

Abstract
Genome-wide association studies (GWAS) have ascertained numerous trait-associated common genetic variants, frequently localized to regulatory DNA. We find that common genetic variation at BCL11A associated with fetal hemoglobin (HbF) level lies in noncoding sequences decorated by an erythroid enhancer chromatin signature. Fine-mapping uncovers a motif-disrupting common variant associated with reduced transcription factor binding, modestly diminished BCL11A expression and elevated HbF. The surrounding sequences function in vivo as a developmental stage-specific lineage-restricted enhancer. Genome engineering reveals the enhancer is required in erythroid but not B-lymphoid cells for BCL11A expression. These findings illustrate how GWAS may expose functional variants of modest impact within causal elements essential for appropriate gene expression. We propose the GWAS-marked BCL11A enhancer represents an attractive target for therapeutic genome engineering for the β-hemoglobinopathies.
GWAS have identified numerous common single nucleotide polymorphisms (SNPs) associated with human traits and diseases. However advancing from genetic association to causal biologic process has been challenging (1). Recent genome-scale chromatin mapping studies have highlighted the enrichment of GWAS variants in regulatory DNA elements, suggesting many causal variants may affect gene regulation (2–6). GWAS of HbF level have identified trait-associated variants at BCL11A (7–12) (see supplementary online text). The transcriptional repressor BCL11A has been validated as a direct regulator of HbF level (13–18). Although constitutive BCL11A deficiency results in embryonic lethality and impaired lymphocyte development (19, 20), erythroid-specific deficiency of BCL11A counteracts developmental silencing of embryonic and fetal globin genes and rescues the hematologic and pathologic features of sickle cell disease (SCD) in mouse models (17).
To further understand how common genetic variation impacts BCL11A, HbF level and β-globin disorder severity, we compared the distribution of the HbF-associated SNPs at BCL11A with DNase I sensitivity, an indicator of chromatin state suggestive of regulatory potential. In primary human erythroblasts, three peaks of DNase I hypersensitivity were observed in intron-2, adjacent to and overlying the HbF-associated variants (Fig. 1A). We term these DNase I hypersensitive sites (DHSs) +62, +58 and +55 based on distance in kb from the transcription start site (TSS) of BCL11A. Brain and B-lymphocytes, two tissues that express high levels, and T-lymphocytes, which do not express BCL11A, showed unique patterns of DNase I sensitivity at the BCL11A locus, with a paucity of hypersensitivity overlying the trait-associated SNPs (Figs. 1A and S1).
Fig. 1. Chromatin state and TF occupancy at BCL11A.

(A) ChIP-seq from human erythroblasts with indicated antibodies. DNase I cleavage density from indicated human tissues. Three erythroid DHSs termed +62, +58 and +55 based on distance in kb from BCL11A TSS. BCL11A transcription from right to left.
(B) ChIP-qPCR from human erythroblasts at BCL11A intron-2. DHSs +62, +58 and +55 boxed. Enrichment at negative (GAPDH, OCT4) and positive control (β-globin LCR HS3 and α-globin HS-40) loci displayed.
(C) Chromosome conformation capture in human erythroblasts using BCL11A promoter as anchor. Error bars indicate s.d.
ChIP-seq demonstrated histone modifications with an enhancer signature overlying the trait-associated SNPs at BCL11A intron-2, including the presence of H3K4me1 and H3K27ac, and absence of H3K4me3 and H3K27me3 marks (Figs. 1A and S1). The major erythroid transcription factors (TFs) GATA1 and TAL1 also occupy this enhancer region. ChIP-qPCR confirmed three discrete peaks of GATA1 and TAL1 binding within BCL11A intron-2, each falling within an erythroid DHS (Fig. 1B). A common feature of distal regulatory elements is long-range interaction with cognate promoters. We evaluated the interactions between the BCL11A promoter and fragments across 250 kb of the BCL11A locus using a chromosome conformation capture assay. The greatest promoter interaction was observed within the region of intron-2 containing the trait-associated SNPs (Fig. 1C).
We hypothesized that the causal trait-associated SNPs could function by modulating critical cis-regulatory elements. Therefore we performed extensive genotyping of SNPs within the three erythroid DHSs +62, +58 and +55 in 1,263 DNA samples from the Cooperative Study of SCD (CSSCD) (21). 1,178 individuals and 38 SNPs were used for association testing (Fig. S2A). Analysis of common variants (MAF > 1%) revealed that rs1427407 in DHS +62 had the strongest association to HbF level (P = 7.23 × 10−50; Figs. 2A and S2B, also see supplementary online text). We identified associations to HbF level within the three DHSs that remained following conditioning on rs1427407 (Figs. 2A and S2B), consistent with the hypothesis that multiple functional SNPs within the composite enhancer act combinatorially to influence BCL11A regulation. The most significant residual association was for rs7606173 in DHS +55 (P = 9.66 × 10−11).
Fig. 2. Regulatory variants at BCL11A.

(A) Genotype data obtained in 1,178 individuals from CSSCD for 38 variants within BCL11A +62, +58 or +55 DHSs. Most highly significant associations to HbF level among common (MAF > 1%) SNPs (n = 10) prior to (rs1427407) or following (rs7606173) conditional analysis on rs1427407. SNP coordinates chromosome 2, build hg19.
(B) Chromatin from erythroblasts of individuals heterozygous for rs1427407, immunoprecipitated by GATA1 or TAL1 and pyrosequenced to quantify the relative abundance of the rs1427407-G allele. Composite half E-box–GATA motif previously identified (23).
(C) gDNA and cDNA from erythroblasts of individuals heterozygous for rs1427407, rs7606173 and rs7569946. Haplotyping demonstrated rs7569946-G, rs1427407-G and rs7606173-C on the same chromosome in each. Pyrosequencing to quantify the relative abundance of the rs7569946-G allele.
The SNP rs1427407 falls within a peak of GATA1 and TAL1 binding (Figs. 1A and 1B). The minor T-allele disrupts the G-nucleotide of a sequence element resembling a half E-box/GATA composite motif [CTG(n9)GATA], a consensus sequence enriched for chromatin bound by GATA1 and TAL1 complexes in erythroid cells (22, 23). We identified five primary erythroblast samples from individuals heterozygous for the major G-allele and minor T-allele at rs1427407 and subjected these samples to ChIP followed by pyrosequencing. As anticipated, we observed an even balance of alleles in the input DNA. However, we detected more frequent binding to the G-allele compared to the T-allele in both the GATA1 and TAL1 immunoprecipitated chromatin samples (Fig. 2B).
As the common synonymous SNP rs7569946 lies within exon-4 of BCL11A, it can be used to discriminate expression of alleles. We identified three primary erythroblast samples doubly heterozygous for the rs1427407–rs7606173 haplotype and rs7569946. For each sample, we determined by molecular haplotyping that the major rs7569946 G-allele was in phase with the low-HbF associated rs1427407–rs7606173 G–C haplotype (Table S4) (24, 25). Pyrosequencing revealed that whereas the alleles were balanced in genomic DNA (gDNA), significant imbalance was observed in complementary DNA (cDNA) with 1.7-fold increased expression of the low-HbF linked G-allele of rs7569946 (Fig. 2C, also see supplementary online text).
To understand the context within which these apparent regulatory trait-associated SNPs play their role, we explored the function of the harboring composite element. We cloned a 12.4 kb (+52.0–64.4 kb from TSS) human gDNA fragment containing the three erythroid DHSs to assay enhancer potential in a murine transgenic lacZ reporter assay (Fig. S4). Endogenous BCL11A shows abundant expression throughout the developing central nervous system with much lower expression observed in the fetal liver (26). In contrast, we observed in the transgenic embryos reporter gene expression largely confined to the fetal liver, the site of definitive erythropoiesis, with weaker expression noted in the central nervous system (Fig. 3A).
Fig. 3. The GWAS-marked BCL11A enhancer is sufficient for adult-stage erythroid expression.
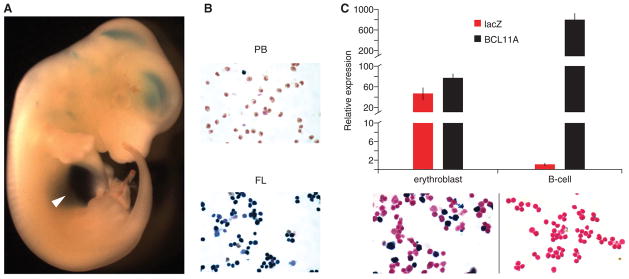
(A) A 12.4-kb fragment of BCL11A intron-2 (+52.0–64.4 kb from TSS) was cloned to a lacZ reporter construct. Transient transgenic mouse embryo from 12.5 dpc X-gal stained. Arrowhead indicates liver.
(B) Cell suspensions isolated from peripheral blood (PB) and fetal liver (FL) of stable transgenic embryos at 12.5 dpc X-gal stained.
(C) Sorted erythroblasts and B-lymphocytes from young adult stable transgenic mice subject to X-gal staining or RNA isolation followed by RT-qPCR. Gene expression normalized to GAPDH and expressed relative to T-lymphocytes. Error bars indicate s.d.
A characteristic feature of globin gene and BCL11A expression is developmental regulation (see supplementary online text). In stable transgenic BCL11A +52.0–64.4 reporter lines at 12.5 dpc, circulating primitive erythrocytes failed to stain for X-gal whereas definitive erythroblasts in fetal liver robustly stained positive (Fig. 3B). Endogenous BCL11A was expressed at 10.4-fold higher levels in B-lymphocytes as compared to erythroblasts. LacZ expression was restricted to erythroblasts and not observed in B-lymphocytes (Fig. 3C). These results indicate that the GWAS-marked BCL11A intron-2 regulatory sequences are sufficient to specify developmentally-restricted, erythroid-specific gene expression.
We aimed to disrupt the enhancer to investigate its requirement for BCL11A expression. Since there are no suitable adult-stage human erythroid cell lines, we turned to the mouse erythroleukemia (MEL) cell line. We observed an orthologous enhancer signature at intron-2 of mouse Bcl11a indicated by sequence homology, erythroid-specific DNase I hypersensitivity, characteristic histone marks and GATA1/TAL1 occupancy (Fig. S6) (22, 27). Sequence-specific nucleases can produce small chromosomal deletions via NHEJ-mediated repair (28). We engineered TALENs to introduce double-strand breaks to flank the orthologous 10 kb Bcl11a intron-2 sequences carrying the erythroid enhancer chromatin signature (Fig. S7A). Three unique clones were isolated that had undergone biallelic excision of the intronic segment (Figs. S7 and S8, also see supplementary online text). BCL11A transcript was profoundly reduced in the absence of the orthologous erythroid composite enhancer (Fig. 4A). BCL11A protein expression was not detectable in the enhancer-deleted clones (Fig. 4B). In the absence of the BCL11A enhancer, embryonic globin gene derepression was pronounced, with the ratio of embryonic εy to adult β1/2 globin increased by a mean of 364-fold (Fig. S9).
Fig. 4. The GWAS-marked BCL11A enhancer is necessary for erythroid but dispensable for non-erythroid expression.

(A) Three mouse erythroleukemia (MEL) and two pre-B lymphocyte clones with biallelic deletion of the orthologous Bcl11a erythroid enhancer (Δ50.4–60.4) subject to RT-qPCR.
(B) Immunoblot of Δ50.4–60.4 MEL and pre-B lymphocyte clones.
To examine potential lineage-restriction of the requirement for the +50.4–60.4 kb intronic sequences for BCL11A expression, we evaluated their loss in a non-erythroid context. The same strategy of introduction of two pairs of TALENs to obtain clones with NHEJ-mediated deletion was employed in a pre-B lymphocyte cell line. In contrast to the erythroid cells, BCL11A expression was retained in the Δ50.4–60.4 kb enhancer deleted pre-B cell clones at both the RNA and protein levels (Figs. 4A and 4B). These results indicate the orthologous erythroid enhancer sequences are essential for erythroid gene expression but not required in B-lymphoid cells for integrity of transcription from the Bcl11a locus.
The prior identification of BCL11A as a critical repressor of HbF levels has raised new hope for mechanism-based therapeutic approaches to the β-hemoglobinopathies (29). However, the paradox that genetic variation at BCL11A is common, well-tolerated and disease-protective despite the critical roles of BCL11A in neurogenesis and lymphopoiesis (19, 20, 30) has remained unresolved. Here we demonstrate that the HbF-associated variants localize to an erythroid enhancer of BCL11A. By allele-specific analyses, we show that genetic variation within this enhancer is associated with modest impact on TF binding, BCL11A expression and HbF level. Relatively small effect sizes associated with individual variants may not be surprising given that most single nucleotide substitutions, even within critical motifs, result in only modest loss of enhancer activity (31, 32). In contrast, loss of the BCL11A enhancer results in the absence of BCL11A expression in the erythroid lineage. Most trait-associated SNPs identified by GWAS are noncoding and have small effect size (1, 33). The impact of GWAS-identified SNPs on biological processes is often uncertain. Our findings underscore how a modest influence engendered by an individual noncoding variant neither predicts nor precludes a profound contribution of an underlying regulatory element.
Challenges to inhibiting BCL11A for mechanism-based reactivation of HbF include the supposedly “undruggable” nature of transcription factors (34) and its important non-erythroid functions (20, 30). With recent developments in their efficiency and precision, sequence-specific nucleases can be designed to exquisitely target genomic sequences of interest (35–37). We propose the GWAS-identified enhancer of BCL11A as a particularly promising therapeutic target for genome engineering in the β-hemoglobinopathies. Disruption of this enhancer would impair BCL11A expression in erythroid precursors with resultant HbF derepression, while sparing BCL11A expression in non-erythroid lineages. Rational intervention might mimic common protective genetic variation.
Supplementary Material
Acknowledgments
Thanks to A. Woo, A. Cantor, M. Kowalczyk, S. Burns, J. Wright, J. Snow, J. Trowbridge and members of the Orkin laboratory, particularly C. Peng, P. Das, G. Guo, M. Kerenyi, and E. Baena, for discussions. C. Guo and F. Alt provided the pre-B cell line, A. He and W. Pu the pWHERE lacZ reporter construct, C. Currie and M. Nguyen technical assistance, D. Bates and T. Kutyavin expertise with sequence analysis, R. Sandstrom help with data management, G. Losyev and J. Daley aid with flow cytometry and J. Desimini graphical assistance. L. Yan at EpigenDx (Hopkinton, Massachusetts) conducted the custom pyrosequencing reactions. This work was funded by grants from the Doris Duke Charitable Foundation (#2009089) and Canadian Institute of Health Research (#123382) to G.L.; Amon Carter Foundation, Hyundai Hope on Wheels, NIH, Lucille Packard Foundation to M.H.P.; NIH grants U54HG004594 and U54HG007010 to J.A.S.; and NIH R01HL032259, P01HL032262, and P30DK049216 (Center of Excellence in Molecular Hematology) to S.H.O. D.E.B. is supported by NIDDK Career Development Award K08DK093705. A patent application related to this work was filed by Boston Children’s Hospital, and D.E.B., J.X., and S.H.O. are inventors.
Footnotes
Materials and Methods
References (39–55)
References
- 1.Fugger L, McVean G, Bell JI. N Engl J Med. 2012;367:2370–2371. doi: 10.1056/NEJMp1212285. [DOI] [PubMed] [Google Scholar]
- 2.Ernst J, et al. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paul DS, et al. PLoS Genet. 2011;7:e1002139. doi: 10.1371/journal.pgen.1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maurano MT, et al. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Harst P, et al. Nature. 2012;492:369–375. [Google Scholar]
- 6.ENCODE Project Consortium et al. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menzel S, et al. Nat Genet. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 8.Uda M, et al. Proc Natl Acad Sci U S A. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lettre G, et al. Proc Natl Acad Sci U S A. 2008;105:11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuinoon M, et al. Hum Genet. 2010;127:303–314. doi: 10.1007/s00439-009-0770-2. [DOI] [PubMed] [Google Scholar]
- 11.Solovieff N, et al. Blood. 2010;115:1815–1822. doi: 10.1182/blood-2009-08-239517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatnagar P, et al. J Hum Genet. 2011;56:316–323. doi: 10.1038/jhg.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sankaran VG, et al. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 14.Xu J, et al. Genes Dev. 2010;24:783–798. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, et al. Proc Natl Acad Sci U S A. 2013;110:6518–6523. doi: 10.1073/pnas.1303976110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sankaran VG, et al. Nature. 2009;460:1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, et al. Science. 2011;334:993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esteghamat F, et al. Blood. 2013;121:2553–2562. doi: 10.1182/blood-2012-06-434530. [DOI] [PubMed] [Google Scholar]
- 19.Liu P, et al. Nat Immunol. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 20.Yu Y, et al. J Exp Med. 2012;209:2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farber MD, Koshy M, Kinney TR. J Chronic Dis. 1985;38:495–505. doi: 10.1016/0021-9681(85)90033-5. [DOI] [PubMed] [Google Scholar]
- 22.Soler E, et al. Genes Dev. 2010;24:277–289. doi: 10.1101/gad.551810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kassouf MT, et al. Genome Res. 2010;20:1064–1083. doi: 10.1101/gr.104935.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner DJ, Hurles ME. Nat Protoc. 2009;4:1771–1783. doi: 10.1038/nprot.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyson J, Armour JA. BMC Genomics. 2012;13:693-2164-13-693. doi: 10.1186/1471-2164-13-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leid M, et al. Gene Expr Patterns. 2004;4:733–739. doi: 10.1016/j.modgep.2004.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalczyk MS, et al. Mol Cell. 2012;45:447–458. doi: 10.1016/j.molcel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 28.Lee HJ, Kim E, Kim JS. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer DE, Kamran SC, Orkin SH. Blood. 2012;120:2945–2953. doi: 10.1182/blood-2012-06-292078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.John A, et al. Development. 2012;139:1831–1841. doi: 10.1242/dev.072850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patwardhan RP, et al. Nat Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melnikov A, et al. Nat Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frazer KA, Murray SS, Schork NJ, Topol EJ. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 34.Koehler AN. Curr Opin Chem Biol. 2010;14:331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 36.Joung JK, Sander JD. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Oost J. Science. 2013;339:768–770. doi: 10.1126/science.1234726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.