

Abstract
Traditional lead optimization projects involve long synthesis and testing cycles, favoring extensive structure−activity relationship (SAR) analysis and molecular design steps, in an attempt to limit the number of cycles that a project must run to optimize a development candidate. Microfluidic-based chemistry and biology platforms, with cycle times of minutes rather than weeks, lend themselves to unattended autonomous operation. The bottleneck in the lead optimization process is therefore shifted from synthesis or test to SAR analysis and design. As such, the way is open to an algorithm-directed process, without the need for detailed user data analysis. Here, we present results of two synthesis and screening experiments, undertaken using traditional methodology, to validate a genetic algorithm optimization process for future application to a microfluidic system. The algorithm has several novel features that are important for the intended application. For example, it is robust to missing data and can suggest compounds for retest to ensure reliability of optimization. The algorithm is first validated on a retrospective analysis of an in-house library embedded in a larger virtual array of presumed inactive compounds. In a second, prospective experiment with MMP-12 as the target protein, 140 compounds are submitted for synthesis over 10 cycles of optimization. Comparison is made to the results from the full combinatorial library that was synthesized manually and tested independently. The results show that compounds selected by the algorithm are heavily biased toward the more active regions of the library, while the algorithm is robust to both missing data (compounds where synthesis failed) and inactive compounds. This publication places the full combinatorial library and biological data into the public domain with the intention of advancing research into algorithm-directed lead optimization methods.
Keywords: Lead optimization, MMP-12 inhibitors, genetic algorithm, microfluidic chemistry
Using biological data in “real time” to drive a chemistry optimization program was suggested over 10 years ago by several groups.1−7 At GlaxoSmithKline (GSK), we have retained an interest in such approaches for a number of years and have made several attempts to drive traditional lead generation or lead optimization projects in this fashion. However, several factors contributed to only incomplete results. The traditional make/test cycle can be very long for anything but the most straightforward chemistry. This is compounded by the fact that the algorithms tend to suggest small numbers of noncombinatorial products. The extended cycle times provide plenty of time for reflection and analysis, which will inevitably compete with the suggestions of the algorithm, particularly in the early stages. In addition, other external factors come into play, such as structure−activity relationship (SAR) from related series, which may make the current template of less interest to the program.
A microfluidic-based chemistry and biology platform8 providing autonomous operation addresses many of these issues and is ideally suited to a real-time biology-driven optimization. Such systems offer the advantage of rapid synthesis under controlled conditions, followed by almost immediate measurement of biological response. When guided by the appropriate software tools, such platforms lend themselves to unattended autonomous 24/7 operation. The process iterates over the “μ-COSM” (collection of steps and materials), using the SAR generated at each iteration to design the choice of reactant and reactions for subsequent cycles. The ultimate goal is to discover the optimum product(s) accessible from each μ-COSM in the minimum time.
We have successfully implemented the individual components of such a system.9−13 However, traditional medicinal chemistry-based SAR analysis becomes the bottleneck when cycle times of minutes can be achieved through automation. Thus, for the system to operate effectively, an efficient design algorithm is required to drive each iteration.
To facilitate development and validation of algorithms to drive the autonomous selection process ideally requires access to a full combinatorial data set of reasonable scope. However, such data sets are relatively rare. Thus, to validate our approach and to demonstrate the concept of autonomous optimization, a large-scale experiment was undertaken with the following goals:
-
(1)
To establish a test environment for evaluating the performance of microfluidic platforms, under development for rapid synthesis and assay of compounds, by providing high-quality compound samples made and purified by conventional processes and high-quality assay data (in conventional plate-based assays) to act as standards against which to compare the corresponding output from the microfluidic assay platform.
-
(2)
To provide a test environment in which to evaluate algorithms for potential autonomous compound selection, by running in real time through 10 generations of synthesis and assay using conventional processes, guided by a suitable optimization algorithm (see below).
-
(3)
To provide a uniquely complete data set against which to assess the effectiveness of the above and other algorithms for iterative lead optimization, by synthesizing (through several different routes appropriate to the R groups), purifying, analyzing, and assaying a full 50 × 50 sulfonamide array, using conventional processes.
In this paper, we describe the results of this experiment, as well as details of a genetic algorithm optimizer (GAO) developed specifically to drive our microfluidic system. The chemical structures of a 50 × 50 library, synthesized in a conventional manner, are disclosed with associated QC and biological data, thus providing a unique and valuable data set for further exploration and algorithmic development.
The GAO used to drive each iteration cycle is similar to previous publications,1,2 and the general scheme is illustrated in Figure 1. However, we have incorporated a number of important additional features. In the language of the GA, each reagent is an allele in a specific gene (product). The complete library, or genome, is the collection of genes. The population is the set of molecules (individuals) that have been, or are to be, made. To start the process, the user provides the algorithm with the genome and various optimization parameters. The algorithm generates the starting population (each molecule being a combination of alleles from each gene), and the compounds are synthesized and tested. The population is updated with the screening results (scores), and a new population is generated using genetic operators. The process is repeated until convergence or a user-defined number of iterations have been reached. Here, we highlight several important features of the implementation that distinguish it from previous approaches, with in-depth details provided in the Supporting Information.
Figure 1.
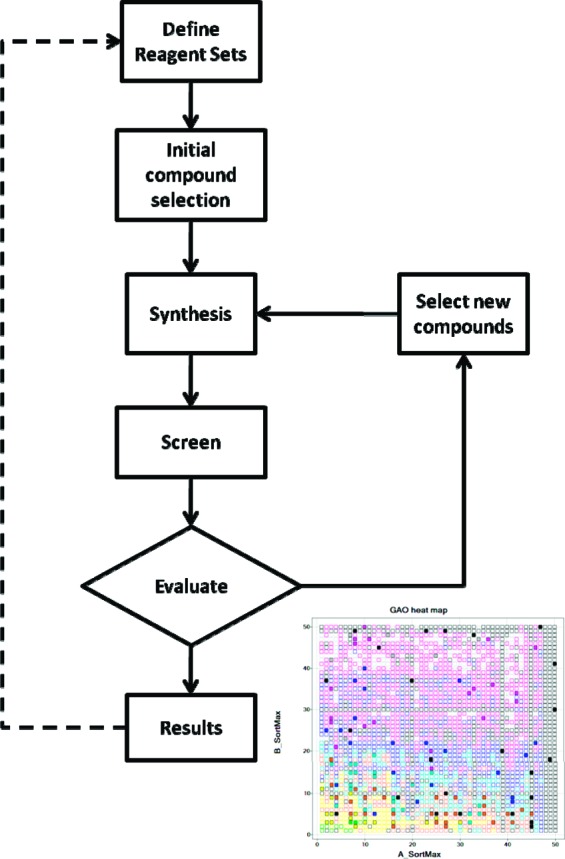
Outline of the GAO process.
Any experimental process, be that synthesis or assay, will be prone to errors. Similarly, certain molecules may not be synthesizable. Thus, a novel aspect of the algorithm described here is to allow for these potential errors in the optimization process. At each iteration, those individuals that were selected to be a parent are considered for retesting. If the parent has been tested less than a user-defined number of times, then it will be flagged for retesting.
A proportion of random individuals can be a good way of maintaining diversity within the population. Within the context of a virtual array, it is possible to identify regions of the space that have been poorly sampled. Thus, as an alternative to the random operator, a seek operator was implemented to identify such regions and focus the random generation there.
If GAO requests a test and there are no results available for the test, then the user can do one of three things: add no entry for the test in to the fitness file, add an entry with fitness data, or add an entry with “<NULL_FITNESS>” as the fitness entry.
In all cases, GAO will keep the individual for retesting and will request that it be tested again at some future generation. All untested individuals will be stored in the population file as individuals without fitness data. If the user wishes to indicate that the test cannot be performed and that GAO should not request a retest, then the user marks the individual with “<NULL_INDIV>” as the fitness.
The algorithm knows nothing about the chemical composition of the individual reagents (alleles) or the products. Thus, we have implemented the ability to define similarity between alleles. The similarity is defined in terms of distance and need not be symmetric. In addition, alleles may be grouped. Two alleles that are not in the same group have a similarity of zero. An allele may belong to more than one group. There are many ways of defining similarity between reagents and clustering or grouping compounds, and the implementation leaves it up to the user to decide on the most appropriate criteria to use, based upon the problem at hand. For instance, a group of heterocycles may belong to both a group of aromatic ring substituents and a group of hydrogen bond-accepting substituents. The similarity between products (genes) rather than between the reagents (individual alleles) can also be defined. When enabled, similarity and grouping lead to modified selection conditions for crossover and mutation that take account of the molecular or reagent similarity in addition to the similarity in biological response.
If the user has defined an initial set of groupings or similarities, how do these relate to the actual biological data obtained? Is it possible to learn appropriate groupings and thus generate an implied SAR from the data? To answer these questions, options have been added to re-evaluate the similarity levels associated within the groups at regular intervals during the optimization. The algorithm uses the standard deviation of fitness within the group to redefine the group similarity.
To test the GAO, we used data from an in-house combinatorial array of 7 amines by 80 acids on a template designed in a drug discovery program. The compounds had pIC50 values varying from 4 to 8.8. Data were not available on 24 members of the array, and these were set to NULL_INDIV (see above). To make the experiment more realistic, an additional 100 amines and 100 acids were selected at random from an in-house reagent database and used to expand the library to 19260 compounds. Activity data for the additional compounds were set to a random number on the interval 3.5−4.
The GAO was run with default settings and either in optimization mode (crossover, 40%; mutation, 60%) or as a random walk. The algorithm was run to completeness, that is, until all products had been selected, and the results averaged over 50 independent runs (different random number seeds). The optimization performance is shown in Figure 2, for various generation sizes: 5, 10, 15, 20, 25, 30, and 50 (where generation size is the number of molecules selected in that generation). The score represents the average activity of the top 10 most active molecules in the population at that point. The optimization works well compared to the random walk with the optimization runs converging after about 2000 molecules over all generation sizes.
Figure 2.
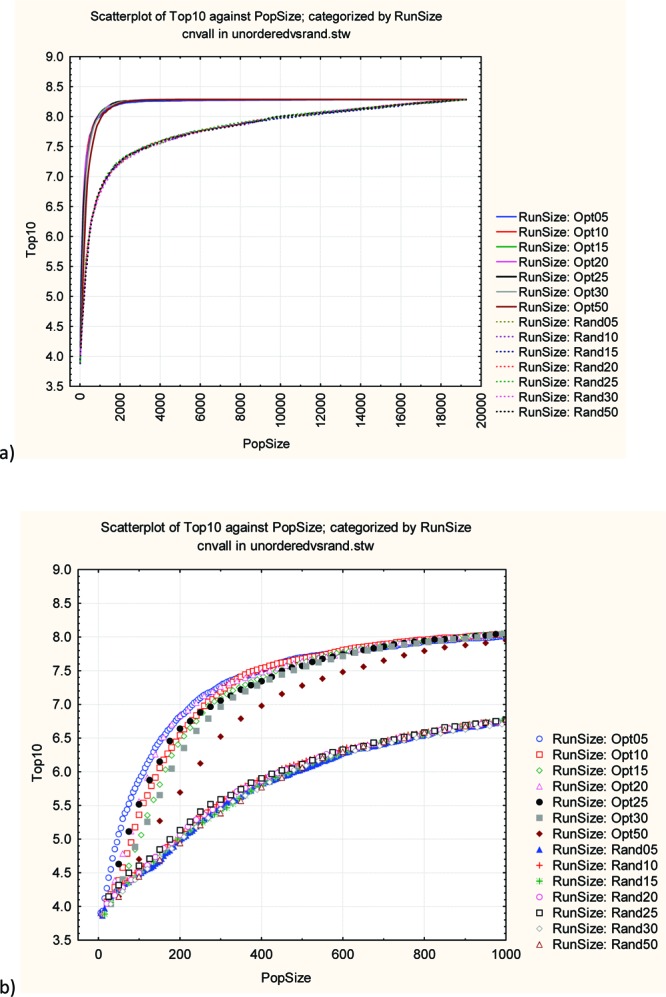
Mean activity of the top 10 most active compounds selected so far as a function of population size. The results represent the average of 50 runs at each generation size. (a) Overall performance with the optimization runs as solid lines and the random runs as dotted lines. (b) The early part of the plot up to a population size of 1000. The random runs all overlap in the lower curve.
Looking more closely at the early part of the curve (Figure 2b), it can be seen that generation size has only a minor impact on optimization performance except for a generation size of 50. The algorithm remembers and can use all the data generated, not just that from the last generation (see Supporting Information). The results of this retrospective analysis suggest that the number of iterations is more important than generation size per se. Smaller generation sizes allow more efficient use of the information.
The results above show that the GAO can identify small islands of activity efficiently from large compound sets. In fact, the algorithm performs equivalently to a designed subset synthesized as a combinatorial array. It is thus ideally suited to the problem at hand, which is to drive a microfluidic synthesis and testing system for lead optimization where the cycle times mean that many iterations are possible. These results gave us sufficient confidence to validate further the approach by applying the algorithm in a prospective setting.

MMP-12 is a target of therapeutic importance, and in-house high-throughput screening (HTS) had identified a series of biaryl sulfonamides represented by 1, amenable to array synthesis. Thus, this target and series were chosen for the experiment. In accordance with the first goal above, compounds were synthesized and screened iteratively off platform using conventional methods as described in the Supporting Information. The experiment involved two independent synthetic efforts. The first was focused on the synthesis of a complete 50 × 50 array with subsequent biological testing (see the Supporting Information). The initial reagent pool was selected with diversity in mind to allow exploration of as comprehensive a chemical space as possible, while still maintaining a reasonable physicochemical profile. The reagents were selected with the aid of the ADEPT system,14 an in-house web-based application that provides tools for identifying available reagents, calculating properties, refining the reagent list, and enumerating virtual libraries. An important implication of the reagent selection approach is that different chemistries were necessary in the synthesis of the full library, such that a traditional combinatorial chemistry approach to the full array would not be feasible. Similar issues will arise when attempting to move this chemistry to the microfluidic platform, not so much because of the large number of reaction schemes, as they can be implemented, but because of a need to determine a priori the most appropriate chemistry for any particular product. It is interesting to note that the retest capabilities of the GAO provide one approach to this problem and, in principle at least, the GAO could learn from successful reactions for related reagents.
In a second independent effort, compounds were synthesized in accordance with the results of the GAO selections. Learning from the retrospective analysis, we chose to synthesize 14 compounds per generation. GAO was run with crossover, 50%; mutation, 40%; and random, 10%. To avoid the scenario of early generations merely searching for active start points, the first generation was seeded with two compounds that were known to be active (A04B02 and A28B02), while the remaining 12 were selected at random by GAO. No structural knowledge (such as clustering and grouping features of GAO) was used in this preliminary experiment, although they would be anticipated to give more rapid optimization. In total, there were 10 iterations of synthesis and testing. All results are shown with respect to the data from the full array for consistency, although the correlation between the results was very high.
Detailed results for the full library are given in the Supporting Information, together with an indication of the generation from the GAO where applicable. The heat map for the full library is shown in Figure 3. The reagents have been sorted by their maximum pIC50 value. An alternative view of the data is given in Figure 4. This presents a histogram of the proportion of compounds within a defined activity range, comparing the full library (red bars) to the GAO-optimized selection (blue bars). From these figures, it is clear that the GAO has indeed optimized the selection, favoring the higher activity molecules within the data set. For example, considering a cutoff of pIC50 > 6, the GAO compound set has 68/140 (48.5%) of compounds in this activity range as compared to just 21.3% of the full array. Of course, while the enrichment is good, not all of the most potent compounds have been suggested within the limited set of compounds. Is this a problem? We would argue not. Clearly, this question only has value once all compounds have been made (benefit of hindsight). We have already shown in the previous experiment that, with enough iterations, GAO will locate all active compounds significantly more efficiently than a random walk. However, what is more important is that any subsequent lead optimization is focused on the most relevant region of chemistry space. From Figure 3, it can be seen that SAR is tighter around R2, and this is evident from the GAO output. Nine out of 10 top-ranked reagents for R2 (sorting by most active compound) have been covered with compounds having pIC50 > 6. The “missing” reagent is B07, phenyl, which was not selected at all during the limited number of iterations used here. However, both p-Br and p-Me phenyl are included in the GAO set, thus providing sufficient information to focus a further iteration.
Figure 3.
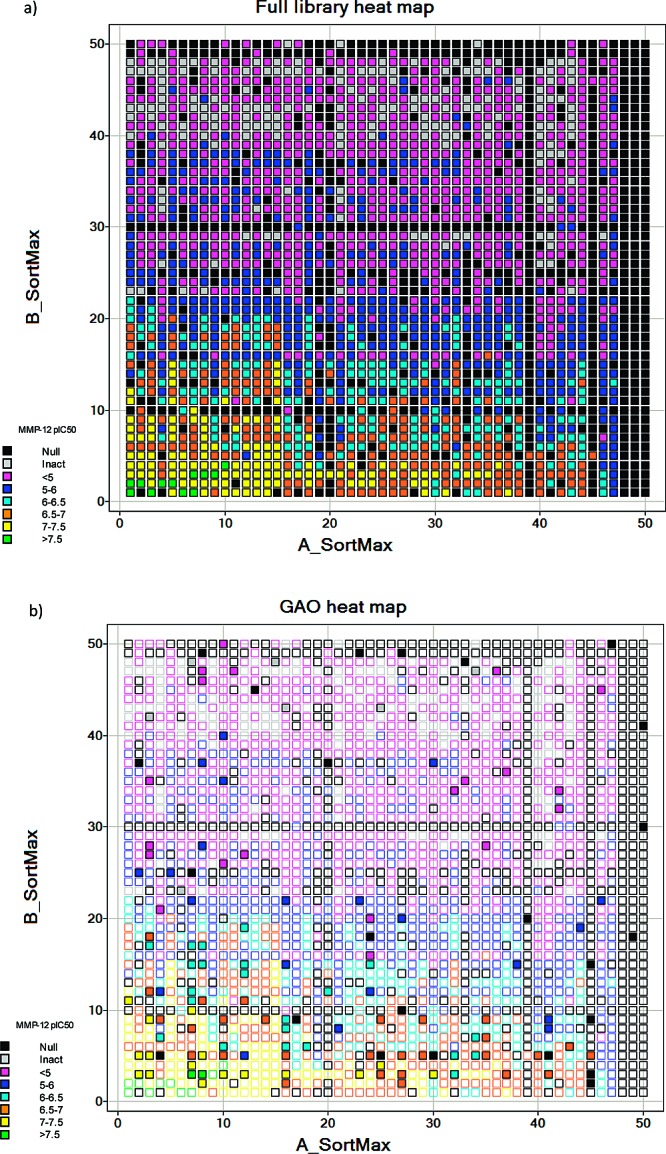
Activity heat maps for the full library and the GAO-optimized library. (a) MMP-12 pIC50 heat map for the full library. (b) Heat map for the GAO library, solid squares; superimposed on the full library heat map, open squares. The reagents are sorted by the activity of the most potent compound containing the reagent.
Figure 4.
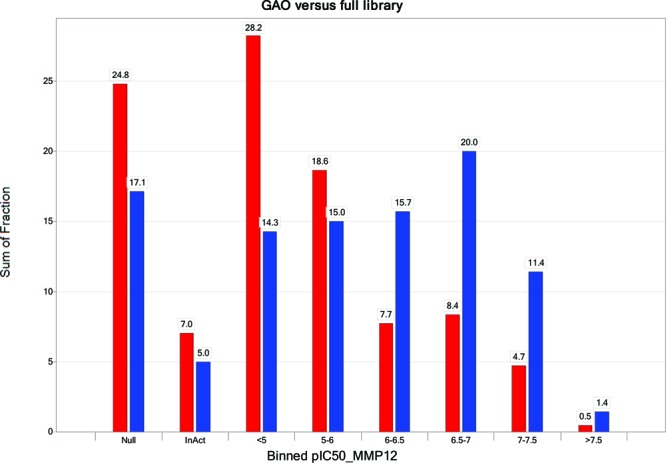
Proportion of compounds sampled by GAO (blue bars) as compared to the full library (red bars) as a function of MMP12 pIC50.
The results illustrate that the GAO is a robust optimization process. Overall, 25% of the full library (620 compounds) could not be synthesized, as shown in Figure 4 (red bars). A further 7% (176 compounds) were inactive in the assay. Thus, nearly a third of the library provided no or limited SAR. Figure 5 shows the activity ranges for each of the 10 generations. As noted above, GAO can use the information from all previous iterations so it is the fitness of the library as a whole that should improve, which may be less apparent in any particular generation (e.g., if all of the most potent compounds have already been made). Nevertheless, individual generations can provide interesting information on the progress of the optimization. In the first generation, three of the molecules could not be made, and a further two were inactive on assay. The numbers of nonsynthesizable and inactive compounds decrease over the next few generations and then increases again as the GAO looks for new areas to explore.
Figure 5.
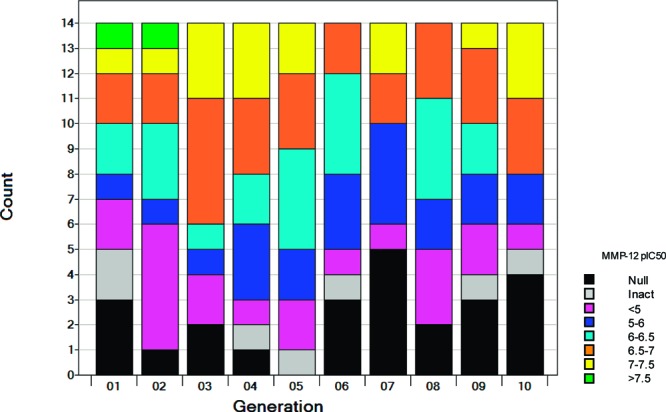
Activity ranges for each generation of the GAO.
In conclusion, the implementation of a microfluidic-based platform for compound optimization requires a combination of robust technology and informatics to support the process. The cycle times achievable on a microfluidic platform mean that alternatives to traditional medicinal chemistry optimization strategies need to be explored as otherwise the interpretation of the data and resulting decision-making becomes the bottleneck. In this paper, we have described an optimization procedure, GAO, that is based on the principles of a genetic algorithm. GAO incorporates novel features that make it particularly suited to the task. It is robust to missing data and can suggest that compounds are to be remade and retested. In a prospective experiment involving optimization of a series of compounds against MMP-12, compounds were synthesized and tested using conventional synthetic and screening methodology according to the GAO suggestions in cycles of 14 compounds. The full library of 2500 compounds was synthesized independently to provide evidence that the GAO did indeed optimize and find potent molecules. In just 10 cycles, or 140 compounds submitted for synthesis, a thorough sampling of the active region of chemical space was achieved, and active compounds were identified for further investigation. The chemical structures of the full library are disclosed with associated QC and biological data, thus providing a unique and valuable data set for further exploration and algorithmic development.
This proof of principle experiment illustrates that optimization algorithms used in many other disciplines and industries are applicable to medicinal chemistry, once the bottlenecks of synthesis and screening are overcome. Although this optimization experiment was conducted on a single parameter (MMP-12 inhibition), the underlying methods may readily be adapted to direct multiobjective optimization.15
Acknowledgments
We acknowledge Emma Vickerstaffe, Caroline Winn, and Charlotte Griffiths-Jones who synthesized the compounds and Michelle Heathcote and Theresa Pell who performed the assays.
Supporting Information Available
Full experimental procedures for synthesizing the compounds described, reagent lists, QC and NMR data on key compounds, and the full experimental data on the 50 × 50 array. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Weber L.; Wallbaum S.; Broger C.; Gubernator K. Optimization of the Biological Activity of Combinatorial Compound Libraries by a Genetic Algorithm. Angew. Chem., Int. Ed. Engl. 1995, 34, 2280–2282. [Google Scholar]
- Singh J.; Ator M. A.; Jaeger E. P.; Allen M. P.; Whipple D. A.; Soloweij J. E.; Chowdhary S.; Treasurywala A. M. Application of Genetic Algorithms to Combinatorial Synthesis: A Computational Approach to Lead Identification and Lead Optimization. J. Am. Chem. Soc. 1996, 118, 1669–1676. [Google Scholar]
- Yokobayashi Y.; Ikebukuro K.; McNiven S.; Karube I. Directed Evolution of Trypsin Inhibiting Peptides Using a Genetic Algorithm. J. Chem. Soc., Perkin Trans. 1 1996, 1, 2435–2439. [Google Scholar]
- Illgen K.; Enderle T.; Broger C.; Weber L. Simulated Molecular Evolution in a Full Combinatorial Library. Chem. Biol. 2000, 7, 433–441. [DOI] [PubMed] [Google Scholar]
- Kampenhausen S.; Holtge N.; Wirsching F.; Morys-Wortmann C.; Reister D.; Goetz R.; Thurk M.; Schweinhorst A. A Genetic Algorithm for the Design of Molecules with Desired properties. J. Comput.-Aided Mol. Des. 2002, 16, 551–567. [DOI] [PubMed] [Google Scholar]
- Weber L. Evolutionary Combinatorial Chemistry: Application of Genetic Algorithms. Drug Discovery Today 1998, 3, 379–385. [Google Scholar]
- Brauer S.; Almstetter M.; Antuch W.; Behnke D.; Taube R.; Furer P.; Hess S. Evolutionary Chemistry Approach toward Finding Novel Inhibitors of the Type 2 Diabetes Target Glucose-6-phosphate Translocase. J. Comb. Chem. 2005, 7, 218–226. [DOI] [PubMed] [Google Scholar]
- Hong J.; Edel J. B.; deMello A. J. Micro- and Nanofluidic Systems for High-throughput Biological Screening. Drug Discovery Today 2009, 14, 134–146. [DOI] [PubMed] [Google Scholar]
- Wong Hawkes S. Y. F.; Chapela M. J. V.; Montembault M. Leveraging the Advantages Offered by Microfluidics to Enhance the Drug Discovery Process. QSAR Comb. Sci. 2005, 24, 712–721. [Google Scholar]
- Hughes I.; Warrington B. H.; Wong Y. F. Microfluidic System. WO-2004089533 A1, 2004.
- Hughes I.; Warrington B. H.; Wong Y. F. A Method for Controlling System Having Microfluidic Channel Structure. WO-2006038014 A1, 2006.
- Hoyle C. K.; Pell T.; Hawkes S. Y. F. W.; Warrington B. H. Flow/microfluidic System-based Assays for the Effect of a Drug/compound on a Target. WO-2007021815 A2, 2007.
- Warrington B. H.; Hoyle C. K.; Pell T.; Pardoe D. A. Microfluidic System and Methods Using Microchannels with Fluorinated or fluorous inner surfaces. WO-2007021813 A2, 2007.
- Leach A. R.; Bradshaw J.; Green D. V. S.; Hann M. M.; Delany J. J. Implementation of a System for Reagent Selection and Library Enumeration, Profiling and Design. J. Chem. Inf. Comput. Sci. 1999, 39, 1161–1172. [DOI] [PubMed] [Google Scholar]
- Gillet V. J.; Willett P.; Fleming P. J.; Green D. V. S. Designing Focused Libraries Using MoSELECT. J. Mol. Graphics Modell. 2002, 20, 491–498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.