

Abstract
Integration of human immunodeficiency virus type 1 DNA into an infected cell genome is one of the key steps of the viral replication cycle. Therefore, viral enzyme integrase, which realizes the integration, represents an attractive and validated target for the development of new antiviral drugs. In this paper, the anti-integrase activity of a series of conjugates of single-stranded oligonucleotides with hydrophobic molecules was tested, and the structure–activity relationships were also analyzed. Both oligonucleotide and hydrophobic parts of the conjugates influenced the inhibitory potency. Conjugates of 11-mer phosphorothioate oligonucleotides with 6-carboxy-4,7,2′,4′,5′,7′-hexachlorofluorescein (HEX) were found to be the most efficient inhibitors (IC50 = 20 nM) and might be considered as lead compounds for further development of integrase inhibitors.
Keywords: HIV-1 integrase, inhibitor, xanthene, oligonucleotide conjugate
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency syndrome (AIDS), which is still a global public health problem that warrants the constant search for new inhibitors of HIV replication. Current treatments generally referred to as highly active antiretroviral treatments (HAART) combine several active drugs that target one or several key steps of the HIV life cycle. Until recently, HAART consisted of a combination of protease inhibitors, non-nucleoside reverse transcriptase inhibitors, and nucleoside reverse transcriptase inhibitors.1 In 2003, an inhibitor of viral fusion with the cell membrane, enfuvirtide,2 was added to the pharmacopeia. Recent progress was made in two directions: first, designing of CCR5 coreceptor ligands such as Maraviroc that impairs the viral entry and, second, development of inhibitors of the third viral enzyme, integrase (IN).3
HIV-1 IN is a 32 kDa viral protein that catalyzes a covalent insertion of viral DNA produced by reverse transcription of the viral genomic RNA into the chromosomes of infected cells. Once integrated, the provirus persists in the host cell and serves as a template for the transcription of viral genes and replication of the viral genome, leading to the production of new viruses. Several specific inhibitors of the integration step, including raltegravir, elvitegravir, and S/GSK1349572, have shown great promise, ensuring rapid recognition of IN inhibitors (INIs) as an important new class of antiretroviral drugs. However, as with other antiretroviral drugs, resistance to INIs has emerged both in vitro and in patients failing to respond to treatment, justifying the search for novel inhibitors. Viral DNA is inserted into the host cell DNA in a series of distinct steps.4−7 Following reverse transcription of the viral genome, IN binds both extremities of the viral DNA, forming a nucleoprotein complex that constitutes the core of the preintegration complex (PIC). Within the PIC, IN catalyzes 3′-processing, cleaving the terminal dinucleotides from both 3′-ends of the viral DNA. Once the PIC has migrated into the nucleus of the infected cell, IN mediates a strand transfer reaction in which the viral DNA is inserted into the host cell DNA. Thus, in infected cells, IN remains associated with the viral DNA from the time of its synthesis until its insertion into the cell genome. This IN/DNA complex turned out to be the most pertinent target for IN inhibitors as demonstrated by the potency of the specific strand transfer inhibitors.
Earlier, we have shown that 11-mer oligonucleotides conjugated with hydrophobic molecules inhibit the catalytic activity of recombinant HIV-1 IN.8,9 These compounds may be considered as promising IN inhibitors due to their capacity to interact with and to disrupt IN complex with DNA.8 To address the role of the hydrophobic moiety, we have tested the inhibitory activity of the oligonucleotide GGTTTTTGTGT (11D) conjugated with such molecules as acridine8 or xanthenes [fluorescein, tetramethylrhodamine (TAMRA), and eosin] as well as cholesterol, thiochrome, and oleic acid (Ole).9 Conjugates with xanthenes were found to be the most active inhibitors. In addition, fluorescein replacement by eosin (tetrabromofluorescein) resulted in a 6-fold increase of the inhibitory effect, thus demonstrating the importance of substituents in xanthenes. Interestingly, pyranodipyrimidines having a scaffold similar to xanthenes were reported as efficient integration inhibitors.10 Although the oligonucleotide sequence of the conjugates was found to have a limited influence on their action, shortening the oligonucleotide length led to a strong decrease of their inhibitory potency.9 Altogether, these results have shown that both parts of the conjugates strongly influence their inhibitory effect. Of note, the anti-IN activity has been also found for conjugates of various modified dinucleotides and intercalators, with the inhibition potency also depending on both parts of the compounds.11
In this study, we carried out step-by-step modifications of both oligonucleotide and hydrophobic parts of the oligonucleotide conjugates and tested the influence of these modifications on the inhibitory effect. This structure–activity relationship (SAR) analysis was aimed at the determination of the structural features of the conjugates responsible for IN inhibition to optimize the inhibitor structure and the method of its synthesis.
As previously shown,8 the binding of the conjugates to the IN/DNA complex and its subsequent disruption leads to a strong inhibition of the 3′-processing. Thus, the conjugates potency was monitored by quantifying their capability to inhibit this reaction. We have earlier shown that the inhibitory activity of the oligonucleotide conjugates does not depend on whether the hydrophobic moiety is attached to the 3′- or the 5′-end.9 Nevertheless, to choose an optimal method for the hydrophobic molecule attachment to the oligonucleotide 11D, we studied the influence of the linkage between eosin and fluorescein and 11D on the conjugate inhibitory activity (Figure 1). Eosin was attached to the 3′-end of 11D either through a flexible linker (11D-E) or directly to the 2′-amino group in the terminal nucleoside (11D-E2). Fluorescein was attached to the 5′-end similarly to eosin (FAM2-11D) or through a more long and rigid hydroxyprolinol moiety (FAM1-11D). Conjugates containing the same hydrophobic moiety showed comparable inhibitory potency; IC50 was about 50 nM for 11D-E and 11D-E2 and 300 nM for 11D-FITC, FAM1-11D, and FAM2-11D. Therefore, the structure of the linkage between both parts of the conjugates does not affect their capability to inhibit HIV-1 IN.
Figure 1.

Structures of residues conjugated to 2′-deoxy- (11D) and 2′-O-methyl- (11M) oligonucleotides.
The synthesis of the most efficient inhibitor 11D-E is rather laborious and tedious, thus interfering with its further development for pharmacological use. Eosin is destroyed during oligonucleotide deprotection under basic conditions, and as a result, it cannot be coupled to oligonucleotides during the automatic synthesis. Therefore, an additional step of eosin coupling to 3′-amino oligonucleotides followed by HPLC purification is obligatory for the conjugate synthesis. To avoid this additional step, we replaced eosin by a set of xanthenes that can be easily attached to oligonucleotides during automatic synthesis (Figure 1) and then tested the ability of these novel conjugates to inhibit IN. Because the mode of the hydrophobic molecule and 11D joining does not affect the anti-IN activity, we used corresponding 5′-modifying agents to simplify the conjugate synthesis and purification. Thus, most of the compounds were attached at the 5′-end of 11D. The SARs in these new compounds were investigated by coupling xanthene derivatives with different substituents such as 6-carboxyfluorescein (FAM), 6-carboxy-4,7,2′,7′-tetrachlorofluorescein (TET), 6-carboxy-4,7,2′,4′,5′,7′-hexachlorofluorescein (HEX), 2,4,5,7-tetrachloro-3,6-dihydroxy-9-(2′,5′-dichloro-4′,6′-dicarboxyphenyl)acridine (HEXAcr),12 and 4′,5′-dichloro-2′,7′-dimethoxyfluorescein (JOE) to 11D (Figure 1). Additionally, conjugates containing 3′-oleic acid (Ole) were synthesized by using CPG support with attached oleic acid.9
All of these conjugates were tested for their ability to inhibit the IN catalytic activity in 3′-processing and compared to the conjugate containing eosin (Table 1). Complete substitution of bromine by chlorine on the triple ring fragment had no influence on the conjugate inhibitory activity. Indeed, conjugates with HEX and HEXAcr showed the same inhibitory potency as the 11D-E compound. A change in the triple ring fragment resulting from the oxygen substitution by nitrogen (HEX-11D → HEXAcr-11D) did not affect the inhibitory activity. Compounds containing TET and JOE showed a 6-fold decrease in 3′-processing inhibition as compared to 11D-E and demonstrated the same ability to inhibit IN as FAM-11D. Therefore, the presence of two chlorine atoms is not sufficient to improve the inhibitory potency. Moreover, we may conclude that the location of chlorine atoms has no influence on the IN inhibition; only the number of halogen atoms on the triple ring fragment is important. Continuing SAR studies of the hydrophobic groups, we studied conjugates of the oligonucleotide 11D with two hydrophobic molecules. The simultaneous presence of fluorescein and oleic acid in the conjugates significantly improved their inhibitory potency when compared to the monosubstituted compounds (Table 1, compare compounds FAM-11D-Ole and 11D-FAM-Ole with FAM-11D and 11D-Ole). This effect, which did not depend on the fluorescein location, might result from an enhanced hydrophobicity of the bisubstituted compounds.
Table 1. Effect of the Hydrophobic Residues Conjugated to the Oligonucleotide GGTTTTTGTGT on the Inhibition of 3′-Processing by the Conjugate.
| modification | 3′-processing IC50 (nM)a |
|---|---|
| 11D-3′-E | 50 ± 7 |
| 5′-FAM1-11D | 320 ± 50 |
| 5′-FAM2-11D | 310 ± 50 |
| 11D-3′-FITC | 315 ± 50 |
| 5′-HEX-11D | 55 ± 10 |
| 5′-HEXAcr-11D | 52 ± 8 |
| 5′-TET-11D | 290 ± 40 |
| 5′-JOE-11D | 310 ± 35 |
| 11D-3′-Ole | 200 ± 25 |
| 5′-FAM-11D-3′-Ole | 20 ± 5 |
| 11D-3′-FAM-Ole | 23 ± 4 |
The conjugate inhibitory activity was tested in a standard assay by using the recombinant Mg2+-competent IN (50 nM) and 21-mer DNA duplex (3 nM) mimicking U5 sequence of viral DNA. The change in the amount of processed substrate in the presence of increasing conjugate concentrations was monitored by polyacrylamide gel electrophoresis analysis, and a concentration dependency curve was constructed for each inhibitor and used for the calculation of the IC50 value.
We have earlier shown that the shortening of the oligonucleotide part in the FITC-containing conjugates reduces their inhibitory potency.10 Here, we synthesized conjugates of eosine with oligodeoxynucleotides GG(T)n, where n = 8–13, and tested their inhibitory effect. A conjugate of the 10-mer turned out to be twice less efficient than the 11-mer compound, whereas conjugates of longer oligonucleotides showed about the same level of 3′-processing inhibition (Figure 2). We can therefore conclude that the 11-mer oligonucleotides have the optimal length to act as the efficient inhibitor.
Figure 2.

Effect of the oligonucleotide length on the anti-IN activity of oligodeoxynucleotides GG(T)n 3′-linked with eosine.
A decrease of the conjugate inhibitory potency resulted from the oligonucleotide shortening as well as a weak effect of the oligonucleotide sequence on the inhibitory activity10 allowed us to hypothesize that the main role of the oligonucleotide fragment relies on the electrostatic contacts with Lys and Arg residues of IN. To check this assumption, we changed the oligonucleotide charge without altering its length by replacing phosphodiester groups with methylphosphonates (Figure 3). Three conjugates were synthesized containing three methylphosphonates at different positions: near 3′-end, near 5′-end, and uniformly distributed along the oligonucleotide chain (Table 2, compounds 11DX-E). All of the conjugates inhibited IN approximately 2-fold less efficiently than 11D-E. Thus, the removal of three negative charges produced the same effect as the removal of one nucleotide bearing one negative charge (Figure 2). This indicates that the total amount of nucleotides, but not only the charge, is important for the inhibitory activity. Moreover, the same activity of conjugates 11DX-E-1, 11DX-E-2, and 11DX-E-3 shows that the negative charge distribution is not important for interactions with IN. Furthermore, we replaced all or several phosphodiester groups with phosphorothioates (Figure 3) and found that this modification improved inhibitory activity (Table 2). This could be explained by both the increased localization of the negative charge on the sulfur atoms when compared to oxygens in phosphodiesters and the higher hydrophobic properties of phosphorothiates than those of phosphodiesters. Similarly to the methylphosphonate modification, localization of phosphorothioates along the oligonucleotide chain had no effect on the IN inhibition. Moreover, partial or total phosphodiester substitution by phosphorothioates resulted in the same IC50 values (Table 2).
Figure 3.

Modifications of the sugar–phosphate backbone.
Table 2. Effect of the Oligonucleotide Structure on the Inhibition of 3′-Processing by the Conjugate.
| modification | oligonucleotide 5′ → 3′ | 3′-processing IC50 (nM) |
|---|---|---|
| 11D-E | GGTTTTTGTGT-E | 50 ± 7 |
| 11DX-E-1 | GGTTTTTxGTxGTx-E | 100 ± 15 |
| 11DX-E-2 | GGTxTxTxTTGTGT-E | 110 ± 20 |
| 11DX-E-3 | GGTxTTxTTGTxGT-E | 100 ± 15 |
| 11DS-E-1 | GsGsTsTsTsTsTsGsTsGsTs-E | 18 ± 4 |
| 11DS-E-2 | GGTTTTTsGTsGTs-E | 20 ± 2 |
| 11DS-E-3 | GGTsTsTsTTGTGT-E | 22 ± 6 |
| 11DS-E-4 | GGTsTTTsTGTsGT-E | 24 ± 7 |
| HEX-11D | HEX-GGTTTTTGTGT | 55 ± 10 |
| HEX-11-ddR | HEX-(ddR)10T | 1200 ± 70 |
| HEX-11D-ddR-1 | HEX-(ddR)6TGTGT | 280 ± 35 |
| HEX-11D-ddR-2 | HEX-GGTT(ddR)6T | 250 ± 30 |
| HEX-11-PD | HEX-(PD)10T | 1300 ± 90 |
| HEX-11D-PD-1 | HEX-(PD)6TGTGT | 250 ± 25 |
| HEX-11D-PD-2 | HEX-GGTT(PD)6T | 310 ± 45 |
| 11M-E | mGmGmUmUmUm-UmUmGmUmGmU-E | 48 ± 8 |
| HEX-11M | HEX-mGmGmUmUmUm-UmUmGmUmGmU | 52 ± 10 |
| HEX-11MS | HEX-mGsmGsmUsmUsm-UsmUsmUsmGsmUsmGsmUs | 19 ± 4 |
| HEX-11MS-Ole | HEX-mGsmGsmUsmUsm-UsmUsmUsmGsmUsmGsmUs-Ole | 20 ± 3 |
Because we found that the oligonucleotide shortening significantly decreased the conjugate inhibitory potency, we continued the SAR studies of the oligonucleotide structure to determine the role of heterocyclic bases. A series of HEX conjugates containing 1,2-dideoxyribose (ddR) or 1,3-propanediol (PD) instead of 10 or 6 native nucleosides was synthesized (Table 2). These compounds had the same number of phosphates as HEX-11D, and in the case of 1,2-dideoxyribose modification, the conjugates retained the same length and rigid structure of the sugar–phosphate backbone. 1,3-Propanediol-containing compounds have a flexible structure. The absence of heterocyclic bases significantly reduced the inhibitory activity of the conjugates (Table 2). The substitution of six nucleotides by both 1,2-dideoxyribose and 1,3-propanediol resulted in the 5–6-fold increase of IC50, whereas the absence of 10 bases led to the 24-fold increase of IC50. Similarly to the modifications of the internucleotide phosphates described above, there was no correlation between the anti-IN activity and the localization of the modified residues in the oligonucleotide; compounds HEX-11D-ddR-1 and HEX-11D-ddR-2 as well as HEX-11D-PD-1 and HEX-11D-PD-2 demonstrated approximately the same effect (Table 2). Thus, the presence of heterocyclic bases is obligatory for the efficient inhibition of IN activity. They might be involved in interactions with positively charged amino acids, which are favored by the base accessibility in the single-stranded oligonucleotide. Interestingly, Arg and Lys may be involved in contacts with all of the nucleotide bases,13 and this fact can explain why the IN inhibition by the oligonucleotide conjugates is not sequence-specific.
2′-O-Methylation is widely used to increase the oligonucleotide resistance to nucleases in subsequent cellular experiments. Both eosin (11M-E) and Hex (HEX-11M) derivatives of 2′-OMe oligonucleotide have shown inhibitory activity identical to 2′-deoxy variants (Table 2), so we used them in further studies.
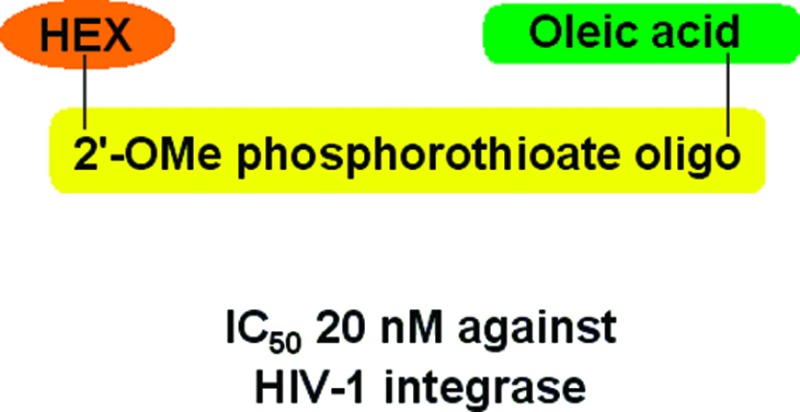
Encouraged by the improved potency of the conjugates containing phosphorothioates and bisubstituted compounds, we synthesized a conjugate of 2′-O-methyl-oligonucleotide GGUUUUUGUGU containing all phosphorothioate internucleotide linkages (11MS) with HEX at the 5′-end (HEX-11MS) and another one with additional 3′-oleic acid (HEX-11MS-Ole) (Figure 4A). Inhibition of 3′-processing by HEX-11MS-Ole in the typical assay is shown at Figure 4B. Surprisingly, the inhibitory activity of both compounds was comparable (Table 2). Thus, attachment of oleic acid to the efficient inhibitor HEX-11MS did not further improve its potency, while its attachment to a middling inhibitor FAM-11D (IC50 about 300 nM) provided more than 10-fold enhancement of 3′-processing inhibitory activity (Table 2, FAM-11D-Ole). Similarly, replacement of all internucleotide phosphodiesters with phosphorothioates (compound 11DS-E-1) did not additionally increase the inhibitory potency when compared to conjugates containing only 3 phosphorothioates (Table 2). It should be noted that IC50 for the best of our inhibitors is not less than 20 nM. Considering the fact that it takes an IN dimer to accomplish a 3′-processing14 and that the IN concentration used is about 50 nM, we can assume that the concentration of the catalytically active IN dimers does not exceed 20–25 nM and thus corresponds to IC50 for the best of our inhibitors. It allows us to hypothesize that, to be active, the conjugate must bind all IN subunits to prevent their interactions with the substrate DNA.
Figure 4.

HEX-11MS-Ole - a lead inhibitor of HIV-1 integrase. (A) Structure of HEX-11MS-Ole. (B) Dependence of the relative efficiency of 3′-processing on the concentrations of HEX-11MS-Ole. Reaction efficiency in the absence of the inhibitor is considered as 100%.
In conclusion, our SAR studies of oligonucleotide conjugates with hydrophobic molecules indicate that (1) eosin can be replaced by HEX since HEX-containing conjugates show the same anti-IN activity, while their synthesis is more practical and efficient; (2) phosphorothioate oligonucleotides are favored for the conjugate preparation due to their enhanced inhibitory potency and higher stability to cell nucleases; (3) both internucleotide phosphates and nucleic bases are necessary; and (4) additional attachment of oleic acid to HEX-containing phosphorothioates does not enhance their inhibitory activity but might be useful to improve their cell penetration, distribution, and stability to nucleases.
Supporting Information Available
Experimental procedures and analytical data (HPLC and MALDI-MS) for all conjugated oligonucleotides. This material is available free of charge via the Internet at http://pubs.acs.org.
The work was supported by the CNRS (International Associated Laboratory—LIADOV), the Russian Foundation for the Basic Science (Grants 09-04-93108-CNRSL and 09-04-93112-CNRSL), grant from the President of Russian Federation (MK-4821.2011.4), and contract with the Russian Federation Government (16.512.11.2193).
Supplementary Material
References
- Pommier Y.; Johnson A. A.; Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discovery 2005, 4, 236–248. [DOI] [PubMed] [Google Scholar]
- De Clercq E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discovery 2007, 6, 1001–1018. [DOI] [PubMed] [Google Scholar]
- Marchand C.; Maddali K.; Metifiot M.; Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr. Top. Med. Chem. 2009, 9, 1016–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman F. D.; Craigie R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: Specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A.; Mizuuchi K.; Craigie R. HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [DOI] [PubMed] [Google Scholar]
- Farnet C. M.; Haseltine W. A. Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J. Virol. 1991, 65, 1910–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky M. I.; Sharova N.; Dempsey M. P.; Stanwick T. L.; Bukrinskaya A. G.; Haggerty S.; Stevenson M. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 6580–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinskaya M.; Romanova E.; Volkov E.; Deprez E.; Leh H.; Brochon J. C.; Mouscadet J. F.; Gottikh M. HIV-1 integrase complexes with DNA dissociate in the presence of short oligonucleotides conjugated to acridine. Biochemistry 2004, 43, 8735–8743. [DOI] [PubMed] [Google Scholar]
- Prikazchikova T. A.; Volkov E.; Zubin E. M.; Romanova E.; Gottikh M. HIV-1 integrase inhibition by modified oligonucleotides: optimization of the inhibitor structure. Mol. Biol. (Moscow) 2007, 41, 130–138. [PubMed] [Google Scholar]
- Pannecouque C.; Pluymers W.; Van Maele B.; Tetz V.; Cherepanov P.; De Clercq E.; Witvrouw M.; Debyser. Z. New class of HIV integrase inhibitors that block viral replication in cell culture. Curr. Biol. 2002, 12, 1169–1177. [DOI] [PubMed] [Google Scholar]
- Aubert Y.; Chassignol M.; Roig V.; Mbemba G.; Weiss J.; Meudal H.; Mouscadet J.-F.; Asseline U. Synthesis and anti-HIV-1 integrase activity of modified dinucleotides. Eur. J. Med. Chem. 2009, 44, 5029–5044. [DOI] [PubMed] [Google Scholar]
- Chuvilin A. N.; Serebryakova M. V.; Smirnov I. P.; Pozmogova G. E. Byproduct with altered fluorescent properties is formed during standard deprotection step of hexachlorofluorescein labeled oligonucleotides. Bioconjugate Chem. 2009, 20, 1441–1443. [DOI] [PubMed] [Google Scholar]
- Lejeune D.; Delsaux N.; Charloteaux B.; Thomas A.; Brasseur R. Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins 2005, 61, 258–271. [DOI] [PubMed] [Google Scholar]
- Faure A.; Calmels C.; Desjobert C.; Castroviejo M.; Caumont-Sarcos A.; Tarrago-Litvak L.; Litvak S.; Parissi V. HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 2005, 33, 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.