

Abstract
Iron(III) (oxyhydr)oxides can represent the dominant microbial electron acceptors under anoxic conditions in many aquatic environments, which makes understanding the mechanisms and processes regulating their dissolution and transformation particularly important. In a previous laboratory-based study, it has been shown that 0.05 mM thiosulfate can reduce 6 mM ferrihydrite indirectly via enzymatic reduction of thiosulfate to sulfide by the sulfur-reducing bacterium Sulfurospirillum deleyianum, followed by abiotic reduction of ferrihydrite coupled to reoxidation of sulfide. Thiosulfate, elemental sulfur, and polysulfides were proposed as reoxidized sulfur species functioning as electron shuttles. However, the exact electron transfer pathway remained unknown. Here, we present a detailed analysis of the sulfur species involved. Apart from thiosulfate, substoichiometric amounts of sulfite, tetrathionate, sulfide, or polysulfides also initiated ferrihydrite reduction. The portion of thiosulfate produced during abiotic ferrihydrite-dependent reoxidation of sulfide was about 10% of the total sulfur at maximum. The main abiotic oxidation product was elemental sulfur attached to the iron mineral surface, which indicates that direct contact between microorganisms and ferrihydrite is necessary to maintain the iron reduction process. Polysulfides were not detected in the liquid phase. Minor amounts were found associated either with microorganisms or the mineral phase. The abiotic oxidation of sulfide in the reaction with ferrihydrite was identified as rate determining. Cysteine, added as a sulfur source and a reducing agent, also led to abiotic ferrihydrite reduction and therefore should be eliminated when sulfur redox reactions are investigated. Overall, we could demonstrate the large impact of intermediate sulfur species on biogeochemical iron transformations.
INTRODUCTION
Iron(III) (oxyhydr)oxides are important for various processes in the environment. Potentially toxic trace metalloids and metals such as arsenic, lead, and cadmium sorb to the surface of iron(III) (oxyhydr)oxides and are removed from the aqueous phase (1–5). Also, the availability of nutrients such as phosphate can be limited by adsorption (6, 7). During reductive dissolution, substances adsorbed to the surface of iron(III) (oxyhydr)oxides are released.
Microorganisms play an important role in the reduction of iron(III) (oxyhydr)oxides. Several bacterial species, such as Geobacter and Shewanella spp., can grow by using iron(III) minerals as electron acceptors. Either the bacteria are in direct contact with poorly soluble iron(III) (oxyhydr)oxides and transfer electrons directly to the minerals (8) or different electron-shuttling compounds such as flavins, humic substances, or quinones can transfer electrons from the cells to the iron(III) (oxyhydr)oxides (9–12).
The occurrence of sulfur is another factor that is of importance for biogeochemical redox processes related to iron. Especially in its reduced state, sulfur is highly reactive with iron. Hydrogen sulfide (H2S) can lead to the abiotic reductive dissolution of iron oxides (13) with the consequent release of adsorbed substances. However, sulfur does not necessarily have to be present as sulfide to lead to the reduction of iron oxides. A wide range of sulfur-reducing bacteria exist that can use the different oxidized sulfur species as electron acceptors and therefore contribute to the formation of sulfide, which can then react abiotically with iron oxides (14). Sulfidogenesis by sulfate-reducing bacteria was found to lead to iron mineral transformation and arsenic mobilization (15, 16). Straub and Schink (17) also investigated this reaction pathway of sulfur-mediated iron(III) mineral reduction in experiments with Sulfurospirillum deleyianum and ferrihydrite. The sulfur-reducing bacterium S. deleyianum is not able to use ferric iron as an electron acceptor (18) but can reduce thiosulfate (S2O32−), elemental sulfur (S0), and sulfite (SO32−). Sulfate (SO42−) cannot be reduced (19, 20). When offering the bacteria 0.05 mM thiosulfate as an electron acceptor, Straub and Schink (17) observed a reduction of 6 mM ferrihydrite. On the basis of this finding, they proposed a shuttling mechanism whereby thiosulfate is microbially reduced to sulfide, which is then reoxidized by ferrihydrite reduction. Their data suggested that sulfur had cycled between the bacteria and iron up to 60 times. Thiosulfate, elemental sulfur, or polysulfides (Sn2−) were proposed by Straub and Schink (17) to complete the sulfur cycle, but the identity of the reoxidized sulfur species was not revealed until now.
In the present study, we therefore investigated the process of electron shuttling between S. deleyianum and ferrihydrite in more detail with a focus on sulfur speciation. The main goal was to identify the reoxidized sulfur species that are produced by the abiotic reaction of sulfide and ferrihydrite. To this end, the experiment was split into its reductive (microbial) and oxidative (abiotic) reactions. We then repeated the experiments done by Straub and Schink (17) but also tested sulfur species other than thiosulfate and analyzed the sulfur species present.
MATERIALS AND METHODS
Bacterial cultivation.
A culture of S. deleyianum (DSM 6946) was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen. Freshwater medium, which contained 4.4 mM KH2PO4, 5.6 mM NH4Cl, 0.7 mM CaCl2 · 2H2O, and 2.0 mM MgSO4 · 7H2O or, for a sulfate-free medium, 2.0 mM MgCl2 · 6H2O, was prepared in a Widdel flask and autoclaved. After cooling under an atmosphere of N2-CO2 (90/10 [vol/vol]), the medium was supplied with bicarbonate buffer, autoclaved under N2-CO2 (90/10 [vol/vol]) to a final concentration of 30 mM, and 1 ml/liter each of a sterile vitamin, a trace element (SL-10), and a selenite tungstate solution was added (21). The pH was adjusted to 7 by the addition of anoxic and sterile 1 M HCl. The medium was used to fill serum bottles that had been washed with 1 M HCl and distilled water prior to autoclaving. The headspace was flushed with N2-CO2 (90/10 [vol/vol]), and the bottles were sealed with butyl rubber stoppers and aluminum crimps. For standard cultivation, the medium was supplied with 20 mM fumarate (C4H2Na2O4; Merck) as an electron acceptor, 20 mM formate (CHNaO2; Merck) as an electron donor, 20 mM acetate (C2H3NaO2; Sigma-Aldrich) as a carbon source, and 0.5 mM l-cysteine (C3H7NO2S; Roth) as both a sulfur source and a reducing agent. All solutions were prepared in ultrapure water (Merck Millipore, 18.2 mΩ/cm, 3 ppb total organic carbon at 25°C), either autoclaved or sterilely filtered with 0.2-μm cellulose acetate filters (Membrex; membraPure), and stored in serum bottles under N2. The cultures were incubated at 28°C in the dark.
Preparation of ferrihydrite suspensions.
Ferrihydrite was synthesized as described by Schwertmann and Cornell (22) and Raven et al. (23). A total of 20 g of iron(III) nitrate nonahydrate [Fe(NO3)3 · 9H2O; Acros Organics] was suspended in ultrapure water. The pH was adjusted to 7.3 by adding 1 M KOH. The suspension was centrifuged and washed with ultrapure water four times. The wet pellet was resuspended in ultrapure water to a final concentration of about 0.5 M ferric iron [Fe(III)], transferred to a serum bottle, which was deoxygenized by flushing with N2, and stored at 8°C in the dark. Ferrihydrite suspensions were used for experiments within 2 months after preparation.
Experimental setup and sampling.
All experiments were carried out with serum bottles (30, 50, or 100 ml), which were standing upright for the whole duration of the experiment and were filled with medium (30, 25, or 100 ml). For the experiments, the standard concentrations of thiosulfate, cysteine, acetate, and formate, at 0.1, 2, 5, and 10 mM, respectively, were the same as in the study of Straub and Schink (17). Experimental variations included different thiosulfate and cysteine concentrations, as well as replacement of thiosulfate with other sulfur compounds. Note that the concentrations of sulfur-containing substances are referred to as moles equivalent of S, in contrast to the paper of Straub and Schink (17). Stock solutions were either autoclaved or sterilely filtered and stored in serum bottles under N2. Solutions of sodium thiosulfate pentahydrate (Na2O3S2 · 5H2O), sodium sulfite (Na2SO3 [anhydrous]; Fluka), potassium tetrathionate (K2S4O6; Sigma-Aldrich), sodium sulfide nonahydrate (Na2S · 9H2O; Sigma-Aldrich), and potassium polysulfide (K2Sx; Sigma-Aldrich) were prepared in ultrapure water. The latter two were prepared inside a Coy anoxic glove box (N2-H2 [95/5, vol/vol]). The solution of l-cystine (C6H12N2O4S2; Applichem) used first was also prepared in ultrapure water. As cystine is not very soluble in water, the solution for a further experiment was prepared by dissolving cystine in 5 M NaOH, diluting it with ultrapure water, and adjusting the pH to 9 by adding 1 M HCl.
The standard nominal ferrihydrite concentration was 5 mM, as described by Straub and Schink (17), to avoid the formation of magnetite, which was observed at higher concentrations (24). As particles settled very fast in ferrihydrite suspension, it was difficult to add exactly the same amount of ferrihydrite to each serum bottle. Therefore, the initial ferrihydrite concentrations varied from approximately 3 to 5 mM. A standard inoculum size of 1% was chosen instead of the 0.01% used by Straub and Schink (17) to shorten the initial lag phase and to observe mineral transformations in shorter times. Comparative experiments were also done with a 0.01% inoculum.
Samples were taken from the serum bottles inside the glove box with a needle and a syringe, which was flushed and filled with N2-CO2 (90/10 [vol/vol]) to replace the volume of liquid removed with the same volume of gas. Also inside the glove box, samples were filtered with 0.2-μm cellulose-acetate filters (Membrex; membraPure), acidified for iron analysis, derivatized for polysulfide analysis, and mixed with 2% (wt/vol) zinc acetate dihydrate [Zn(CH3COO)2 · 2H2O; Grüssing] to precipitate dissolved sulfide.
Analytical methods. (i) Iron.
Ferrous iron [Fe(II)] and total iron [Fe(tot)] were determined photometrically by the ferrozine assay (25). Therefore, unfiltered samples were diluted in 1 M HCl 2-fold for Fe(II) determination and 20-fold for Fe(tot) determination. After Fe mineral dissolution, samples were centrifuged for 5 min at 15,000 rpm and hydroxylamine hydrochloride (NH2OH · HCl [Acros Organics], 10% [wt/vol] in 1 M HCl) was added to Fe(tot) samples to reduce all iron to Fe(II) before adding the ferrozine reagent (0.1% [wt/vol] ferrozine [C20H13N4NaO6S2 · H2O; Acros Organics]–50% ammonium acetate [C2H7NO2; Acros Organics]). The absorbance at 570 nm of the purple ferrozine-Fe(II) complex was measured with a microplate reader (Infinite 200 PRO; TECAN). Fe(III) concentrations were calculated by subtracting Fe(II) concentrations from Fe(tot) concentrations.
Over the course of ferrihydrite reduction, we observed significant color changes of the suspensions from brown to black. These black suspended particles immediately dissolved in 1 M HCl and had a characteristic hydrogen sulfide smell, suggesting that they were ferrous sulfides and not magnetite. As iron concentrations were quantified from unfiltered samples, such suspended iron precipitates were codetermined with truly dissolved iron species. However, we observed that in most experimental setups with ferrihydrite and bacteria, the Fe(tot) concentration in the samples taken decreased over time (data not shown) and whitish-gray precipitates were sticking to the bottoms of the serum bottles. These precipitates were soluble in 1 M HCl, and test measurements showed that they contained 93 to 101% Fe(II) (data not shown). Straub and Schink (17) also observed white to gray precipitates, which were assumed to be precipitates of Fe(II) and carbonate (siderite) or phosphate (vivianite). Overall, this means that Fe(II) concentrations determined in solution accounted only for aqueous iron and ferrous sulfides and not for ferrous carbonates or phosphates. To quantify the total amount of ferrous iron [Fe(II) total] formed, the missing amount of Fe(tot), i.e., the difference between the initial and actually measured values of Fe(tot), was added to the respective values of Fe(II) determined in solution, assuming that the missing Fe was precipitated as Fe(II) carbonates and phosphates at the glass wall.
(ii) Sulfide.
The analysis of sulfide in filtered samples was performed photometrically by the methylene blue method (26). Absorption at 660 nm was measured (Hach Lange DR 3800).
(iii) Polysulfides and elemental sulfur.
Polysulfide analysis was performed by the method of Kamyshny et al. (27), which is based on the conversion of labile inorganic polysulfides into more stable dimethylpolysulfanes. This is achieved by derivatization with methyl trifluoromethanesulfonate (methyl triflate, CF3SO2OCH3; Sigma-Aldrich) in a water-methanol (H3COH, high-performance liquid chromatography [HPLC] gradient grade; VWR) mixture. In the present study, derivatization was done with both unfiltered and filtered samples. For the filtered samples, 800 μl of methanol was placed in a 1.5-ml HPLC vial to which 200 μl of filtered sample and 6 μl of methyl triflate were added simultaneously. For the unfiltered samples, derivatization was performed in 2-ml Eppendorf centrifuge tubes with 1.5 times the amount of each reagent. After derivatization, these solutions were also filtered into 1.5-ml HPLC vials. Samples were stored in a freezer until analysis. Dimethylpolysulfanes were analyzed by HPLC (Merck Hitachi L-2130 pump, L-2200 autosampler, and L-2420 UV-VIS detector) and separated by gradient elution over a C18 column (Bischoff Waters Spherisorb, ODS2, 5 μm, 250 by 4.6 mm) by a modification of the method of Rizkov et al. (28). Initially, the solvent consisted of 70% methanol and 30% ultrapure water at a flow rate of 1 ml/min. The methanol concentration was increased to 80% for 10 min, kept constant for 15 min, increased to 100% for 10 min, kept constant for 10 min, decreased to 70% for 5 min, and kept constant for the last 10 min. The injection volume was 100 μl, and detection was performed at a wavelength of 230 nm. Concentrations of dimethyl disulfide and dimethyl trisulfide were determined by the use of commercially available standards (C2H6S2 [Acros Organics], C2H6S3 [Acros Organics]). For quantification of polysulfides from dimethyl tetrasulfide to dimethyl octasulfide, a dimethylpolysulfane mixture was synthesized as described by Rizkov et al. (28). Separation of the mixture was performed by preparative chromatography with a reversed-phase C18 column [Phenomenex SphereClone ODS(2), 5 μm, 250 by 10 mm], an eluent of 50% acetonitrile (C2H3N, HPLC gradient grade; Roth) and 50% formic acid (CH2O2; Grüssing), and a flow rate of 5 ml/min. The individual dimethylpolysulfanes in the collected fractions were oxidized to sulfate in a microwave (MarsXpress; CEM) with hydrochloric and nitric acids at 200°C for 10 min. The resulting sulfate concentrations were analyzed by inductively coupled plasma mass spectrometry (ICPMS, XSeries2; Thermo Fisher) and used for quantification.
The method of polysulfide derivatization in methanol with subsequent HPLC analysis was also suitable for the detection of elemental sulfur. For preparation of calibration standards, elemental sulfur powder (S, reagent grade, purified by sublimation, up to a 100-mesh particle size, powder; Sigma-Aldrich) was dissolved in dichloromethane (CH2Cl2; Sigma-Aldrich) and diluted in the same methanol-water mixture (5:1 ratio) that was used for derivatization.
(iv) Thiosulfate.
Filtered samples for thiosulfate measurement were flash frozen in liquid nitrogen and stored in a freezer until analysis, which was performed with the above-mentioned HPLC system by a modification of the protocol published by Steudel et al. (29). For separation, a reversed-phase C18 column (Grace GraceSmart, RP18, 5 μm, 150 by 4.6 mm) was used. Elution was performed with two alternative eluent compositions, once with 0.002 M tetra-n-butylammonium hydroxide [(CH3CH2CH2CH2)4N(OH); Fluka] and 0.001 M sodium carbonate (Na2CO3; Merck) in 85% ultrapure water and 15% acetonitrile (C2H3N, HPLC gradient grade; Roth), with the pH adjusted to 7.7 by the addition of 1 M HCl and once—to achieve more reliable eluent preparation and hence more stable analytical conditions—with 0.002 M tetrabutylammonium dihydrogen phosphate {(CH3CH2CH2CH2)4N[OP(OH)2O]; Sigma-Aldrich} in 85% ultrapure water and 15% acetonitrile. The flow rate was 1 ml/min, the injection volume was 10 μl, and the detection wavelength was 215 nm.
(v) Sulfate.
Sulfate was measured by anion-exchange chromatography–ICPMS in filtered samples, which were flash frozen in liquid nitrogen and kept in a freezer until analysis (30). The injection volume was 100 μl, and separation was achieved with an anion-exchange column (Dionex IonPac, AG-16/AS-16, 4 mm) and elution with a gradient of 0.02 to 0.1 M NaOH at a flow rate of 1.2 ml/min. Sulfate was detected by ICPMS (XSeries2, Thermo-Fisher) as SO+ (m/z 48).
Genome sequence analysis.
Comparative analysis of the finished S. deleyianum genome was performed with the integrated microbial genomes (IMG) platform and the tools provided therein (31).
RESULTS AND DISCUSSION
As a summary of our studies we propose a revised model for the involvement of different sulfur species in electron shuttling between S. deleyianum and ferrihydrite compared to the model previously published by Straub and Schink (17). Our model is shown in Fig. 1, and the individual reactions will be discussed in the following sections.
FIG 1.

Proposed mechanisms of sulfur cycle-mediated ferrihydrite reduction by S. deleyianum. Thiosulfate, sulfide, and iron are in bold because they are the main reactants in the cycle. The numbered processes are discussed in the text.
Separating the reduction of thiosulfate to sulfide by S. deleyianum from the abiotic oxidation of sulfide by ferrihydrite.
To gain insights into the electron-shuttling processes in the ferrihydrite-S. deleyianum cultures, the experiment was split into reductive (microbial) and oxidative (abiotic) reactions. The first set of experiments was conducted with S. deleyianum but without the addition of iron(III) minerals, and the second was conducted abiotically with ferrihydrite.
Microbial reduction of thiosulfate by S. deleyianum.
In the absence of ferrihydrite, S. deleyianum reduced 2 and 0.1 mM thiosulfate completely to sulfide (see Fig. SI-1 in the supplemental material; reaction 1a in Fig. 1), which is in accordance with the literature (19, 20). According to IMG genome annotation (31), thiosulfate reduction to sulfide is mediated by a Phs-like thiosulfate reductase complex (gene loci Sdel_0269, Sdel_0270, and Sdel_0271) via the non-thiol-dependent thiosulfate disproportionation pathway. Elemental sulfur was measureable between days 2 and 7 in unfiltered and filtered samples (see Table SI-1 in the supplemental material), which means that in the absence of iron(III) minerals, elemental sulfur seems to be stable in solution. From days 2 to 7, thiosulfate was completely reduced to sulfide by S. deleyianum. Elemental sulfur concentrations were highest on day 2 but accounted for less than 1% of the initial thiosulfate concentrations and decreased until day 7. From day 7 on, polysulfides were detectable as S62− in unfiltered samples of both experiments with 2 and 0.1 mM thiosulfate (see Table SI-1). With concentrations of ≤0.012 mM in the experiment with 2 mM thiosulfate, the amount of S62− came to 0.6% of the initial thiosulfate concentration. In the experiment with 0.1 mM thiosulfate, the respective percentage of S62− was 17% at maximum.
Polysulfides can form from the reaction between elemental sulfur and sulfide, which was shown to be the end product of thiosulfate reduction by S. deleyianum. Hedderich et al. (32) came to the conclusion that polysulfides occur as intermediate species in the process of sulfur respiration. This seems to be the case not only with elemental sulfur but also with thiosulfate as an electron acceptor. Finding polysulfides only in unfiltered and not in filtered samples indicates that polysulfides are associated with the microorganisms, either bound to the cell surface or occurring inside the cells. To sum up, elemental sulfur and polysulfides do form as intermediate sulfur species in the microbial thiosulfate reduction process, but the final product in solution is sulfide exclusively.
Abiotic oxidation of sulfide coupled to ferrihydrite reduction.
In the absence of S. deleyianum, sulfide, which was added to a 5 mM ferrihydrite suspension in 10 steps of 0.01 mM at intervals of 1 h, reduced ferrihydrite (reaction 1b in Fig. 1) and was itself oxidized mainly to elemental sulfur (Fig. 2; reaction 1c in Fig. 1). We hypothesize that elemental sulfur was bound to the surface of the iron minerals, as it was detectable only in unfiltered samples. Without the addition of cysteine (Fig. 2A), elemental sulfur was detected after 4.9 h and the concentration increased continuously to 0.037 mM. The thiosulfate concentration also increased after 6.2 h but to a lesser extent than the elemental sulfur concentration (reaction 1d in Fig. 1). Polysulfides occurred in only insignificant amounts in unfiltered samples. As no darkening of the suspension was observed, the formation of FeS could be excluded. Sulfate was detectable in minor amounts over the whole course of the experiment. The oxidation of sulfide to sulfate stops the process of electron shuttling between the microorganisms and ferrihydrite (reaction 1e, Fig. 1). Genome analysis shows that a sulfate adenylyltransferase mediating the conversion of sulfate to adenylylsulfate is encoded in the S. deleyianum genome (gene locus Sdel_1709) but an adenylylsulfate reductase is absent. Thus, S. deleyianum is unable to perform dissimilatory sulfate reduction, which is also in accordance with previous reports (19, 20). In this experiment, the addition of 0.1 mM sulfide led to the reduction of 0.14 mM ferrihydrite.
FIG 2.
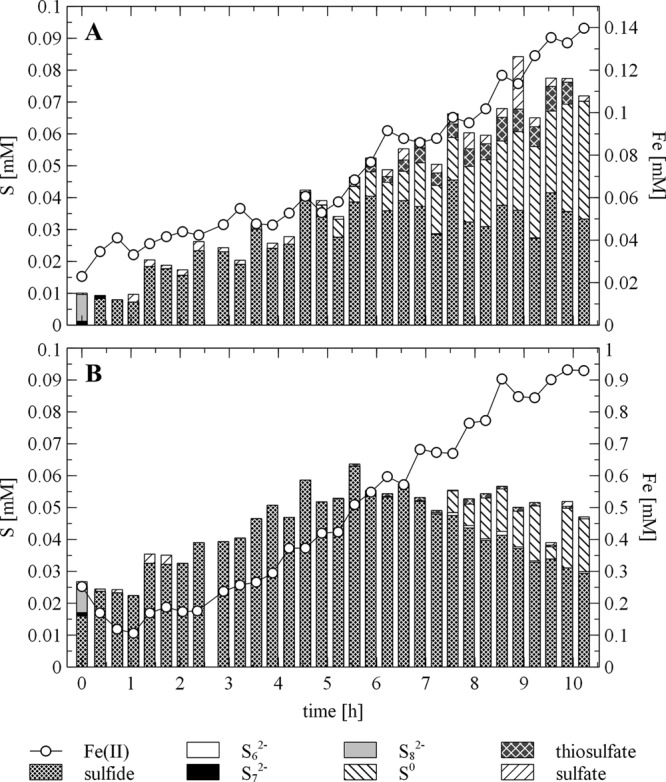
Abiotic Fe(II) production from ferrihydrite and sulfur speciation in freshwater medium to which 0.1 mM sulfide was added in 10 steps at intervals of 1 h beginning at 0.4 h. Concentrations of polysulfides and elemental sulfur (S0) were detectable only in unfiltered samples. Experiment A was conducted without cysteine, and experiment B was conducted with 2 mM cysteine. Note the different scales for Fe(II) on the secondary y axes. Sulfate data for experiment B from 2.1 to 4.2 h and from 5.6 to 9.2 h are missing.
In the second abiotic experiment, where sulfide was added in the presence of 2 mM cysteine to a 5 mM ferrihydrite suspension (Fig. 2B), 0.93 mM Fe(II) was produced. The suspension gradually turned black, suggesting that FeS was formed and sulfide was removed from solution (reaction 2a in Fig. 1). This process was also obvious from the sum of all of the sulfur species, which increased until 5.6 h and then decreased and at the end was two-thirds of the sum of the sulfur species in the experiment without cysteine. Cysteine seemed to enforce the precipitation of FeS by keeping sulfide in the reduced state. The production of elemental sulfur began later and occurred to a lesser extent than in the experiment without cysteine. Polysulfides, thiosulfate, and sulfate occurred in insignificant amounts. Sulfide was already measurable at the beginning of the experiment, i.e., before the first addition of sulfide. This is an analytical artifact because cysteine also yields a sulfide signal in the methylene blue measurement. A comparison of literature data shows that the identity of oxidized sulfur species produced by the reaction of sulfide and ferrihydrite seems to differ, depending on the experimental conditions. In experiments with ferrihydrite-coated sand and gaseous H2S, FeS (ca. 67%) and elemental sulfur (ca. 33%) were found as main products detected under anoxic conditions (33). In artificial seawater, Poulton (34) found that in the reaction with ferrihydrite, dissolved sulfide was oxidized mainly to elemental sulfur and that reduced Fe(II) was associated with the oxide surface to a large extent. Up to 15% of the total Fe(II) was found as FeS at pH 7.5. Under estuarine conditions, the reaction of hydrous ferric iron oxides and aqueous sulfide produced 86% elemental sulfur, including polysulfide sulfur, and 14% thiosulfate (35). In experiments with ferrihydrite, sulfate, and sulfate-reducing bacteria, elemental sulfur was also found as a major oxidation product attached to the iron mineral surface. However, sulfur speciation in solution was dominated by sulfate (36). In all of these studies, elemental sulfur was analyzed directly at the surface or after extraction of unfiltered samples or the filter itself. Nevertheless, elemental sulfur could also be detected in solution as a product of the reaction between ferrihydrite and H2S at low pH (37). In the present study, elemental sulfur was found as a major oxidation product but only in unfiltered samples. Straub and Schink (17) postulated that elemental sulfur, polysulfides, and/or thiosulfate could serve as electron shuttles between S. deleyianum and ferrihydrite, but they did not identify the oxidized sulfur species. Since they also observed ferrihydrite reduction when the iron mineral and the microorganisms were spatially separated, the electron-shuttling compound would have to be present in the aqueous phase. In contrast, our finding that elemental sulfur was attached to the mineral surface indicates that iron reduction is possible only with direct contact of the microorganisms and the iron mineral.
The roles of individual sulfur species in iron(III) mineral reduction by S. deleyianum. (i) The role of thiosulfate as an electron acceptor for S. deleyianum.
Straub and Schink (17) showed that 5 mM ferrihydrite was reduced within 35 days in an experiment with 0.05 mM thiosulfate (equivalent to 0.1 mM S), 2 mM cysteine, and a 0.01% S. deleyianum inoculum. For comparison, we conducted experiments with different concentrations of thiosulfate and different inoculum amounts (Table 1). Ferrihydrite reduction was fastest with 2 mM thiosulfate and a 1% inoculum. The inoculum amount had a greater influence than the thiosulfate concentration on the rate of iron(III) mineral reduction. In all of the experiments, complete reduction of ferrihydrite within 29 days was observed. In the experiments with 2 mM thiosulfate, black mineral particles formed that did not disappear until the end of the experiment. In the experiment with 0.1 mM thiosulfate and a 1% inoculum, the suspension also turned darker in the beginning and then gradually became clearer until complete colorlessness in the end. With 0.1 mM thiosulfate and a 0.01% inoculum, it took longer until the suspension was completely clear. As processes of iron reduction seemed to be qualitatively similar in systems with 0.1 mM thiosulfate and a 1 or 0.01% inoculum, the greater inoculum amount was chosen for further experiments to accelerate the reactions observable in the experiments.
TABLE 1.
Reduction of Fe(III) in ferrihydrite in incubations of S. deleyianum
| Thiosulfate concn (mM) | % inoculum | % Fe(III) reduction in expt 1, 2a |
|---|---|---|
| 2 | 1 | 100, 95 |
| 0.1 | 1 | 91, 86 |
| 2 | 0.01 | 76, 62 |
| 0.1 | 0.01 | 38, 32 |
Bacteria were supplied with 2 mM cysteine as a sulfur source and a reductant, 5 mM acetate as a carbon source, and 10 mM formate as an electron donor. Results obtained in duplicate experiments after 14 days are presented.
(ii) Kinetics of thiosulfate consumption.
After the addition of 2 mM thiosulfate to suspensions with 5 mM ferrihydrite and a 1% inoculum, parallel reduction of Fe(III) and thiosulfate was observable (Fig. 3A). In the first phase, up to day 3, only small amounts of Fe(II) were produced. Up to day 6, thiosulfate and Fe(III) were depleted more rapidly, while the suspension turned completely black, indicating the formation of FeS (reaction 2a, Fig. 1). Almost no free sulfide was detectable in filtered samples, which means that precipitation of FeS or oxidation of sulfide by the reaction with ferrihydrite must have been faster than reduction of thiosulfate by the microorganisms. Small amounts of polysulfides were found in unfiltered samples, suggesting that they are associated with the microorganisms or are attached to the mineral. We have recently found evidence that polysulfides attached to the surface of iron(III) (oxyhydr)oxides are precursors of pyrite formation (51)—a similar process might have occurred in the present experiment. Simultaneously with polysulfides, elemental sulfur concentrations increased rapidly between days 5 and 6 and then were stable until the end. As already seen in the abiotic experiment, elemental sulfur was the main oxidation product of sulfide but accounted for only 10% of the initial thiosulfate concentration. For complete ferrihydrite reduction, polysulfides, elemental sulfur, or sulfide from FeS precipitations (reaction 2b, Fig. 1) had to act as a recyclable sulfur reservoir. When polysulfides and elemental sulfur had reached maximum concentrations (at day 5.7, Fig. 3A), the process of Fe removal from suspension started, as the amount of Fe(II) in suspension remained stable while the calculated amount of total Fe(II) increased. At the bottoms of the bottles, whitish gray precipitations formed, which we assume were siderite or vivianite (reaction 2c in Fig. 1). At that time point, no sulfide seemed to have been left to precipitate with Fe(II). Apparently, an equilibrium between Fe(II) in suspension and surface-bound polysulfides and elemental sulfur had been established.
FIG 3.
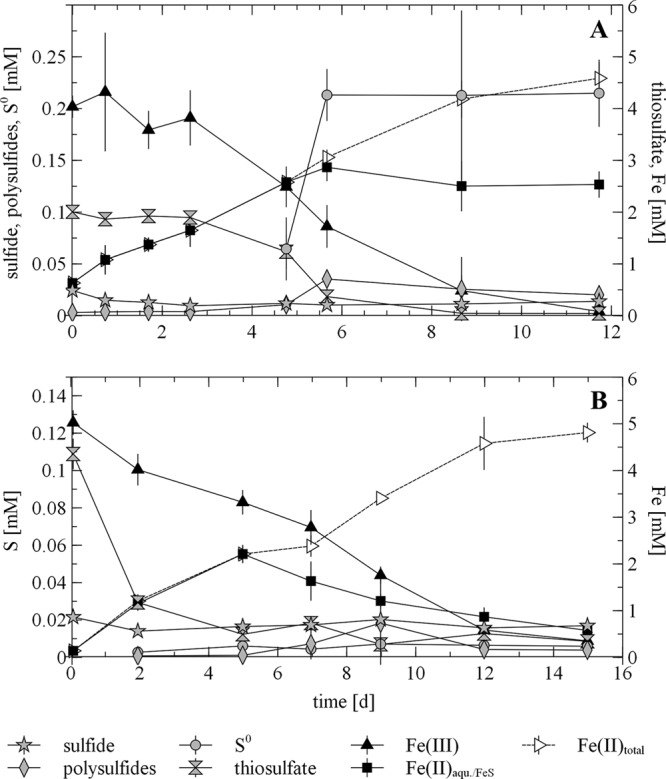
Reduction of Fe(III) in ferrihydrite in incubations of S. deleyianum supplied with 5 mM acetate as a carbon source, 10 mM formate as an electron donor, and 2 mM (A) or 0.1 mM (B) thiosulfate as an electron acceptor. Error bars represent standard deviations based on three replicates.
When only 0.1 mM thiosulfate was used (Fig. 3B), the thiosulfate was consumed almost completely within 5 days, whereas iron(III) mineral reduction took 15 days. After depletion of the initially added amount of thiosulfate, no reincrease of the thiosulfate concentration was measurable. As shown before, thiosulfate was only a minor oxidation product. Therefore, either no thiosulfate was produced during sulfide oxidation or thiosulfate reduction by the microorganisms was faster than abiotic sulfide oxidation. Again, almost no sulfide was detectable in solution but seems to have immediately precipitated as FeS, since the suspension was already black on day 2. Hence, a dynamic equilibrium between abiotic sulfide oxidation and microbial reduction of the oxidized sulfur species seemed to have been established. The hypothesis that sulfide from FeS precipitates was reused (reaction 2b in Fig. 1) is also supported by the finding that Fe(II) concentrations in suspension decreased from day 5 on, when all of the initially added thiosulfate was reduced. The residual Fe(II) was then precipitated as vivianite or siderite (reaction 2c, Fig. 1). Elemental sulfur was measurable in unfiltered samples from day 2 on in much smaller amounts than in the experiment with 2 mM thiosulfate. Concentrations remained stable until the end of the experiment. Polysulfides also occurred in much smaller amounts than in the experiment with 2 mM thiosulfate. Overall, significant amounts of polysulfides and different species were detectable only in experiments with 2 mM thiosulfate.
(ii) The roles of other sulfur species as electron shuttles.
Apart from thiosulfate, also sulfide, polysulfides, sulfite, tetrathionate, and cystine were tested as initiators of the process of electron shuttling between S. deleyianum and ferrihydrite (Fig. 4). The fact that the rates of iron(III) mineral reduction were very comparable in the experiments with the reduced sulfur species (sulfide) and the oxidized sulfur species (thiosulfate, sulfite, and tetrathionate) revealed the rapid kinetics of microbial reduction of the different sulfur species. Thus, not the reductive part of the electron-shuttling process but rather the abiotic oxidation of sulfide seems to be rate limiting.
FIG 4.
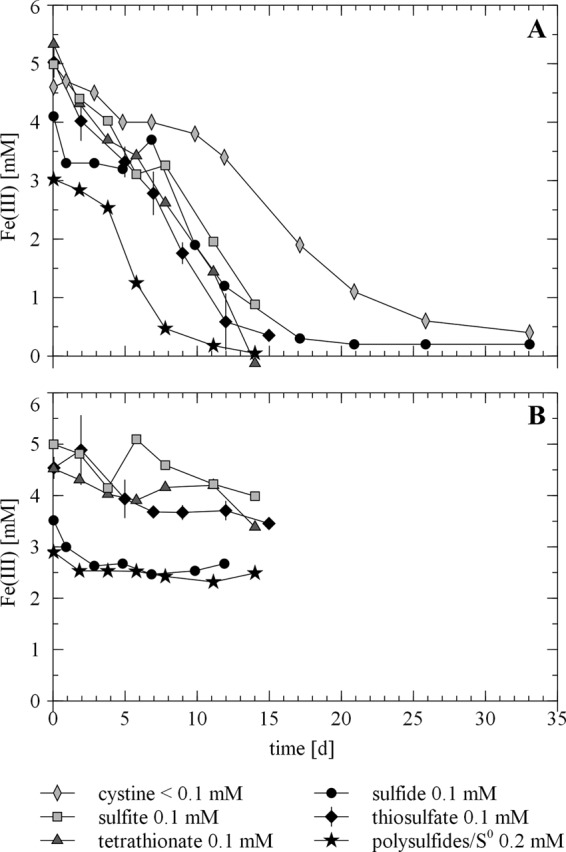
Reduction of Fe(III) in ferrihydrite in incubations of S. deleyianum (A) supplied with 5 mM acetate as a carbon source, 2 mM cysteine as a sulfur source and reductant, 10 mM formate as an electron donor, and different sulfur species as electron acceptors and in an abiotic control experiment (B). Error bars for the experiments with 2 mM cysteine and 0.1 mM thiosulfate represent standard deviations based on three replicates.
(a) Sulfide or polysulfides.
The addition of sulfide or polysulfides to the bottles led to an immediate blackening of the suspensions and hence the formation of FeS. Abiotically, 0.5 and 1.4 mM Fe(II) were measurable directly at the beginning of the experiments with ferrihydrite amended with 0.1 mM sulfide and polysulfides, respectively. Nevertheless, not more than 2 mM Fe(III) was reduced in any of the abiotic experiments. In all biotic experiments, complete Fe(III) reduction was observable, with the fastest reaction kinetics for polysulfides, followed by thiosulfate, tetrathionate, sulfide, sulfite, and finally cystine. The reduction of polysulfides was expected, since according to IMG genome annotation (31), dissimilatory sulfur or polysulfide reduction to hydrogen sulfide is mediated by a sulfur reductase complex containing a NrfD-like subunit C (gene loci Sdel_0265, Sdel_0266, and Sdel_0267). The particularly fast reduction of polysulfides (reaction 3a in Fig. 1) can either be triggered by additional elemental sulfur impurities (up to 0.1 mM) in the commercial K2Sx standard or be linked to their apparent association with the microorganisms as described before, facilitating electron transfer processes.
(b) Sulfite.
Sulfite could be used as efficiently as thiosulfate (reaction 3b in Fig. 1), which is in accordance with earlier studies (19, 20). However, this observation is in contrast to the study of Straub and Schink (17), who could not detect any assimilation or dissimilation of sulfite. Comparative genome analyses revealed the presence of a mccA-type gene encoding a new emerging type of terminal sulfite reductase (Sdel_703) with high sequence similarity to Wolinella succinogenes MccA (blastp: 94% positives). Sulfite reduction activity of MccA was shown in vitro and/or in vivo for W. succinogenes and Shewanella oneidensis MR-1 (recently renamed SirA) (38, 39). The entire mcc gene cluster is present in the S. deleyianum genome (gene loci Sdel_0697 through Sdel_705), including the genes mccABCD and ccsA1, the genes for a two-component regulatory system named MccR/MccS located upstream, and two genes encoding hypothetical proteins downstream. In addition to the octaheme cytochrome c sulfite reductase MccA (gene locus Sdel_703), the genes for the following are essential for sulfite respiration, as shown for W. succinogenes: the predicted iron-sulfur protein MccC (gene locus Sdel_701), the putative quinol dehydrogenase MccD (a member of the NrfD/PsrC family, gene locus Sdel_0700), and a peptidyl-prolyl cis-trans isomerase named MccB (gene locus Sdel_702).
(c) Tetrathionate.
Apparently, tetrathionate can also serve as an electron acceptor for S. deleyianum (reaction 3c in Fig. 1), which was, to the best of our knowledge, never shown before. On the basis of genome analysis, we would assume that tetrathionate is likely to be reduced to thiosulfate by a TsdA-type tetrathionate reductase in S. deleyianum. TsdA was first described in Allochromatium vinosum as a novel diheme cytochrome c with thiosulfate dehydrogenase activity (40, 41). In Campylobacter jejuni 81116 (42), a homologue of TsdA shows bifunctional activity, acting as both a thiosulfate dehydrogenase and a tetrathionate reductase. The similarity between the TsdA protein sequences of S. deleyianum (gene locus Sdel_0259) and C. jejuni (gene locus C8j_0815) was moderately high (blastp: 68% positives). Neither a homologue of the gene for tetrathionate reductase subunit A (TtrA) from Salmonella enterica serovar Typhimurium nor a homologue of the gene for octaheme tetrathionate reductase Otr from Shewanella oneidensis MR-1 is present in the genome of S. deleyianum.
(d) Cysteine.
Cysteine was added to the medium as a sulfur source and a reducing agent as proposed by Straub and Schink (17). Nevertheless, genome analysis reveals that S. deleyianum is an l-cysteine prototroph that is able to convert l-serine via O-acetyl-l-serine (gene locus Sdel_1290 encoding a serine O-acetyltransferase) to l-cysteine (gene locus Sdel_1232 encoding a cysteine synthase). In Escherichia coli, this pathway is regulated by strong feedback inhibition of the final product, l-cysteine, and transcriptional regulation (43). Genes encoding l-cystine oxidoreductases (EC 1.8.1.6) for the synthesis and degradation of l-cystine could not be found in the S. deleyianum genome. Cysteine degradation to pyruvate is mediated by a putative C-S lyase (gene locus Sdel_1447) with high similarity to a PatB-like protein from S. barnesii SES-3 (accession no. YP_006404370 with 83% identical amino acid sites and 90% positives). The PatB protein of Bacillus subtilis is a proven C-S lyase that exhibits both cystathionine beta-lyase and cysteine desulfhydrase activities in vitro (44).
Concerning the electron-shuttling process, it has to be considered that cysteine is a redox-sensitive compound itself. Electron shuttling via cysteine and cystine has been shown for Shewanella species with the Fe(III)-containing clay mineral smectite (45) and for Geobacter sulfurreducens with ferrihydrite as a terminal electron acceptor (46). For S. deleyianum, Straub and Schink (17) excluded a shuttling mechanism via cysteine-cystine, as no ferrihydrite was reduced after the addition of cystine. Instead, they assumed that cysteine only served to protect the reduced sulfur species from oxidation.
We tested cystine, the oxidized form of cysteine, as a possible electron shuttle and found that cystine indeed could also serve as an electron acceptor for S. deleyianum (reaction 4a in Fig. 1) but showed the lowest rate of iron(III) mineral reduction. This might be attributable to a methodological problem. Because cystine is not readily soluble, when it is dissolved in pure water (graph in Fig. 4A), its true concentration might be lower than the nominal one. However, in another experiment, cystine was dissolved in 5 M NaOH prior to adjustment of the experimental pH, which led to complete dissolution. With 0.1 mM cysteine and 0.1 mM cystine, 0.3 mM Fe(III) was reduced within 15 days, whereas 1.8 mM was reduced in the respective experiment with thiosulfate (see Table SI-2 in the supplemental material). This revealed that shuttling via cystine was not as efficient as that via thiosulfate.
(iii) The influence of cysteine on the process of electron shuttling between S. deleyianum and ferrihydrite.
As discussed in the previous section, the presence of cystine seemed to have a certain influence on ferrihydrite reduction. Therefore, we also determined the effect of cysteine on ferrihydrite reduction by S. deleyianum in the presence of 0.1 mM thiosulfate (Fig. 5). Abiotically, up to 1.7 mM ferrihydrite was reduced when 2 mM cysteine was available. Either this could result from an abiotic reaction of cysteine with ferrihydrite (reaction 4b, Fig. 1), which would exclude any abiotic influence of thiosulfate, or cysteine reduced thiosulfate abiotically to sulfide (reaction 4c, Fig. 1), which then reacted with ferrihydrite. From the literature, it is known that sulfide can be produced by the reaction of thiosulfate with cysteine (47). On the basis of the experiments in this study, it cannot be decided whether, in the presence of thiosulfate, cysteine reduced ferrihydrite directly or indirectly via the reduction of thiosulfate. Without cysteine and with only 0.1 mM thiosulfate, 1.1 mM ferrihydrite was reduced in biotic experiments. Obviously, S. deleyianum could use thiosulfate as an electron acceptor, as was also confirmed by Straub and Schink (17). Since there was no cysteine in the system in the aforementioned experiment, thiosulfate must have also served as a sulfur source for the microorganisms. The addition of 0.1 mM cysteine increased the kinetics of ferrihydrite reduction significantly. A further increase was observable after the addition of 0.5 and 2 mM cysteine.
FIG 5.
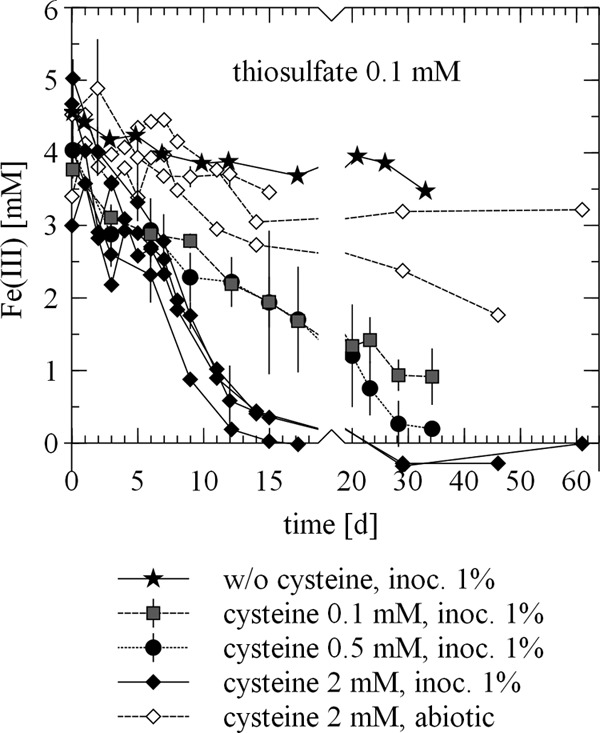
Reduction of Fe(III) in ferrihydrite in an abiotic control experiment and in incubations of S. deleyianum supplied with 5 mM acetate as a carbon source, 10 mM formate as an electron donor, and 0.1 mM thiosulfate as an electron acceptor. Error bars represent standard deviations based on three replicates. For the setup with 2 mM cysteine and a 1% inoculum, results of six experiments are shown, one experiment with three replicates and three additional independent experiments. For the abiotic setup with 2 mM cysteine, results of three independent experiments are shown. w/o, without; inoc., inoculum.
Finally, the effect of cysteine alone on ferrihydrite reduction by S. deleyianum was investigated. Straub and Schink (17) considered the abiotic reduction of ferrihydrite by cysteine negligible. In our control experiments without cysteine, we observed that no Fe(III) was reduced (Fig. 6). With 4 mM cysteine, complete reduction of 4 mM ferrihydrite was observed (Fig. 6). This could be attributed principally to the abiotic reaction of cysteine with ferrihydrite, since one electron is transferred per cysteine molecule in the oxidation of cysteine to cystine (reaction 4c in Fig. 1) and one electron per molecule is also needed for the reduction of ferrihydrite (reaction 1b in Fig. 1). In the two abiotic control experiments, 2 mM cysteine was able to reduce around 1.2 mM ferrihydrite within 46 and 61 days (Fig. 6). This is in line with previous studies in which iron was already shown to be reduced by cysteine under various conditions (48–50). In fact, even Straub and Schink (17) observed a reduction of 1 to 2 mM Fe(III) with 2 mM cysteine and a 0.01% inoculum over 35 days. In three experiments with 2 mM cysteine in the presence of S. deleyianum (1% inoculum), we determined an average ferrihydrite reduction of 2.33 ± 0.70 mM within 33 to 61 days (Fig. 6). The fact that slightly more iron was reduced in the experiment with a 1% inoculum than in the abiotic experiment (Fig. 6) and the results of the experiment with cystine described before indicate that the electron shuttling by the redox couple cysteine-cystine is possible but not very efficient compared to that by other sulfur species and thus plays a minor role in iron(III) reduction by S. deleyianum. Overall, the decisive effect of cysteine on ferrihydrite reduction is that it is an abiotic reductant of ferrihydrite itself or oxidized sulfur species that then can react with ferrihydrite.
FIG 6.
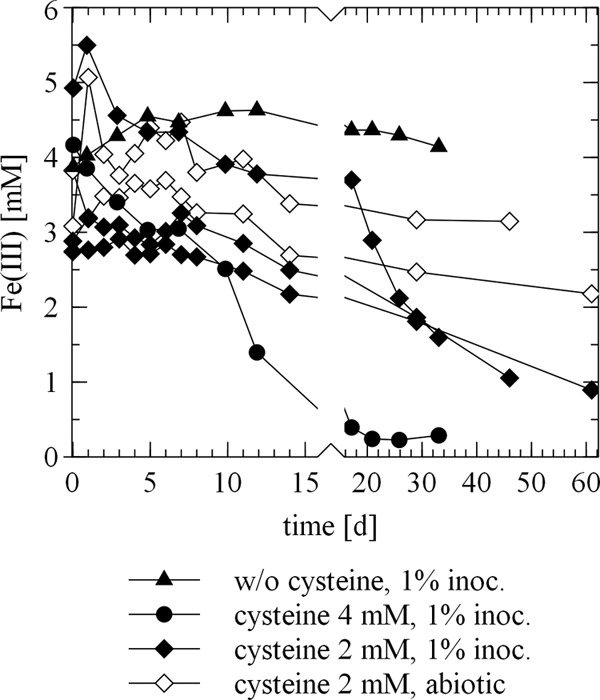
Reduction of Fe(III) in ferrihydrite in an abiotic control experiment and in incubations of S. deleyianum supplied with 5 mM acetate as a carbon source and 10 mM formate as an electron donor. The experiment with 2 mM cysteine and a 1% inoculum was done in triplicate, and the abiotic one with 2 mM cysteine was done in duplicate. w/o, without; inoc., inoculum.
Conclusions.
The present study brings further insights into the role of sulfur species during ferrihydrite reduction specifically by S. deleyianum in the presence of reactive sulfur compounds. We observed complete microbial reduction of thiosulfate to sulfide with elemental sulfur and polysulfides as intermediate products. The main sulfur species, which formed during abiotic reoxidation of sulfide by iron reduction, was elemental sulfur attached to the surface of the iron mineral. Therefore, sulfur electron shuttling seems to be localized on the iron(III) mineral surface and direct physical contact between S. deleyianum and ferrihydrite seems to be necessary to complete the electron-shuttling cycle. No free sulfide could be detected in the presence of iron(III), because of both fast sulfide reoxidation by iron(III) and the formation of FeS, which acted as a recyclable sulfide reservoir.
Small amounts of sulfite, tetrathionate, thiosulfate, or polysulfides accelerated iron(III) mineral reduction considerably and reduced iron(III) to iron(II) in stoichiometric excess. Microbial sulfur reduction was so fast that no difference in iron reduction was observed whether sulfur species were initially applied in the oxidized (sulfite, tetrathionate, thiosulfate) or the reduced (sulfide) state.
Cysteine was found to have a significant influence on ferrihydrite reduction. It can react abiotically with ferrihydrite or oxidized sulfur species or may serve to a minor extent together with cystine as an electron shuttle between microorganisms and ferrihydrite. Cysteine should be eliminated from microbial experiments when sulfur redox reactions are to be investigated, especially since we found that S. deleyianum grows on thiosulfate as the only sulfur source as well.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funding by the German Research Foundation for the research group etrap (electron transfer processes in anoxic aquifers) (FOR 580, PLA 302/7-1), as well as financial support for a Ph.D. scholarship to Regina Lohmayer from the State of Bavaria according to the Bayerisches Eliteförderungsgesetz (BayEFG).
Footnotes
Published ahead of print 14 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04220-13.
REFERENCES
- 1.Petersen W, Wallmann K, Schroer S, Schroeder F. 1993. Studies on the adsorption of cadmium on hydrous iron(III) oxides in oxic sediments. Anal. Chim. Acta 273:323–327. 10.1016/0003-2670(93)80172-H [DOI] [Google Scholar]
- 2.Dong DM, Nelson YM, Lion LW, Shuler ML, Ghiorse WC. 2000. Adsorption of Pb and Cd onto metal oxides and organic material in natural surface coatings as determined by selective extractions: new evidence for the importance of Mn and Fe oxides. Water Res. 34:427–436. 10.1016/S0043-1354(99)00185-2 [DOI] [Google Scholar]
- 3.Bennett B, Dudas MJ. 2003. Release of arsenic and molybdenum by reductive dissolution of iron oxides in a soil with enriched levels of native arsenic. J. Environ. Eng. Sci. 2:265–272. 10.1139/s03-028 [DOI] [Google Scholar]
- 4.Phuengprasop T, Sittiwong J, Unob F. 2011. Removal of heavy metal ions by iron oxide coated sewage sludge. J. Hazard. Mater. 186:502–507. 10.1016/j.jhazmat.2010.11.065 [DOI] [PubMed] [Google Scholar]
- 5.Hohmann C, Winkler E, Morin G, Kappler A. 2010. Anaerobic Fe(II)-oxidizing bacteria show As resistance and immobilize As during Fe(III) mineral precipitation. Environ. Sci. Technol. 44:94–101. 10.1021/es900708s [DOI] [PubMed] [Google Scholar]
- 6.Wang XM, Liu F, Tan WF, Li W, Feng XH, Sparks DL. 2013. Characteristics of phosphate adsorption-desorption onto ferrihydrite: comparison with well-crystalline Fe (hydr)oxides. Soil Sci. 178:1–11. 10.1097/SS.0b013e31828683f8 [DOI] [Google Scholar]
- 7.Heckman K, Welty-Bernard A, Vazquez-Ortega A, Schwartz E, Chorover J, Rasmussen C. 2013. The influence of goethite and gibbsite on soluble nutrient dynamics and microbial community composition. Biogeochemistry 112:179–195. 10.1007/s10533-012-9715-2 [DOI] [Google Scholar]
- 8.Nevin KP, Lovley DR. 2000. Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by Geobacter metallireducens. Appl. Environ. Microbiol. 66:2248–2251. 10.1128/AEM.66.5.2248-2251.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Canstein H, Ogawa J, Shimizu S, Lloyd JR. 2008. Secretion of flavins by Shewanella species and their role in extracellular electron transfer. Appl. Environ. Microbiol. 74:615–623. 10.1128/AEM.01387-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kappler A, Benz M, Schink B, Brune A. 2004. Electron shuttling via humic acids in microbial iron(III) reduction in a freshwater sediment. FEMS Microbiol. Ecol. 47:85–92. 10.1016/S0168-6496(03)00245-9 [DOI] [PubMed] [Google Scholar]
- 11.Roden EE, Kappler A, Bauer I, Jiang J, Paul A, Stoesser R, Konishi H, Xu HF. 2010. Extracellular electron transfer through microbial reduction of solid-phase humic substances. Nat. Geosci. 3:417–421. 10.1038/ngeo870 [DOI] [Google Scholar]
- 12.Li XM, Liu L, Liu TX, Yuan T, Zhang W, Li FB, Zhou SG, Li YT. 2013. Electron transfer capacity dependence of quinone-mediated Fe(III) reduction and current generation by Klebsiella pneumoniae L17. Chemosphere 92:218–224. 10.1016/j.chemosphere.2013.01.098 [DOI] [PubMed] [Google Scholar]
- 13.Afonso MD, Stumm W. 1992. Reductive dissolution of iron(III) (hydr)oxides by hydrogen sulfide. Langmuir 8:1671–1675. 10.1021/la00042a030 [DOI] [Google Scholar]
- 14.Li YL, Vali H, Yang J, Phelps TJ, Zhang CL. 2006. Reduction of iron oxides enhanced by a sulfate-reducing bacterium and biogenic H2S. Geomicrobiol. J. 23:103–117. 10.1080/01490450500533965 [DOI] [Google Scholar]
- 15.Burton ED, Johnston SG, Bush RT. 2011. Microbial sulfidogenesis in ferrihydrite-rich environments: effects on iron mineralogy and arsenic mobility. Geochim. Cosmochim. Acta 75:3072–3087. 10.1016/j.gca.2011.03.001 [DOI] [Google Scholar]
- 16.Burton ED, Johnston SG, Planer-Friedrich B. 2013. Coupling of arsenic mobility to sulfur transformations during microbial sulfate reduction in the presence and absence of humic acid. Chem. Geol. 343:12–24. 10.1016/j.chemgeo.2013.02.005 [DOI] [Google Scholar]
- 17.Straub KL, Schink B. 2004. Ferrihydrite-dependent growth of Sulfurospirillum deleyianum through electron transfer via sulfur cycling. Appl. Environ. Microbiol. 70:5744–5749. 10.1128/AEM.70.10.5744-5749.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luijten M, Weelink SAB, Godschalk B, Langenhoff AAM, van Eekert MHA, Schraa G, Stams AJM. 2004. Anaerobic reduction and oxidation of quinone moieties and the reduction of oxidized metals by halorespiring and related organisms. FEMS Microbiol. Ecol. 49:145–150. 10.1016/j.femsec.2004.01.015 [DOI] [PubMed] [Google Scholar]
- 19.Schumacher W, Kroneck PMH, Pfennig N. 1992. Comparative systematic study on “spirillum” 5175, Campylobacter and Wolinella species—description of “spirillum” 5175 as Sulfurospirillum deleyianum gen. nov., spec. nov. Arch. Microbiol. 158:287–293. 10.1007/BF00245247 [DOI] [Google Scholar]
- 20.Wolfe RS, Pfennig N. 1977. Reduction of sulfur by spirillum 5175 and syntrophism with Chlorobium. Appl. Environ. Microbiol. 33:427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Widdel F, Bak F. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p 3352–3378 In Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K.-H. (ed), The prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications, 2nd ed, vol IV Springer, Berlin, Germany [Google Scholar]
- 22.Schwertmann U, Cornell RM. 2000. Iron oxides in the laboratory—preparation and characterization, 2nd ed. Wiley VCH Weinheim, Germany [Google Scholar]
- 23.Raven KP, Jain A, Loeppert RH. 1998. Arsenite and arsenate adsorption on ferrihydrite: kinetics, equilibrium, and adsorption envelopes. Environ. Sci. Technol. 32:344–349. 10.1021/es970421p [DOI] [Google Scholar]
- 24.Piepenbrock A, Dippon U, Porsch K, Appel E, Kappler A. 2011. Dependence of microbial magnetite formation on humic substance and ferrihydrite concentrations. Geochim. Cosmochim. Acta 75:6844–6858. 10.1016/j.gca.2011.09.007 [DOI] [Google Scholar]
- 25.Stookey LL. 1970. Ferrozine—a new spectrophotometric reagent for iron. Anal. Chem. 42:779–781. 10.1021/ac60289a016 [DOI] [Google Scholar]
- 26.Cline JD. 1969. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14:454–458. 10.4319/lo.1969.14.3.0454 [DOI] [Google Scholar]
- 27.Kamyshny A, Ekeltchik I, Gun J, Lev O. 2006. Method for the determination of inorganic polysulfide distribution in aquatic systems. Anal. Chem. 78:2631–2639. 10.1021/ac051854a [DOI] [PubMed] [Google Scholar]
- 28.Rizkov D, Lev O, Gun J, Anisimov B, Kuselman I. 2004. Development of in-house reference materials for determination of inorganic polysulfides in water. Accredit. Qual. Assur. 9:399–403. 10.1007/s00769-004-0788-z [DOI] [Google Scholar]
- 29.Steudel R, Holdt G, Gobel T. 1989. Ion-pair chromatographic-separation of inorganic sulphur anions including polysulphide. J. Chromatogr. 475:442–446. 10.1016/S0021-9673(01)89701-6 [DOI] [Google Scholar]
- 30.Planer-Friedrich B, London J, McCleskey RB, Nordstrom DK, Wallschlager D. 2007. Thioarsenates in geothermal waters of Yellowstone National Park: determination, preservation, and geochemical importance. Environ. Sci. Technol. 41:5245–5251. 10.1021/es070273v [DOI] [PubMed] [Google Scholar]
- 31.Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang JH, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40:D115–D122. 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedderich R, Klimmek O, Kroger A, Dirmeier R, Keller M, Stetter KO. 1998. Anaerobic respiration with elemental sulfur and with disulfides. FEMS Microbiol. Rev. 22:353–381. 10.1111/j.1574-6976.1998.tb00376.x [DOI] [Google Scholar]
- 33.Cantrell KJ, Yabusaki SB, Engelhard MH, Mitroshkov AV, Thornton EC. 2003. Oxidation of H2S by iron oxides in unsaturated conditions. Environ. Sci. Technol. 37:2192–2199. 10.1021/es020994o [DOI] [PubMed] [Google Scholar]
- 34.Poulton SW. 2003. Sulfide oxidation and iron dissolution kinetics during the reaction of dissolved sulfide with ferrihydrite. Chem. Geol. 202:79–94. 10.1016/S0009-2541(03)00237-7 [DOI] [Google Scholar]
- 35.Pyzik AJ, Sommer SE. 1981. Sedimentary iron monosulfides: kinetics and mechanism of formation. Geochim. Cosmochim. Acta 45:687–698. 10.1016/0016-7037(81)90042-9 [DOI] [Google Scholar]
- 36.Saalfield SL, Bostick BC. 2009. Changes in iron, sulfur, and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems. Environ. Sci. Technol. 43:8787–8793. 10.1021/es901651k [DOI] [PubMed] [Google Scholar]
- 37.Peiffer S, Gade W. 2007. Reactivity of ferric oxides toward H2S at low pH. Environ. Sci. Technol. 41:3159–3164. 10.1021/es062228d [DOI] [PubMed] [Google Scholar]
- 38.Kern M, Klotz MG, Simon J. 2011. The Wolinella succinogenes mcc gene cluster encodes an unconventional respiratory sulphite reduction system. Mol. Microbiol. 82:1515–1530. 10.1111/j.1365-2958.2011.07906.x [DOI] [PubMed] [Google Scholar]
- 39.Shirodkar S, Reed S, Romine M, Saffarini D. 2011. The octahaem SirA catalyses dissimilatory sulfite reduction in Shewanella oneidensis MR- 13:1 Environ. Microbiol. 13: 108–115. 10.1111/j.1462-2920.2010.02313.x [DOI] [PubMed] [Google Scholar]
- 40.Hensen D, Sperling D, Truper HG, Brune DC, Dahl C. 2006. Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Mol. Microbiol. 62:794–810. 10.1111/j.1365-2958.2006.05408.x [DOI] [PubMed] [Google Scholar]
- 41.Denkmann K, Grein F, Zigann R, Siemen A, Bergmann J, van Helmont S, Nicolai A, Pereira IAC, Dahl C. 2012. Thiosulfate dehydrogenase: a widespread unusual acidophilic c-type cytochrome. Environ. Microbiol. 14:2673–2688. 10.1111/j.1462-2920.2012.02820.x [DOI] [PubMed] [Google Scholar]
- 42.Liu YW, Denkmann K, Kosciow K, Dahl C, Kelly DJ. 2013. Tetrathionate stimulated growth of Campylobacter jejuni identifies a new type of bi-functional tetrathionate reductase (TsdA) that is widely distributed in bacteria. Mol. Microbiol. 88:173–188. 10.1111/mmi.12176 [DOI] [PubMed] [Google Scholar]
- 43.Kredich NM. 1992. The molecular basis for positive regulation of cys promoters in Salmonella typhimurium and Escherichia coli. Mol. Microbiol. 6:2747–2753. 10.1111/j.1365-2958.1992.tb01453.x [DOI] [PubMed] [Google Scholar]
- 44.Auger S, Gomez MP, Danchin A, Martin-Verstraete I. 2005. The PatB protein of Bacillus subtilis is a C-S-lyase. Biochimie 87:231–238. 10.1016/j.biochi.2004.09.007 [DOI] [PubMed] [Google Scholar]
- 45.Liu D, Dong H, Zhao L, Wang H. 2013. Smectite reduction by Shewanella species as facilitated by cystine and cysteine. Geomicrobiol. J. 31:53–63. 10.1080/01490451.2013.806609 [DOI] [Google Scholar]
- 46.Doong RA, Schink B. 2002. Cysteine-mediated reductive dissolution of poorly crystalline iron(III) oxides by Geobacter sulfurreducens. Environ. Sci. Technol. 36:2939–2945. 10.1021/es0102235 [DOI] [PubMed] [Google Scholar]
- 47.Szczepkowski TW. 1958. Reactions of thiosulphate with cysteine. Nature 182:934–935. 10.1038/182934a0 [DOI] [PubMed] [Google Scholar]
- 48.Cornell RM, Schneider W, Giovanoli R. 1989. Phase-transformations in the ferrihydrite/cysteine system. Polyhedron 8:2829–2836. 10.1016/S0277-5387(00)80544-6 [DOI] [Google Scholar]
- 49.Morrison KD, Bristow TF, Kennedy MJ. 2013. The reduction of structural iron in ferruginous smectite via the amino acid cysteine: implications for an electron shuttling compound. Geochim. Cosmochim. Acta 106:152–163. 10.1016/j.gca.2012.12.006 [DOI] [Google Scholar]
- 50.Amirbahman A, Sigg L, vonGunten U. 1997. Reductive dissolution of Fe(III) (hydr)oxides by cysteine: kinetics and mechanism. J. Colloid Interface Sci. 194:194–206. 10.1006/jcis.1997.5116 [DOI] [PubMed] [Google Scholar]
- 51.Wan M, Shchukarev A, Lohmayer R, Planer-Friedrich B, Peiffer S. The occurrence of surface polysulphides during the interaction between ferric (hydr)oxides and aqueous sulphide. Environ. Sci. Technol., in press [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.