

Introduction
We have seen the increased use of computational approaches to predict drug interactions with human transporters that affect drug disposition and may lead to toxicity. These predominantly ligand-based methods use limited experimental data but provide new insights into structure activity relationships (SARs). The promiscuity of ligand interaction with transporters represents a challenge to computational methods. Development of models capable of identifying new transport substrates and unwanted drug-drug interactions requires novel applications of current computational methods.
The clinical importance of efflux and uptake transporters in drug disposition is widely acknowledged [1–5] and membrane transporter anomalies are the basis for certain clinical disorders. Consequently, increasing attention is being paid to the potential for transport-based toxicity in vivo, and to unwanted drug-drug interactions [6–9] and consequences from polymorphic transport activity [10]. Furthermore, with hundreds of transporters yet to be characterized, the potential exists for many new drug targets to be discovered [8]. Computational models could therefore enable repurposing of already approved drugs [11] as well as predict the potential for, and ultimately preempt undesirable effects [12–14] that are based on drug-transporter interactions.
Where are we now?
Application of computational methods to the study of transporters has typically involved determining the extent to which test compounds inhibit in vitro uptake of a prototypical probe substrate. The test compounds might include a small number of well-characterized model compounds or, in some recent studies, some tens or hundreds of compounds [7, 9, 15, 16]. These data have been used occasionally to generate quantitative structure activity relationships (QSARs) [17, 18], pharmacophores [19–21] or other types of statistical models [8, 16, 22]. Such computational (or in silico) models have also been used for prospective prediction subsequently validated by in vitro testing [7, 9, 15, 23–29] or the use of additional data from literature case reports [6, 30]. However, the application of computational methods to generate models and SARs for transporters is at least a decade behind that of drug metabolizing enzymes, which has much larger datasets available both inside the pharmaceutical industry [31] and outside [32] (e.g. ChEMBL [33] and PubChem [34]). We have proposed through our transporter studies that if transporter research is to achieve parity with that of drug metabolizing enzymes, it will be through the judicious use of these in vitro and in silico (IVIS) approaches as a combined system.
The prevailing axiom for robust QSAR studies is ‘more is better.’ Accurate insight into the molecular determinants that define ligand-transporter relationship is likely to arise from analyses that employ large and structurally rich groups of test ligands (100’s of compounds). To that end, it would be helpful if data from different studies could be effectively combined. Unfortunately, results in different studies are frequently reported in different kinetic forms (e.g., Ki vs. IC50 vs. percent inhibition), so laboratories with an interest in even the same transporter may have difficulty in quantitatively using one another’s data. But an even greater issue is the variability of results obtained by different groups using similar methods. The bases of such differences are not clear, but differences in reported IC50 values for the inhibition of the same substrate by the same compound using the same experimental system frequently vary by as much as 10 to 100-fold [35], which makes pooling of data virtually impossible. Similarly Kt values (and other kinetic values) for the same compound can differ depending on the expression system used (Table 1). Although our own experience with OCT2 and MATE1 transport in both CHO and HEK293 cells has found no substantive difference in kinetics or selectivity for the same compound, a systematic study of this issue is lacking.
Table 1.
Variability of Kt values for hOCT2 with transfection system (data from [35] and our laboratories).
| Compound | Kt (µM) |
Transfection system |
Reference |
|---|---|---|---|
| Dopamine | 1400 | cc | [53] |
| 390 | oo | [54] | |
| Histamine | 940 | cc | [53] |
| 1300 | oo | [54] | |
| MPP | 19 | oo | [55] |
| 7.8 | cc | [56] | |
| 12 | cc | Unpublished | |
| 3.1 | cc | [57] | |
| 12.3 | Unpublished | ||
| 19.5 | cc | [58] | |
| Norepinephrine | 1500 | cc | [53] |
| 1900 | oo | [54] | |
| Serotonin | 290 | cc | [53] |
| 80 | oo | [54] | |
| TEA | 27 | cc | [59] |
| 76 | oo | [55] | |
| 109 | cc | [60] | |
| 20 | cc | [61] | |
| 46 | cc | [17] | |
cc = cultured cell (HEK293 or CHO), oo = oocyte, MPP = 1-methyl-4-phenylpyridinium, TEA = tetraethylammonium.
What will it take to expand modeling of ligand-transporter interaction?
We suggest that scaling up the current transporter research paradigm will involve a two-pronged approach. First is the development of high-throughput protocols that can interrogate the effect on activity of a target transporter of structurally rich cohorts of test compounds, thereby producing large, ‘internally consistent’ (i.e., comparable) data sets. We also suggest that, from a practical point of view, the efficiency of this approach will benefit from initial studies that use relatively small numbers of compounds to build an initial computational model that is then used to prioritize additional compounds for testing (Figure 1). This ‘bootstrapping’ approach described above can scale but it will need the modeling software to be available and require efforts for sharing QSAR models in a standardized fashion [36]. While there are efforts to standardize how data and models are stored, queried and exchanged, sharing of ‘open source’ transporter models that are created represents a challenge but appears feasible [37] and should be pursued.
Figure 1.
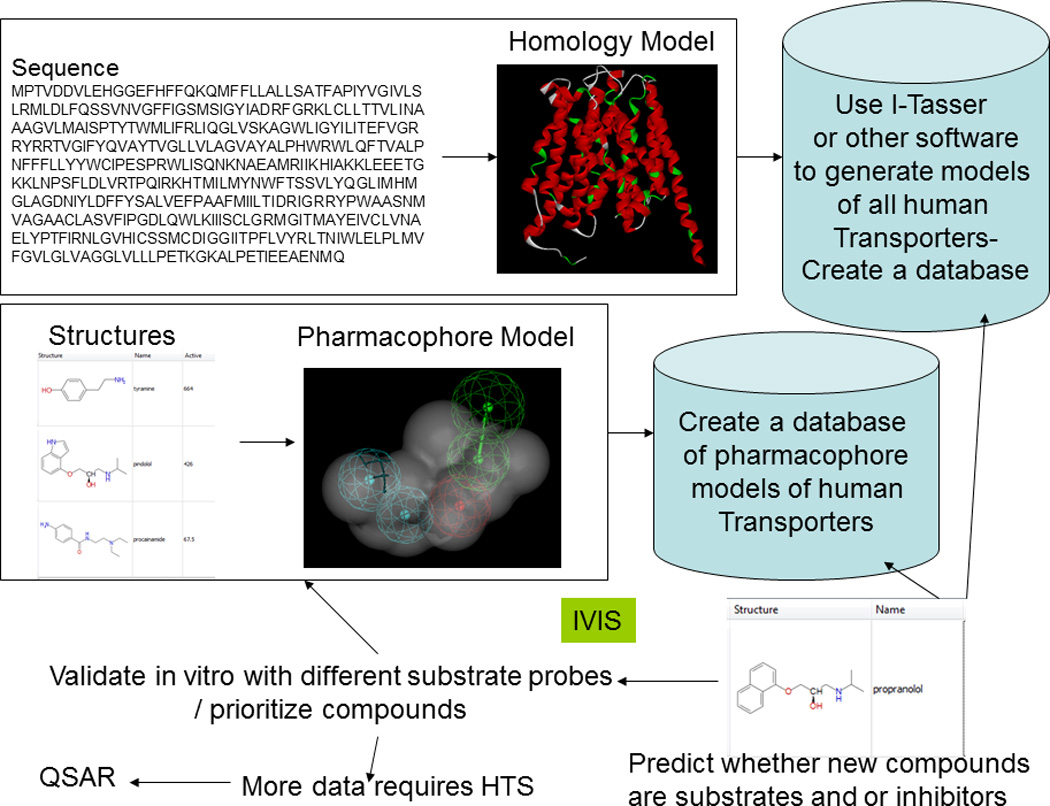
Using in vitro and in silico (IVIS) methods to predict whether a compound is a substrate or inhibitor of a human transporter.
We have previously described how computational transporter models tend to evolve [38] as the amount of inhibition data increases. Such evolution nominally follows the pathway of simple molecule alignments, pharmacophore, QSAR, machine learning models, protein simulation, homology (comparative) modeling, docking and ultimately X-ray crystallography. We are not suggesting that a crystal structure represents, by itself the final static conformation of a transporter due to the complexities of binding/ protein flexibility. P-gp required two decades to traverse this route. Clearly, we cannot afford this time span for every human transporter to be characterized. Especially when computational models for transporter substrates may have the added benefit of assisting in probe substrate identification and selection, in directing mutagenesis studies and in facilitating protein modeling efforts that, in combination, will accelerate our progress.
What can we do to predict transporter substrates?
As noted above, the current modeling approach typically involves assessing the degree of inhibition, produced by a set of test compounds, of substrate uptake by a target transporter. The focus on ‘inhibition’ reflects the fact that (i) it is easy to measure, and (ii) there are comparatively few substrates in which the uptake can be conveniently measured (e.g. via scintillation counting). There are few examples where both substrate and inhibitor models have been generated for a single transporter (e.g., [6, 39, 40]). Our early work on P-gp is perhaps an exception [20] and these models have continued to be used by us to make predictions [25, 41]. Can we differentiate between inhibitors and substrates for transporters using such models? The answer (in at least some cases) appears to be yes. An example is our recent work on hOCTN2 inhibitor pharmacophores [7, 23]; all had at least one hydrophobic feature, whereas the separately developed hOCTN2 substrate pharmacophore has none [6]. These differences between OCTN2 substrate and inhibitor pharmacophores may point to important interactions on the protein that differentiate substrates and inhibitors. In turn, these models may be useful for targeting transporters, for example, by defining which L-carnitine mimics are transported and with what affinity. Currently, the database of human OCTN2 substrates is very small [6] so an exhaustive SAR analysis is not possible. But these early stage models could enable us to search small molecule databases such as virtual compounds or L-carnitine mimic libraries to identify additional compounds for testing, and hence expand our knowledge of the SAR and test hypotheses. The excluded volumes in the substrate pharmacophore [6] could also help to limit the range of molecules that can map the three key features on L-carnitine.
Promiscuity of Ligand Binding – what do the models mean?
During more than a decade of constructing computational models for drug transporters we have made several observations. Initially we suggested there was considerable promiscuity of ligand interaction with some proteins, especially those like P-gp that have affinity for a diverse range of hydrophobic molecules [42]. There is a growing understanding that ligand interaction with multidrug binding proteins is unlikely to be restricted to classical competition between substrates/inhibitors for a single common binding site [43]. Instead, the interaction of multidrug transporters with their structurally diverse cohort of substrates/inhibitors may involve what is more accurately viewed as a binding surface containing multiple, potentially overlapping, binding sites [43]. Consequently, structurally distinct transport probes may well display different inhibitory profiles for the same battery of test compounds. Evidence for this view is found in a recent review that listed an extensive literature analysis of hOCT2 inhibitors and their substrate probes (Table 2) [35]. For example, cimetidine shows consistently different IC50 values depending on the identity of the transported probe (e.g., ASP vs. MPP+).
Table 2.
Substrate-dependent IC50 or Ki values for inhibition of hOCT2 by cimetidine (data from [35] and our laboratories).
| Cimetidine Ki/IC50 (µM) |
Substrate | Substrate concentration (µM) |
Reference |
|---|---|---|---|
| 14 | Amil | 1 | [62] |
| 36 | ASP | 1 | [63] |
| 23 | ASP | 20 | [44] |
| 26 | ASP | 1 | [62] |
| 27 | Crea | 5 | [64] |
| 1380 | Et | 1 | [57] |
| 510 | Met | 10 | [65] |
| 110 | MPP | 0.01 | Unpublished |
| 142 | MPP | 15 | Unpublished |
| 120 | MPP | 0.01 | [58] |
| 70.4 | NBD-MTMA | 10 | Unpublished |
| 70 | TEA | 5 | [17] |
Amil = Amiloride, ASP = 4-(4-(dimethylamino)styryl)-N-methylpyridinium, Crea = Creatinine, Et = ethidium, Met = metformin, MPP = 1-methyl-4-phenylpyridinium, NBD-MTMA = [2-(4-nitro-2,1,3-benzoxadiazol-7-yl)aminoethyl]trimethylammonium, TEA = tetraethylammonium. Bold = molecules highlighted to show for the same probe substrate the comparable IC50 or Ki
In this light, pharmacophores may represent a ‘statistical average’ of ligand interaction and the use of different substrate probes may result in different inhibition pharmacophores for the same transporter. We have provided some preliminary evidence to support this using published data for 6 inhibitors of MATE1-mediated ASP transport [44] to create a pharmacophore that differed markedly with one generated based on inhibition of MPP+ transport [15]. While this dataset is small it immediately brought to mind the situation with the enzyme CYP3A4 which requires the use of multiple distinct substrates in order to obtain a reliable measure of potential for a compound to cause drug-drug interactions [45, 46]. Further evidence for differences in pharmacophores might be seen based on other factors, such as different training set size, stereoselectivity and different cell lines expressing the transporter. All of these parameters may further compound comparing models, as suggested by pharmacophores for hOCT1 from 3 different groups (Supplemental Table 1).
The future
So far much of the modeling we have described is ligand-based but there have been efforts to generate protein homology models for different transporters (reviewed earlier [38, 47– 49]). How can we expand these efforts too? Certainly there is a wealth of comparative protein structure prediction software like I-Tasser [50] (and many others). These could be used to build transporter models de novo that could then be validated experimentally by site-directed mutagenesis, or in vitro testing. Such protein models could be used to dock known substrates and inhibitors (Figure 1) which, in turn, might help in validation (e.g., MRP4 [51]). Simple software scripting of transporter (and potential mutant) sequences to run them through such protein modeling resources would be a viable option. This approach in turn could be used to create a database of transporter models that could be used for docking by any researcher. Such an approach in parallel with ligand-based efforts [30] would provide some insight as to whether a new drug was likely to be a substrate or inhibitor for a transporter (the principal goal of such studies). It may be possible to create a simple to use software interface for the program, whereby the scientist could submit a small molecule structure or file with multiple structures, and this would be run against selected proteins and the results returned upon completion. With recent efforts to develop mobile applications for drug discovery [52] it might be possible to do this on a mobile device (such as a smartphone, or tablet computer) accessing the models on the cloud, and retrieve a score for potential interactions with different transporters. We expect in the future an increased use of structural models in combination with ligand-based methods to rationalize SAR. To get to this point will require investment to develop the ligand and protein models and make them accessible to all. We need to obtain more efficiently information on human drug transporters so we can reliably predict drug interactions and it is imperative that we fund such IVIS studies.
Supplementary Material
Acknowledgments
We gratefully acknowledge the many students and collaborators that have facilitated much of the work described in this commentary. JEP was supported in part by National Institutes of Health Grant DK67530. SHW was supported by the National Institutes of Health National Institutes of (i) Diabetes and Digestive and Kidney Diseases [Grant 1R01DK080801; and NRSA award DK752422], (ii) Environmental Health Sciences [Grant 5P30ES006694], and (iii) Heart Lung and Blood [Grant 5T32HL07249]. We apologize for omitting any of our colleagues papers due to the limited space.
References
- 1.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010 Mar;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan Q. Pharmacogenomics and systems biology of membrane transporters. Mol Biotechnol. 2005;29:75–87. doi: 10.1385/MB:29:1:75. [DOI] [PubMed] [Google Scholar]
- 3.Koepsell H. Polyspecific organic cation transporters: their functions and interactions with drugs. Trends Pharmacol Sci. 2004 Jul;25(7):375–381. doi: 10.1016/j.tips.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Pauli-Magnus C, Meier PJ. Pharmacogenetics of hepatocellular transporters. Pharmacogenetics. 2003;13:189–198. doi: 10.1097/00008571-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003 Jan 10;1609(1):1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 6.Ekins S, Diao L, Polli JE. A Substrate Pharmacophore for the Human Organic Cation/Carnitine Transporter Identifies Compounds Associated with Rhabdomyolysis. Molecular pharmaceutics. 2012 Feb 28;9:905–913. doi: 10.1021/mp200438v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diao L, Ekins S, Polli JE. Quantitative Structure Activity Relationship for Inhibition of Human Organic Cation/Carnitine Transporter. Molecular pharmaceutics. 2010 Sep 29;7:2120–2130. doi: 10.1021/mp100226q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bahadduri PM, Polli JE, Swaan PW, Ekins S. Targeting drug transporters - combining in silico and in vitro approaches to predict in vivo. Methods Mol Biol. 2010;637:65–103. doi: 10.1007/978-1-60761-700-6_4. [DOI] [PubMed] [Google Scholar]
- 9.Zheng X, Ekins S, Rauffman J-P, Polli JE. Computational models for drug inhibition of the Human Apical Sodium-dependent Bile Acid Transporter. Molecular pharmaceutics. 2009;6:1591–1603. doi: 10.1021/mp900163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekins S, Abramovitz DL. A systems biology view of drug transporters. In: Ecker G, Chiba P, editors. Transporters as drug carriers; Structure, Function, Substrates. Weinheim: Wiley-VCH; 2009. pp. 365–385. [Google Scholar]
- 11.Ekins S, Williams AJ, Krasowski MD, Freundlich JS. In silico repositioning of approved drugs for rare and neglected diseases. Drug Disc Today. 2011;16:298–310. doi: 10.1016/j.drudis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Zhang EY, Knipp GT, Ekins S, Swaan PW. Structural biology and function of solute transporters: implications for identifying and designing substrates. Drug Metab Rev. 2002;34:709–750. doi: 10.1081/dmr-120015692. [DOI] [PubMed] [Google Scholar]
- 13.Zhang EY, Phelps MA, Cheng C, Ekins S, Swaan PW. Modeling of active transport systems. Adv Drug Del Rev. 2002;54:329–354. doi: 10.1016/s0169-409x(02)00007-8. [DOI] [PubMed] [Google Scholar]
- 14.Ekins S, Waller CL, Swaan PW, Cruciani G, Wrighton SA, Wikel JH. Progress in predicting human ADME parameters in silico. J Pharmacol Toxicol Methods. 2000;44(1):251–272. doi: 10.1016/s1056-8719(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 15.Astorga B, Ekins S, Morales M, Wright SH. Molecular Determinants of Ligand Selectivity for the Human Multidrug And Toxin Extrusion Proteins, MATE1 and MATE-2K. The Journal of pharmacology and experimental therapeutics. 2012 Mar 14;341(3):743–755. doi: 10.1124/jpet.112.191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsson P, Pedersen JM, Norinder U, Bergstrom CA, Artursson P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharmaceutical research. 2009 Aug;26(8):1816–1831. doi: 10.1007/s11095-009-9896-0. [DOI] [PubMed] [Google Scholar]
- 17.Suhre WM, Ekins S, Chang C, Swaan PW, Wright SH. Molecular Determinants of Substrate/Inhibitor Binding to the Human and Rabbit Renal Organic Cation Transporters, hOCT2 and rbOCT2. Molecular pharmacology. 2005;67(1067–1077) doi: 10.1124/mol.104.004713. [DOI] [PubMed] [Google Scholar]
- 18.Bednarczyk D, Ekins S, Wikel JH, Wright SH. Influence of molecular structure of substrate binding to the human organic cation transporter, hOCT1. Molecular pharmacology. 2003;63:489–498. doi: 10.1124/mol.63.3.489. [DOI] [PubMed] [Google Scholar]
- 19.Ekins S, Kim RB, Leake BF, Dantzig AH, Schuetz E, Lan LB, et al. Three dimensional quantitative structure-activity relationships of inhibitors of P-glycoprotein. Molecular pharmacology. 2002;61:964–973. doi: 10.1124/mol.61.5.964. [DOI] [PubMed] [Google Scholar]
- 20.Ekins S, Kim RB, Leake BF, Dantzig AH, Schuetz E, Lan LB, et al. Application of three dimensional quantitative structure-activity relationships of P-glycoprotein inhibitors and substrates. Molecular pharmacology. 2002;61:974–981. doi: 10.1124/mol.61.5.974. [DOI] [PubMed] [Google Scholar]
- 21.Chang C, Pang KS, Swaan PW, Ekins S. Comparative Pharmacophore Modeling of Organic Anion Transporting Polypeptides:A Meta-analysis of Rat Oatp1a1 and OATP1B1. The Journal of pharmacology and experimental therapeutics. 2005;314:533–541. doi: 10.1124/jpet.104.082370. [DOI] [PubMed] [Google Scholar]
- 22.Matsson P, Englund G, Ahlin G, Bergstrom CA, Norinder U, Artursson P. A global drug inhibition pattern for the human ATP-binding cassette transporter breast cancer resistance protein (ABCG2) The Journal of pharmacology and experimental therapeutics. 2007 Oct;323(1):19–30. doi: 10.1124/jpet.107.124768. [DOI] [PubMed] [Google Scholar]
- 23.Diao L, Ekins S, Polli JE. Novel Inhibitors of Human Organic Cation/Carnitine Transporter (hOCTN2) via Computational Modeling and In Vitro Testing. Pharmaceutical research. 2009 May 13;26:1890–1900. doi: 10.1007/s11095-009-9905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swaan PW, Bensman T, Bahadduri PM, Hall MW, Sarkar A, Bao S, et al. Bacterial peptide recognition and immune activation facilitated by human peptide transporter PEPT2. Am J Respir Cell Mol Biol. 2008 Nov;39(5):536–542. doi: 10.1165/rcmb.2008-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang C, Bahadduri PM, Polli JE, Swaan PW, Ekins S. Rapid Identification of P-glycoprotein Substrates and Inhibitors. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:1976–1984. doi: 10.1124/dmd.106.012351. [DOI] [PubMed] [Google Scholar]
- 26.Ekins S, Johnston JS, Bahadduri P, D'Souzza VM, Ray A, Chang C, et al. In Vitro And Pharmacophore Based Discovery Of Novel hPEPT1 Inhibitors. Pharmaceutical research. 2005;22:512–517. doi: 10.1007/s11095-005-2505-y. [DOI] [PubMed] [Google Scholar]
- 27.Nies AT, Hofmann U, Resch C, Schaeffeler E, Rius M, Schwab M. Proton pump inhibitors inhibit metformin uptake by organic cation transporters (OCTs) PloS one. 2011;6(7):e22163. doi: 10.1371/journal.pone.0022163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho HT, Pan Y, Cui Z, Duan H, Swaan PW, Wang J. Molecular analysis and structure-activity relationship modeling of the substrate/inhibitor interaction site of plasma membrane monoamine transporter. The Journal of pharmacology and experimental therapeutics. 2011 Nov;339(2):376–385. doi: 10.1124/jpet.111.184036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang C, Swaan PW, Ngo LY, Lum PY, Patil SD, Unadkat JD. Molecular requirements of the human nucleoside transporters hCNT1, hCNT2, and hENT1. Molecular pharmacology. 2004;65(3):558–570. doi: 10.1124/mol.65.3.558. [DOI] [PubMed] [Google Scholar]
- 30.Bikadi Z, Hazai I, Malik D, Jemnitz K, Veres Z, Hari P, et al. Predicting P-glycoprotein-mediated drug transport based on support vector machine and three-dimensional crystal structure of P-glycoprotein. PloS one. 2011;6(10):e25815. doi: 10.1371/journal.pone.0025815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zientek M, Stoner C, Ayscue R, Klug-McLeod J, Jiang Y, West M, et al. Integrated in silico-in vitro strategy for addressing cytochrome P450 3A4 time-dependent inhibition. Chem Res Toxicol. 2010 Mar 15;23(3):664–676. doi: 10.1021/tx900417f. [DOI] [PubMed] [Google Scholar]
- 32.Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, et al. In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environmental health perspectives. 2010 Apr;118(4):485–492. doi: 10.1289/ehp.0901392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ChEMBL. [cited; Available from: http://www.ebi.ac.uk/chembldb/index.php. [Google Scholar]
- 34.The PubChem Database. [cited; Available from: http://pubchem.ncbi.nlm.nih.gov/
- 35.Nies AT, Koepsell H, Damme K, Schwab M. In: Organic cation transporters (OCTs, MATEs), In vitro and In vivo evidence for the importance in drug therapy. Fromm MF, Kim RB, editors. Drug Transporters Berlin: Springer-Verlag; 2011. pp. 105–167. [DOI] [PubMed] [Google Scholar]
- 36.Spjuth O, Willighagen EL, Guha R, Eklund M, Wikberg JE. Towards interoperable and reproducible QSAR analyses: Exchange of datasets. Journal of cheminformatics. 2010;2(1):5. doi: 10.1186/1758-2946-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta RR, Gifford EM, Liston T, Waller CL, Bunin B, Ekins S. Using open source computational tools for predicting human metabolic stability and additional ADME/TOX properties. Drug metabolism and disposition: the biological fate of chemicals. 2010;38:2083–2090. doi: 10.1124/dmd.110.034918. [DOI] [PubMed] [Google Scholar]
- 38.Ekins S, Ecker GF, Chiba P, Swaan PW. Future directions for drug transporter modelling. Xenobiotica; the fate of foreign compounds in biological systems. 2007 Oct;37(10):1152–1170. doi: 10.1080/00498250701646341. [DOI] [PubMed] [Google Scholar]
- 39.Rais R, Acharya C, Mackerell AD, Polli JE. Structural determinants for transport across the intestinal bile acid transporter using C-24 bile acid conjugates. Molecular pharmaceutics. 2010 Dec 6;7(6):2240–2254. doi: 10.1021/mp100233v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yates CR, Chang C, Kearbey JD, Yasuda K, Schuetz EG, Miller DD, et al. Structural determinants of P-glycoprotein-mediated transport of glucocorticoids. Pharmaceutical research. 2003 Nov;20(11):1794–1803. doi: 10.1023/b:pham.0000003377.39548.f6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.