

Abstract
Despite almost universal implementation of reno-protective therapies over the last 25 years, risk of ESRD in type 1 diabetes (T1D) is not decreasing, and ESRD remains the major cause of excess morbidity and premature mortality [1]. Such a state of affairs prompts a call to action. In this review we re-evaluated the proteinuria-centric model of diabetic nephropathy and showed its deficiencies. On the basis of the extensive studies that we have been conducting in the population of the Joslin Clinic, we propose that progressive renal decline, not abnormalities in urinary albumin excretion, should be considered as the major feature of disease processes leading to ESRD in T1D.
Etiology of diabetic nephropathy should be reconsidered in light of our new findings so our perspective can be broadened regarding new therapeutic targets available for interrupting the progressive renal decline in T1D. Reduction in the rate of GFR loss, not reduction of AER, should become the measure for evaluating the effectiveness of new therapeutic interventions. We need new accurate methods for early diagnosis of patients at risk of progressive renal decline or, better yet, for detecting in advance which patients will have rapid, moderate or minimal rate of progression to ESRD.
Keywords: type 1 diabetes, kidney complications, Proteinuria, progressive renal decline
Since there is no animal model that mimics progressive diabetic nephropathy in humans, there is a great need to understand the etiology of this devastating complication through clinical, epidemiological and genetic studies.
Prof. Hirofumi Makino introducing Dr. Krolewski as the speaker at the 2013 JSN meeting.
Proteinuria-centric model of diabetic nephropathy
Already in 1836 Bright postulated that proteinuria could reflect a serious renal disease specific to diabetes [2]. Almost one hundred years later Kimmelstiel and Wilson observed nodular glomerular intercapillary lesions in autopsy reports of long-standing patients with diabetes and heavy proteinuria which they considered to be morphological lesions specific to diabetic nephropathy [3]. In the 1970s radioimmunoassay techniques were developed that permitted the quantitative measurement of the minutely elevated urinary albumin levels that distinguished microalbuminuria (MA) (typically AER between 30 and 299 μg per minute), from the elevations of albumin detectable by urinary dipstick method that define proteinuria (typically AER ≥ 300 μg per minute) [4]. During the 1980s longitudinal studies were conducted in patients with Type 1 diabetes (T1D) which used the new method of measuring urinary albumin. These studies found a 60 to 85 percent risk of progression of MA to proteinuria/impaired renal function within 6-14 years of follow-up [5-7]. These findings together with the previous observations provided a foundation for so-called proteinuria-centric model of natural history of diabetic nephropathy.
This model developed in 1990s postulated that in diabetes an increase in urinary albumin excretion rate (AER) that reflects glomerular abnormalities precedes the development of impaired renal function and ESRD. In this model, the onset of MA can be considered a manifestation of an initiating disease process that leads to proteinuria, and the latter is followed by GFR loss that eventually results in end-stage renal disease (ESRD) [8]. The sequential occurrence of the above stages has been referred to as progressive diabetic nephropathy.
Shortcomings of the proteinuria model
Clinical and epidemiological studies conducted during the last decade have shown that the development of elevated urinary excretion of albumin leading to proteinuria and GFR loss or renal decline are two separable manifestations of diabetic nephropathy, rather than two successive stages of one disease process. Abnormal urinary albumin excretion waxes and wanes (progresses and regresses), and renal decline is usually progressive and leads to ESRD. While the two manifestations can progress in parallel, changes in one are not well-correlated with changes in the other. As a result, some patients have abnormal urinary albumin excretion that progresses, regresses or simply persists, and all the while their renal function remains stable. On the other hand, renal decline is initiated in patients with normoalbuminuria (NA) or MA and it is progressive (we refer to it as progressive renal decline) regardless of the variation in urinary albumin excretion. Following is a summary of the above studies.
Microalbuminuria is a poor predictor of progressive diabetic nephropathy
Frequent Remission of Microalbuminuria to Normoalbuminuria
Reviewing and analyzing studies on the natural history of MA, Caramori et al. noticed a frequent remission of MA to NA [9]. However, it remained uncertain whether such remission occurred simply because of short-term change in urinary albumin excretion or it was a biological process which takes several or more years [9,10]. We investigated this issue in the 1st Joslin Study on Natural History of Microalbuminuria. In that study, 386 patients with persistent MA were observed for the occurrence of remission to NA during a six-year follow-up [11]. Analysis of the trends in urinary albumin excretion rate revealed that values of albumin excretion during repeated measurements were not random – rather, a clear pattern of remission over several years was observed. This remission was independent from treatment with ACE inhibitors. Our findings proved that remission of MA was a real biological phenomenon and not a consequence of short-term random changes in urinary albumin excretion. In the 1st Joslin study, the six-year cumulative incidence of remission to NA was remarkably high (cumulative incidence 59%; 95 CI, 54 – 64%) [11]. In a large proportion of patients this remission to NA persisted for 4 years or more [11].
The subsequently published results of the EURODIAB Prospective Complications Study confirmed the frequency of MA remission that was observed in the 1st Joslin study [12]. Of 352 subjects who had MA in the single baseline measurement, over 50% were found to have NA on two measurements taken at termination of the mean follow-up of seven years. Recently published results obtained from the DCCT/EDIC study showed almost identical results as the findings obtained in our 1st Joslin study [13]. The summary of the results obtained in the above studies is shown in Table 1.
Table 1.
Frequency of Microalbuminuria Remission and Progression in Three Recent Observational Studies.
| Study and Reference | Number of patients with microalbuminuria | Mean Follow-up | Cumulative Incidence of Remission to normoalbuminuria | Cumulative Incidence of Progression to Proteinuria |
|---|---|---|---|---|
| 1ST Joslin Study (2003) (11) | 386 | 6 years | 59 percent (54 to 64) | 27 percent (22 to 32)* |
| EURODIAB (2005) (12) | 352 | 7 years | 51 percent | 14 percent* |
| DCCT/EDIC (2011) (13) | 325 | 13 years | 40 percent (36 to 48)** | 28 percent (23 to 33)** |
12-year cumulative incidence (unpublished data)
10-year cumulative incidence
Numbers in brackets represent the 95 percent confidence intervals.
Infrequent Progression of Microalbuminuria to Proteinuria
Further support for the concept that MA is a functional abnormality, rather than a critical step in the disease process leading to ESRD, is its infrequent progression to proteinuria. Rather than being in the range of 60 to 85 percent as originally reported [5-7], studies published in the 1990s showed 10-year cumulative risks of progression to proteinuria in the range of 25 to 30 percent [9,14,15]. In the 1st Joslin Study the cumulative risk of progression from MA to proteinuria during 6 years of follow-up was 15% and 23% during 12 years (see Table 1) [12, 16].
Multiple clinical trials demonstrated the effectiveness of ACE inhibitors both in reducing urinary albumin excretion and in delaying the progression of MA to overt proteinuria [17-22]. However, those effects were small and not all studies showed them [23]. The question arises whether the introduction of the reno-protective drugs during the 1990s into the care of patients with diabetes might have changed the natural history of MA and could account for the strikingly different results obtained in the three landmark studies from the 1980s which included a total of 30 patients [5-7] and the more contemporary studies that comprised more than a thousand of MA patients [11-13]. Interestingly, in the 1st Joslin Study only a limited number of patients with MA were treated with ACE inhibitors and the effect of these drugs had minimal or no impact on frequency of remission of MA to NA or its progression to proteinuria [11,16].
Progressive renal decline in patients with normo- and microalbuminuria
Over the last 20 years several studies showed un-coupling of the development of MA/proteinuria and the occurrence of progressive renal decline [24-26]. Recently, we examined this issue in great detail in the 2nd Joslin Study on Natural History of Microalbuminuria [27]. Two cohorts of T1D patients were included; 286 patients with NA and 248 with MA. All patients were enrolled between 2003 and 2006, had normal renal function at baseline (eGFRcr-cys > 60 ml/min/1.73m2) and were followed for 4-10 years. Characteristics of the study groups are summarized in Table 2. At baseline, the MA group had higher HbA1c, higher blood pressure, and was more frequently treated with reno-protective drugs that the NA group. While the distribution of AER in the NA group was uniform across the normal range (<30 μg/min), the majority of the distribution in the MA group was in the low abnormal range (25th, 50th and 75th percentiles were 44, 65, and 116μg/min, respectively).
Table 2. Characteristics of the study groups included in the 2nd Joslin Study on Natural History of Microalbuminuria in T1D (Data adapted from ref. 27).
| Normoalbuminuria | Microalbuminuria | |
|---|---|---|
| Baseline characteristics | N=286 | N=248 |
| Male | 43% | 62% |
| Duration of DM (y) | 20 [14, 29] | 24 [15, 30] |
| Age (y) | 41 [30, 48] | 42 [34, 49] |
| HbA1c in% | 8.0 [7.4, 8.8] | 8.3 [7.5, 9.3] |
| Systolic Blood Pressure | 118 [110, 127] | 122 [114, 130] |
| ACE & ARBs Rx | 33% | 74% |
| AER μg/min | 16 [12, 22] | 65 [44, 116] |
| Baseline serum markers | ||
| Uric Acid (mg/dL) | 4.2 [3.7, 5.0] | 4.9 [4.2, 5.9] |
| TNFR-1 (pg/mL) | 1300 [1094, 1516] | 1536 [1291, 1870] |
| TNFR-2 (pg/mL) | 2073 [1717, 2525] | 2374 [1997, 2988] |
| Baseline Renal Function | ||
| Serum cystatin C (mg/L) | 0.65 [0.58, 0.72] | 0.66 [0.60, 0.78] |
| Serum creatinine (mg/dL) | 0.76 [0.65, 0.86] | 0.79 [0.69, 0.90] |
| eGFR(cr-cys) (ml/min/1.73m2) | 113 [102, 123] | 112 [96, 122] |
| Follow-up results: | ||
| Duration of Follow-up (y) | 8.0 [6.0, 8.7] | 7.9 [5.5, 9.2] |
| # of serum samples | 5 [4, 7] | 7 [5, 9] |
| eGFRcr-cys loss (%/y) | -1.5 [-2.4, -0.8] | -2.2 [-4.1, -1.2] |
| Renal decline (eGFRcr-cys loss 3.3% or more per year) | 10% (28 decliners) | 35% (86 decliners) |
| 8-yr Cumulative Risk of CKD≥3 | 6% | 22% |
Data are percents or medians [25th, 75th percentiles]
During 4-10 year follow-up, serial measurements of serum creatinine and cystatin C were performed and were used to derive trajectories of eGFRcr-cys over time. The annual rate of eGFRcr-cys change was used to classify patients to those with stable renal function (non-decliners) and to those with progressive renal decline (decliners). Progressive renal decline, defined as continuous rate of eGFRcr-cys loss ≥3.3% per year, was present in 10% of the NA group and 35% in the MA group. Figure 1 illustrates the trajectories of eGFRcr-cys in 28 patients with progressive renal decline who at baseline had NA and normal renal function. Two features deserve emphasis. First, the majority of trajectories of eGFRcr-cys loss over time were approximately linear. Two deviated visibly by accelerating the rate of decline (trajectories in red). One of these patients has already reached ESRD and the other soon will. Second, the slopes of eGFRcr-cys loss varied tremendously, ranging from a loss of 3.3% to 21% per year. The fastest decliners, including the two already mentioned, will reach ESRD within 5-15 years, while the slowest might take 25-30 years. During follow-up half of these decliners developed MA and three progressed to proteinuria. Patients with NA and with eGFRcr-cys loss less than 3.3%/year were considered as non-decliners (n=258) and they are not shown.
Figure 1.
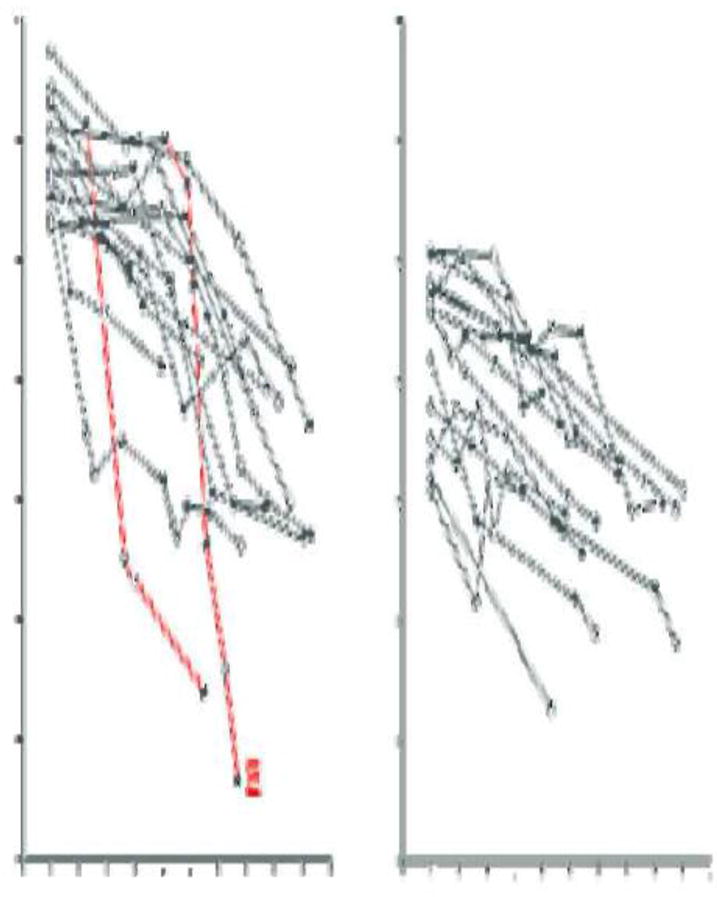
eGFRcr-cys trajectories in T1D patients with normoalbuminuria and progressive renal decline (eGFRcr-cyst loss ≥3.3%/year) during 4-10 years of follow-up. The trajectories are plotted in patients with baseline eGFRcr-cys 105 ml/min/1.73m2 and above in Panel A, and in patients with baseline eGFRcr-cys below 105 ml/min/1.73m2 in Panel B. Lines in red indicate presence of macroalbuminuria.
E – end stage renal disease. (Figure adapted from ref. 27)
eGFRcr-cys trajectories in 86 decliners in the MA group in the 2nd Joslin study (Table 2) are not presented. However, the features of these trajectories were similar to that of decliners with NA, i.e. the majority of trajectories were linear and varied with regard to rate of eGFRcr-cys loss. Instead we showed trajectories of renal function changes in a group of 79 patients with normal renal function, who developed new MA in the early 1990s and subsequently were followed for 10 years during the 1st Joslin Study [28]. Renal function variation over time in these patients was determined by serial measurements of serum cystatin C. The trajectories of eGFRcys are shown in Figure 2.
Figure 2.
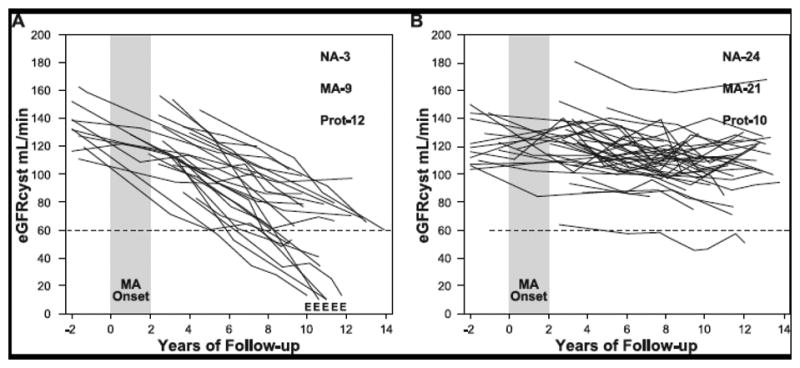
Trajectories of renal function changes in patients with T1D and new onset microalbuminuria who were followed for 12 years.
MA onset – 2 year interval during which multiple determinations of ACR became elevated; E – date when ESRD was diagnosed; eGFRcys – glomerular filtration rate estimated from serial measurements of serum cystatin C. At the end of follow-up numbers of patients with various categories of AER are reported: NA – normoalbuminuria, MA - microalbuminuria, Prot – proteinuria.
Panel A shows patients with early progressive renal decline (decliners). eGFRcys slopes in these patients were -3.3% min/year or faster.
Panel B shows patients with stable renal function (non-decliners). eGFRcys slopes of these patients were slower than 3.3% min/year. Figure adapted from reference #28 and supplemented with unpublished data about patients who developed ESRD.
In this study 24 (30%) patients developed progressive renal decline (defined as eGFRcys loss ≥3.3% per year). Their trajectories are shown in Figure 2A [28]. Renal decline was present at the time or soon after the development of MA and the significant rate of eGFRcys loss continued to be constant during the subsequent follow-up. Similarly to decliners with NA (Figure 1), the decline within individuals in this group of decliners was linear and could be well represented by a simple regression slope. Also, the rates of eGFR loss per year varied widely among individuals. Within 10 years of follow-up, almost half (10 patients) reached CKD stage ≥3 and in 5 patients the eGFRcyst decline was so rapid that they progressed to ESRD. The rest of the decliners will most likely reach CKD stage 3 during the next 10 years of follow-up, assuming their trajectories of eGFR decline remain linear. For comparison Figure 2B shows eGFRcys trajectories in non-decliners. Most of the trajectories were linear and horizontal. A few patients had variation in eGFRcys over time but renal function at the end of follow-up was the same as at baseline.
It is interesting that the distribution of AER abnormalities during the 2-year interval when new onset MA occurred was not different among patients who subsequently had renal decline and those in whom renal function remained stable [28]. During follow-up one can see un-coupling of changes in AER and changes in eGFRcys. Only half of the decliners developed proteinuria. In contrast among non-decliners half had either persistent MA or progressed to proteinuria.
Progressive renal decline in patients with proteinuria
Recently we examined the trajectories of renal function changes over time in patients included in the Joslin cohort of patients with T1D and proteinuria [29]. We used serial measurements of serum creatinine to estimate eGFRcre. In this cohort, 240 patients entered the study with normal eGFRcre (above 60 ml/min/1.73m2) and were followed for 5 to 18 years. Using previously developed statistical criteria, the trajectories were classified as linear in 70% of patients, “mildly non-linear” in 17%, and “strongly non-linear” in only 13%. In the last two categories there was an equal proportion of those who had accelerated and de-accelerated trajectories of eGFRcre loss. The examples of linear eGFRcre trajectories are shown in Figure 3. The rates of eGFRcre loss in the three patients ranged from very rapid (-52.5 ml/min/1.73m2/year, panel A) to moderate (-7.5 ml/min/1.73m2/year, panel B), and to minimal (-0.9 ml/min/1.73m2/year, panel C). These trajectories are similar to the eGFRcre-cys, or eGFRcys trajectories shown in Figures 1 and 2.
Figure 3.

Examples of trajectories of changes in renal function in patients with T1D and proteinuria. For the patient in Panel 3A, eGFRcre loss was 52.5 ml/min/1.73m2/year and renal function progressed from normal to ESRD within 2 years. For the patient in Panel 3B, eGFRcre loss was 7.5 ml/min/1.73m2/year and renal function progressed from normal to ESRD within 18 years. For the patient in panel 4C, eGFRcre loss was 0.9 ml/min/1.73m2/year, and renal function is estimated to remain stable during the next 30 years.
Examples were selected from a large cohort of patients with proteinuria who entered observation with normal renal function and were followed for 5-18 years (Adapted from ref. 29)
To gain an appreciation of the distribution of the overall rate of annual eGFR loss in the whole group of 240 patients, we extracted the linear component of each trajectory as a simple slope. Figure 4 shows distribution of such slopes. A few individuals had a positive slope, i.e. improved in renal function at the end of follow-up (n=25), but the majority had negative slopes, i.e. declining renal function. The median (25th, 75th percentiles) was -2.9 (-7.1, -1.27) ml/min/1.73m2/year. Reflecting the long tail of negative values, the 5th percentile was -16.8 and the minimum -70.5 ml/min/1.73m2/year. Assuming that the slopes would not change in the future, one may expect that half of the patients are not at risk of ESRD during their life time. In the other half, patients are at risk of developing ESRD but time to onset would vary between less than two years to more than 25 years.
Figure 4.
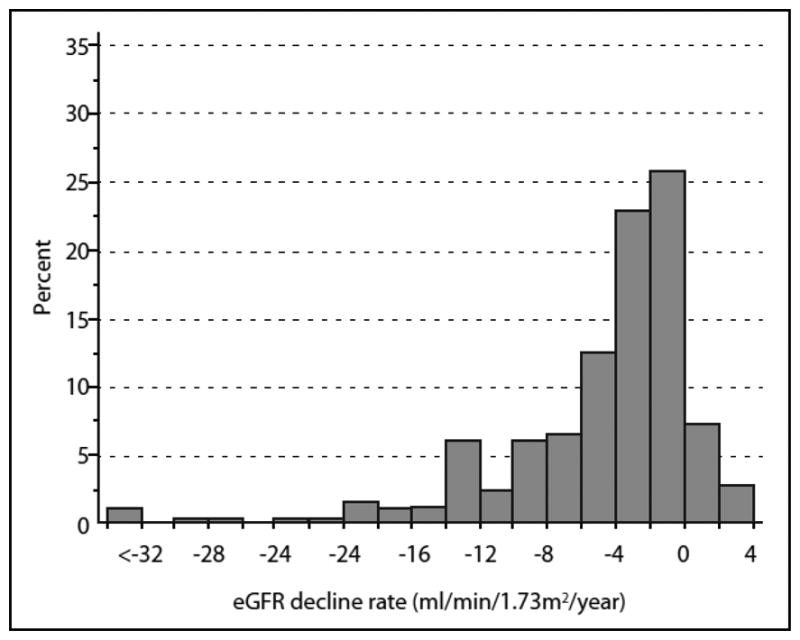
Distribution of slopes of eGFRcre changes in 240 subjects with proteinuria who entered the study with normal renal function and were followed for 5 to 18 years. The three patients in the interval <-32 had slopes -52.5, -56.4 and -70.5 ml/min/1.73m2/year. Adapted from ref. 29.
Progressive Renal Decline as the Major Feature of Diabetic Nephropathy
The data presented in Figures 1-3 unequivocally demonstrated that the progressive renal decline is the primary manifestation of diabetic nephropathy and best represents the disease process underlying the development of ESRD in T1D. These observations do not support the proteinuria-centric model of diabetic nephropathy in which renal decline is a late consequence of proteinuria [8].
These apparent discrepancies prompted us to propose a new model of diabetic nephropathy in T1D, which is outlined in a diagram in Figure 5. Progressive renal decline is represented as a one-directional process superimposed upon the natural course of abnormal urinary albumin excretion. The latter, as discussed earlier, can regress, stay the same or progress. On the other hand renal decline, once initiated, seems to progress relentlessly to ESRD albeit at very variable rate. Overall, about 10%, 25% and 50% have progresssive renal decline in patients with NA, MA and proteinuria respectively. It needs to be noticed that deliners with NA are at increased risk of MA, therefore a study group ascertained for MA by definition will be always enriched for decliners. Similarly decliners with MA have a high rate of progression to proteinuria, therefore, a study group ascertained for proteinuria will be enriched a great deal for decliners. Overall in T1D, the lifetime risk of ESRD, the end of progressive renal decline that starts in patients with NA (see Figure 1), is estimated to be 10-15%. However, the cases occur over a long span of T1D duration (15th to 40th year of diabetes) [30].
Figure 5.
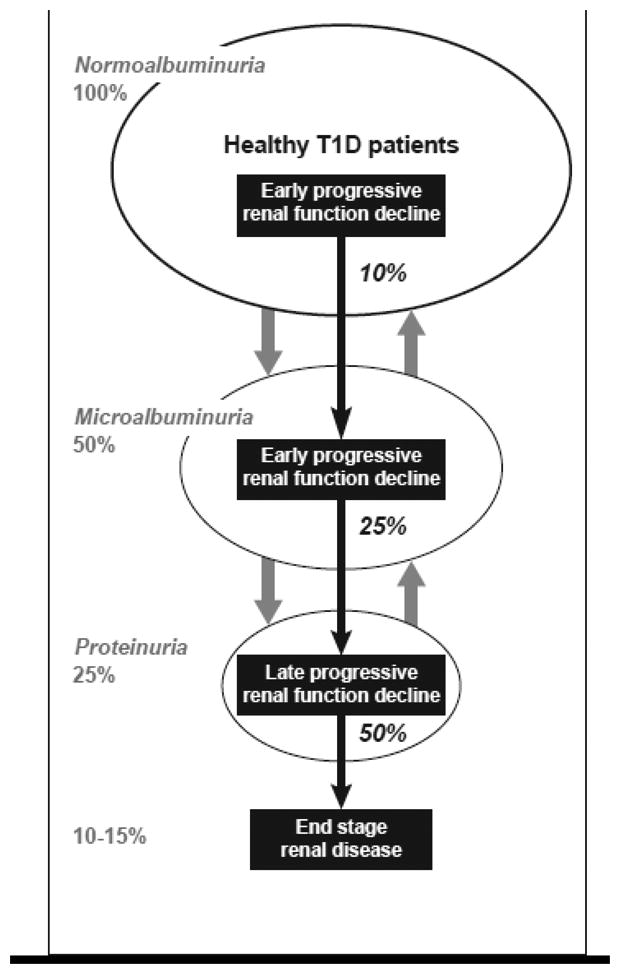
New model of diabetic nephropathy in T1D. Urinary albumin excretion increases in progressively smaller subsets and also regresses, while progressive renal decline develops early in a subset of normoalbuminurics, microalbuminurics and proteinurics and almost always progresses to ESRD. Percentages in italics indicate proportion of patients with progressive renal decline in the subgroups of normoalbuminurics, microalbuminurics and proteinurics accordingly. (Adapted from ref. 53).
Mechanisms of progressive renal decline
Presently, the putative mechanisms underlying progressive renal decline in T1D are not known. To advance our understanding of these mechanisms and taking into account the salient features of progressive renal decline, several questions must be answered. First, we need to know in which kidney compartment/tissue/cells the disease process that underlies progressive renal decline seen in patients with NA or MA (see Figure 1, and Figure 2A) is initiated. Theoretically, this process may take place in glomerula, tubules, interstitium, or in vasculature. Second, the nature of the putative mechanisms that are responsible for the progressive GFR loss is unclear. It is possible that early GFR loss (in the range of normal renal function) is due to some (yet unknown) functional changes and the late GFR loss (CKD stage 3-5) is due to morphological lesions in some putative kidney compartments/tissues/cells. However, the rates of GFR loss during the early and late phases of progressive renal decline are similar. This suggests that the mechanisms may also be similar. Third, since trajectories of GFR loss over time seems to be linear (stable) within individuals but very variable among individuals, one may postulate a role of genetic factors as determinants of progressive renal decline (29). A search for these factors may unravel pathways involved in the etiology of progressive renal decline.
Lack of an animal model that would mimic progressive renal decline in human diabetes and the lack of kidney biopsy specimens leave us with clinical and epidemiological studies to investigate the above questions. Identifying systemic risk factors, genetic markers or biomarkers specifically associated with risk of progressive renal decline should help us understand the mechanisms of progressive renal decline in T1D.
Systemic factors and serum markers and progressive renal decline
We have been searching for the determinants of the progressive renal decline in patients with NA and MA who were enrolled into the 2nd Joslin Study [27]. The baseline clinical characteristics and concentrations of serum markers in the study groups are presented in Table 2 above. Figure 6 shows plots of the risk of renal decline according to these characteristics.
Figure 6.
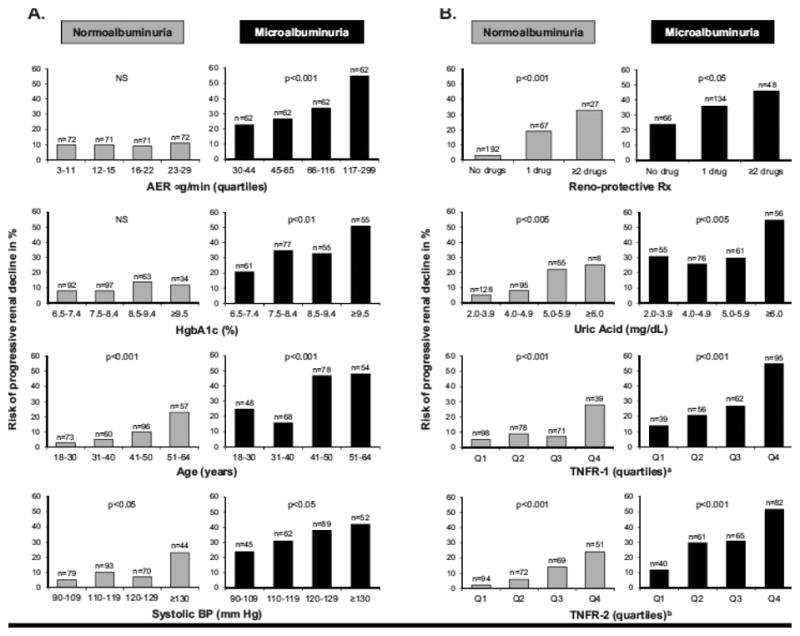
Risk of progressive renal decline according to categories of baseline clinical characteristics (Panel A) and serum markers (Panel B) and according to study groups. Data from the 2nd Joslin Study on Natural History of Microalbuminuria in T1D. Figure reprinted from ref. 27.
a cut points for circulating TNFR1 for 25th, 50th, and 75th percentiles were:1173, 1394, 1685 pg/ml
b cut points for circulating TNFR2 for 25th, 50th, and 75th percentiles were: 1810, 2186, 2690 pg/ml
The risk of renal decline according to quartiles of baseline AER (top graph in Panel A) was flat (around 10%) across quartiles in the NA group. In MA, the risk of renal decline was significantly higher than in NA and increased from 23% in the lowest quartile to 55% in the highest. For HbA1c, the increase risk was moderate in the NA group but strong in the MA group (Panel A). The risk of decline increased with age in both groups (Panel A) and increased similarly with duration of T1D because of its co-linearity with age (data not shown). Risk of decline also increased with systolic blood pressure in both groups (Panel A) and increased as the number of prescribed reno-protecting treatments increased (Panel B).
Plots of the risk of renal decline according to three serum markers are shown in Panel B. In both study groups, elevated serum uric acid increased the risk of renal decline, and the effect of an increasing serum TNFR1 or TNFR2 was even more striking. The effects of both TNFRs were very similar and the information in them redundant. The independent effects of all of these variables on risk of renal decline were confirmed in multiple logistic analyses. We did not find any association between risk of progressive renal decline in either study group and baseline serum levels of other markers such as TNFa (free), IL-6, IL-8, IP-10, MCP-1, VCAM, ICAM, Fas and FasL [27].
Recognizing that multiple clinical factors and serum markers contribute in a similar way to renal decline in NA and in MA strengthens the hypothesis that progressive renal decline is the primary clinical abnormality of diabetic nephropathy. Although the exact mechanisms through which these factors contribute to early progressive renal decline are not clear at present, we discussed some mechanisms with regard to the three serum markers.
Elevated serum uric acid has pro-inflammatory properties and may act as either a pro-oxidant or anti-oxidant molecule depending on the circumstances [31]. In vivo, rats rendered hyperuricemic by means of a uricase inhibitor develop an afferent arteriolopathy that decreases luminal diameter and produces renal ischemia, leading to glomerulosclerosis and tubulointerstitial fibrosis [32]. Similar histological changes occur in humans with gouty nephropathy [33]. Lowering serum uric acid concentration with allopurinol attenuates these histological and functional changes, although this effect may be due in part to reduced oxidative stress resulting from xanthine-oxidase inhibition [31]. It is intriguing to postulate that lowering serum uric acid in patients at risk of renal decline may be an effective intervention to reduce risk of renal decline in T1D [34]. Preparations for such a clinical trial are underway [35].
Our previous studies described the strong effect of serum concentrations of TNFR1 or TNFR2 on the risk of advanced stages of renal decline such as CKD stage 3 or ESRD [36,37]. The recent study in NA and MA extends demonstration of their effects to onset of the process of renal decline itself [27]. We do not know how elevated concentrations of TNFRs may initiate renal decline and lead to renal failure. However, we excluded some hypotheses. For example we showed that serum TNFα is not involved directly or indirectly, through regulation of serum TNFRs, in the development of renal decline. Furthermore, we showed that serum concentration of several chemokines and circulating adhesion molecules (IL-8, IP-10, MCP-1, VCAM, ICAM as potential downstream effectors of TNFRs) were not associated with renal decline either [27]. An intriguing finding of the present study is the negative interaction between serum uric acid and TNFR1 on the risk of renal decline in both NA and MA, meaning that the risk of renal decline for patients with elevated serum uric acid and serum TNFR1 is less than the sum of the individual risks associated with the two predictors. This suggests that the predisposing effects of serum uric acid and TNFR1 converge on some common pathway that cannot be further activated by one factor if it has already been turned on by the other. The nature of this putative pathway is unknown at this time.
Urinary markers of inflammation and progressive renal decline
Although glomerulopathy has been assumed to be the major contributor to the pathogenesis of diabetic nephropathy [8,38], a growing body of evidence suggests that tubulointerstitial injury mediated through an inflammation process may also contribute to the development of diabetic nephropathy and particularly to progressive renal decline [39]. In 1991 Bohle et al. were the first to draw attention to the presence of tubulointerstitial injury in the human diabetic kidney and its strong association with renal failure [40]. Based on a large collection of renal biopsies from humans with diabetic nephropathy, they demonstrated the presence of infiltrates of monocytes, macrophages and T cells in the interstitium similar to those seen in chronic glomerulonephritides. Several subsequent kidney biopsy studies showed cross-sectional associations between severity of diabetic nephropathy measured as GFR and presence of specific inflammatory markers [41-44].
To follow these findings Wolkow et al. investigated the role of chemokinases in urine in the development of early progressive renal decline in patients with T1D and new onset of MA who were followed for 8-12 years during the 1st Joslin Study [45]. Trajectories of GFRcys in these patients were presented in Figure 2 above. We assayed 23 cytokines/chemokines in urine specimens but only five of them (IL-6, IL-8, MCP-1, IP-10 and MIP-1δ) were detected in the majority of patients with diabetes. These markers were then measured in urine specimens obtained 2-4 years after onset of MA in the two study groups presented in Figure 2. There were 28 patients with new onset MA who had progressive renal decline (Figure 2A) during follow-up and 43 patients with onset of MA who had stable renal function during follow-up (Figure 2B). As a reference group we used 74 patients with NA and stable renal function during 8-12 years follow-up. The results of the study are presented in Table 3. Urinary concentrations of all five inflammatory markers were significantly higher in decliners than in the other groups. It is important to note that results in the non-decliners are identical despite one group having MA and the other remaining with NA. A multiplicity of elevations in decliners characterized the association best. In multivariate analysis elevation of two or more cytokines/chemokines in urine specimens was strongly associated with future risk of early progressive renal decline (OR= 5.4; 95% C.I.: 1.9,15.6). In contrast, concentration of CRP, IL-8, and MIP-1δ in serum did not differ between decliners and non-decliners.
Table 3. Urinary concentrations of chemokines and cytokines according to study groups (from 1st Joslin Study on natural History of Microalbuminuria, see Figure 2 above).Table adapted from ref. 45).
| Normoalbuminurics | Microalbuminurics | |||
|---|---|---|---|---|
| Markers | Renal function | |||
| Reference Group | Non-Decliners | Decliners | ||
| N= 74 | N= 43 | N= 28 | p-value* | |
| Concentrations adjusted for urinary creatinine in pg/mg creatinine | ||||
| IL-6 | 0.9 (0.3, 2.0) | 1.1 (0.4, 1.8) | 1.6 (0.6, 15) | 0.078 |
| IL-8 | 1.0 (0.3, 4.9) | 0.8 (0.1, 3.9) | 15 (3.1, 79) | 0.0001 |
| IP-10 | 5.1 (3.1, 60) | 5.3 (2.6, 66) | 49 (6.9, 228) | 0.0021 |
| MCP-1 | 49 (25, 90) | 59 (32, 89) | 95 (55, 198) | 0.0054 |
| MIP-1δ | 50 (16, 97) | 48 (10, 91) | 80 (48, 155) | 0.0215 |
Data are median (25th, 75th percentile)
Kruskal-Wallis test of the null hypothesis that all three groups are from the same distribution.
In conclusion, urinary concentrations of inflammatory markers IL-6, IL-8, MCP-1, IP-10 and MIP-1δ are elevated in patients with MA who are at risk of progressive renal decline. Our results are consistent with previous work in experimental models and observational studies in humans that implicated inflammation in the development and progression of renal injury in diabetes [46-48]. However, we refined the characterization of its role in humans with T1D by demonstrating that elevated levels of markers of low level inflammation in urine are not associated with MA per se but are specific for early progressive renal decline [45].
None of the covariates available in this study such as age, duration of diabetes, glycemic control, urinary albumin excretion, and treatment with ACE inhibitors accounted for the associations. Therefore, the nature of the factors determining the elevated concentrations of urinary pro-inflammatory chemokines and cytokine in T1D remains unknown.
An explanation for the findings reported by Wolkow et al. [45] is that kidney cells, primarily tubular, are the source of the elevated urinary concentrations of these markers. Although, the nature of the stimulus to synthesize these chemokines in tubular cells is unknown, it might originate from the glomerular filtrate. We hypothesize that impairment of the glomerular filtration barrier (evidenced by the presence of MA) permits injurious serum proteins or growth factors to leak into the urinary space. These putative factors, which we refer to as toxic urinary proteins (txUPs), may stimulate proximal tubular cells to secrete chemokines/cytokines and other stress proteins indicating tubular damage that leads to tubular atrophy, interstitial fibrosis and early GFR loss. Recently it was demonstrated in animal studies that tubular damage initiates a disease process that leads to inflammation, loss of blood vessels, interstitial fibrosis and glomerulosclerosis [49].
Toxic urinary proteins and renal decline
Recently Wanic et al. tested the above hypothesis [50]. Archived baseline urine specimens from patients described in Figure 2 were used. Urine specimens from five decliners (Figure 2A) and five non-decliners (Figure 2B) were pooled and used in vitro experiments. Human proximal tubular cells (HK-2 cells) were grown in serum-free medium enriched with pooled urines from decliners or non-decliners. Genome-wide expression profiles were determined in extracted mRNA from the two sets of HK-2 cells. We found that pooled urines from decliners induced differential expression of 312 genes. There were 119 up-regulated genes. Their biologic processes were enriched for defense response, responses to other organisms, regulation of cellular processes, or response to stress or stimulus, and programmed cell death. There were 195 down-regulated genes. They were disproportionately represented in biological processes for regulation of metabolic processes, nucleic acid metabolic processes, cellular response to stress and macromolecule biosynthesis.
The set of up-regulated genes in HK-2 cells reported by Wanic et al. overlapped significantly with sets of over-expressed genes in tubular and interstitial compartments of kidney biopsies from patients with advanced diabetic nephropathy [50, 51] (see Figure 7). The overlap shown by the area in yellow, included genes encoding chemokines and cytokines (including those reported by Wolkow et al.). Overlap of down-regulated genes was no more than expected by chance. In conclusion, molecular and biological processes in tubules and interstitium seen in advanced diabetic nephropathy can be induced in vitro by exposure to urine from patients with MA who subsequently developed progressive renal decline, presumably due to putative txUPs which filtered to the urinary space. The nature of these putative txUPs is unknown at present.
Figure 7.

Overlap of the up-regulates genes in HK-2 cells in response to urines from Decliners and the corresponding sets of genes in tubular and interstitial compartments of kidney biopsies obtained from patients with advanced diabetic nephropathy [P-value <10-9 for overlap with data of Woroniecka et al. (51) and <10-4 for overlap with data of Schmid et al. (43)]. Figure adapted from ref. 50.
Progressive Renal Decline: How to diagnose it?
An important message conveyed by Figure 5 is that the majority of patients with NA and a large proportion of those with MA and proteinuria will never develop ESRD. They have elevated risk of death unrelated to ESRD, but its excess risk is only one-tenth of the excess risk of death seen among those who develop ESRD [1]. On the other hand, among those with renal decline the rate of GFR loss varies widely (Figure 1-4). Therefore, physicians face not only the challenge of distinguishing patients who will remain with stable renal function for their lifetime from patients who will have progressive renal decline, but also the challenge within the latter group of identifying rapid, moderate and slow decliners and estimating the time to onset of ESRD (examples in Figure 3 panels A and B)
Several legacy markers are used to diagnose diabetic nephropathy in T1D, including measurements of levels of hemoglobin A1C (HbA1c, exposure), concentration of urinary albumin excretion (supposedly early disease process) and concentration of serum creatinine (late disease process). The last two markers used cross-sectionally (during one patient visit) indicate presence/absence or extent of diabetic nephropathy. When these markers are used in prospective epidemiological studies, they quantify risk of progression to ESRD or risk of deaths. However, as discussed earlier their utility to predict these outcomes in individual patients is limited. They have some specificity to diagnose patients at risk but they are hopelessly unspecific to predict future renal decline and progression to ESRD. The usage of legacy markers will continue until new, more specific markers are discovered and introduced to the clinical practice.
During the last several years, intensive research has been underway to find new markers to more reliably diagnose patients at risk of renal decline and progression to ESRD. Serum concentration of cystatin C emerged as a candidate diagnostic marker that might be a more accurate indicator of impaired renal function than serum creatinine. Only recently we have shown that one determination of serum cystatin C in patients with diabetes and proteinuria provides better risk stratification of subsequent ESRD than determination of serum creatinine obtained at the same time [52].
Just recently, we showed that serum concentrations of TNF receptors 1 or 2 (TNFR1, TNFR2) are very good predictors of future development of CKD stage≥3 in T1D patients [36]. We reported similar findings regarding the relationship between circulating TNFRs and risk of ESRD during 12 years of follow-up in patients with T2D [37].
Progressive renal decline and considerations for therapeutic trials
New effective therapies are desperately needed to reduce risk of ESRD in T1D as it was discussed in the recent issue of Seminars in Nephrology [53]. In designing therapies for T1D it is important to put aside the previous proteinuria-centric model of diabetic nephropathy and fully appreciate the features of progressive renal decline. Following we discuss several implications of this switch.
First, the wide variation in rates of renal decline (between -50 and -5 ml/min/1.73m2/year) illustrated in Figure 4 may be determined by multiple mechanisms, which may be identified with the help of modern genetics, proteomics, and metabolomics platforms. Prediction of the rate of progression will not only help in stratification of patients according to risk of ESRD but will also direct development/selection of specific therapies to prevent or delay the onset of ESRD. Such personalized approaches are being developed in other fields [54-56].
Second, patients with the fastest renal decline (example in Figures 3A), referred to as rapid progressors, may be suitable for more aggressive therapies that have received little consideration so far. The ability to recognize their imminent risk of ESRD and high post-ESRD mortality could justify taking strong measures such as pancreas transplant [57], pre-emptive kidney transplant [58], cellular therapies [59] or aggressive new pharmacological therapies. The latter approach has been practiced in cancer therapies.
Third, the effectiveness of new therapies against progressive renal decline cannot be evaluated in clinical trials that use changes in urinary albumin excretion as an outcome. A much more reliable outcome measure is a change in the rate of renal decline or postponement of the time to events such as CKD stage 3, doubling of serum creatinine or ESRD. The efficiency of a study design based on either of the latter outcomes is significantly reduced (and cost significantly increased) by including patients who are non-decliners or have a slow rate of renal decline. Recruitment of non-decliners or slow decliners like the patients in Figure 3C would be counterproductive despite their having proteinuria, hypertension or impaired renal function. This points to the importance of having markers/algorithms to determine, on the basis of one or few baseline measurements, which patients are decliners (rapid, moderate, or slow) and which are non-decliners. At this time, measurements of circulating levels of TNFR1 or TNFR2 seem to be the best markers to achieve such a goal [36,37]. However, to increase the accuracy of prediction, baseline TNFR1 or TNFR2 needs to be combined with baseline levels of AER and eGFR.
Acknowledgments
This study was supported by the following grants: NIH grants DK-41526 and DK676381 and JDRF grants 1-2008-1018 and 17-2013-8 to A.S. Krolewski; and Diabetes Research Center - Joslin, Pilot and Feasibility Grant, P30DK036836 to M.A. Niewczas.
Footnotes
This article was presented at the 56th Annual Meeting of the Japanese Society of Nephrology.
Conflict of interest: The authors have declared that no Conflict of interest exists.
References
- 1.Rosolowsky ET, Skupien J, Smiles AM, Niewczas MA, Roshan B, Stanton R, Eckfeldt JH, Warram JH, Krolewski AS. Risk of ESRD in Type 1 Diabetes Remains High in spite of Renoprotection. J Am Soc Nephrol. 2011;22(3):545–53. doi: 10.1681/ASN.2010040354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bright R. Cases and observations illustrative of renal disease accompanied with the secretion of albuminous urine. Guy Hosp Rep. 1836;1:338–400. [PMC free article] [PubMed] [Google Scholar]
- 3.Kimmelstiel P, Wilson C. Intercapillary lesions in the glomeruli of the kidney. Am J Pathol. 1936;12:83–96. [PMC free article] [PubMed] [Google Scholar]
- 4.Miles DW, Mogensen CE, Gundersen HJG. Radioimmunoassay for urinary albumin using a single antibody. Scand J Clin Lab Invest. 1970;26:5–11. doi: 10.3109/00365517009049206. [DOI] [PubMed] [Google Scholar]
- 5.Parving HH, Oxenbøll B, Svendsen PA, Christiansen JS, Andersen AR. Early detection of patients at risk of developing diabetic nephropathy. A longitudinal study of urinary albumin excretion. Acta Endocrinol (Copenh) 1982;100(4):550–5. doi: 10.1530/acta.0.1000550. [DOI] [PubMed] [Google Scholar]
- 6.Viberti GC, Jarrett RJ, Keen H. Microalbuminuria as prediction of nephropathy in diabetics. Lancet. 1982;2(8298):611. doi: 10.1016/s0140-6736(82)90688-2. [DOI] [PubMed] [Google Scholar]
- 7.Mogensen CE, Christensen CK, Christensen Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med. 1984;311(2):89–93. doi: 10.1056/NEJM198407123110204. [DOI] [PubMed] [Google Scholar]
- 8.Parving HH, Mauer M, Ritz E. Diabetic Nephropathy. In: Brenner BM, editor. The Kidney. 7th. Philadelphia: Elsevier; 2004. pp. 1777–1818. [Google Scholar]
- 9.Caramori ML, Fioretto P, Mauer M. The need for early predictors of diabetic nephropathy risk: is albumin excretion rate sufficient? Diabetes. 2000;49:1399–408. doi: 10.2337/diabetes.49.9.1399. [DOI] [PubMed] [Google Scholar]
- 10.Tabaei BP, Al-Kassab AS, Ilag LL, Zawacki CM, Herman WH. Does microalbuminuria predict diabetic nephropathy? Diabetes Care. 2001;24(9):1560–6. doi: 10.2337/diacare.24.9.1560. [DOI] [PubMed] [Google Scholar]
- 11.Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med. 2003;348(23):2285–93. doi: 10.1056/NEJMoa021835. [DOI] [PubMed] [Google Scholar]
- 12.Giorgino F, Laviola L, Cavallo Perin P, Solnica B, Fuller J, Chaturvedi N. Factors associated with progression to macroalbuminuria in microalbuminuric Type 1 diabetic patients: the EURODIAB Prospective Complications Study. Diabetologia. 2004;47(6):1020–8. doi: 10.1007/s00125-004-1413-8. [DOI] [PubMed] [Google Scholar]
- 13.de Boer IH, Rue TC, Cleary PA, Lachin JM, Molitch ME, Steffes MW, Sun W, Zinman B, Brunzell JD, DCCT/EDIC Research Group. White NH, Danis RP, Davis MD, Hainsworth D, Hubbard LD, Nathan DM. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Arch Intern Med. 2011;171(5):412–20. doi: 10.1001/archinternmed.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsblom CM, Groop PH, Ekstrand A, Groop LC. Predictive value of microalbuminuria in patients with insulin-dependent diabetes of long duration. BMJ. 1992;305(6861):1051–3. doi: 10.1136/bmj.305.6861.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudberg S, Persson B, Dahlquist G. Increased glomerular filtration rate as a predictor of diabetic nephropathy--an 8-year prospective study. Kidney Int. 1992;41(4):822–8. doi: 10.1038/ki.1992.126. [DOI] [PubMed] [Google Scholar]
- 16.Ficociello L, Perkins BA, Silva KH, Finkelstein DM, Ignatowska-Switalska H, Gaciong Z, Cupples LA, Aschengrau A, Warram JH, Krolewski AS. Progression from microalbuminuria to proteinuria in individuals with type 1 diabetes treated with angiotensin converting enzyme inhibitors. Clin J Am Soc Nephrol. 2007;2(3):461–9. doi: 10.2215/CJN.03691106. [DOI] [PubMed] [Google Scholar]
- 17.Mathiesen ER, Hommel E, Giese J, Parving HH. Efficacy of captopril in postponing nephropathy in normotensive insulin dependent diabetic patients with microalbuminuria. BMJ. 1991;303(6794):81–7. doi: 10.1136/bmj.303.6794.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viberti G, Mogensen CE, Groop LC, Pauls JF. Effect of captopril on progression to clinical proteinuria in patients with insulin-dependent diabetes mellitus and microalbuminuria. European Microalbuminuria Captopril Study Group. JAMA. 1994;271(4):275–9. [PubMed] [Google Scholar]
- 19.Laffel LM, McGill JB, Gans DJ. The beneficial effect of angiotensin-converting enzyme inhibition with captopril on diabetic nephropathy in normotensive IDDM patients with microalbuminuria. North American Microalbuminuria Study Group. Am J Med. 1995;99(5):497–504. doi: 10.1016/s0002-9343(99)80226-5. [DOI] [PubMed] [Google Scholar]
- 20.Randomised placebo-controlled trial of lisinopril in normotensive patients with insulin-dependent diabetes and normoalbuminuria or microalbuminuria. The EUCLID Study Group. Lancet. 1997;349(9068):1787–92. [PubMed] [Google Scholar]
- 21.O'Hare P, Bilbous R, Mitchell T, O' Callaghan CJ, Viberti GC. Ace-Inhibitor Trial to Lower Albuminuria in Normotensive Insulin-Dependent Subjects Study Group. Low-dose ramipril reduces microalbuminuria in type 1 diabetic patients without hypertension: results of a randomized controlled trial. Diabetes Care. 2000;23(12):1823–9. doi: 10.2337/diacare.23.12.1823. [DOI] [PubMed] [Google Scholar]
- 22.Crepaldi G, Carta Q, Deferrari G, Mangili R, Navalesi R, Santeusanio F, Spalluto A, Vanasia A, Villa GM, Nosadini R. Effects of lisinopril and nifedipine on the progression to overt albuminuria in IDDM patients with incipient nephropathy and normal blood pressure. The Italian Microalbuminuria Study Group in IDDM. Diabetes Care. 1998;21(1):104–10. doi: 10.2337/diacare.21.1.104. [DOI] [PubMed] [Google Scholar]
- 23.Bojestig M, Karlberg BE, Lindström T, Nystrom FH. Reduction of ACE activity is insufficient to decrease microalbuminuria in normotensive patients with type 1 diabetes. Diabetes Care. 2001;24(5):919–24. doi: 10.2337/diacare.24.5.919. [DOI] [PubMed] [Google Scholar]
- 24.Tsalamandris C, Allen TJ, Gilbert RE, Sinha A, Panagiotopoulos, Cooper MF, Jerums G. Progressive decline in renal function in diabetic patients with and without albuminuria. Diabetes. 1994;43:649–55. doi: 10.2337/diab.43.5.649. [DOI] [PubMed] [Google Scholar]
- 25.Retnakaran R, Cull CA, Thorne KI, Adler AI, Holman RR, UKPDS Study Group Risk factors for renal dysfunction in type 2 diabetes. U.K. Prospective Diabetes Study 74. Diabetes. 2006;55:1832–9. doi: 10.2337/db05-1620. [DOI] [PubMed] [Google Scholar]
- 26.Perkins BA, Ficociello LH, Roshan B, Warram JH, Krolewski AS. In patients with Type 1 diabetes and new onset micro-albuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int. 2010;77:57–64. doi: 10.1038/ki.2009.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krolewski AS, Niewczas MA, Skupien J, Gohda T, Smiles A, Eckfeldt JH, Doria A, Warram JH. Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes Care. 2013 Aug 12; doi: 10.2337/dc13-0985. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merchant ML, Perkins BA, Boratyn GM, Ficociello LH, Wilkey DW, Barati MT, Bertram CC, Page GP, Rovin BH, Warram JH, Krolewski AS, Klein JB. Urinary peptidome may predict renal function decline in type 1 diabetes and microalbuminuria. J Am Soc Nephrol. 2009;20(9):2065–74. doi: 10.1681/ASN.2008121233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skupien J, Warram JH, Smiles AM, Niewczas MA, Gohda G, Pezzolesi MG, Cantarovich D, Stanton R, Krolewski AS. Early Renal Function Decline Predicts Risk of ESRD: 5-18 year Follow-up of Patients with Type 1 Diabetes and Proteinuria. Kidney Int. 2012;82(5):589–97. doi: 10.1038/ki.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krolewski M, Eggers PW, Warram JH. Magnitude of end-stage renal disease in IDDM: a 35 year follow-up study. Kidney Int. 1996;50(6):2041–6. doi: 10.1038/ki.1996.527. [DOI] [PubMed] [Google Scholar]
- 31.Yu MA, Sánchez-Lozada LG, Johnson RJ, Kang DH. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens. 2010;28(6):1234–42. [PubMed] [Google Scholar]
- 32.Mazzali M, Kanellis J, Han L, Feng L, Xia YY, Chen Q, Kang DH, Gordon KL, Watanabe S, Nakagawa T, Lan HY, Johnson RJ. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol. 2002;282(6):F991–F997. doi: 10.1152/ajprenal.00283.2001. [DOI] [PubMed] [Google Scholar]
- 33.Talbott JH, Terplan KL. The kidney in gout. Medicine. 1960;39:405–67. [PubMed] [Google Scholar]
- 34.Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–9. doi: 10.1053/j.ajkd.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Doria A, Niewczas M, Fiorina P. Can existing drugs approved for other indications retard renal function decline in patients with Type 1 diabetes and nephropathy. Semin Nephrol. 2012;32(5):437–44. doi: 10.1016/j.semnephrol.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gohda T, Niewczas MA, Ficociello LH, Walker WH, Skupien J, Rosetti F, Cullere X, Johnson AC, Crabtree G, Smiles AM, Mayadas TN, Warram JH, Krolewski AS. Circulating TNF Receptors 1 and 2 predict stage 3 of CKD in Type 1 diabetes. J Am Soc Nephrol. 2012;23(3):507–15. doi: 10.1681/ASN.2011060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niewczas MA, Gohda T, Skupien J, Smiles AM, Walker WH, Rosetti F, Cullere X, Eckfeldt JH, Doria A, Mayadas TN, Warram JH, Krolewski AS. Circulating TNF Receptors 1 and 2 Predict ESRD in Type 2 diabetes. J Am Soc Nephrol. 2012;23(3):507–15. doi: 10.1681/ASN.2011060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74:1143–55. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999;56:1627–37. doi: 10.1046/j.1523-1755.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- 40.Bohle A, Wehrmann M, Bogenschutz O, Batz C, Muller CA, Muller GA. The pathogenesis of chronic renal failure in diabetic nephropathy. Investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187:251–9. doi: 10.1016/s0344-0338(11)80780-6. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki D, Miyazaki M, Naka R, Koji T, Yagame M, Jinde K, Endoh M, Nomoto Y, Sakai H. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes. 1995;44:1233–8. doi: 10.2337/diab.44.10.1233. [DOI] [PubMed] [Google Scholar]
- 42.Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, Takeda SI, Takasawa K, Yoshimura M, Kida H, Kobayashi KI, Mukaida N, Naito T, Matsushima K, Yokoyama H. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 2000;58:1492–9. doi: 10.1046/j.1523-1755.2000.00311.x. [DOI] [PubMed] [Google Scholar]
- 43.Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, Nitsche A, Kiss E, Bleich M, Grone HJ, Nelson PJ, Schlondorff D, Cohen CD, Kretzler M. European Renal cDNA Bank (ERCB) Consortium: Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55:2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 44.Morcos M, Sayed AA, Bierhaus A, Yard B, Waldherr R, Merz W, Kloeting I, Schleicher E, Mentz S, Abd el Baki RF, Tritschler H, Kasper M, Schwenger V, Hamann A, Dugi KA, Schmidt AM, Stern D, Ziegler R, Haering HU, Andrassy M, van der Woude F, Nawroth PP. Activation of tubular epithelial cells in diabetic nephropathy. Diabetes. 2002;51:3532–44. doi: 10.2337/diabetes.51.12.3532. [DOI] [PubMed] [Google Scholar]
- 45.Wolkow PP, Niewczas MA, Perkins B, Ficociello LH, Lipinski B, Warram JH, Krolewski AS. Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. J Am Soc Nephrol. 2008;19:789–97. doi: 10.1681/ASN.2007050556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol. 2000;11:152–76. doi: 10.1681/ASN.V111152. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int. 2001;59:1626–40. doi: 10.1046/j.1523-1755.2001.0590051626.x. [DOI] [PubMed] [Google Scholar]
- 48.Anders HJ, Vielhauer V, Schlondorff D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int. 2003;63:401–15. doi: 10.1046/j.1523-1755.2003.00750.x. [DOI] [PubMed] [Google Scholar]
- 49.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82:172–83. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wanic K, Krolewski B, Ju W, Placha G, Niewczas MA, Walker W, Warram JH, Kretzler M, Krolewski AS. Transcriptome analysis of Proximal Tubular Cells (HK-2) exposed to Urines of Type 1 Diabetes Patients at Risk of Early Progressive Renal Function Decline. PLoS One. 2013;8(3):e57751. doi: 10.1371/journal.pone.0057751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–69. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krolewski AS, Warram JH, Forsblom C, Smiles A, Thorn L, Skupien J, Harjutsalo V, Stanton R, Eckfeldt JH, Inker LA, Groop PH. Serum concentration of cystatin C and risk of ESRD in diabetes. Diabetes Care. 2012;35(11):2311–6. doi: 10.2337/dc11-2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krolewski AS, Bonventre JV. High risk of ESRD in type 1 diabetes: new strategies are needed to retard progressive renal function decline. Semin Nephrol. 2012;32(5):407–14. doi: 10.1016/j.semnephrol.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Degome EM, Rivera G, Lilly SM, Usman MH, Mohler ER. Personalized vascular medicine; individualizing drug therapy. Vasc Med. 2011;16(5):391–404. doi: 10.1177/1358863X11422251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chokrungvaranon N, Deer J, Reaven PD. Intensive glycemic control and cardiovascular disease; are there patients who may benefit? Postgrad Med. 2011;123(6):114–23. doi: 10.3810/pgm.2011.11.2501. [DOI] [PubMed] [Google Scholar]
- 56.Higgins MJ, Baselga J. Targeted therapies for breast cancer. J Clin Invest. 2011;121(10):3797–803. doi: 10.1172/JCI57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cantarovich D, Perrone V. Pancreas transplant as treatment to arrest renal function decline in patients with type 1 diabetes and proteinuria. Semin Nephrol. 2012;32(5):432–6. doi: 10.1016/j.semnephrol.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 58.Pavlakis M, Kher A. Pre-emptive kidney transplantation to improve survival in patients with Type 1 diabetes and imminent risk of ESRD. Semin Nephrol. 2012 Sep;32(5):505–11. doi: 10.1016/j.semnephrol.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 59.Gilbert RE, Zhang Y, Yuen DA. Cell therapy for diabetic nephropathy: Is the future, now? Semin Nephrol. 2012;32(5):486–93. doi: 10.1016/j.semnephrol.2012.07.012. [DOI] [PubMed] [Google Scholar]