

Abstract

We report the identification of 13 (INCB3284) as a potent human CCR2 (hCCR2) antagonist. INCB3284 exhibited an IC50 of 3.7 nM in antagonism of monocyte chemoattractant protein-1 binding to hCCR2, an IC50 of 4.7 nM in antagonism of chemotaxis activity, an IC50 of 84 μM in inhibition of the hERG potassium current, a free fraction of 58% in protein binding, high selectivity over other chemokine receptors and G-protein-coupled receptors, and acceptable oral bioavailability in rodents and primates. In human clinical trials, INCB3284 exhibited a pharmacokinetic profile suitable for once-a-day dosing (T1/2 = 15 h).
Keywords: CCR2, chemokine, antagonist, hERG, oral absorption
Macrophages, which are derived from monocytes, a subset of leukocytes, are well-characterized mediators of tissue destruction. These cells can be activated to secrete proinflammatory cytokines such as TNFα and IL-1β, tissue-degrading enzymes such as MMPs, and other chemokines that mediate the influx of other inflammatory cells.1 Excessive recruitment of these cells to sites of inflammation leads to significant tissue destruction and contributes to the morbidity of chronic inflammatory and autoimmune diseases. The trafficking of monocytes/macrophages to sites of inflammation is believed to be predominantly mediated by monocyte chemoattractant protein-1 (MCP-1, CCL2) through interaction with its specific receptor, CCR2, which is a member of the super family of seven-transmembrane G-protein-coupled receptors (GPCRs) and is predominantly expressed on monocytes. Binding of MCP-1 to CCR2 induces chemotaxis, resulting in directed migration of monocytes/macrophages to disease sites where MCP-1 expression is elevated.2 Studies in rodent models have demonstrated the critical role of MCP-1/CCR2 in inflammatory and autoimmune diseases and strongly suggest that CCR2 is an attractive therapeutic target.3 As a result, inhibition of CCR2 has emerged as a novel therapeutic approach for pharmaceutical research, and a number of potent small molecule CCR2 antagonists have been identified.4−10
We have reported the discovery of a novel series of CCR2 antagonists through rational design.10 Our structure−activity relationship (SAR) studies on that series of compounds led to the identification of INCB3344 (Figure 1), a potent, selective, and orally bioavailable antagonist of human and murine CCR2 (hCCR2 and mCCR2). INCB3344 has been used as a tool compound for target validation in rodent models because of its potent inhibitory activity toward murine CCR2, its selectivity over other homologous chemokine receptors, and its good pharmacokinetics profile but was not suitable as a clinical candidate due to its moderate hERG activity (IC50 = 13 μM) as assessed using a dofetilide binding assay, which did not meet our criteria in hERG binding activity. In addition, INCB3344 was an inhibitor of CYP3A4.
Figure 1.

Structure of INCB3344.
In the course of SAR studies on the INCB3344 series in an attempt to identify a clinical candidate, we discovered that removal of the ethoxy at the 3-position on the pyrrolidine in the INCB3344 series as in 1 (Figure 2) resulted in a significant loss in mCCR2 activity but retained the hCCR2 activity as in 2 (Figure 2). An R configuration at the 3-position on the pyrrolidine as shown in 2 is required for activity as the S enantiomer of 2 displayed an IC50 of >1 μM in hCCR2 MCP-1 assay. Although this des-ethoxy series is not superior to the INCB3344 series by comparison of 2 with 1 in hCCR2 activity and hERG binding activity, the 1,3-disubstituted pyrrolidine core in this series offers an advantage over the 1,3,4-trisubstituted pyrrolidine core in the INCB3344 series from a synthetic point of view as the former core structure, (R)-3-aminopyrrolidine, has only one chiral center and is commercially available, while the latter core structure, trans-4-amino-3-ethoxypyrrolidine, has two chiral centers and is not commercially available. These considerations prompted us to take 2 as a lead compound for modifications.
Figure 2.

Like INCB3344, 2 exhibited moderate dofetilide hERG binding activity with an IC50 of 13.2 μM, which exceeded our criteria. Because 2 has a cLogP of 4.08, we reasoned that the moderate hERG binding activity of 2 could be ascribed to the hydrophobicity of the molecule. While the impact on CCR2 binding could not be predicted, if the cLogP is decreased, it is likely that the hERG activity could be reduced,11,12 The high cLogP of 2 is attributed to the two hydrophobic phenyl rings on the left-hand and right-hand sides. Our previous SAR studies had demonstrated that the trifluoromethylphenyl residue on the right-hand side is critical to the CCR2 binding affinity. For this reason, we targeted the phenyl residue on the left-hand side for modifications in an attempt to increase the polarity of the molecule. cLogP calculations revealed that the cLogP of 2 can be dramatically lowered down if the phenyl residue on the left-hand side is replaced with a heteroaromatic ring such as a pyridyl (cLogP = 2.09). Thus, SAR studies at the 4-position of the cyclohexyl were carried out by replacing the phenyl ring with different heteroaryl rings. The heterocycles that we investigated include pyridine, pyrimidine, pyridazine, and pyrazine. As shown in Table 1, the three pyridyl regioisomers 3−5 displayed similar hCCR2 activity and, more importantly, were very potent hCCR2 antagonists with low nanomolar activities in the MCP-1 and chemotaxis assays, demonstrating the tolerability of a polar heteroaromatic ring at the 4-position of the cyclohexyl ring for hCCR2 activity. As expected, these polar analogues exhibited weak hERG binding activity, with an IC50 of >30 μM, meeting our criteria for advancing to mouse pharmacokinetic (PK) screens. When administered orally to mice, the pyridin-2-yl analogue 3 was well absorbed, with a high AUC of 2812 nM·h at a dose of 10 mg/kg, while the pyridin-3-yl analogue 4 and the pyridine-4-yl analogue 5 exhibited very low blood levels at the same dose. The excellent PK profile of 3 prompted us to conduct a patch clamp assay. Disappointingly, it exhibited 57% inhibition of the hERG potassium current at a concentration of 10 μM (IC50 < 10 μM), which did not meet our criteria (IC50 > 10 μM) in the hERG patch clamp activity for a clinical candidate.
Table 1. Heteroaryl Analogues 3−14.
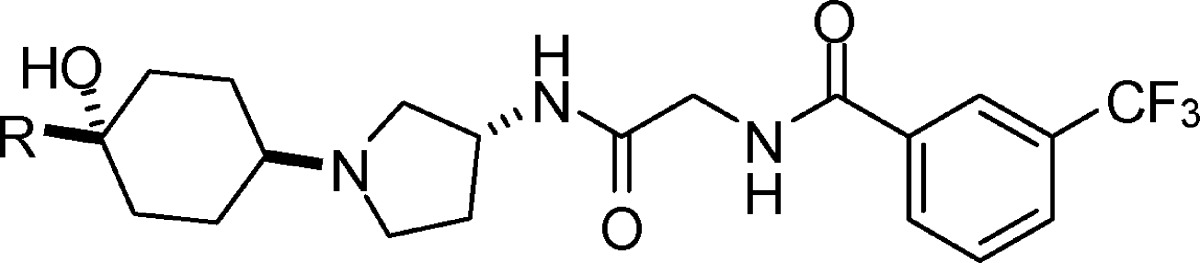
| IC50 (nM) |
||||||
|---|---|---|---|---|---|---|
| compd | R | MCP-1a,b | CTXa,c | hERG bindingd | patch clamp at 10 μMe (%) | mouse oral AUCf (nM·h) |
| 3 | pyridin-2-yl | 3.2 (10) | 3.2 (8) | >30 | 57 | 2812 |
| 4 | pyridin-3-yl | 2.6 (16) | 3.9 (12) | >30 | 50 | |
| 5 | pyridin-4-yl | 4.3 (2) | 3.0 (2) | >30 | 100 | |
| 6 | pyrimidin-2-yl | 10.3 (10) | 9.3 (9) | >30 | 25 | 1442 |
| 7 | pyrimidin-4-yl | 7.8 (22) | 7.5 (30) | >30 | 7 | 930 |
| 8 | pyrimidin-5-yl | 14 (2) | 20 (2) | |||
| 9 | pyridazin-3-yl | 7.3 (8) | 5.8 (6) | >30 | 155 | |
| 10 | pyridazin-4-yl | 4.9 (4) | 7.2 (2) | >30 | 80 | |
| 11 | pyrazin-2-yl | 10.9 (22) | 10.4 (30) | >30 | 7 | 1550 |
| 12 | 6-Me-pyridin-3-yl | 2.4 (2) | 5.6 (2) | >30 | 127 | |
| 13 | 6-MeO-pyridin-3-yl | 3.7 (30) | 4.7 (30) | >30 | 7.7 | 617 |
| 14 | 6-EtO-pyridin-3-yl | 3.2 (10) | 2.7 (6) | >30 | 287 | |
Averaged values were given from numbers of determinations indicated in parentheses. Standard deviations are within 30% of the measured value. For assay protocols, see ref (10).
Antagonism of MCP-1 binding to hCCR2.
Antagonism of chemotaxis activity.
Dofetilide hERG binding activity from two determinations.
Inhibition of hERG potassium current at 10 μM from single determination of patch clamp assay.
Normalized mouse oral AUC at 10 mg/kg from cassette studies (three animals each cassette).
By comparison with the pyridyl analogues 3−5, the two-nitrogen-containing heteroaryl analogues 6−11 displayed slightly weaker hCCR2 activity in both the MCP-1 and the chemotaxis assays (Table 1). The pyrimidin-5-yl analogue 8 was not potent enough in CCR2 activity, while the pyrimidin-2-yl (6) and pyrazin-2-yl (11) analogues were just potent enough to meet our criteria (IC50 < 10 nM in MCP-1 and chemotaxis assays) for further profiling. Because compounds 6 and 7 and 9−11 exhibited weak dofetilide hERG binding activity (IC50 > 30 μM), they were subjected to PK screens in mice. High plasma levels were observed for the two pyrimidine compounds 6 and 7 and the pyrazine compound 11, while the two pyridazine compounds 9 and 10 were poorly absorbed in mice. When subjected to patch clamp assays, strikingly, 6, 7, and 11 exhibited weak inhibition of the hERG potassium current. Of special note were 7 and 11, which displayed only 7% inhibition at a concentration of 10 μM in the hERG patch clamp assay, representing the first two compounds from this series with good oral absorption in mice concurrent with weak inhibition (<10% at 10 μM) of the hERG potassium current.
To obtain an insight into the huge difference in oral exposure between the 2-pyridyl analogue 3 and the 3-pyridyl analogue 4, ADME studies were carried out on 3 and 4 and revealed that 4 had a much lower permeability and more profound N-oxidation at the pyridyl nitrogen than 3 (data not shown). We reasoned that these differences could be ascribed to the steric effect on the pyridyl nitrogen. The pyridyl nitrogen in 3 is sterically shielded by the cyclohexyl residue. In contrast, the pyridyl nitrogen in 4 is more exposed with a likely impact on transcellular permeation and an increased susceptibility to oxidation as compared to the 2-pyridyl nitrogen. To shield the pyridyl nitrogen in 4 from solvation and oxidation, we introduced a methyl group at the 6-position of the pyridyl to provide 12. This modification maintained the hCCR2 potency and the weak hERG binding activity but resulted in a little improvement in mouse oral exposure, with only 2.5-fold enhancement in the area under the curve (AUC). It is likely that the methyl group is not bulky enough to shield the pyridyl nitrogen from solvation and oxidation. In contrast, introduction of a methoxy group at the 6-position of the pyridyl in 4 to provide a less basic pyridyl residue not only maintained the hCCR2 potency and the weak dofetilide binding activity but improved the oral exposure from an AUC of 50 nM·h at 10 mg/kg in 4 to an AUC of 617 nM·h at the same dose in 13, a 12-fold enhancement. When subjected to a patch clamp assay, remarkably, compound 13 exhibited only 7.7% inhibition of the hERG potassium current at a concentration of 10 μM, representing another potent CCR2 antagonist with acceptable oral exposure in mice concurrent with weak hERG activity (<10% at 10 μM) in the patch clamp assay. Increasing the length of the alkoxy group in 13 by replacing the methoxy with ethoxy (14) maintained the hCCR2 activity but led to a lower blood level (AUC = 287 nM·h at 10 mg/kg) in mice.
Given its superior profile in a combination of hCCR2 potency, mouse oral absorption, and hERG patch clamp activity, compound 13 (INCB3284) was selected for further in vitro and in vivo evaluations. INCB3284 is a potent hCCR2 antagonist with IC50 values of 3.7 nM in antagonism of MCP-1 binding to hCCR2 and 4.7 nM in antagonism of chemotaxis activity. It potently inhibited CCR2-mediated signaling events such as intracellular calcium mobilization and ERK phosphorylation with IC50 values of 6 and 2.6 nM, respectively. Cerep screens revealed that INCB3284 is a selective CCR2 inhibitor, showing no significant inhibitory activity at a concentration of 1 μM when tested against a panel of >50 ion channels, transporters, chemokine receptors including CCR1, CCR3, CCR5, CXCR3, and CXCR5, and additional GPCRs. In the hERG patch clamp assay, INCB3284 inhibited hERG potassium current with an IC50 of 84 μM. In protein binding, INCB3284 had a high free fraction (58%) in human serum at concentrations of 1 and 10 μM. When incubated with human liver microsomes, INCB3284 exhibited a moderate intrinsic clearance. However, studies with recombinant CYP isozymes showed that INCB003284 is a substrate for CYP3A4 and CYP2D6. Recombinant CYP3A4 metabolized INCB003284 at the C−N bond between the pyrrolidine nitrogen and the cyclohexyl carbon to its N-dealkylated metabolite, while recombinant CYP2D6 metabolized INCB3284 at the C−O bond of the methoxy group between the methyl and the 6-hydroxypyridyl oxygen to the des-methyl metabolite. When INCB3284 was incubated with human S9 with or without NADPH and the cofactor glutathione, no glutathione adducts were detected. INCB003284 is not a CYP inhibitor, with IC50 values of >25 μM against five major CYP isozymes CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. INCB3284 is not a CYP inducer at concentrations up to 10 μM as measured in the luminometric luciferase assay.
The pharmacokinetics of INCB3284 was investigated in rats, dogs, cynomolgus monkeys, and chimpanzees (Table 2). Following iv administration of INCB3284, the total systemic clearance was high in rats but low in dogs, cynomolgus monkeys, and chimpanzees. The apparent steady state volume of distribution (Vss) followed the same trend as in clearance, with high Vss in rats and low Vss in dogs, cynomolgus monkeys, and chimpanzees. As a result, INCB003284 had a short to moderate half-life after iv administration ranging from 2.2 to 3.8 h. When administered orally, a similar half-life was observed in rats and dogs, while slightly longer half-life was achieved in monkeys and chimpanzees. The oral bioavailability ranged from 13% in cynomolgus monkeys to 31% in dogs.
Table 2. Pharmacokinetic Parameters of INCB003284.
| rats | dogs | monkeys | chimpanzees | ||
|---|---|---|---|---|---|
| iv (two animals in each species) | dose (mg/kg) | 5 | 5 | 5 | 1 |
| CL (L/h/kg) | 3.4 ± 0.3 | 0.57 ± 0.02 | 0.82 ± 0.04 | 0.35 ± 0.01 | |
| Vss | 10.5 ± 0.5 | 1.6 ± 0.1 | 0.7 ± 0.2 | 0.94 ± 0.03 | |
| T1/2 (h) | 2.8 ± 0.2 | 3.5 ± 0.1 | 2.2 ± 0.3 | 3.8 ± 0.2 | |
| po (two animals in each species) | dose (mg/kg) | 10 | 30 | 30 | 4 |
| Cmax (nM) | 573 ± 120 | 7813 ± 560 | 2633 ± 320 | 1122 ± 750 | |
| Tmax (h) | 4.0 ± 0.6 | 1.5 ± 0.5 | 1.5 ± 0.3 | 2.0 ± 0 | |
| AUC (nM·h) | 3657 ± 560 | 33,283 ± 1215 | 9078 ± 980 | 4942 ± 3388 | |
| T1/2 (h) | 2.9 ± 0.3 | 3.3 ± 0.7 | 5.1 ± 0.5 | 5.3 ± 1.0 | |
| F % | 20 | 31 | 13 | 23 |
As outlined in Scheme 1, this series of compounds were synthesized starting from the commercially available (R)-1-benzyl-3-aminopyrrolidine 15. Coupling of 15 with the glycine derivative 16 using isobutyl chloroformate followed by removal of the benzyl group by hydrogenation produced the pyrrolidine intermediate 18. Reductive amination of 18 with 4,4-disubstituted cyclohexanone 19 using sodium triacetoxyborohydride as the reducing agent provided the final compounds as a mixture of two isomers with a ratio of about 3:2. The two isomers were separated by silica gel chromatography. The major isomers were characterized by NMR to be trans isomers with the hydroxyl at the 4-position on cyclohexyl trans to the pyrrolidin-1-yl residue at the 1-position on the cyclohexyl and were the active isomers 3−14. The cis isomers were inactive (<50% inhibition) at a concentration of 1 μM in the MCP-1 assay. The stereochemistry at the cyclohexyl was further confirmed by X-ray crystallography on the tartaric acid salt of 3. The X-ray crystal structure revealed that the cyclohexyl assumes a chair conformation with pyridin-2-yl at the equatorial position and hydroxyl and pyrrolidin-1-yl at the axial positions. There is no intramolecular hydrogen bond formed between the hydroxyl proton and the pyridyl nitrogen in the crystal structure as the hydroxyl group and the pyridyl nitrogen are situated at the opposite directions on the cyclohexane plane.
Scheme 1. Synthesis of Compounds 3−14.

Reagents and conditions: (a) Isobutyl chloroformate, NMM, THF, 76%. (b) H2, Pd(OH)2, MeOH, 95%. (c) Na(OAc)3BH, THF.
A general route to the synthesis of the ketone intermediate 19 is presented in Scheme 2A. Lithiation of a (un)substituted bromoheterocycle (R-Br, 20) using n-butyl lithium at −78 °C followed by addition of 1,4-cyclohexanedione mono-ethylene ketal 21 gave rise to the ketal intermediate 22. Treatment of 22 with aqueous HCl converted the ketal to the corresponding ketone 19. For some of the heterocycles, bromine-substituted heterocycles were not commercially available or lithiation of bromoheterocycles did not produce the desired products. For these reasons, alternative synthetic routes were developed as shown in Scheme 2B−D. The 4-hydroxy-4-(pyrimidin-4-yl)cyclohexanone 19a and 4-hydroxy-4-(pyrazin-2-yl)cyclohexanone 19b were prepared from 5-bromopyrimidine 23 (Scheme 2B) and 2,6-dichloropyrazine 25 (Scheme 2C), respectively. Treatment of 23 or 25 with lithium diisopropylamide (LDA) followed by quenching with 21 provided the bromine-containing ketal 24 or the chlorine-containing ketal 26. Hydrogenation in the presence of triethylamine to remove the bromine in 24 or chlorine in 26 followed by acid treatment to hydrolyze the ketal furnished the ketones 19a and 19b. The two pyridazine derivatives 19c and 19d were prepared from pyridazine 27 (Scheme 2D). Treatment of 27 with lithium 2,2,6,6-tetramethylpiperidinide followed by addition of 21 gave rise to a mixture of two regioisomers 28a and 28b. After separation by chromatography, the two isomers were treated with acid to provide the corresponding ketones 19c and 19d.
Scheme 2. Synthesis of Ketones.

Reagents and conditions: (a) n-Butyllithium, THF, −78 °C. (b) (1) 2 N HCl, H2O, THF; (2) Na2CO3, pH 8. (c) LDA, THF, −78 °C, 21, 20% yield for 24, 25% yield for 26. (d) H2, Pd/C, Et3N, MeOH. (e) Lithium 2,2,6,6-tetramethylpiperidinide, THF, −78 °C, 21, 12% yield for 28a, 10% yield for 28b.
The reductive amination to produce INCB3284 from its corresponding ketone 19e and the pyrrolidine intermediate 18 have been investigated extensively. It was discovered that the 3:2 ratio of the trans isomer (INCB3284) versus the cis isomer obtained using sodium triacetoxyborohydride as the reducing agent could be improved to 4:1 when the reductive amination was conducted by hydrogenation in the presence of aluminum oxide using palladium on carbon as the catalyst. Without chromatography, the trans isomer (INCB3284) could be enriched to a ratio of trans:cis = 98:2 by crystallization of the crude product with methanesulfonic acid. The crystalline salt obtained was basified with NaOH, and the resulting free base was subjected to another crystallization using methanesulfonic acid, affording pure crystalline salt of INCB3284 bismesylate (Scheme 3).
Scheme 3. Synthesis of INCB3284 Bismesylate.

Reagents and conditions: (a) H2, Pd/C, Al2O3, CH2Cl2, trans:cis = 4:1. (b) 2 × MeSO3H, EtOH, EtOAc, trans:cis = 98:2, 60% yield. (c) NaOH, CH2Cl2. (d) 2 × MeSO3H, EtOH, EtOAc, trans:cis = 99.8:0.2, 90% yield.
In summary, we took 2 as a lead compound for modifications in an attempt to optimize its moderate hERG activity. SAR studies were carried out on the left-hand side in 2 by replacing the phenyl ring with a variety of heteroaromatic rings, leading to the identification of 13 (INCB3284). INCB3284 exhibited a balanced profile in potent hCCR2 activity, high selectivity, weak hERG activity, high free fraction in protein binding, and acceptable oral bioavailability in rats, dogs, cynomolgus monkeys, and chimpanzees, meeting our criteria for a clinical candidate. The tolerated safety profile from GLP toxicology studies in rodents and primates justified its advancement into human clinical trials. Phase I and phase II clinical studies revealed that INCB3284 exhibited a PK profile suitable for once-a-day dosing (T1/2 = 15 h).
Acknowledgments
We thank Lynn Leffet, Karen Gallagher, Patricia Feldman, Bitao Zhao, Yanlong Li, Robert Collins, Xiaomei Gu, and Peng Pan for technical assistance.
Supporting Information Available
Experimental procedures for the synthesis of 13, characterization data for 1−14, and X-ray crystal structure of the tartaric acid salt of 3. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Kinne R. W.; Brauer R.; Stuhlmuller B.; Palombo-Kinne E.; Burmester G. Macrophages in rheumatoid arthritis. Arthritis Res. 2000, 2, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C. A.; Proudfoot A. E. The chemokine system: Novel broad-spectrum therapeutic targets. Curr. Opin. Pharmacol. 2001, 1, 417–424. [DOI] [PubMed] [Google Scholar]
- Feria M.; Diaz-Conzalez F. The CCR2 receptor as a therapeutic target. Expert Opin. Ther. Pat. 2006, 16, 49–57. [Google Scholar]
- Xia M.; Sui Z. Recent development in CCR2 antagonists. Expert Opin. Ther. Pat. 2009, 19, 295–303. [DOI] [PubMed] [Google Scholar]
- Pease J. E.; Horuk R. Chemokine receptor antagonists: Part I. Expert Opin. Ther. Pat. 2009, 19, 39–58. [DOI] [PubMed] [Google Scholar]
- Struthers M.; Pasternak A. CCR2 antagonists. Curr. Top. Med. Chem. 2010, 10, 1278–1298. [DOI] [PubMed] [Google Scholar]
- Pasternak A.; Goble S. D.; Struthers M.; Vicario P. P.; Ayala J. M.; Di Salvo J.; Kilburn R.; Wisniewski T.; DeMartino J. A.; Mills S. G.; Yang L. Discovery of a potent and orally bioavailable CCR2 and CCR5 dual antagonist. ACS Med. Chem. Lett. 2010, 1, 14–18. [Google Scholar]
- Cherney R. J.; Mo R.; Meyer D. T.; Voss M. E.; Yang M. G.; Santella J. B. III; Duncia J. V.; Lo Y. C.; Yang G.; Miller P. B.; Scherle P. A.; Zhao Q.; Mandlekar S.; Cvijic M. E.; Barrish J. C.; Decicco C. P.; Carter P. H. γ-Lactams as glycinamide replacements in cyclohexane-based CC chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 2425–2430. [DOI] [PubMed] [Google Scholar]
- Peace S.; Philp J.; Brooks C.; Piercy V.; Moores K.; Smethurst C.; Watson S.; Gaines S.; Zippoli M.; Mookherjee C.; Ife R. Identification of a sulfonamide series of CCR2 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3961–3964. [DOI] [PubMed] [Google Scholar]
- Xue C.-B.; Wang A.; Meloni D.; Zhang K.; Kong L.; Feng H.; Glenn J.; Huang T.; Zhang Y.; Cao G.; Anand R.; Zheng C.; Xia M.; Han Q.; Robinson D. J.; Storace L.; Shao L.; Li M.; Brodmerkel C. M.; Covington M.; Scherle P.; Diamond S.; Yeleswaram S; Vaddi K.; Newton R.; Hollis G.; Friedman S.; Metcalf B. Discovery of INCB3344, a potent, selective and orally bioavailable antagonist of human and murine CCR2. Bioorg. Med. Chem. Lett. 2010, 20, 7473–7478. [DOI] [PubMed] [Google Scholar]
- Aronov A. M. Predictive in silico modeling for hERG channel blockers. Drug Discovery Today 2005, 10, 149–155. [DOI] [PubMed] [Google Scholar]
- Farid R.; Day T.; Friesner R. A.; Pearlstein R. A. New insight about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.