standard Deviations.
| day 3 | day 6 | |||
|---|---|---|---|---|
| no. of neurites/cell | average neurite length (μm) | no. of neurites/cell | average neurite length (μm) | |
| GFP | 3.4 ± 0.81 | 74.2 ± 14.32 | nca | nca |
| Tmod1 | 3.2 ± 0.91 | 64.4 ± 15.8 | 3.1 ± 1.22 | 101.4 ± 24.1 |
| Tmod1[1–159] | 1.4 ± 0.58 | 34.7 ± 14.1 | nca | nca |
| Tmod1[DDD] | 0.7 ± 0.58 | 14.2 ± 12.69 | nca | nca |
| mChFP | 3.1 ± 0.63 | 72.1 ± 17.38 | nca | nca |
| Tmod2 | 1.1 ± 0.75 | 19.2 ± 10.6 | 0.9 ± 0.78 | 26.5 ± 16.1 |
| Tmod2[1–162] | 0.9 ± 0.64 | 22.9 ± 11.42 | 1.3 ± 0.71 | 78.4 ± 19 |
| Tmod1/Tmod2 | 1.57 ± 0.51 | 29.2 ± 14.7 | nca | nca |
| Tmod2/Tmod1 | 3.17 ± 0.63 | 57.5 ± 16.8 | 2.2 ± 0.98 | 112.3 ± 22.3 |
No noticeable change observed relative to day 3 measurements.
Surprisingly, after NGF-induced differentiation for 6 days, we found that while cells overexpressing GFP-Tmod1 differentiated normally and cells overexpressing mChFP-Tmod2 failed to differentiate as previously observed after 3 days, a limited but significant number (approximately 20%) of cells overexpressing the mChFP-Tmod2/Tmod1 chimera or the mChFP-Tmod2[1–162] truncated mutant were able to grow at least one elongated neurite (Figure 9a). The number of neurites per cell, the average length of neurites, and the length of the longest neurites are shown in Figure 9b. Overexpression of the mChFP-Tmod2/Tmod1 chimera resulted in a 1.5-fold decrease in the number of neurites (2.2 ± 0.98 neurites/cell) compared to the number seen with GFP-Tmod1, 3.1 ± 1.22 neurites/cell. However, the average length (112.3 ± 22.3 μm) and the length of the longest neurites (140.2 ± 19.6 μm) matched GFP-Tmod1 controls, 101.4 ± 24.1 and 133.1 ± 18.4 μm, respectively. In cells overexpressing the mChFP-Tmod2[1–162] truncated mutant, while the number of neurites per cell remains significantly lower than in cells overexpressing GFP-Tmod1, the length (average and maximum) of neurites becomes significantly higher (78.4 ± 19 μm) than in cells overexpressing mChFP-Tmod2 (26.5 ± 16.1 μm). These data suggested that the Tmod2 LRR domain strongly inhibits neurite formation and growth in the later steps of neuronal differentiation, and its deletion or replacement with Tmod1’s LRR domain, on the other hand, can facilitate a partial recovery neurite growth and elongation in the later steps of neuronal differentiation. PC12 cells are not recognized as developing axons; however, the later appearance of single long neurites is reminiscent of axon differentiation.
Figure 9.

Comparison of the effect of Tmod1 and Tmod2 mutants on PC12 cell differentiation. (a) Confocal imaging of PC12 cells overexpressing wild-type GFP-Tmod1, wild-type mChFP-Tmod2, the mChFP-Tmod2/Tmod1 chimera, and the mChFP-Tmod2[1–162] truncated mutant after NGF-induced differentiation for 6 days. The bar is 50 μm. (b) Bar graphs showing the quantification of the number of neurites per cell, the average length of neurites, and the length of the longest neurite in PC12 cells overexpressing the different Tmod1 and Tmod2 constructs: Tmod1 (n = 42), Tmod2 (n = 33), Tmod2/Tmod1 (n = 40), and Tmod2[1–162] (n = 28). Error bars represent the standard deviation (*p < 0.04, and ***p < 0.0005; one-way ANOVA).
Discussion
In our PC12 cell experiments, we set out to investigate the relative impacts of the functional domains of Tmod1 and Tmod2 on neuritogenesis and begin to narrow down the roles of different sites in Tmods. The work presented here provides new insights into the role of Tmods in neurite formation previously reported.12,26 The overexpression of Tmod2 in PC12 cells inhibits neurite formation, while the overexpression of Tmod1 does not significantly alter the number of neurites per cell or the length of neurites.26 One might expect that overexpression of pointed end stabilizing proteins would interfere with actin turnover, thereby interfering with neurite outgrowth, yet the overexpression of Tmod1 does not impact neurite outgrowth. Fath et al. hypothesized that Tmod2’s actin sequestering ability caused the inhibition of neurite outgrowth by Tmod2;12 however, co-overexpression of Tmod1 and Tmod2 resulted in a normal phenotype,26 which challenges this hypothesis. This suggests that the ratio of Tmod1 to Tmod2 is more important than the absolute expression.
Results of overexpressing the N-terminal domains of Tmod1 and Tmod2 indicate that Tmod’s N-terminal domain, which is responsible for capping actin filaments in the presence of TMs, is inhibitory for neurite formation (Table 1 and Figure 10). Mutations in Tmod1’s LRR domain, which alter its tertiary structure, have the same inhibitory effect. The LRR domain is currently attributed to Tmod’s actin sequestering and/or nucleation ability21,48 and is important for targeting.19 Our findings suggest that the LRR domain of Tmod1 counteracts inhibition of neurite formation by Tmod1’s N-terminal half.
Figure 10.
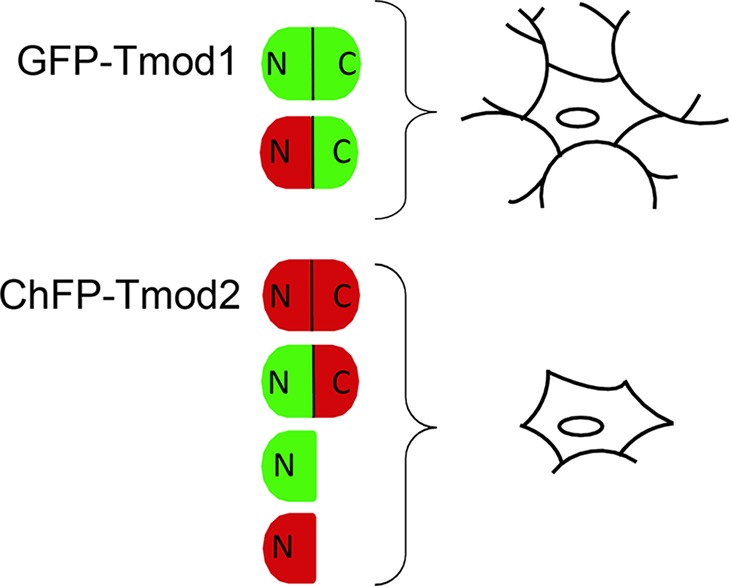
Visual summary of PC12 cell experiments.
Fath et al. observed the expression of Tmod1 and Tmod2 during N2a differentiation and found that Tmod2 has a nearly constant expression level during development and that Tmod1’s level of expression increased by 74% after differentiation had been induced for 24 h.12 If an increasing level of expression of Tmod1 counteracts the inhibition by Tmod2 and neurites begin to form, then regulation of Tmod expression may play a pivotal role in regulating neurite formation.
Highly ordered subcompartments within neuronal growth cones that have different localizations of actin-associated proteins have been observed.49 Tmods are known to have isoform specific interactions with TMs,50 which suggests that different Tmod isoforms could stabilize actin filaments coated by different TM isoforms. The N-terminal domain is responsible for this function; however, by destroying the LRR domain, we could be disrupting the targeting of Tmods to the subcompartments with different TM actin filaments and thereby disrupting the spatially specific actin/TM organization. The targeting may be realized through the LRR domain’s interaction with proteins other than actin.
The binding partners of Tmod’s LRR domain are still poorly understood. Some evidence that Tmod binds to nebulin and thymosin β-10 has been presented.51,52 Several nebulin isoforms have been shown to be expressed in the brain.53 Recently, nebulin–Tmod binding in muscle cells has been challenged by microscopic techniques,54 which brings into question the relevance of the possible interaction between the two proteins. Moreover, when the blot overlay experiments with biotinylated Tmod as described in ref (51) were performed using the M1–M3 nebulin fragment instead of full-length nebulin, no interaction was detected with the fragment. A strong interaction with a high-molecular mass contaminant of the nebulin sample was observed (A. S. Kostyukova, unpublished data). Other methods such as cross-linking, native gel electrophoresis, and CD also did not show interaction. Because of the inconsistencies in these results, additional experiments should be conducted to verify or refute this interaction between Tmods and nebulin and then explore its relevance in neurons.
Another possible binding partner for Tmod is thymosin β-10, which has been shown to be strongly expressed during development and decreases after birth.55−58 Using truncations, Rho et al. suggest that there is a Tmod–thymosin interaction site within the first two repeats of the LRR domain52 (Figure 1, denoted with an asterisk). Our Tmod1[DDD] mutations are outside of this region; however, it is possible that these mutations might alter Tmod–thymosin interactions and contribute to phenotypic changes observed in our experiments. Although there has been no experimentally determined isoform-dependent difference in Tmod–thymosin interactions, the sequence is only 55% identical and 62% similar to those of tropomodulin isoforms, which keeps this door of reasoning open.
To investigate the impact of Tmod’s LRR domain while maintaining the TM-dependent actin capping function, two chimeras were produced with the N-terminal and C-terminal halves switched between isoforms. In these experiments, we found recovery of neurite formation when Tmod1’s LRR domain is expressed with either isoform’s N-terminal domain; however, Tmod2’s LRR domain had no apparent impact. These data support our conclusion that Tmod LRR domains have isoform specific impacts on neurite formation. We used limited proteolysis, CD, and MDS to investigate structural differences between the LRR domains of Tmod1, Tmod1[DDD], and Tmod2. Altogether, these results indicate that Tmod1[DDD] and Tmod2 are more disordered and flexible than Tmod1.
From limited proteolysis data, we conclude that the lowered stability is localized at the C-terminal end of the LRR domain. The MD simulations predict that the sixth α-helices in the triple mutant and Tmod2 are offset relative to the Tmod1 control, which agrees with our data on limited proteolysis. These structural changes may correlate with isoform specific functions of Tmods’ LRR domains that result in the observed phenotypic differences.
Our experiments suggest that the different domains of Tmod isoforms play different roles in neurite formation, possibly implicating the role of Tmod targeting in neurite outgrowth. These results suggest two directions of study: (1) to check localization of the Tmod fragments and localization in neurons and (2) to further delve into the roles of individual functional sites of Tmod isoforms. From these continued studies, we aim to further elucidate the roles of Tmods in neural development.
Acknowledgments
We thank Dr. Inna Krieger for differential scanning fluorimetry measurements, Dr. Dmitri Tolkatchev for discussion, and the University of Medicine and Dentistry of New Jersey Neuroscience Summer Undergraduate Research Program that allowed C.P. to work in the laboratory of A.S.K.
Author Contributions
L.G. and K.G. contributed equally to this work. N.M. and C.P. also contributed equally to this work.
This work was supported by National Institutes of Health Grants GM081688 to A.S.K. and AR062279 to E.P.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- da Silva J. S.; Dotti C. G. (2002) Breaking the neuronal sphere: Regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 3, 694–704. [DOI] [PubMed] [Google Scholar]
- Hur E. M.; Saijilafu; Zhou F. Q. (2012) Growing the growth cone: Remodeling the cytoskeleton to promote axon regeneration. Trends Neurosci. 35, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitriol E. A.; Zheng J. Q. (2012) Growth cone travel in space and time: The cellular ensemble of cytoskeleton, adhesion, and membrane. Neuron 73, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez T. M.; Letourneau P. C. (2014) Actin dynamics in growth cone motility and navigation. J. Neurochem. 129, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M. F.; Laurent V.; Santolini J.; Melki R.; Didry D.; Xia G. X.; Hong Y.; Chua N. H.; Pantaloni D. (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J. Cell Biol. 136, 1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A.; Pennise C. R.; Fowler V. M. (1999) Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting ADP·Pi-actin to ADP-actin at all pointed filament ends. J. Biol. Chem. 274, 34637–34645. [DOI] [PubMed] [Google Scholar]
- Almenar-Queralt A.; Lee A.; Conley C. A.; Ribas de Pouplana L.; Fowler V. M. (1999) Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J. Biol. Chem. 274, 28466–28475. [DOI] [PubMed] [Google Scholar]
- Watakabe A.; Kobayashi R.; Helfman D. M. (1996) N-tropomodulin: A novel isoform of tropomodulin identified as the major binding protein to brain tropomyosin. J. Cell Sci. 109(Part 9), 2299–2310. [DOI] [PubMed] [Google Scholar]
- Fowler V. M. (1987) Identification and purification of a novel Mr 43,000 tropomyosin-binding protein from human erythrocyte membranes. J. Biol. Chem. 262, 12792–12800. [PubMed] [Google Scholar]
- Cox P. R.; Zoghbi H. Y. (2000) Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics 63, 97–107. [DOI] [PubMed] [Google Scholar]
- Krieger I.; Kostyukova A.; Yamashita A.; Nitanai Y.; Maeda Y. (2002) Crystal structure of the C-terminal half of tropomodulin and structural basis of actin filament pointed-end capping. Biophys. J. 83, 2716–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath T.; Fischer R. S.; Dehmelt L.; Halpain S.; Fowler V. M. (2011) Tropomodulins are negative regulators of neurite outgrowth. Eur. J. Cell Biol. 90, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyukova A.; Maeda K.; Yamauchi E.; Krieger I.; Maeda Y. (2000) Domain structure of tropomodulin: Distinct properties of the N-terminal and C-terminal halves. Eur. J. Biochem. 267, 6470–6475. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J.; Kostyukova A. S.; Hitchcock-DeGregori S. E. (2005) Structure and tropomyosin binding properties of the N-terminal capping domain of tropomodulin 1. Biophys. J. 88, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyukova A. S.; Tiktopulo E. I.; Maeda Y. (2001) Folding properties of functional domains of tropomodulin. Biophys. J. 81, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyukova A. S.; Rapp B. A.; Choy A.; Greenfield N. J.; Hitchcock-DeGregori S. E. (2005) Structural requirements of tropomodulin for tropomyosin binding and actin filament capping. Biochemistry 44, 4905–4910. [DOI] [PubMed] [Google Scholar]
- Kostyukova A. S.; Choy A.; Rapp B. A. (2006) Tropomodulin binds two tropomyosins: A novel model for actin filament capping. Biochemistry 45, 12068–12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B.; Kajava A. V. (2001) The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11, 725–732. [DOI] [PubMed] [Google Scholar]
- Tsukada T.; Kotlyanskaya L.; Huynh R.; Desai B.; Novak S. M.; Kajava A. V.; Gregorio C. C.; Kostyukova A. S. (2011) Identification of residues within tropomodulin-1 responsible for its localization at the pointed ends of the actin filaments in cardiac myocytes. J. Biol. Chem. 286, 2194–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyukova A. S.; Hitchcock-DeGregori S. E. (2004) Effect of the structure of the N terminus of tropomyosin on tropomodulin function. J. Biol. Chem. 279, 5066–5071. [DOI] [PubMed] [Google Scholar]
- Yamashiro S.; Speicher K. D.; Speicher D. W.; Fowler V. M. (2010) Mammalian tropomodulins nucleate actin polymerization via their actin monomer binding and filament pointed end-capping activities. J. Biol. Chem. 285, 33265–33280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning P.; O’Neill G.; Hardeman E. (2008) Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol. Rev. 88, 1–35. [DOI] [PubMed] [Google Scholar]
- Gunning P. W.; Schevzov G.; Kee A. J.; Hardeman E. C. (2005) Tropomyosin isoforms: Divining rods for actin cytoskeleton function. Trends Cell Biol. 15, 333–341. [DOI] [PubMed] [Google Scholar]
- Schevzov G.; Bryce N. S.; Almonte-Baldonado R.; Joya J.; Lin J. J.; Hardeman E.; Weinberger R.; Gunning P. (2005) Specific features of neuronal size and shape are regulated by tropomyosin isoforms. Mol. Biol. Cell 16, 3425–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curthoys N. M.; Freittag H.; Connor A.; Desouza M.; Brettle M.; Poljak A.; Hall A.; Hardeman E.; Schevzov G.; Gunning P. W.; Fath T. (2013) Tropomyosins induce neuritogenesis and determine neurite branching patterns in B35 neuroblastoma cells. Mol. Cell. Neurosci. 58C, 11–21. [DOI] [PubMed] [Google Scholar]
- Moroz N.; Guillaud L.; Desai B.; Kostyukova A. S. (2013) Mutations changing tropomodulin affinity for tropomyosin alter neurite formation and extension. PeerJ 1, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox P. R.; Fowler V.; Xu B.; Sweatt J. D.; Paylor R.; Zoghbi H. Y. (2003) Mice lacking tropomodulin-2 show enhanced long-term potentiation, hyperactivity, and deficits in learning and memory. Mol. Cell. Neurosci. 23, 1–12. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Dierssen M.; Toran N.; Pollak D. D.; Chen W. Q.; Lubec G. (2011) A gel-based proteomic method reveals several protein pathway abnormalities in fetal Down syndrome brain. J. Proteomics 74, 547–557. [DOI] [PubMed] [Google Scholar]
- Yang J. W.; Czech T.; Felizardo M.; Baumgartner C.; Lubec G. (2006) Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids 30, 477–493. [DOI] [PubMed] [Google Scholar]
- Sussman M. A.; Sakhi S.; Tocco G.; Najm I.; Baudry M.; Kedes L.; Schreiber S. S. (1994) Neural tropomodulin: Developmental expression and effect of seizure activity. Dev. Brain Res. 80, 45–53. [DOI] [PubMed] [Google Scholar]
- Chen A.; Liao W. P.; Lu Q.; Wong W. S.; Wong P. T. (2007) Upregulation of dihydropyrimidinase-related protein 2, spectrin α II chain, heat shock cognate protein 70 pseudogene 1 and tropomodulin 2 after focal cerebral ischemia in rats: A proteomics approach. Neurochem. Int. 50, 1078–1086. [DOI] [PubMed] [Google Scholar]
- Iwazaki T.; McGregor I. S.; Matsumoto I. (2006) Protein expression profile in the striatum of acute methamphetamine-treated rats. Brain Res. 1097, 19–25. [DOI] [PubMed] [Google Scholar]
- Kostyukova A. S.; Hitchcock-Degregori S. E.; Greenfield N. J. (2007) Molecular basis of tropomyosin binding to tropomodulin, an actin-capping protein. J. Mol. Biol. 372, 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjellqvist B.; Hughes G. J.; Pasquali C.; Paquet N.; Ravier F.; Sanchez J. C.; Frutiger S.; Hochstrasser D. (1993) The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 14, 1023–1031. [DOI] [PubMed] [Google Scholar]
- Bjellqvist B.; Basse B.; Olsen E.; Celis J. E. (1994) Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis 15, 529–539. [DOI] [PubMed] [Google Scholar]
- Wilkins M. R.; Gasteiger E.; Bairoch A.; Sanchez J. C.; Williams K. L.; Appel R. D.; Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552. [DOI] [PubMed] [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Wang B., Hayik S., Roitberg A., Seabra G., Wong K. F., Paesani F., Vanicek J., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A. and Kollman P. A. (2010) AMBER 11, University of California, San Francisco. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle Mesh Ewald: An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- Hawkins G. D.; Cramer C. J.; Truhlar D. G. (1996) Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 100, 19824–19839. [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. (1984) Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Ryckaert A. J.; Cicotti G.; Berendsen H. J. C. (1977) Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dyanmics of n-Alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Das K. P.; Freudenrich T. M.; Mundy W. R. (2004) Assessment of PC12 cell differentiation and neurite growth: A comparison of morphological and neurochemical measures. Neurotoxicol. Teratol. 26, 397–406. [DOI] [PubMed] [Google Scholar]
- Ohuchi T.; Maruoka S.; Sakudo A.; Arai T. (2002) Assay-based quantitative analysis of PC12 cell differentiation. J. Neurosci. Methods 118, 1–8. [DOI] [PubMed] [Google Scholar]
- Schimmelpfeng J.; Weibezahn K. F.; Dertinger H. (2004) Quantification of NGF-dependent neuronal differentiation of PC-12 cells by means of neurofilament-L mRNA expression and neuronal outgrowth. J. Neurosci. Methods 139, 299–306. [DOI] [PubMed] [Google Scholar]
- Krieger I.; Kostyukova A. S.; Maeda Y. (2001) Crystallization and preliminary characterization of crystals of the C-terminal half fragment of tropomodulin. Acta Crystallogr. D57, 743–744. [DOI] [PubMed] [Google Scholar]
- Fischer R. S.; Yarmola E. G.; Weber K. L.; Speicher K. D.; Speicher D. W.; Bubb M. R.; Fowler V. M. (2006) Tropomodulin 3 binds to actin monomers. J. Biol. Chem. 281, 36454–36465. [DOI] [PubMed] [Google Scholar]
- Dent E. W.; Gupton S. L.; Gertler F. B. (2011) The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harbor Perspect. Biol. 3, DOI: 10.1101/cshperspect.a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Shah S. P.; Gritsyna Y.; Hitchcock-DeGregori S. E.; Kostyukova A. S. (2011) Systematic analysis of tropomodulin/tropomyosin interactions uncovers fine-tuned binding specificity of intrinsically disordered proteins. J. Mol. Recognit. 24, 647–655. [DOI] [PubMed] [Google Scholar]
- McElhinny A. S.; Kolmerer B.; Fowler V. M.; Labeit S.; Gregorio C. C. (2001) The N-terminal end of nebulin interacts with tropomodulin at the pointed ends of the thin filaments. J. Biol. Chem. 276, 583–592. [DOI] [PubMed] [Google Scholar]
- Rho S. B.; Chun T.; Lee S. H.; Park K.; Lee J. H. (2004) The interaction between E-tropomodulin and thymosin β-10 rescues tumor cells from thymosin β-10 mediated apoptosis by restoring actin architecture. FEBS Lett. 557, 57–63. [DOI] [PubMed] [Google Scholar]
- Laitila J.; Hanif M.; Paetau A.; Hujanen S.; Keto J.; Somervuo P.; Huovinen S.; Udd B.; Wallgren-Pettersson C.; Auvinen P.; Hackman P.; Pelin K. (2012) Expression of multiple nebulin isoforms in human skeletal muscle and brain. Muscle Nerve 46, 730–737. [DOI] [PubMed] [Google Scholar]
- Castillo A.; Nowak R.; Littlefield K. P.; Fowler V. M.; Littlefield R. S. (2009) A nebulin ruler does not dictate thin filament lengths. Biophys. J. 96, 1856–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpintero P.; Franco del Amo F.; Anadon R.; Gomez-Marquez J. (1996) Thymosin β10 mRNA expression during early postimplantation mouse development. FEBS Lett. 394, 103–106. [DOI] [PubMed] [Google Scholar]
- Hall A. K.; Hempstead J.; Morgan J. I. (1990) Thymosin β10 levels in developing human brain and its regulation by retinoic acid in the HTB-10 neuroblastoma. Mol. Brain Res. 8, 129–135. [DOI] [PubMed] [Google Scholar]
- Hall A. K. (1991) Developmental regulation of thymosin β10 mRNA in the human brain. Mol. Brain Res. 9, 175–177. [DOI] [PubMed] [Google Scholar]
- Lin S. C.; Morrison-Bogorad M. (1990) Developmental expression of mRNAs encoding thymosins β4 and β10 in rat brain and other tissues. J. Mol. Neurosci. 2, 35–44. [DOI] [PubMed] [Google Scholar]