

Abstract
Host inflammatory responses against pathogenic organisms can be abrogated by commensals; however, the molecular mechanisms by which pathogenesis is prevented are still poorly understood. Previous studies demonstrated that administration of a single dose of Bacillus subtilis prevented disease and inflammation by the enteric mouse pathogen Citrobacter rodentium, which causes disease similar to the human pathogen enteropathogenic E. coli. No protection was observed when an exopolysaccharide (EPS)-deficient mutant of B. subtilis was used, suggesting that EPS are the protective factor. Here, we isolated and characterized EPS, and showed that they also prevent C. rodentium-associated intestinal disease after a single injection. Protection is TLR4-dependent, since EPS-treated TLR4 KO mice developed disease. Further, protection could be conveyed to wild-type (wt) mice by adoptive transfer of macrophage-rich peritoneal cells from EPS-treated mice. We found that EPS specifically bind peritoneal macrophages and because mice lacking MyD88 signaling in myeloid cells were not protected by EPS, we conclude that bacterial EPS prevent colitis in a TLR4-dependent manner that requires myeloid cells. These studies provide a simple means of preventing intestinal inflammation caused by enteric pathogens.
Introduction
The gastrointestinal microbiota contributes to the development and maintenance of the host immune system. One benefit of a healthy microbiota is protection from colitis induced by enteric pathogens as well as by inflammatory agents such as dextran sulfate or 2,4,6-trinitrobenzene sulfonic acid (1, 2). Although much work has been done to identify specific bacteria that prevent colitis, many questions remain about the mechanisms by which these bacteria elicit a protective response. We previously showed that a single oral dose of Bacillus subtilis protects mice from disease induced by the enteric pathogen Citrobacter rodentium (1), which shares many characteristics with the human pathogen enteropathogenic E. coli. Symptoms of infection include diarrhea, systemic increases in proinflammatory cytokines, and altered colonic architecture, such as crypt hyperplasia, goblet cell depletion, and infiltration of immune cells, including neutrophils and T cells. However, mice administered B. subtilis in addition to C. rodentium display no evidence of diarrhea, have normal levels of proinflammatory cytokines, and normal colonic architecture (1).
During infection, C. rodentium disrupts the intestinal barrier (3), resulting in translocation of lumenal contents and activation of the host pattern recognition receptors (PRRs), which include Toll like receptors (TLRs). TLRs recognize conserved motifs of microbial proteins (e.g., flagella), lipids (e.g., LPS), and nucleic acids (e.g., CpG) as well as host Danger-Associated Molecular Patterns (4). Activation of TLRs results in translocation of NF-κB to the nucleus, production of chemokines and cytokines, and ultimately recruitment of immune cells to the site of infection (4). This inflammatory cascade is needed to clear the pathogen, but it also damages the host tissues (5). For example, MyD88 KO mice do not develop colonic hyperplasia or recruit neutrophils, but succumb to infection. In contrast, most immunocompetent strains of mice clear C. rodentium 3 to 4 weeks post-infection.
B. subtilis is a gram positive spore-forming bacterium present in the gastrointestinal tract of both humans and mice (8, 9). Several groups report that select probiotic strains of B. subtilis relieve the symptoms associated with antibiotic-associated diarrhea and irritable bowel syndrome in human patients; however the mechanisms of protection have not been well established (6, 8). In a previous study, we showed that an exopolysaccharide (EPS) mutant failed to prevent C. rodentium-associated disease, suggesting that EPS are the bacterial components mediating protection (10). EPS are secreted heterogeneous structures comprised primarily of carbohydrates which not only sometimes coat bacteria, but are major components of the biofilm matrix. The role of EPS during pathogen infection is well appreciated. For example, pathogenic S. aureus are coated with an EPS-containing capsule which prevents phagocytosis and allows adherence of the bacteria to host tissues and subsequent immune evasion (11). Less understood is the role of bacterial EPS during probiosis. EPS may be important for probiotic or commensal organisms to establish and maintain an intestinal niche which could prevent pathogen colonization. Alternatively, gut metabolism of EPS could contribute to short chain fatty acid synthesis, a process that regulates intestinal permeability (12). Interestingly, a few groups have demonstrated that EPS suppress disease by modulating the host inflammatory response via TLR2 signaling (13, 14). Collectively, these studies suggest that bacterial EPS, such as those produced by B. subtilis, could prevent intestinal disease using one or more of several different mechanisms, including alteration of pathogen colonization, reduction of gut permeability, and/or immunomodulation of the host response. We show here that B. subtilis treatment did not alter pathogen colonization, nor prevent disruption of the epithelium, and we hypothesized that protection by B. subtilis EPS is a result of host immune modulation. After purifying EPS and showing that they mediate protection, we identified host immune cells that bind EPS, and further showed that protection requires TLR4 and MyD88-signaling myeloid cells. Further, cells from wild-type (wt) and TLR4 KO mice were adoptively transferred to naïve wt mice to test if these cells conveyed protection from enteric disease caused by C. rodentium and to identify which cells utilize TLR4. These studies identify bacterial polysaccharides which after a single injection, have the capacity to prevent colitis in an infectious disease model in a TLR4-dependent manner.
Materials and methods
Reagents and mice
Anti-F4/80 (clone BM8), anti-CD11b (clone M1/70) were obtained from BioLegend (San Diego, CA); donkey anti-rabbit Ig was obtained from Jackson laboratory (Bar Harbor, ME). All other reagents were purchased from Sigma unless otherwise noted. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Usage Committee at Loyola University Medical Center, Maywood, IL. Specific pathogen-free C57Bl/6, MyD88 KO, and TLR4 KO founders were purchased from Jackson Laboratory (Bar Harbor, ME). Mice lacking MyD88 in myeloid cells and epithelial cells were generated by crossing a Lyz2-Cre or Villin-Cre transgenic mouse, respectively, to a MyD88 floxed mouse as previously described (3). Mice utilized for these experiments (4 to 8 weeks of age) were bred at Loyola University Chicago. Sterile standard chow and tap water were given to mice ad libitum.
Bacterial and spore preparation
Wildtype B. subtilis 3610 spores were germinated via exhaustion as described (4). On the day of administration, B. subtilis spores were washed with ice cold water, re-suspended in 100 μL of PBS, and administered to mice via oral gavage. For infection studies, C. rodentium ATCC 51459 was cultured for 16 hours in LB medium, washed once in PBS, and an infectious dose was resuspended in 100 μL of sterile PBS for administration to mice by oral gavage. MyD88 KO and epithelial MyD88 deficient mice received 107 CFUs; all other mouse strains received 5 × 108 CFUs of pathogen.
In vivo imaging of C. rodentium
As previously described (5), C. rodentium ICC180 (C. rodentium lux+) was grown overnight at 37°C in LB and orally gavaged into C57Bl/6 mice (~5 ° × 108 CFU per mouse). Assessment of bioluminescence (photons s−1 cm−2 sr−1) in living animals was measured using the IVIS100 system (Xenogen Corporation, Alameda, CA). A photograph (grayscale reference image) was taken under low illumination prior to quantification of photons emitted from C. rodentium ICC180 (medium binning, 5 minute exposure) using the software program Living Image (Xenogen). A pseudocolor heat map image representing light intensity (blue [least intense] to red [most intense]) was generated using Living Image software and superimposed over the grayscale reference image.
C. rodentium colonization
C. rodentium colonization was assessed in fresh fecal samples homogenized in 500 μL sterile 20% glycerol in PBS. For mucosal studies, colonic fecal contents were removed and the tissue flushed with sterile PBS. The colon was homogenized in 2 mL sterile 20% glycerol in PBS. Serial dilutions were cultured on selective MacConkey plates for 16 hrs at 37°C; only colonies that displayed the characteristic pink center surrounded by a white rim (C. rodentium) were counted. Colonization was calculated and expressed as CFUs per gram feces.
Exopolysaccharide preparation
Exopolysaccharides were isolated from B. subtilis DS991 (sinRtasA mutant), a strain that produces and secretes large amounts of EPS; material from this strain is designated EPS+ (6). As a control, we used DS5187 (sinRtasAepsH mutant), a strain that does not produce EPS (6) and material from this strain is referred to as EPS−. EPS were isolated as described (6). Briefly, stationary phase supernatants were mixed with an equal volume of 100% EtOH at 4°C for 90 minutes to precipitate the EPS. The precipitant was pelleted (15000×g, 4°C, 20 minutes), washed in PBS, and re-supended in 0.1 M Tris. Samples were digested with DNase (67 μg/mL) and RNase (330 μg/mL) at 37°C; after 1 hr, proteinase K (40 μg/mL) was added and samples were incubated at 55°C for 1 hr. EPS was EtOH precipitated, resuspended in 0.1M Tris pH 8, and further purified by gel filtration on an S1000 column in 0.1 M Tris pH 8 and then de-salted by dialysis. EPS was quantified by a colorimetric phenol sulfuric acid assay using serial dilutions of fructose as standard (7). Sample purity was assessed by immunoelectrophoresis and western blot analysis using anti-EPS antiserum.
Composition and linkage analysis of EPS
These analyses were performed at the Complex Carbohydrate Research Center (U. Georgia) (8). GC/MS analysis of TMS methyl glucosides was performed on an Agilent 7890A GC interfaced to a 5975C MSD, using an Agilent DB-1 fused silica capillary column (30m × 0.25 mm ID) and linkages were determined on an Agilent 7890A GC interfaced to a 5975C MSD (mass selective detector, electron impact ionization mode); separation was performed on a Supelco 2380 fused silica capillary column (30m × 0.25 mm ID).
Generation of EPS Specific Antibodies
A New Zealand White rabbit was immunized by intramuscular and subcutaneous injection of 100μg EPS in TiterMax Gold adjuvant. Three weeks post primary immunization, the rabbit was boosted with 100μg of EPS in adjuvant. Eight days later, serum was collected. Antibody to EPS was detected by western blot analysis using donkey anti-rabbit (H&L)-HRP (Jackson Labs) antibodies as secondary antibody, and by immunoelectrophoresis followed by staining with Coomassie Brilliant blue to visualize antigen/antibody arcs of precipitation.
Study design
B. subtilis spores (109 in 100 μL PBS, orally administered) or 200 μL of EPS (1 mM in 0.1 M Tris, i.p.) or hydraluronic acid (PBS, i.p.) were administered to mice 24 hours prior to infection with C. rodentium by oral gavage. Age and gender matched mice were utilized for each experiment. To assess disease, all mice were euthanized 10 days post infection (dpi) and tissues collected, except for the MyD88 KO mice which were euthanized 9 dpi. These days were chosen because at these times the pathogen is well established in each strain and colitis is evident (9–12). Before euthanization, blood was collected. Serum KC levels were assessed by ELISA (R&D Systems, Minneapolis, MN). To assess diarrhea, feces were examined and scored 1 to 4 (13): No diarrhea (hard, dry pellets) were scored as 1; slightly soft stool (mild diarrhea) = 2; very soft stool (moderate diarrhea) = 3; and unformed stool (severe diarrhea) = 4. Distal colons were collected and processed for histological analysis as follows: Colons were fixed overnight in 10% formalin-buffered phosphate, dehydrated through an alcohol gradient, cleared with xylene and infiltrated with paraffin. Tissues were sectioned longitudinally at 4 μM and stained with hematoxylin and eosin (H&E). Epithelial hyperplasia in the distal colon was determined from images of each colon taken with a Leica DM IRB microscope equipped with MagnaFire charge-coupled device camera as described (14). Five well-oriented crypt heights/mouse were measured from 2–3 regions.
Assessment of EPS binding to Cells
Peritoneal cells were obtained from mice (4 to 6 weeks of age) injected i.p. with 5 mL DMEM (10% fetal bovine serum). After lysing red blood cells, cells were incubated with EPS, washed and then incubated with anti-F4/80 (clone BM8), anti-CD11b (clone M1/70), or anti-EPS followed by donkey anti-rabbit Ig as secondary antibody. Fluorescence intensity was assessed by flow cytometry.
Assessment of EPS-induced cytokine production
Peritoneal cells were obtained from euthanized mice (4 to 6 weeks of age) injected i.p. with 5 mL DMEM (10% fetal bovine serum). After lysing red blood cells, cells were incubated with EPS (5, 15, or 30 μg/mL), LPS (100 ng/mL) or Pam3Cys4 (100 ng/mL) and supernatant was collected at 2 and 6 hours for measurement of KC and TNFα, respectively, by ELISA. As a control, the same volume of material from the non-EPS producing strain was utilized (30 μg/mL).
Transfer studies
Peritoneal cells were isolated from mice (4–6 weeks of age) injected i.p. with 5 mL DMEM (10% fetal bovine serum) 2 to 3 days post treatment with EPS (i.p.). Cells (6 × 104) were injected (300 μL, i.p.) into naïve mice (4–6 weeks of age) at +1, −1, and −3 dpi with C. rodentium.
Statistical analysis
All experiments were performed a minimum of three times and analyzed using the Student’s t test. Error bars denote SEM. Differences were considered statistically significant if p < 0.05.
Results
Effect of B. subtilis on C. rodentium colonization and pathogen-induced gut leakiness
B. subtilis could prevent disease by altering pathogen adherence and/or colonization, by maintaining epithelial barrier integrity, or by changing the host inflammatory response. To test if pathogen colonization was altered in the presence of B. subtilis, we utilized a lux+ strain of C. rodentium in combination with IVIS as well as traditional plating techniques. Mice were given B. subtilis (109 CFU) followed 24 hr later by C. rodentium (5 × 108 CFU) by oral gavage; and using IVIS, we detected the lux+ C. rodentium during the course of disease. We found that administration of B. subtilis did not change the localization or quantity of fluorescence of C. rodentium (Fig S1). We also assessed the quantity of adherent and lumenal C. rodentium by plating colonic (adherent) and fecal (lumenal) samples and did not observe any differences when mice were treated with B. subtilis (Fig 1A and B). These data suggest that B. subtilis does not protect mice by altering the localization, adherence, or density of the pathogen.
Figure 1. Colonization and gut-induced leakiness in C. rodentium-infected mice after treatment with B. subtilis.
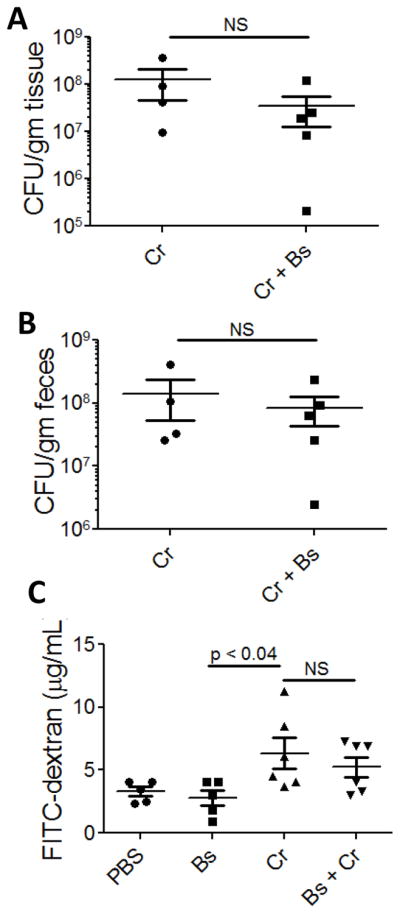
Mucosal (A) and lumenal (B) colonization of C. rodentium 11 dpi. FITC-dextran in serum of mice 11 dpi with lux+ C. rodentium (C). Cr, C. rodentium-infected mice; Cr + Bs, mice treated with B. subtilis 24 hr prior to C. rodentium; PBS, phosphate-buffered saline. The results are averages from 2 independent experiments and a total of 5–6 mice were assessed for each group. No statistical difference was found between C. rodentium-infected mice and those that received B. subtilis prior to infection.
To test if B. subtilis prevents disease by maintaining epithelial barrier integrity, we orally administered FITC-dextran to mice and then assessed the serum for fluorescence. If B. subtilis functions by preventing epithelial damage, then we expected to detect little to no FITC-dextran in serum. However, we found that mice infected with C. rodentium as well as those that received B. subtilis prior to pathogen infection had increased quantities of serum FITC-dextran (6.3 μg/mL and 5.2 μg/mL, respectively) when compared to PBS-treated control mice (3.3 μg/mL) (Fig 1C). These data suggest that B. subtilis does not protect from C. rodentium-induced colitis by preventing pathogen-induced disruption of the epithelium.
Analysis of EPS composition and structure
Since an epsH mutant, which does not produce EPS, failed to protect mice from C. rodentium-induced disease (9), we hypothesized that EPS may have immunomodulatory activity. To begin to test this idea, we first isolated EPS and analyzed its structure. EPS were purified from the sinRtasA mutant (DS991), which overproduces and secretes EPS into the supernatant (EPS+); as a control, supernatant of the sinRtasAepsH mutant (DS5187), which is unable to synthesize EPS (6), was subjected to the same purification process (EPS−). The purity of EPS were assessed by immunoelectrophoresis and western blot analysis using rabbit anti-EPS antiserum. By immunoelectrophoresis, we observed only a single precipitation arc (Fig 2A); no bands were observed with pre-immune serum or with the EPS− material (data not shown). By western blot analysis, we observed only a single band of the expected size (~300 kDa) produced by the EPS+ strain (Fig S2). The OD280 and OD260 of purified EPS at a concentration of 1 mg/ml was 0.091 and 0.013, respectively, indicating that EPS were contaminated by little to no protein or nucleic acid.
Figure 2. Assessment of the B. subtilis exopolysaccharides on C. rodentium-associated disease 10 days post-infection dpi of wt mice.

A) Immunoelectrophoresis analysis of purified EPS (arrow points to precipitation arc). B) Average colonic crypt heights from each treatment group. Serum KC levels (C), and evidence of diarrhea (D) were also used as disease markers. Results are averages from at least three independent experiments; a total of 5–12 mice were assessed for each group. PBS, phosphate-buffered saline; EPS+, exopolysaccharide from B. subtilis strain DS991; EPS−, material from B. subtilis strain DS5187; Cr, C. rodentium. Representative images of H&E stained colons from wildtype mice (100X). Images are representative of mice that received EPS from DS991 prior to C. rodentium infection (E), or material from the non-EPS producing strain DS5187 prior to pathogen infection (F). Representative images from myeloid MyD88 KO mice (G) and epithelial MyD88 KO mice (H) treated with EPS prior to infection with C. rodentium.
The structure of purified EPS was analyzed by gas chromatography/mass spectrometry at the Complex Carbohydrate Research Center (U. Georgia), and the carbohydrate portion was found to be primarily mannose (88%) and glucose (11.9%) (Table I). Further structural analysis to determine the carbohydrate linkages revealed that the primary linkages are 2,6-mannose (31.8%), terminal mannose (29.9%), 3-mannose (15%), 2-mannose (4.7%), 6-mannose (4.7%), 6-glucose (3.7%) and terminal glucose (3.5%) (Table II); these data are consistent with the compositional analysis which indicates that mannose is the primary component of EPS.
Table I.
Analysis of EPS composition.
| Glycosyl residue | Mass (μg) | Mol %1 |
|---|---|---|
| Ribose | n.d. | - |
| Arabinose | n.d. | - |
| Rhamnose | n.d. | - |
| Fucose | n.d. | - |
| Xylose | n.d. | - |
| Glucuronic Acid | n.d. | - |
| Galacturonic acid | n.d. | - |
| Mannose | 250.6 | 88.0 |
| Galactose | n.d. | - |
| Glucose | 33.9 | 11.9 |
| N-Acetyl Galactosamine | n.d. | - |
| N-Acetyl Glucosamine | 0.2 | 0.1 |
| N-Acetyl Mannosamine | n.d. | - |
| Σ = | 284.7 | 100 |
Values are expressed as mole percent of total carbohydrate. The total percentage may not add to exactly 100% due to rounding.
Table II.
Linkage analyses of EPS by gas chromatograph and mass spectroscopy.
| Glycosyl Linkage Residue EPS % Present | EPS % Present |
|---|---|
| 2-Rhamnopyranosyl residue (2-Rha) | 0.1 |
| Terminal Mannopyransosyl residue (t-Man) | 29.9 |
| Terminal Glucopyranosyl residue (t-Glc) | 3.5 |
| 3 linked Glucopyranosyl residue (3-Gle) | 0.2 |
| 2 linked Mannopyranosyl residue (2-Man) | 4.7 |
| 3 linked Mannopyranosyl residue (3-Man) | 15.0 |
| 2 linked Glucopyranosyl residue (2-Glc) | 0.3 |
| 4 linked Mannopyranosyl residue (4-Man) | 0.4 |
| 6 linked Mannopyranosyl residue (6-Man) | 4.7 |
| 6 linked Glucopyranosyl residue (6-Glc) | 3.7 |
| 4 linked Glucopyranosyl residue (4-Glc) | 1.3 |
| 2,3 linked Mannopyranosyl residue (2,3-Man) | 0.3 |
| 3,4 linked Mannopyranosyl residue (3,4-Man) | 0.1 |
| 2,4 linked Mannopyranosyl residue (2,4-Man) | 0.2 |
| 4,6 linked Mannopyranosyl residue (4,6-Man) | 0.2 |
| 3,6 linked Glucopyranosyl residue (3,6-Glc) | 0.3 |
| 3,6 linked Mannopyranosyl residue (3,6-Man) | 0.4 |
| 2,6 linked Mannopyranosyl residue (2,6-Man) | 31.8 |
| 4,6 linked Glucopyranosyl residue (4,6-Glc) | 0.6 |
| 2,6 linked Glucopyranosyl residue (2,6-Glc) | 0.6 |
| 2,3,6 linked Mannopyranosyl residue (2,3,6-Man) | 0.5 |
| 2,4,6 linked Mannopyranosyl residue (2,4,6-Man) | 0.6 |
| 2,3,4,6 linked Mannopyranosyl residue (2,3,4,6-Man) | 0.5 |
| 4 linked N-acetyl Glucosamine (4-GlcNAc) | 0.1 |
Effect of B. subtilis EPS on C. rodentium-associated disease
To test if EPS are sufficient to prevent disease, we administered purified EPS i.p. to wt mice, and 24 hr later infected them with C. rodentium. Disease was assessed 10 dpi by examining the colon, serum, and feces. Mice that received EPS displayed no evidence of disease (Fig 2B, C, D and E), whereas mice that received material from the non-EPS producing strain (EPS−), or no treatment other than C. rodentium, had altered colonic architecture (Fig 2B, E, and F), increased levels of pro-inflammatory KC (Fig 2C), and diarrhea (Fig 2D). These data indicate that EPS from B. subtilis are sufficient to protect wt mice from inflammation after infection with C. rodentium.
Role of MyD88 and TLR4 in B. subtilis-mediated protection
Bacterial carbohydrates are ligands for many host PRRs, including C-type lectins and TLRs, which are MyD88-dependent. C. rodentium-induced crypt hyperplasia is dependent on MyD88 signaling (10) and because we observed that B. subtilis and EPS suppressed crypt hyperplasia, we hypothesized that B. subtilis could mediate protection via this signaling pathway. Because MyD88 KO mice are highly susceptible to C. rodentium and succumb to disease 3–6 days post infection (10, 15), we titrated the C. rodentium inoculum and found a minimal dose (107 CFU) for which all mice developed disease (soft stool) at 5–7 dpi, similar to that observed with wt mice. After infection of MyD88 KO mice with C. rodentium (107 CFU), mice lost weight (8–9 dpi), failed to clear the pathogen, and succumbed to disease by 11 dpi; administration of B. subtilis did not protect mice (data not shown). We conclude that MyD88 signaling plays a role in B. subtilis-mediated protection of C. rodentium-induced colitis.
To identify the relevant MyD88-dependent TLR needed for protection in our model, we began to test individual TLR KO mice for susceptibility to C. rodentium after EPS treatment and started with TLR4 KO. EPS-treated TLR4 KO mice infected with C. rodentium showed evidence of disease including crypt hyperplasia, elevated serum KC, and diarrhea comparable to infected animals without EPS (Fig 3A–C). As expected, neither material from the (EPS−) strain nor B. subtilis spores protected TLR4 KO mice from disease induced by the enteric pathogen (data not shown). These data suggest that EPS mediate protection via TLR4.
Figure 3. Assessment of C. rodentium-associated disease in EPS or TLR4 agonist-treated TLR4 KO or wt mice.

Quantification by ELISA of pro-inflammatory KC in serum of TLR4 KO mice infected with C. rodentium (Cr) with or without EPS (EPS+); PBS and EPS+ are negative controls (A). Summary of colonic crypt heights from each treatment group (B). Diarrhea (C) also served as a disease marker. Results are averages from at least three independent experiments; a total of 5–10 mice were assessed for each group. (D–F): Wt mice were treated with 50, 100, or 150 μg of the TLR4 agonist hyaluronic acid (HA) prior to infection and then assessed for disease 10 dpi. Serum KC was measured by ELISA (D), colonic crypt heights from each treatment group were measured (E), and diarrhea (F) also served as a disease marker. Results are averages from at two independent experiments; a total of 4–5 mice were assessed for each group.
Because TLR4 is required for EPS-mediated protection, we tested if a TLR4 agonist, hyaluronic acid was sufficient to prevent C. rodentium-associated disease. Mice were injected with hyaluronic acid (i.p.) prior to infection with C. rodentium and disease was assessed 10 dpi. Hyaluronic acid did not protect mice at any of the concentrations tested (Fig 3D–F), indicating that a TLR4 agonist is not capable of, or sufficient for preventing disease. These data suggest that EPS does not act as a TLR4 agonist, but instead, may prevent disease by antagonizing TLR4.
Identification of EPS-binding cells
Because i.p. administration of EPS prevents C. rodentium-induced colitis, we searched by flow cytometry for peritoneal cells that bind EPS. We found that EPS bind cells in the granulocyte gate, with little to no binding to cells in the lymphocyte gate (Fig 4A and B). Over 70% of cells in the granulocyte gate are F4/80+CD11b+ macrophages and we found that EPS bind nearly all of these peritoneal macrophages (4C and D). While macrophages are “sticky” and readily bind polysaccharides, we think EPS binding is specific because EPS did not bind splenic macrophages, murine macrophage-like RAW264.7 cells, or human monocytoid THP-1 cells (data not shown). EPS bound peritoneal macrophages from TLR4 KO mice (Fig 4D), indicating that while EPS-mediated protection requires TLR4 signaling, EPS either do not bind directly to TLR4 on the peritoneal macrophages, or EPS bind to both TLR4 and another receptor.
Figure 4. Flow cytometric analysis of EPS-binding to peritoneal cells from wt and TLR4 KO mice.

FSC vs. SSC (A); granulocyte and lymphocyte binding to EPS (B) - gray peak is negative isotype control; staining of wt or TLR4 KO F4/80+CD11b+ gated cells with EPS (C and D). Fluorescence intensity represents EPS binding. Data shown are from one of three independent experiments.
Effect of EPS on cytokine production by wt and TLR4 KO peritoneal cells
We examined the effect of EPS on peritoneal cells in vitro by incubating EPS with wt or TLR4 KO peritoneal cells and examining cytokine production by ELISA. We observed that even at high concentrations EPS did not induce KC or TNFα production by wt or TLR4 KO peritoneal cells (Fig 5). As expected, wt, but not TLR4 KO, peritoneal cells produced KC and TNFα when incubated with the TLR4 agonist (LPS) and all cells produced pro-inflammatory cytokines in response to a TLR2 agonist (Pam3Cys4). We also used ELISA to test for production of IL-10 by peritoneal cells from EPS-treated mice, but found no evidence that EPS induced production of IL-10 (data not shown). These data indicate that EPS does not induce a pro-inflammatory response by peritoneal cells. Similarly, we have no evidence that an IL-10-mediated anti-inflammatory response is stimulated.
Figure 5. ELISA analysis of cytokines induced by in vitro culture of EPS with peritoneal cells from wt and TLR4 KO mice.

Peritoneal cells were incubated with EPS (EPS+) (30 μg/ml), material from the non-EPS producing strain (EPS−) (30 μg/mL), LPS (100 ng/mL), Pam3Cys4 (100 ng/mL), or without addition (sham). Results are averages from three independent experiments. ND, not detectable
Effect of myeloid-specific MyD88 deletion on EPS-mediated protection
Since EPS bound peritoneal macrophages and because MyD88 was required for protection, we hypothesized that mice lacking MyD88 in myeloid cells would be susceptible to C. rodentium-induced disease after treatment with EPS. We titrated the amount of C. rodentium needed to induce disease and determined that 5 × 108 CFUs, the same infectious dose used with wt mice, was sufficient to induce disease and that at lower doses not all mice were colonized successfully with the pathogen. Mice were treated with EPS (i.p.), and as hypothesized, these mice developed disease (Fig 6 and 2G), including elevated serum KC, crypt hyperplasia, and diarrhea. We also tested if EPS could protect mice lacking MyD88 signaling in epithelial cells from C. rodentium and no disease was observed in these mice (Fig 6 and 2H), demonstrating that the requirement for MyD88 in myeloid cells is specific. We conclude that MyD88 signaling by myeloid cells is required for EPS-mediated protection.
Figure 6. Assessment of C. rodentium-associated disease in EPS-treated mice lacking MyD88 in myeloid or epithelial cells.

Myeloid MyD88 KO and epithelial MyD88 KO mice were treated with EPS (EPS+) (i.p.) 1 day prior to infection with C. rodentium (Cr) and disease was assessed 10 dpi. Injection with PBS and EPS+ alone served as negative controls. Serum KC levels (A), colonic crypt height (B), and diarrhea (C) were used as disease markers. Results are averages from at least two independent experiments and a total of 2–5 mice were assessed for each group.
Effect of adoptively-transferred EPS-treated peritoneal cells on development of C. rodentium-induced disease
Because we observed that i.p. administration of EPS prevented disease and because EPS bound peritoneal macrophages, we hypothesized that peritoneal cells from an EPS-treated mouse could convey protection to naïve mice infected with C. rodentium. Peritoneal cells were collected by lavage 2–3 days after i.p. injection with EPS+ or EPS−, and 6 × 104 cells were injected i.p. into recipient mice on −1, 1, and 3 dpi. Disease was assessed 10 dpi, and we found no evidence of disease in mice that received peritoneal cells from EPS+-treated mice (Fig 7A–C) when compared to PBS control mice (Fig 2B–D). In contrast, crypt hyperplasia, elevated KC, and diarrhea were evident in mice that received peritoneal cells treated with material from the non-EPS producing B. subtilis strain (EPS−) (Fig. 7A–C). These data indicate that following treatment with EPS, cells within the peritoneal cavity can suppress inflammation and that this protective effect is only observed with cells from EPS+-treated mice. At the time of transfer, peritoneal cells did not have detectable EPS bound, but instead, the cells bound freshly added EPS (similar to that shown in Fig 4C and D). We hypothesize that following EPS administration, EPS are internalized or degraded by host cells and that the protection observed after transfer of peritoneal cells is not due to native EPS transferred with the cells, but instead to cells that were activated by the EPS injection.
Figure 7. Assessment of disease after transfer of peritoneal cells from EPS-treated wt and TLR4 KO mice to C. rodentium-infected wt mice.

Donor wt mice were treated with EPS+ or EPS− material (i.p.) 2–3 days before peritoneal cells (6 × 104) were transferred i.p. to naïve recipient wt (A–C) mice 1 day prior to, 1 dpi and 3 dpi with C. rodentium. Peritoneal cells from wt or TLR4 KO mice similarly treated with EPS+ were transferred i.p. into naïve wt or TLR4 KO mice (D–F). Disease was assessed for all mice 10 dpi; serum KC (A and D), crypt hyperplasia (B and E), and diarrhea (C and F) were used as disease markers. KC quantification, crypt height and diarrhea scores can be compared to uninfected (PBS-treated) mice shown in Figure 2B–D. Results are averages from at least three independent experiments and a total of 6–10 mice were assessed for each group.
Because TLR4 signaling is necessary for protection by EPS, we tested if peritoneal cells require TLR4 signaling. TLR4 KO and wt mice were treated with EPS, and donor peritoneal cells were transferred into wt or TLR4 KO recipients with the expectation that if TLR4-signaling is required by peritoneal cells to mediate protection, then EPS-treated peritoneal cells from mice lacking TLR4 will not protect wt mice from pathogen-associated disease. As predicted, we found that EPS-treated TLR4 KO peritoneal cells did not protect wt mice from disease as evidenced by elevated serum KC, crypt hyperplasia, and diarrhea (Fig 7D–F and PBS controls in Fig 2B–D). In contrast, TLR4 KO recipient mice were protected by injection of EPS-treated peritoneal cells from wt mice. These data confirm the requirement of TLR4 in our model and suggest that peritoneal cells utilize TLR4 to mediate protection.
Discussion
The peritoneal cavity contains a variety of host immune cells, the most numerous of which are macrophages (~30–50%) and B cells (~40%) (16). To identify the cells that contribute to protection in our model, we searched for cells that bind EPS and found that they bind peritoneal F4/80+CD11b+ macrophages, suggesting a role for macrophages in EPS-mediated protection. Transfer of total peritoneal cells from an EPS-treated mouse was sufficient to protect naive mice from C. rodentium-induced enteric inflammation. In contrast, cells from a mouse treated with the (EPS−) material did not protect mice from disease, demonstrating that EPS are required for protection. This protection requires TLR4-signaling, and because EPS do not protect mice that lack MyD88 in myeloid cells, the TLR4-dependent immunosuppressive cells in the peritoneal cavity are likely macrophages.
TLR signaling during C. rodentium infection is complex; some TLRs contribute to host defense and others promote host damage (10–12, 15, 17). Previous studies using MyD88 KO mice infected with C. rodentium demonstrate that TLR signaling is required for neutrophil recruitment, for limiting C. rodentium translocation, and for epithelial cell repair (10, 15). Interestingly, these mice do not develop crypt hyperplasia (10), suggesting that TLR signaling drives this inflammatory process. Additionally, during C. rodentium infection, TLR4 signaling promotes disease and inflammation rather than host defense, since development of disease is slightly delayed in TLR4 KO mice, yet these mice clear the pathogen and recover similarly to wt mice (12). This previous study suggests that suppression of TLR4 signaling could alleviate disease and our data suggest EPS may antagonize TLR4 signaling, since a TLR4 agonist was not capable of protecting mice from disease and EPS do not induce peritoneal cells to produce inflammatory cytokines which one would expect of a TLR agonist. Interestingly, peritoneal macrophages bind EPS, but not directly to TLR4 because EPS binds peritoneal macrophages from TLR4 KO mice. EPS could bind to a protein that is part of the TLR4 signaling complex, such as the ligand binding accessory proteins RP105, CD14, CD36, GD1b, or Dectin-1, and suppress TLR4 signaling, as has been shown for RP105 (18, 19). Alternatively, because EPS are comprised primarily of mannose, EPS may bind to mannose-binding receptors which could cooperate with TLR to initiate an immune response, as described for pathogenic staphylococcyl infections (20).
Other studies have demonstrated that EPS from commensals, Bacteroides fragilis, and Bifidobacterium breve can also prevent colitis. Immunomodulatory EPS produced by B. breve, modulate host B cell responses and promote this commensal’s colonization (21); however, the structure of these EPS are currently unknown. Polysaccharide A (PSA) produced by B. fragilis, is comprised of a repeating tetrasaccharide moiety and is approximately 110–130 kDa (22); it has free carboxyl, phosphate and amino groups that contribute to its zwitterionic nature. PSA is processed by dendritic cells and presented to T cells in an MHCII dependent manner and induces anti-inflammatory IL-10 producing Treg cells (2, 22, 23). In contrast, B. subtilis EPS are comprised of 3 sugars, mannose (88%), glucose (11.9%) and N-acetyl-glucosamine (<0.1%), bind macrophages and are larger than PSA (>250 kDa). Based on the carbohydrate analysis, the structure of B. subtilis EPS is significantly different than that of B. fragilis PSA, and they likely modulate the host immune response differently.
The probiotic C. butyricum promotes development of anti-inflammatory IL-10-producing F4/80+CD11b+CD11cint macrophages which are critical for preventing DSS-induced colitis (1). In this case, the active bacterial molecules have not been identified. Although protection by B. subtilis EPS may be mediated by macrophages, the mechanism must be different than by B. fragilis or C. butyricum because they require TLR2 signaling whereas EPS requires TLR4 signaling. Collectively, our results and previous studies highlight the importance of selective modulation of TLR by commensal and probiotic bacteria to maintain intestinal homeostasis of CD4+ Treg cells and macrophages.
We do not know if EPS causes an anti-inflammatory response, e.g., production of IL-10 or other anti-inflammatory cytokines as is the case with B. fragilis and C. butyricum, or whether it inhibits induction of the inflammatory response initiated by C. rodentium infection. Because in preliminary studies we do not find evidence of increased anti-inflammatory cytokines after administration of EPS or B. subtilis, we hypothesize that EPS functions by altering macrophages in a TLR4-dependent manner to generate suppressor M2-like macrophages which upon injection into wt recipient mice, prevent the inflammatory response caused by C. rodentium. Future experiments are needed to elucidate the mechanisms by which EPS and peritoneal macrophages prevent C. rodentium-induced colitis.
How can i.p. injection of macrophages suppress inflammation at a distant mucosal site? We hypothesize that peritoneal macrophages convey protection by one or both of two mechanisms. First, they could secrete a soluble immunosuppressive factor that modulates other immune cells. Alternatively, select peritoneal macrophages may migrate to the colon and suppress pathogen-induced colonic inflammation similar to that observed by Fraga-Silva et al (24) who demonstrated that peritoneal macrophages migrate to areas of fungal infections.
Oral administration of B. subtilis provides protection, but administration of EPS by oral gavage does not protect against C. rodentium-induced colitis (data not shown). We showed previously, that protection by B. subtilis requires it to be motile (9), and it may be that motile B. subtilis localizes to a particular niche in the gut and secretes a concentrated quantity of EPS, whereas administered by oral gavage, EPS do not reach this niche. Alternatively, EPS delivered by oral gavage may be degraded in the stomach before they can suppress inflammation. We know that B. subtilis can prevent C. rodentium-associated disease when administered up to 3 dpi, a time at which disease has already begun (data not shown), which suggests to us that EPS may suppress systemic inflammation and be a successful therapeutic in other inflammation models.
Polysaccharides are not the only bacterial molecules with potent immunomodulatory activity. Commensal DNA, sphingolipids from B. fragilis, as well as proteins and phospholipids from L. rhamnosus modulate the host immune system to suppress inflammation (25–27). These studies and ours indicate that commensals produce a variety of factors to maintain immune homeostasis with their host, but studies to identify these important compounds and elucidate their mechanisms of action are in their infancy.
In summary, we identified EPS as the protective agents of B. subtilis, and showed that purified EPS, but not similarly treated material from an EPS− strain, prevent inflammatory disease induced by the enteric pathogen, C. rodentium. EPS-mediated protection requires TLR4 signaling; and although TLR signaling is known to regulate pathogen colonization and intestinal permeability, the protective effects of EPS seem to be a result of immunomodulation. Adoptive transfer studies demonstrate that TLR4 signaling on macrophage-rich peritoneal cells is required for EPS-mediated protection. Consistent with the idea that macrophages mediate protection in our model, we show EPS bind peritoneal macrophages and that mice with MyD88-deficient myeloid cells are not protected by EPS. This study highlights how a single dose of purified bacterial molecules, such as EPS, can impact the host immune responses during infection with an enteric pathogen.
Supplementary Material
Acknowledgments
The CCRC at UGA is funded by DOE and this work was supported by NIH R21 AI 098187 and NIH F32 DK 92054.
We would like to thank all the members of the Knight lab for their thoughtful comments and insights.
Footnotes
Conflicts of interest: The authors have no conflicts of interest.
Works Cited
- 1.Hayashi A, Sato T, Kamada N, Mikami Y, Matsuoka K, Hisamatsu T, Hibi T, Roers A, Yagita H, Ohteki T, Yoshimura A, Kanai T. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe. 2013;13:711–722. doi: 10.1016/j.chom.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gais P, Reim D, Jusek G, Rossmann-Bloeck T, Weighardt H, Pfeffer K, Altmayr F, Janssen KP, Holzmann B. Cutting edge: Divergent cell-specific functions of MyD88 for inflammatory responses and organ injury in septic peritonitis. J Immunol. 2012;188:5833–5837. doi: 10.4049/jimmunol.1200038. [DOI] [PubMed] [Google Scholar]
- 4.D’Arienzo R, Maurano F, Mazzarella G, Luongo D, Stefanile R, Ricca E, Rossi M. Bacillus subtilis spores reduce susceptibility to Citrobacter rodentium-mediated enteropathy in a mouse model. Res Microbiol. 2006;157:891–897. doi: 10.1016/j.resmic.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Wiles S, Pickard KM, Peng K, MacDonald TT, Frankel G. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect Immun. 2006;74:5391–5396. doi: 10.1128/IAI.00848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guttenplan SB, Blair KM, Kearns DB. The EpsE flagellar clutch is bifunctional and synergizes with EPS biosynthesis to promote Bacillus subtilis biofilm formation. PLoS Genet. 2010;6:e1001243. doi: 10.1371/journal.pgen.1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S, Lee YC. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal Biochem. 2005;339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 8.York WS, Darvill AG, McNeil M, Stevenson TT, Albersheim P. Isolation and characterization of plant cell walls and cell-wall components. Methods Enzymol. 1985;118:3–40. [Google Scholar]
- 9.Jones SE, Knight KL. Bacillus subtilis-mediated Protection from Citrobacter rodentium-associated enteric disease requires espH and functional flagella. Infect Immun. 2012 doi: 10.1128/IAI.05843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson DL, Ma C, Bergstrom KS, Huang JT, Man C, Vallance BA. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol. 2008;10:618–631. doi: 10.1111/j.1462-5822.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- 11.Gibson DL, Ma C, Rosenberger CM, Bergstrom KS, Valdez Y, Huang JT, Khan MA, Vallance BA. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol. 2008;10:388–403. doi: 10.1111/j.1462-5822.2007.01052.x. [DOI] [PubMed] [Google Scholar]
- 12.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun. 2006;74:2522–2536. doi: 10.1128/IAI.74.5.2522-2536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pant N, Marcotte H, Brussow H, Svensson L, Hammarstrom L. Effective prophylaxis against rotavirus diarrhea using a combination of Lactobacillus rhamnosus GG and antibodies. BMC Microbiol. 2007;7:86. doi: 10.1186/1471-2180-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu X, Vallance BA, Boyer L, Bergstrom KS, Walker J, Madsen K, O’Kusky JR, Buchan AM, Jacobson K. Saccharomyces boulardii ameliorates Citrobacter rodentium-induced colitis through actions on bacterial virulence factors. Am J Physiol Gastrointest Liver Physiol. 2008;294:G295–306. doi: 10.1152/ajpgi.00173.2007. [DOI] [PubMed] [Google Scholar]
- 15.Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol. 2007;179:566–577. doi: 10.4049/jimmunol.179.1.566. [DOI] [PubMed] [Google Scholar]
- 16.Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan MA, Bouzari S, Ma C, Rosenberger CM, Bergstrom KS, Gibson DL, Steiner TS, Vallance BA. Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect Immun. 2008;76:1410–1422. doi: 10.1128/IAI.01141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Inhibition of TLR-4/MD-2 signaling by RP105/MD-1. J Endotoxin Res. 2005;11:363–368. doi: 10.1179/096805105X67300. [DOI] [PubMed] [Google Scholar]
- 20.Ip WK, Takahashi K, Moore KJ, Stuart LM, Ezekowitz RA. Mannose-binding lectin enhances Toll-like receptors 2 and 6 signaling from the phagosome. J Exp Med. 2008;205:169–181. doi: 10.1084/jem.20071164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fanning S, Hall LJ, Cronin M, Zomer A, MacSharry J, Goulding D, Motherway MO, Shanahan F, Nally K, Dougan G, van Sinderen D. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc Natl Acad Sci U S A. 2012;109:2108–2113. doi: 10.1073/pnas.1115621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazmanian SK, Kasper DL. The love-hate relationship between bacterial polysaccharides and the host immune system. Nat Rev Immunol. 2006;6:849–858. doi: 10.1038/nri1956. [DOI] [PubMed] [Google Scholar]
- 23.Kalka-Moll WM, Tzianabos AO, Bryant PW, Niemeyer M, Ploegh HL, Kasper DL. Zwitterionic polysaccharides stimulate T cells by MHC class II-dependent interactions. J Immunol. 2002;169:6149–6153. doi: 10.4049/jimmunol.169.11.6149. [DOI] [PubMed] [Google Scholar]
- 24.Fraga-Silva TF, Venturini J, de Arruda MS. Trafficking of phagocytic peritoneal cells in hypoinsulinemic-hyperglycemic mice with systemic candidiasis. BMC Infect Dis. 13:147. doi: 10.1186/1471-2334-13-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An D, Na C, Bielawski J, Hannun YA, Kasper DL. Membrane sphingolipids as essential molecular signals for Bacteroides survival in the intestine. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4666–4671. doi: 10.1073/pnas.1001501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouladoux N, Hall JA, Grainger JR, dos Santos LM, Kann MG, Nagarajan V, Verthelyi D, Belkaid Y. Regulatory role of suppressive motifs from commensal DNA. Mucosal Immunol. 2012;5:623–634. doi: 10.1038/mi.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebeer S, Vanderleyden J, De Keersmaecker SC. Genes and molecules of lactobacilli supporting probiotic action. Microbiol Mol Biol Rev. 2008;72:728–764. doi: 10.1128/MMBR.00017-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.