

Abstract
Cellular processes, such as the digestion of macromolecules, phosphate acquisition, and cell motility, require bacterial secretion systems. In Bacillus subtilis, the predominant protein export pathways are Sec (generalized secretory pathway) and Tat (twin-arginine translocase). Unlike Sec, which secretes unfolded proteins, the Tat machinery secretes fully folded proteins across the plasma membrane and into the medium. Proteins are directed for Tat-dependent export by N-terminal signal peptides that contain a conserved twin-arginine motif. Thus, utilizing the Tat secretion system by fusing a Tat signal peptide is an attractive strategy for the production and export of heterologous proteins. As a proof of concept, we expressed green fluorescent protein (GFP) fused to the PhoD Tat signal peptide in the laboratory and ancestral strains of B. subtilis. Secretion of the Tat-GFP construct, as well as secretion of proteins in general, was substantially increased in the ancestral strain. Furthermore, our results show that secreted, fluorescent GFP could be purified directly from the extracellular medium. Nonetheless, export was not dependent on the known Tat secretion components or the signal peptide twin-arginine motif. We propose that the ancestral strain contains additional Tat components and/or secretion regulators that were abrogated following domestication.
INTRODUCTION
Protein secretion is the process by which a cell expends energy to export a protein through at least one membrane to various locations of the cell exterior. Bacteria have evolved numerous protein secretion systems that differ in the number and identity of proteins they secrete and the number of membranes that the cargo proteins must transit (1, 2). Although many protein secretion systems are specific determinants of bacterial virulence, others, like the Sec (generalized secretory) and Tat (twin-arginine translocase) secretion systems, are fundamental export pathways that are conserved in all domains of life (3–5). Whereas the Sec system secretes proteins that are in their unfolded state, the Tat pathway exclusively secretes fully folded, active proteins from the cell (6–10). How the various protein export components specifically recognize their protein substrates is complex. Comparing the Sec and Tat secretion systems, there are differences in the signal peptides that target proteins for secretion and the complexes that mediate transport of cellular proteins to the cell exterior.
The signal peptides that are characteristic of the Sec and Tat secretion systems are similar in that they exhibit three features: an N-terminal positively charged region, a hydrophobic α-helical region, and a C-terminal region that may contain a proteolytic cleavage site (11–13). When comparing Sec and Tat signal peptides, the Tat signal peptides have a longer leader sequence prior to the hydrophobic α-helical helix, an α-helical region that is less hydrophobic (14), and more basic residues before the α-helical region, including two consecutive arginine residues (RR; sometimes RK) (14–19). The subtle differences between the two types of signal peptides are enough to dictate which secretion pathway, either Sec or Tat, is used to direct protein export.
Tat transport systems are composed of multiprotein complexes that are embedded within, and secrete fully folded proteins through, the plasma membrane (4). Precisely how the Tat system accommodates the variable sizes and diverse shapes of cargo proteins remains unclear, but it is thought to be dependent on the structural organization of the transport complex (4). The canonical Tat transport complex, such as the one found in Gram-negative Escherichia coli, is composed of at least three subunits: TatA, TatB, and TatC (20–23). TatC is a multipass transmembrane protein that interacts with TatB and recognizes the substrate signal peptide (24–28). Once bound, the TatBC export protein complex is thought to recruit TatA protomers to create an active translocon (29–32). The extent of TatA polymerization may dictate the size of the active export channel (21, 32). The precise role of TatB is unclear; however, this subunit does interact with the signal peptide following substrate binding (24, 25, 33). In certain bacteria, a TatA homologue, called TatE, is present and carries out a function that is redundant with TatA (34). It is not known which subunit consumes the proton-motive force to power protein secretion (35, 36).
Although most bacteria utilize only a single set of Tat proteins, the laboratory strain of Bacillus subtilis (PY79) encodes two sets of Tat homologues, named TatAdCd and TatAyCy. The TatAd and TatAy proteins function in a manner analogous to that of both the E. coli TatA and TatB proteins, whereas TatCd and TatCy function in a manner analogous to that of TatC (37–41). The Tat system in B. subtilis encodes a fifth Tat protein of unknown function, called TatAc, but does not encode a homologue of the E. coli TatB protein (42–45). Despite containing two Tat pathways, the repertoire of identified Tat-secreted proteins in B. subtilis is relatively limited (46, 47). PhoD, an alkaline phosphatase, is secreted via TatAdCd, whereas YwbN, a protein of unknown function, and QcrA, a component of the cytochrome c complex, are secreted via TatAyCy (42, 43, 47, 48). It is not known if additional proteins are secreted by these systems.
Secretion of proteins via the Tat system is advantageous because proteins fold and mature in the host cytoplasm before being secreted directly into the medium, but development of a heterologous Tat secretion system in B. subtilis has been challenging (6–10). Previous work has shown that expressing green fluorescent protein (GFP) fused to the TMAO reductase (TorA) Tat signal peptide in E. coli resulted in Tat-dependent secretion of folded GFP into the periplasm (49) and to the extracellular medium if the B. subtilis TatAdCd exporter was expressed in place of the native E. coli TatABC exporter (50). In contrast, expressing a similar TorA-GFP construct in B. subtilis resulted in the secretion of unfolded or nonfluorescent GFP (8). Alternatively, expressing GFP fused to E. coli Tat signal peptides derived from either AmiA, DmsA, or MdoD in B. subtilis resulted in Tat-independent secretion under high-salinity conditions (51). It was inferred that Tat-independent secretion of GFP was mediated by the Sec pathway due to the similarities in signal peptide structure and accounting for the transport of unfolded protein (51). Finally, it has been demonstrated that the E. coli AppA protein can be secreted from B. subtilis when fused to the PhoD signal peptide (PhoDSP) (52).
In this work, we show that when GFP is fused to the native B. subtilis PhoDSP and expressed in the 3610 ancestral strain of B. subtilis, folded, fluorescent GFP can be secreted directly into the extracellular medium. Although our GFP construct was designed to contain the hallmarks of a Tat-secreted protein, export of folded GFP was not dependent on the canonical Tat machinery components or the twin-arginine motif in the signal peptide. Our data are not consistent with spurious recognition and secretion through the constitutive Sec pathway. We hypothesize that the ancestral strain of B. subtilis encodes an as-yet-unidentified export pathway and/or export regulator that may have been lost during domestication of the laboratory strain.
MATERIALS AND METHODS
Growth conditions for strain construction.
B. subtilis strains were grown in Luria-Bertani (LB; 10 g tryptone, 5 g yeast extract, 5 g NaCl per liter) broth or on LB plates fortified with 1.5% Bacto agar at 37°C. When appropriate, antibiotics were included at the following concentrations: 10 μg/ml tetracycline, 100 μg/ml spectinomycin, 5 μg/ml chloramphenicol, 5 μg/ml kanamycin, and 1 μg/ml erythromycin plus 25 μg/ml lincomycin (termed MLS, for macrolides-lincosamides-streptogramin B).
SPP1 phage transduction.
To 0.2 ml of dense culture grown in TY broth (LB broth supplemented after autoclaving with 10 mM MgSO4 and 100 μM MnSO4), serial dilutions of SPP1 phage stock were added and statically incubated for 15 min at 37°C. To each mixture, 3 ml TYSA (molten TY supplemented with 0.5% agar) was added, poured atop fresh TY plates, and incubated at 37°C overnight. Top agar from the plate containing nearly confluent plaques was harvested by scraping into a 50-ml conical tube, vortexed, and centrifuged at 5,000 × g for 10 min. The supernatant was treated with 25 μg/ml DNase I (final concentration) before being passed through a 0.45-μm-pore-size syringe filter and stored at 4°C.
Recipient cells were grown to stationary phase in 2 ml TY broth at 37°C. Cells (0.9 ml) were mixed with 5 μl of SPP1 donor phage stock. Nine milliliters of TY broth was added to the mixture and allowed to stand at 37°C for 30 min. The transduction mixture was then centrifuged at 5,000 × g for 10 min, the supernatant was discarded, and the pellet was resuspended in the remaining volume. The cell suspension (100 μl) was then plated on TY fortified with 1.5% agar, the appropriate antibiotic, and 10 mM sodium citrate.
Strain construction.
All constructs were first introduced into either the domesticated strain PY79 or the cured undomesticated strain DS2569 by natural competence and then transferred to the 3610 background using SPP1-mediated generalized phage transduction (Table 1) (53, 54).
TABLE 1.
Bacterial strains used in this study

In-frame deletions of genes encoding extracellular proteases.
To generate the Δbpr in-frame markerless deletion construct, the region upstream of bpr was PCR amplified using the primer pair 1739/1740 and digested with EcoRI and SalI, and the region downstream of bpr was PCR amplified using the primer pair 1741/1742 and digested with SalI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP311 (55).
To generate the Δvpr in-frame markerless deletion construct, the region upstream of vpr was PCR amplified using the primer pair 1743/1744 and digested with EcoRI and SalI, and the region downstream of vpr was PCR amplified using the primer pair 1745/1746 and digested with SalI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP312.
To generate the ΔnprE in-frame markerless deletion construct, the region upstream of nprE was PCR amplified using the primer pair 1747/1748 and digested with EcoRI and SalI, and the region downstream of nprE was PCR amplified using the primer pair 1749/1750 and digested with SalI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP313.
To generate the ΔaprE in-frame markerless deletion construct, the region upstream of aprE was PCR amplified using the primer pair 1751/1752 and digested with EcoRI and SalI, and the region downstream of aprE was PCR amplified using the primer pair 1753/1754 and digested with SalI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP314.
To generate the Δepr in-frame markerless deletion construct, the region upstream of epr was PCR amplified using the primer pair 1755/1756 and digested with EcoRI and XhoI, and the region downstream of epr was PCR amplified using the primer pair 1757/1758 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP315.
To generate the ΔwprA in-frame markerless deletion construct, the region upstream of wprA was PCR amplified using the primer pair 1855/1856 and digested with EcoRI and XhoI, and the region downstream of wprA was PCR amplified using the primer pair 1857/1858 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP319.
To generate the Δmpr in-frame markerless deletion construct, the region upstream of mpr was PCR amplified using the primer pair 1759/1760 and digested with EcoRI and XhoI, and the region downstream of mpr was PCR amplified using the primer pair 1919/1770 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette, to generate pDP320.
Each in-frame markerless deletion plasmid was concatemerized by passage through recA+ E. coli and introduced by natural competence into B. subtilis strain PY79 by single-crossover integration by transformation at the restrictive temperature for plasmid replication (37°C) using MLS resistance as a selection. SPP1 phage-mediated generalized transduction into B. subtilis strain 3610 or an appropriate recipient then transduced the integrated plasmid. To evict the plasmid, the strain was incubated in 3 ml LB broth at a permissive temperature for plasmid replication (22°C) for 14 h and serially diluted and plated on LB agar at 37°C. Individual colonies were patched on LB plates and LB plates containing MLS to identify MLS-sensitive colonies that had evicted the plasmid. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR to determine which isolate had retained the deleted allele. After one in-frame deletion of the protease strain was confirmed, the next deletion construct was introduced. The series of deletions culminated in strain DS6329 containing a series of seven protease deletions. Each deletion was confirmed by PCR length polymorphism.
tat mutants.
The ΔtatCd::kan insertion deletion allele was generated by isothermal assembly using primer pairs 3252/3253, 3254/3255, and 3250/3251 to PCR amplify regions upstream and downstream of tatCd and a kanamycin drug resistance gene (pDG780), respectively, to obtain the insertion deletion construct ITADBK6.
The ΔtatAdCd::kan insertion deletion allele was generated by isothermal assembly using primer pairs 3252/3860, 3254/3255, and 3250/3251 to PCR amplify regions upstream and downstream of tatAdCd and a kanamycin drug resistance gene (pDG780), respectively, to obtain the insertion deletion construct ITASM31 (56, 57).
The ΔtatCy::cat insertion deletion allele was generated by isothermal assembly using primer pairs 3256/3257, 3258/3259, and 3250/3251 to PCR amplify regions upstream and downstream of tatCy and a chloramphenicol drug resistance gene (pAC225), respectively, to obtain the insertion deletion construct ITADBK8.
The ΔtatAyCy::cat insertion deletion allele was generated by isothermal assembly using primer pairs 3256/3861, 3258/3259, and 3250/3251 to PCR amplify regions upstream and downstream of tatAyCy and a chloramphenicol drug resistance gene (pAC225), respectively, to obtain the insertion deletion construct ITASM32 (56, 57).
The ΔtatAc::mls insertion deletion allele was generated by isothermal assembly using primer pairs 3866/3867, 3868/3869, and 3250/3251 to PCR amplify regions upstream and downstream of tatAc and a erythromycin drug resistance gene (pAH52), respectively, to obtain the insertion deletion construct ITASM33 (56, 57).
Inducible constructs.
To generate the inducible amyE::Physpank-PhoDSP spec construct pTM1, a PCR product containing PhoDSP (signal peptide) was amplified from B. subtilis 3610 chromosomal DNA using the 3903/3904 primer pair and cloned into the HindIII and NheI sites of pDR111 containing a spectinomycin resistance cassette, a polylinker downstream of the Physpank promoter, and the gene encoding the LacI repressor between the two arms of the amyE gene (58).
To generate the inducible amyE::Physpank-PhoDSP-GFP spec construct pTM2, a PCR product containing gfp was amplified from B. subtilis strain DS908 chromosomal DNA using the 3069/3070 primer pair, digested with NotI and SalI, and cloned into the NotI and SalI sites of pTM1 (58).
The inducible amyE::Physpank-GFP spec construct pTM7 was generated by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) using primers TM885/TM886 and the pTM2 construct as a template. The inducible amyE::Physpank-AAPhoDSP-GFP spec construct pTM8 was generated by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) using primers TM883/TM884 and the pTM2 construct as a template. The inducible amyE::Physpank-PhoDSP-GFP6×His spec construct pTM9 was generated by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) using primer pairs TM887/TM888 and the pTM2 construct as a template.
To generate the amyE::Physpank-AmyESP-GFP spec construct pTM14, a PCR product containing AmyESP (signal peptide) was amplified from B. subtilis 3610 chromosomal DNA using the TM1013/TM1014 primer pair. The PCR product was used as the primer in a QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) reaction with the pTM7 construct serving as the template.
Growth conditions for GFP secretion under phosphate-limiting conditions.
The indicated strains were grown overnight at 25°C in high-phosphate defined medium (HPDM) [50 mM Tris (pH 7.1), 3.03 mM (NH4)2SO4, 6.8 mM trisodium citrate, 3.04 mM FeCl3, 1 mM MnCl2, 3.5 mM MgSO4, 0.01 mM ZnCl2, 0.5% glucose, 0.05% Casamino Acids, 10 mM l-arginine, and 3.5 mM KH2PO4] (59) supplemented with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The next day, the cells were pelleted at 6,300 × g for 15 min at 25°C, washed three times with low-phosphate defined medium (LPDM) [50 mM Tris (pH 7.1), 3.03 mM (NH4)2SO4, 6.8 mM trisodium citrate, 3.04 mM FeCl3, 1 mM MnCl2, 3.5 mM MgSO4, 0.01 mM ZnCl2, 0.5% glucose, 0.05% Casamino Acids, 10 mM l-arginine, and 0.065 mM KH2PO4] (59), and resuspended in fresh LPDM (optical density at 600 nm [OD600] of 2.0) supplemented with 1 mM IPTG. The resuspended cells were grown at 25°C. At the indicated time points, cell culture was harvested and spun at 16,000 × g for 15 min at 25°C to pellet the cells. The cleared medium was removed and retained for fluorescence and Western blot analysis. GFP fluorescence (excitation at 495 nm, emission at 508 nm) within the cleared medium was measured using a Synergy H1 hybrid plate reader (BioTek, Winooski, VT). Relative fluorescence units were normalized to account for differences in cell density.
Western blot analysis of cell lysates and growth medium.
Cell pellets from 1 ml of cell culture were resuspended in 100 μl of lysis buffer (20 mM Tris [pH 7.0], 10 mM EDTA, 1 mg/ml lysozyme, 10 μg/ml DNase I, 100 μg/ml RNase I, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) and incubated for 30 min at 37°C. Following cell lysis, the lysates were solubilized in reducing SDS sample buffer. The medium harvested from 1 ml of cell culture was precipitated using trichloroacetic acid (TCA). The precipitated medium was solubilized in 50 μl of reducing SDS sample buffer. The solubilized cell lysates and solubilized medium were analyzed by SDS-PAGE and probed with a monoclonal antibody recognizing GFP (Clontech Laboratories, Mountain View, CA) and polyclonal antibodies recognizing SigA (courtesy of Masaya Fujita) and alkaline phosphatase D (PhoD) (courtesy of Joerg Mueller). The volumes of cell lysates and TCA-precipitated medium that were loaded onto the SDS-PAGE gels were normalized to account for differences in cell density.
Fluorescence microscopy.
The indicated strains were grown overnight at 25°C in HPDM supplemented with 1 mM IPTG. The next day, 1 ml of cells was pelleted and resuspended in 100 μl of HPDM. Ten microliters of cell suspension was applied to a microscope slide and immobilized with a coverslip. The cells were imaged for GFP fluorescence using an Olympus 1X71 fluorescence microscope (Olympus, Center Valley, PA). All images were captured with the same magnification and exposure settings. False coloring and background normalization were done equally for each image.
Quantitation of GFP expression levels by quantitative reverse transcription-PCR (qRT-PCR).
The indicated strains were grown overnight at 25°C in HPDM supplemented with 1 mM IPTG. The next day, 5 ml of cell culture was mixed with 750 μl of stop solution (5% phenol diluted in 100% ethanol) and spun at 6,300 × g for 10 min at 4°C. The cell pellet was resuspended in 500 μl of methanol and spun again at 16,000 × g for 1 min at 4°C. The cell pellet was resuspended in 475 μl of lysis buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 0.5 mg/ml lysozyme, and 0.5% SDS) and incubated at 37°C for 45 min. Following cell lysis, 25 μl of 3 M sodium acetate (pH 5.2) and 500 μl of phenol were added to each sample. The samples were incubated for 6 min at 64°C. RNA was isolated from cell lysates using phenol-chloroform extraction followed by ethanol precipitation. Residual DNA was removed using RQ1 DNase (Promega, Madison, WI) by following the manufacturer's protocol. After DNase digestion, RNA was isolated using phenol-chloroform extraction followed by ethanol precipitation. The precipitated RNA was resuspended in 50 μl of RNase-free water.
The RNA prepared as described above (500 ng) was mixed with random hexamer reverse transcription (RT) primers. The samples were incubated at 94°C for 5 min and 70°C for 5 min and then transferred to ice. Once on ice, an RT mixture containing 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 10 mM dithiothreitol, 3 mM MgCl2, 0.5 mM deoxynucleoside triphosphate, RNase inhibitor (GenScript, Piscataway, NJ), and ImProm-II reverse transcriptase (Promega, Madison, WI) was added to each sample. Following addition of the RT mixture, the samples were incubated at 25°C for 5 min, 42°C for 45 min, and 75°C for 15 min.
The cDNA prepared as described above (2 μl) was mixed with 1× Brilliant SYBR green reagent (Stratagene, La Jolla, CA) and 250 nM forward and reverse detection primers (TM860/TM861 for GFP detection or TM858/TM859 for SigA detection) to a final volume of 25 μl per well in a 96-well plate. Multiple qRT-PCR measurements were made for each sample. The fold difference in GFP RNA abundance was calculated using the delta delta comparative threshold cycle method (ΔΔCT). SigA RNA levels were used as the reference. Expression of PhoDSP-GFP RNA was standardized to 1.0.
Purification of secreted GFP6×His.
The B. subtilis strain integrated with PhoDSP-GFP6×His was grown overnight at 25°C in HPDM supplemented with 1 mM IPTG. The next day, the cells were pelleted at 6,300 × g for 15 min at 25°C, washed three times with LPDM, and resuspended in fresh LPDM (OD600 of 2.0) supplemented with 1 mM IPTG. The resuspended cells were grown at 25°C. After 6 h, the medium was harvested by pelleting the cells at 6,300 × g for 15 min at 25°C. The cleared medium was dialyzed into loading buffer (20 mM phosphate [pH 8], 300 mM NaCl, and 10 mM imidazole) and filtered using a 0.2-μm-pore-size syringe filter. The filtered medium was loaded onto a HisTrap FF crude column (1 ml) (GE Healthcare, Piscataway, NJ). Protein was eluted from the column by step gradient using elution buffer (20 mM phosphate [pH 8], 300 mM NaCl, and 300 mM imidazole). GFP fluorescence (excitation at 495 nm, emission at 508 nm) within each fraction was measured using a Synergy H1 hybrid plate reader (BioTek, Winooski, VT). The fractions (1 ml) were also precipitated using TCA. The precipitated fractions were solubilized in 50 μl of reducing SDS sample buffer, analyzed by SDS-PAGE, and probed with a monoclonal antibody recognizing GFP (Clontech Laboratories, Mountain View, CA). Equal volumes of each TCA-precipitated fraction were loaded onto the SDS-PAGE gels.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
Five nanograms of the top and bottoms bands of purified GFP6×His were digested with chymotrypsin (Sigma-Aldrich, St. Louis, MO) for 18 h at 37°C. The digests were quenched with 1% formic acid (Sigma-Aldrich, St. Louis, MO). Six microliters of digested protein was loaded onto a C18 reversed-phase trapping column (15-mm, 100-μm-inner-diameter capillary packed with 5-μm Magic C18AQ particles with 200-Å pore sizes; Michrom Bioresources, Auburn, CA) and washed with approximately 20 μl of solvent A (3% acetonitrile, 0.1% formic acid). Peptides were separated by elution through a 15-cm reversed-phase nano-LC column (75-μm-inner-diameter capillary pulled to a tip and packed with 5-μm Magic C18AQ particles with 100-Å pore sizes; Michrom Bioresources) by increasing solvent B (0.1% formic acid in acetonitrile) from 5% to 40% at 250 ml/min over 30 min and electrosprayed directly into the source of an ion trap mass spectrometer, which recorded mass spectra and data-dependent tandem mass spectra of the peptide ions (LCQ Deca XP; ThermoFinnigan, San Jose, CA). Data-dependent tandem mass spectra were recorded by acquiring a precursor mass spectrum followed by two tandem mass spectra of the two most intense ions from the precursor scan (unless excluded by the dynamic exclusion algorithm, in which case the next most abundant ions were selected). Spectra were automatically interpreted using the database searching tool Mascot v1.9 (Matrix Science, Boston, MA) and manually validated.
RESULTS
The ancestral strain of B. subtilis can secrete folded, recombinant GFP.
Previous studies to investigate the capacity of Tat signal peptides to direct Tat-dependent export of functional, heterologous proteins from laboratory strains of B. subtilis achieved limited success (8, 51, 52). Since commonly used laboratory strains are heavily domesticated, we hypothesized that secretion efficiency was strain dependent (60). We determined the amount of secreted protein in the laboratory strain PY79 and the ancestral strain 3610. Each strain was grown overnight in high-phosphate defined medium (HPDM), and the next day the cells were washed and grown in phosphate-limiting or low-phosphate defined medium (LPDM). Phosphate starvation upregulates expression of the TatAdCd transporter, which in turn mediates Tat-dependent secretion (42, 43). After 0 and 6 h of incubation under reduced phosphate conditions, the medium was harvested, TCA precipitated, and analyzed by SDS-PAGE. The ancestral strain showed a dramatic enrichment of extracellular proteins compared to the laboratory strain derivative (Fig. 1).
FIG 1.
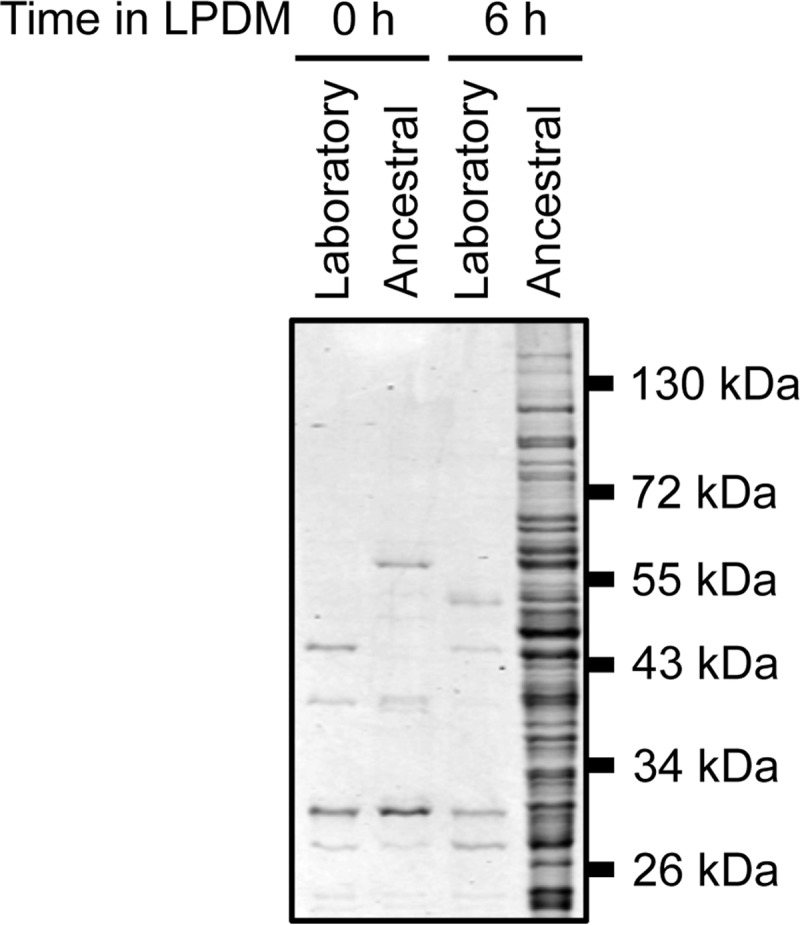
Total protein secretion from laboratory (PY79) and wild-type ancestral (3610) strains of B. subtilis under phosphate-limiting conditions. The indicated strains were grown overnight in HPDM. The next day, the cells were washed and resuspended in LPDM. Medium aliquots were collected at 0 and 6 h after growing in LPDM, precipitated with TCA, and analyzed by SDS-PAGE. The gel was Coomassie stained. Migration of molecular mass standards is indicated to the right of the panel.
Enhanced extracellular protein accumulation can be explained as either increased diversity of secreted proteins or enhanced secretion of extracellular proteases that commensurately increased the number of degradation products. We hypothesized that the ancestral strain background would improve Tat-dependent secretion of heterologous proteins. To investigate Tat-dependent secretion of a heterologous protein, we fused GFP to the Tat signal peptide derived from B. subtilis alkaline phosphatase D (PhoDSP-GFP) (Fig. 2A) (43). PhoDSP-GFP was designed so that no residual amino acids would remain at the N terminus of GFP following cleavage of the signal peptide. The construct was integrated into the B. subtilis genome at the ectopic amyE locus in PY79 and 3610 and expressed from an IPTG-inducible Physpank promoter. The endogenous PhoD and its cognate TatAdCd export machinery, however, are expressed in response to phosphate starvation (42, 43). To determine if PhoDSP-GFP could be secreted under conditions similar to those for PhoD, strains were grown overnight in HPDM. The next day, the cells were washed and resuspended in either HPDM or LPDM and grown for an additional 6 h. PhoDSP-GFP expression and secretion were monitored by Western blot probing with anti-GFP (α-GFP) and anti-PhoD (α-PhoD) antibodies and by GFP fluorescence in the medium (Fig. 2B and C). As a control for cell lysis, cell lysates and supernatants were probed for the housekeeping sigma factor SigA, which is a cytoplasmic and constitutively expressed protein. When expressed in the PY79 laboratory strain, PhoDSP-GFP accumulated intracellularly (Fig. 2B, lysate lanes). Furthermore, the cytoplasm of expressing cells was highly fluorescent (data not shown); however, after growing under phosphate starvation conditions, GFP was not detected in the medium (Fig. 2B and C).
FIG 2.

Protein secretion from laboratory, Δ7 ancestral, and wild-type ancestral strains of B. subtilis under phosphate-limiting conditions. (A) Endogenous PhoD gene arrangement and PhoDSP-GFP construct design. The predicted molecular mass of each protein is indicated in parentheses. (B and D) Expression and secretion of total GFP from laboratory (B) and Δ7 ancestral strains (D). The indicated strains expressing PhoDSP-GFP were grown overnight in HPDM supplemented with 1 mM IPTG. The next day, the cells were washed and resuspended in either HPDM or LPDM supplemented with 1 mM IPTG. After 0 and 6 h in LPDM, the cells were lysed and the medium was precipitated with TCA. The cell lysates and precipitated medium were examined by Western blot probing with α-GFP, α-PhoD, and α-SigA antibodies. The migration of PhoDSP-GFP, PhoD, and SigA bands is indicated to the left of each blot. The migration of molecular mass standards is indicated to the right of each blot. (C and E) Secretion of folded, fluorescent GFP from laboratory and Δ7 ancestral strains. The indicated strains expressing PhoDSP-GFP were grown overnight as described for panels B and D. At the indicated time points, the medium was harvested and GFP fluorescence was measured. Relative fluorescent units were normalized to account for differences in cell density. Results from one representative experiment are shown. (F and G) Expression and secretion of total GFP (F) and secretion of fluorescent GFP (G) from wild-type (WT) ancestral and Δ7 ancestral strains. The indicated strains were grown and the samples were prepared as described for panels B through E. Relative fluorescent units were normalized to account for differences in cell density. Results from one representative experiment are shown.
To maximize the chances of successfully secreting folded GFP, we next introduced the PhoDSP-GFP construct into the 3610 ancestral strain that was simultaneously mutated for all seven known extracellular proteases (here referred to as the Δ7 strain) (61–70). Expression of PhoDSP-GFP in the Δ7 strain resulted in fluorescent protein within the cytoplasm of expressing cells (data not shown). In contrast to the laboratory strain, we observed extracellular accumulation of fluorescent (properly folded) GFP, and protein secretion was enhanced in LPDM compared to its level in HPDM (Fig. 2D and E). The α-PhoD antibody recognized cytoplasmic but not secreted GFP, indicating that signal peptide cleavage was correlated with export. Furthermore, SigA was not detected in the medium, demonstrating that GFP secretion was not due to cell lysis (Fig. 2D). Taken together, we conclude that fluorescent GFP was secreted from the Δ7 strain when fused to a Tat signal peptide.
To examine the relevance of deleting the extracellular proteases, we expressed PhoDSP-GFP in the 3610 ancestral strain that produced each of the seven proteases (wild-type ancestral). Western blot analysis showed that intracellular PhoDSP-GFP (Fig. 2F, lysate lanes) and secreted GFP (Fig. 2F, medium lanes, and G) were present for each strain. Although the presence of the extracellular proteases did not seem to have a significant effect on folded PhoDSP-GFP processing and yield (as determined by fluorescence) (Fig. 2G), the Δ7 strain was used as our preferred expression strain unless otherwise indicated.
Purification and characterization of secreted GFP6×His.
The results shown in Fig. 2 indicated that multiple GFP bands (each differing in electrophoretic mobility) were secreted following PhoDSP-GFP expression under phosphate-limiting conditions. These bands may represent alternative cleavage products of the signal peptide (or of GFP) that arise prior to or concurrent with export. To examine the nature of these bands, we purified and characterized secreted GFP. To aid in purification, we generated a strain that expressed PhoDSP-GFP with a C-terminal His tag (PhoDSP-GFP6×His) (see Fig. S1A in the supplemental material). qRT-PCR, fluorescent images of expressing cells, and Western blot analysis indicated that PhoDSP-GFP6×His was made at lower levels than PhoDSP-GFP (see Table S3 and Fig. S1B and C); however, we still observed multiple GFP6×His species in the lysates and in the medium (see Fig. S1C).
After growing the strain expressing PhoDSP-GFP6×His under phosphate-limiting conditions, the medium was collected, dialyzed, and applied to a Ni2+-agarose column. After several washes, GFP6×His was eluted from the column with increasing amounts of imidazole. When fractions were collected and examined for GFP fluorescence, two prominent fluorescent peaks were observed (Fig. 3A). The fractions that contained maximum fluorescence were precipitated with TCA and analyzed by SDS-PAGE and Western blot analysis (Fig. 3B, upper and lower, respectively). These analyses revealed two GFP bands (top and bottom) that were present in fractions with peak fluorescent intensity and migrated at approximately the same molecular weight as GFP6×His. To our knowledge, this is the first report that folded, fluorescent GFP can be secreted and purified from Gram-positive B. subtilis supernatants.
FIG 3.

Purification of GFP6×His secreted from B. subtilis. The Δ7 3610 strain expressing PhoDSP-GFP6×His was grown overnight in HPDM supplemented with 1 mM IPTG. The next day, the cells were washed and resuspended in LPDM supplemented with 1 mM IPTG. After 6 h in LPDM, the medium was harvested, dialyzed into loading buffer, and applied to a Ni2+-agarose column, and protein was eluted from the column with increasing concentrations of imidazole. (A) GFP fluorescence in fractions collected off the Ni2+ column. (B) The indicated fractions were TCA precipitated and analyzed by SDS-PAGE. The gel was either Coomassie stained (upper) or examined by Western blot probing with an α-GFP antibody (lower). Migration of the top and bottom GFP6×His bands is indicated to the left of each panel. The migration of molecular mass standards is indicated to the right of each panel.
The top and bottom bands of purified GFP6×His, as indicated by their reaction with the α-GFP antibody (Fig. 3B, lower), were excised from the SDS-PAGE gel, digested in-gel with chymotrypsin, and analyzed by LC/MS-MS to identify specific peptides corresponding to GFP6×His (Fig. 4; also see Tables S4 and S5 in the supplemental material). Each sample had ∼50% peptide coverage to GFP6×His. Interestingly, the top band contained peptides that corresponded to the last four amino acids of the PhoD signal peptide and the first six or seven amino acids of GFP (EVNASKGEEL and EVNASKGEELF, respectively) (Fig. 4A; also see Table S4). We conclude that the top band was the result of noncanonical Tat signal peptide cleavage. We did not obtain N-terminal peptides for the lower band, but since the lower band is smaller and fluorescent, it potentially represents most, if not all, GFP.
FIG 4.

Mass spectrometry analysis of purified GFP6×His. The top and bottom bands of purified GFP6×His (Fig. 3B) were isolated and subjected to in-gel digestion with chymotrypsin. The digests were analyzed by LC-MS/MS to identify specific peptides corresponding to GFP6×His. (A and B) Peptide coverages of the top and bottom bands of purified GFP6×His. The sequence identified by LC/MS-MS is in boldface and is underlined within the PhoDSP-GFP6×His sequence.
Secretion of folded GFP can occur in a Tat- and Sec-independent manner.
Our results show that folded, fluorescent GFP was successfully secreted from B. subtilis by fusing an N-terminal Tat signal peptide; however, Tat-independent secretion of a heterologous Tat-tagged protein has been previously reported (51). Thus, we wanted to determine if PhoDSP-GFP was secreted in a Tat-dependent manner. Toward this end, we generated strains that contained individual and double deletions of the two known Tat signal peptide recognition proteins, TatCd and TatCy (24–26, 71). qRT-PCR, fluorescent images of expressing cells, and Western blot analysis indicated that each Tat deletion strain expressed PhoDSP-GFP at similar levels (see Table S3 and Fig. S2 in the supplemental material). After growing under phosphate-limiting conditions, we observed similar levels of secreted GFP (total [Fig. 5A, medium lanes] and folded [Fig. 5B]) for each Tat deletion strain. We also monitored export of endogenous PhoD in the presence or absence of PhoDSP-GFP induction (Fig. 5C; also see Fig. S3). PhoD was expressed under phosphate-limiting conditions, but as previously reported, secretion was blocked specifically when TatCd was absent (43).
FIG 5.

GFP secretion from B. subtilis Tat deletion strains. (A) Secretion of total GFP in single or double Tat mutants. The indicated strains (in a Δ7 3610 background) were grown overnight in HPDM supplemented with 1 mM IPTG. The next day, the cells were washed and resuspended in LPDM supplemented with 1 mM IPTG. At the indicated time points, the cells were lysed and the medium was precipitated with TCA. The cell lysates and precipitated medium were examined by Western blot probing with α-GFP and α-SigA antibodies. (B) Secretion of folded GFP. The indicated strains were grown overnight as described for panel A. At the indicated time points, the medium was harvested and examined for GFP fluorescence. Relative fluorescent units were normalized to account for differences in cell density. Results from one representative experiment are shown. (C) Expression and secretion of endogenous PhoD in the single and double Tat mutants. Samples were prepared as described for panel A. The migration of molecular mass standards is indicated to the right of each blot. (D) Secretion of total GFP in a quintuple Tat mutant in a Δ7 3610 background. Samples were prepared as described for panel A. The cell lysates and precipitated medium were examined by Western blot probing with α-GFP, α-PhoD, and α-SigA antibodies.
We next generated a quintuple mutant that disrupted all of the predicted Tat secretion proteins: TatAd, TatCd, TatAy, TatCy, and TatAc. Despite these deletions, export of total GFP was not abolished and did not appear to be reduced (Fig. 5D, medium lanes). The quintuple deletion also did not affect secretion of folded GFP after 6 h in LPDM (data not shown). Thus, we conclude that GFP secretion was mediated independently of the known Tat pathway; therefore, the rules that govern export of heterologous proteins may differ from those of endogenous PhoD.
It has been hypothesized that one route for GFP secretion is spurious recognition and transport through the Sec pathway (51). Because the Sec pathway is essential for bacterial survival (3, 5), we could not analyze PhoDSP-GFP expression and secretion from strains containing deletions of Sec components. Instead, for comparison, we fused GFP to the Sec signal peptide derived from α-amylase AmyE and expressed the construct from the IPTG-inducible Physpank promoter (called AmyESP-GFP) (72). After growing cells overnight in HPDM supplemented with IPTG, we detected very small amounts of intracellular AmyESP-GFP expression by Western blotting (Fig. 6A and B, lysate lanes). Furthermore, GFP was not found in the medium following expression under phosphate-limiting conditions (Fig. 6A, AmyESP-GFP medium lanes). Thus, we were unable to demonstrate export of folded GFP using the AmyE Sec signal peptide. We also note that enhancement of PhoDSP-GFP secretion under phosphate starvation conditions is incongruent with transport through the constitutively active Sec pathway (Fig. 2D and E). Taking these findings together, we infer that the Tat-independent mechanism of PhoDSP-GFP secretion is also Sec independent.
FIG 6.

Expression of GFP fused to the AmyE Sec signal peptide in B. subtilis. (A) Secretion of total GFP. The indicated strains in a Δ7 3610 background were grown overnight in HPDM supplemented with 1 mM IPTG. The next day, the cells were washed and resuspended in LPDM supplemented with 1 mM IPTG. At the indicated time points, the cells were lysed and the medium was precipitated with TCA. The cell lysates and precipitated medium were examined by Western blot probing with α-GFP, α-PhoD, and α-SigA antibodies. Migration of PhoDSP-GFP and SigA bands is indicated to the left of each blot. The migration of molecular mass standards is indicated to the right of each blot. In panel B, the SDS-PAGE gel was overloaded with sample and the blot was overexposed to visualize the AmyESP-GFP band.
Changing the timing or amount of IPTG induction does not influence the GFP secretion pathway.
Our results show that GFP secretion is (i) enhanced under phosphate starvation conditions and (ii) independent of the canonical Tat export machinery. We hypothesized that our experimental approach contributed to the observed Tat-independent export phenotype. For example, expression of endogenous PhoD and the TatAdCd machinery occurs only in response to phosphate-limiting conditions (42, 43); however, in our system, PhoDSP-GFP expression was induced with IPTG prior to and during phosphate starvation. To determine if the timing and/or amount of IPTG induction influenced which protein secretion pathway was utilized, we used two alternative experimental conditions. In the first experiment, we delayed PhoDSP-GFP expression until after the cells were resuspended in LPDM (Fig. 7A). In the second experiment, we induced PhoDSP-GFP expression with 10-fold less IPTG (in both HPDM and LPDM) throughout the time course (Fig. 7B). Regardless of the experimental conditions, we observed comparable levels of total GFP secretion from the Δ7 and ΔTatCdCy strains (Fig. 7A and B, compare medium lanes of the Δ7 and ΔTatCdCy strains). These results are consistent with Tat-independent export irrespective of the amount or timing of PhoDSP-GFP induction.
FIG 7.

GFP secretion from B. subtilis under modified IPTG induction conditions. (A and B) Secretion of total GFP. The indicated strains in a Δ7 3610 background were grown according to the outlined schematics. After 6 h in LPDM, the cells were lysed and the medium was precipitated with TCA. The cell lysates and precipitated medium were examined by Western blot probing with α-GFP, α-PhoD, and α-SigA antibodies. The migration of PhoDSP-GFP and SigA bands is indicated to the left of each blot. The migration of molecular size standards is indicated to the right of each blot.
As an alternative approach, we also attempted to express the TatAdCd and/or TatAyCy export machinery artificially under an IPTG-inducible promoter in HPDM so the production of PhoDSP-GFP and the transport complexes would occur simultaneously. Overexpression of the Tat machinery components resulted in significant amounts of cell lysis as determined by detection of SigA in the supernatant during Western blot analysis (data not shown). Previous work has shown that overexpressing the B. subtilis TatAdCd exporter in E. coli causes leakage of periplasmic content into the medium, perhaps due to a compromised bacterial membrane (50).
GFP secretion is enhanced by the Tat signal peptide.
Tat signal peptides are characterized by a conserved (but not essential) twin-arginine (RR) motif, which is thought to play a role in cargo recognition during Tat-dependent export (15, 20, 73, 74). To determine if the RR motif was required for protein export in our system, we mutated the conserved arginine residues in PhoDSP to two alanine residues (called AAPhoDSP-GFP) (Fig. 8A). PhoDSP-GFP and AAPhoDSP-GFP expressed to comparable levels (see Table S3 and Fig. S4 in the supplemental material). Consistent with Tat-independent export, we observed similar levels of secreted GFP (total [Fig. 8B, medium lanes] and folded [Fig. 8C]) for the PhoDSP-GFP- and AAPhoDSP-GFP-expressing strains.
FIG 8.

Signal peptide-dependent GFP secretion from B. subtilis. (A) Sequence of the PhoD signal peptide fused to GFP. The mutation site in the twin-arginine motif is indicated with black circles. The GFP construct with no signal peptide is shown below. The predicted molecular mass is indicated in parentheses. (B and D) Secretion of total GFP (B) and endogenous alkaline phosphatase D (PhoD) (D). The indicated strains in a Δ7 3610 background were grown overnight in HPDM supplemented with 1 mM IPTG. The next day, the cells were washed and resuspended in LPDM supplemented with 1 mM IPTG. At the indicated time points, the cells were lysed and the medium was precipitated with TCA. The cell lysates and precipitated medium were examined by Western blot probing with α-GFP and α-SigA antibodies (B) or α-PhoD and α-SigA antibodies (D). Migration of PhoDSP-GFP, GFP, and SigA bands (B) or PhoD and SigA bands (D) is indicated to the left of each blot. The migration of molecular mass standards is indicated to the right of each blot. (C) Secretion of folded GFP. The indicated strains were grown as described for panel B. At the indicated time points, the medium was harvested and examined for GFP fluorescence. Relative fluorescent units were normalized to account for differences in cell density. Results from one representative experiment are shown.
We also expressed untagged GFP to determine how much GFP was secreted in the absence of a Tat or Sec signal peptide (Fig. 8A). We observed reduced secretion of total and folded untagged GFP compared to PhoDSP-GFP (Fig. 8B and C). The reduced extracellular accumulation of untagged GFP likely was not due to greater sensitivity to proteolytic degradation; Western blot analysis did not show lower-molecular-weight GFP degradation products (either in cell lysates or in the medium; data not shown). As expected, expression and secretion of endogenous PhoD only occurred under phosphate-limiting conditions (Fig. 8D). Thus, maximum secretion of folded GFP was enhanced by the PhoD signal peptide; however, the twin-arginine motif, and indeed the signal peptide, was dispensable.
DISCUSSION
B. subtilis is an attractive model for the synthesis and secretion of heterologous proteins, as it is harmless, grows rapidly, and has a facile genetic system. Furthermore, due to its Gram-positive cell architecture, proteins need to transit only a single membrane to be secreted directly into the extracellular medium (75, 76). The Tat pathway, in particular, exports proteins that fold in the cytoplasm; thus, this pathway may allow for the large-scale production of functional proteins that require the reducing environment of the cytoplasm to fold and/or contributions from cytoplasmic cofactors to undergo maturation (6–10). In this work, we demonstrate that folded GFP can be secreted from B. subtilis. GFP was fused to the Tat signal peptide derived from alkaline phosphatase D (PhoDSP-GFP) and secreted in response to phosphate-limiting conditions. Interestingly, PhoDSP-GFP export did not require the known B. subtilis Tat proteins (TatAdCd, TatAyCy, and TatAc) or the signal peptide twin-arginine motif. Importantly, secretion of folded, fluorescent GFP was achieved in the ancestral strain of B. subtilis but not in a laboratory strain derivative.
Domestication of B. subtilis laboratory strains altered many phenotypes found in the ancestral strain, including the loss of biofilm formation, swarming motility, antibiotic synthesis, and polyglutamate production and the gain of high-frequency transformation (54, 77–81). Genome sequence comparisons indicate that the domesticated laboratory strain PY79 descended from the ancestral 3610 strain, and previous studies have shown that some of the phenotypic differences in PY79 can be genetically repaired with 3610 alleles (54, 60, 78–81). Here, we find that the domesticated PY79 variant is highly reduced for protein secretion (Fig. 1). Laboratory strains encode a promoter defect that reduces expression of DegQ, a protein that activates extracellular protease secretion through the DegS/DegU two-component system (79, 82, 83); however, reduction in extracellular protease secretion in the laboratory strain might be predicted to improve extracellular protein accumulation and does not seem to account for the observed reduction. The genetic reason for a generalized decrease in protein secretion in PY79 is unknown. We conclude that the enhanced secretion exhibited by the ancestral 3610 strain is advantageous for protein secretion studies.
We observed export of folded GFP from the ancestral strain even when each of the five genes that encode Tat secretion proteins was deleted from the B. subtilis genome (Fig. 5). Tat-independent secretion of Tat-tagged proteins was reported previously and was attributed to promiscuous cargo recognition by the Sec pathway (51). We propose that Sec likely is not responsible for Tat-independent secretion of Tat-tagged GFP for the following reasons. (i) Although the Tat and Sec signal peptides have casual similarity, there are significant differences that ensure export fidelity (14–19). (ii) Secretion of folded GFP was enhanced under phosphate starvation conditions (Fig. 2D and E), whereas the Sec machinery is constitutively expressed (3, 5). (iii) High levels of fluorescent PhoDSP-GFP accumulated within the cytoplasm of expressing cells (see Fig. S1B, S2A, and S4A in the supplemental material), whereas the Sec machinery requires unfolded cargo. (iv) Fusing a Sec signal peptide to GFP (AmyESP-GFP) resulted in poor protein expression and secretion (Fig. 6). (v) Folded GFP was secreted at low levels when expressed without a Tat signal peptide (Fig. 8B and C). (vi) No evidence was presented to support the hypothesis of promiscuous secretion of folded protein by the Sec system in the original report (51). We infer that an as-yet-undiscovered secretion system is activated under phosphate-limiting conditions (or other cellular stresses) and may be responsible for recognition and secretion of PhoDSP-GFP.
In contrast to PhoDSP-GFP, secretion of endogenous PhoD required the TatAdCd exporter (Fig. 5C; also see Fig. S3 in the supplemental material). When analyzing the exported proteins, extracellular PhoD was present as a single species, whereas PhoDSP-GFP was present as multiple bands (both in cell lysates and in the media), suggesting aberrant proteolysis. Mass spectrometry analysis of secreted GFP6×His revealed evidence for aberrant signal peptide cleavage (Fig. 4), potentially due to improper interactions with an unidentified cellular factor that aids in the identification and cleavage of proteins that are secreted independently of the Tat system. In addition, we found that PhoDSP-GFP secretion did not require the signal peptide twin-arginine motif (Fig. 8B and C). We conclude that the rules that govern secretion of endogenous PhoD and PhoDSP-GFP differ. We infer that the primary and/or tertiary structure information contained outside the signal peptide of PhoD, or other Tat-dependent proteins, is critical for conferring secretion apparatus specificity.
Previous studies have investigated the capacity of Tat signal peptides to direct Tat-dependent export of heterologous proteins from B. subtilis laboratory strains. When GFP was fused to an E. coli-derived Tat signal peptide and expressed in B. subtilis, GFP was secreted into the extracellular medium; however, the exported protein was not properly folded (as determined by a lack of GFP fluorescence in the medium) (8). In contrast, expressing a similar GFP construct in E. coli resulted in Tat-dependent export of folded protein to the periplasm (49). Thus, the mechanism that regulates signal peptide recognition may differ between hosts, perhaps explaining why Tat-dependent secretion of heterologous proteins is generally successful in E. coli but not B. subtilis. In this work, we demonstrated that fusing GFP to the Tat signal peptide derived from endogenous PhoD resulted in secretion of folded protein. The primary reason for our success seems to be due to our use of the undomesticated, ancestral 3610 strain of B. subtilis. We speculate that the ancestral strain contains additional Tat components, regulators, or alternative export systems that were abrogated following domestication.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of our laboratories for critical discussions about this research. We thank Masaya Fujita for providing the SigA antibody and Joerg Mueller for providing the PhoD antibody.
This work received support from NIH grant GM093030 to D.B.K. and NSF grant MCB-1157716 to S. Mukhopadhyay. A.J.S. was supported by Indiana University Genetics, Cellular, and Molecular Sciences training grant T32-GM007757.
Footnotes
Published ahead of print 14 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00335-14.
REFERENCES
- 1.Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM. 2004. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 68:207–233. 10.1128/MMBR.68.2.207-233.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antelmann H, Tjalsma H, Voigt B, Ohlmeier S, Bron S, van Dijl JM, Hecker M. 2001. A proteomic view on genome-based signal peptide predictions. Genome Res. 11:1484–1502. 10.1101/gr.182801 [DOI] [PubMed] [Google Scholar]
- 3.de Keyzer J, van der Does C, Driessen AJ. 2003. The bacterial translocase: a dynamic protein channel complex. Cell. Mol. Life Sci. 60:2034–2052. 10.1007/s00018-003-3006-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10:483–496. 10.1038/nrmicro2814 [DOI] [PubMed] [Google Scholar]
- 5.van Wely KHM, Swaving J, Freudl R, Driessen AJM. 2001. Translocation of proteins across the cell envelope of Gram-positive bacteria. FEMS Microbiol. Rev. 25:437–454. 10.1111/j.1574-6976.2001.tb00586.x [DOI] [PubMed] [Google Scholar]
- 6.Creighton AM, Hulford A, Mant A, Robinson D, Robinson C. 1995. A monomeric, tightly folded stromal intermediate on the ΔpH-dependent thylakoidal protein transport pathway. J. Biol. Chem. 270:1663–1669. 10.1074/jbc.270.4.1663 [DOI] [PubMed] [Google Scholar]
- 7.Hynds PJ, Robinson D, Robinson C. 1998. The sec-independent twin-arginine translocation system can transport both tightly folded and malfolded proteins across the thylakoid membrane. J. Biol. Chem. 273:34868–34874. 10.1074/jbc.273.52.34868 [DOI] [PubMed] [Google Scholar]
- 8.Meissner D, Vollstedt A, van Dijl JM, Freudl R. 2007. Comparative analysis of twin-arginine (Tat)-dependent protein secretion of a heterologous model protein (GFP) in three different Gram-positive bacteria. Appl. Microbiol. Biotechnol. 76:633–642. 10.1007/s00253-007-0934-8 [DOI] [PubMed] [Google Scholar]
- 9.Robinson C, Bolhuis A. 2004. Tat-dependent protein targeting in prokaryotes and chloroplasts. Biochim. Biophys. Acta 1694:135–147. 10.1016/j.bbamcr.2004.03.010 [DOI] [PubMed] [Google Scholar]
- 10.Teter SA, Klionsky DJ. 1999. How to get a folded protein across a membrane. Trends Cell Biol. 9:428–431. 10.1016/S0962-8924(99)01652-9 [DOI] [PubMed] [Google Scholar]
- 11.Luke I, Handford JI, Palmer T, Sargent F. 2009. Proteolytic processing of Escherichia coli twin-arginine signal peptides by LepB. Arch. Microbiol. 191:919–925. 10.1007/s00203-009-0516-5 [DOI] [PubMed] [Google Scholar]
- 12.Thompson BJ, Widdick DA, Hicks MG, Chandra G, Sutcliffe IC, Palmer T, Hutchings MI. 21 June 2010. Investigating lipoprotein biogenesis and function in the model Gram-positive bacterium Streptomyces coelicolor. Mol. Microbiol. 10.1111/j.1365-2958.2010.07261.x [DOI] [PubMed] [Google Scholar]
- 13.Yahr TL, Wickner WT. 2001. Functional reconstitution of bacterial Tat translocation in vitro. EMBO J. 20:2472–2479. 10.1093/emboj/20.10.2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cristobal S, de Gier JW, Nielsen H, von Heijne G. 1999. Competition between Sec- and TAT-dependent protein translocation in Escherichia coli. EMBO J. 18:2982–2990. 10.1093/emboj/18.11.2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berks BC. 1996. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22:393–404. 10.1046/j.1365-2958.1996.00114.x [DOI] [PubMed] [Google Scholar]
- 16.Blaudeck N, Kreutzenbeck P, Freudl R, Sprenger GA. 2003. Genetic analysis of pathway specificity during posttranslational protein translocation across the Escherichia coli plasma membrane. J. Bacteriol. 185:2811–2819. 10.1128/JB.185.9.2811-2819.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogsch E, Brink S, Robinson C. 1997. Pathway specificity for a delta pH-dependent precursor thylakoid lumen protein is governed by a “Sec-avoidance” motif in the transfer peptide and a “Sec-incompatible” mature protein. EMBO J. 16:3851–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi MV, Mann SG, Antelmann H, Widdick DA, Fyans JK, Chandra G, Hutchings MI, Toth I, Hecker M, Loria R, Palmer T. 2010. The twin arginine protein transport pathway exports multiple virulence proteins in the plant pathogen Streptomyces scabies. Mol. Microbiol. 77:252–271. 10.1111/j.1365-2958.2010.07206.x [DOI] [PubMed] [Google Scholar]
- 19.Stanley NR, Palmer T, Berks BC. 2000. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 275:11591–11596. 10.1074/jbc.275.16.11591 [DOI] [PubMed] [Google Scholar]
- 20.Berks BC, Sargent F, Palmer T. 2000. The Tat protein export pathway. Mol. Microbiol. 35:260–274. 10.1046/j.1365-2958.2000.01719.x [DOI] [PubMed] [Google Scholar]
- 21.Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC. 2005. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc. Natl. Acad. Sci. U. S. A. 102:10482–10486. 10.1073/pnas.0503558102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oates J, Barrett CM, Barnett JP, Byrne KG, Bolhuis A, Robinson C. 2005. The Escherichia coli twin-arginine translocation apparatus incorporates a distinct form of TatABC complex, spectrum of modular TatA complexes and minor TatAB complex. J. Mol. Biol. 346:295–305. 10.1016/j.jmb.2004.11.047 [DOI] [PubMed] [Google Scholar]
- 23.Sargent F, Berks BC, Palmer T. 2002. Assembly of membrane-bound respiratory complexes by the Tat protein-transport system. Arch. Microbiol. 178:77–84. 10.1007/s00203-002-0434-2 [DOI] [PubMed] [Google Scholar]
- 24.Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Muller M. 2003. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell 12:937–946. 10.1016/S1097-2765(03)00398-8 [DOI] [PubMed] [Google Scholar]
- 25.Gerard F, Cline K. 2006. Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J. Biol. Chem. 281:6130–6135. 10.1074/jbc.M512733200 [DOI] [PubMed] [Google Scholar]
- 26.Cline K, Mori H. 2001. Thylakoid ΔpH-dependent precursor proteins bind to a cpTatC-Hcf106 complex before Tha4-dependent transport. J. Cell Biol. 154:719–729. 10.1083/jcb.200105149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolhuis A, Mathers JE, Thomas JD, Barrett CM, Robinson C. 2001. TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J. Biol. Chem. 276:20213–20219. 10.1074/jbc.M100682200 [DOI] [PubMed] [Google Scholar]
- 28.Tarry MJ, Schafer E, Chen S, Buchanan G, Greene NP, Lea SM, Palmer T, Saibil HR, Berks BC. 2009. Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc. Natl. Acad. Sci. U. S. A. 106:13284–13289. 10.1073/pnas.0901566106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mori H, Cline K. 2002. A twin arginine signal peptide and the pH gradient trigger reversible assembly of the thylakoid ΔpH/Tat translocase. J. Cell Biol. 157:205–210. 10.1083/jcb.200202048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dabney-Smith C, Mori H, Cline K. 2006. Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J. Biol. Chem. 281:5476–5483. 10.1074/jbc.M512453200 [DOI] [PubMed] [Google Scholar]
- 31.Dabney-Smith C, Cline K. 2009. Clustering of C-terminal stromal domains of Tha4 homo-oligomers during translocation by the Tat protein transport system. Mol. Biol. Cell 20:2060–2069. 10.1091/mbc.E08-12-1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leake MC, Greene NP, Godun RM, Granjon T, Buchanan G, Chen S, Berry RM, Palmer T, Berks BC. 2008. Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc. Natl. Acad. Sci. U. S. A. 105:15376–15381. 10.1073/pnas.0806338105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maurer C, Panahandeh S, Jungkamp AC, Moser M, Muller M. 2010. TatB functions as an oligomeric binding site for folded Tat precursor proteins. Mol. Biol. Cell 21:4151–4161. 10.1091/mbc.E10-07-0585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. 1998. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 17:3640–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alder NN, Theg SM. 2003. Energetics of protein transport across biological membranes. a study of the thylakoid ΔpH-dependent/cpTat pathway. Cell 112:231–242. 10.1016/S0092-8674(03)00032-1 [DOI] [PubMed] [Google Scholar]
- 36.Bageshwar UK, Musser SM. 2007. Two electrical potential-dependent steps are required for transport by the Escherichia coli Tat machinery. J. Cell Biol. 179:87–99. 10.1083/jcb.200702082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnett JP, Eijlander RT, Kuipers OP, Robinson C. 2008. A minimal Tat system from a gram-positive organism: a bifunctional TatA subunit participates in discrete TatAC and TatA complexes. J. Biol. Chem. 283:2534–2542. 10.1074/jbc.M708134200 [DOI] [PubMed] [Google Scholar]
- 38.Blaudeck N, Kreutzenbeck P, Muller M, Sprenger GA, Freudl R. 2005. Isolation and characterization of bifunctional Escherichia coli TatA mutant proteins that allow efficient tat-dependent protein translocation in the absence of TatB. J. Biol. Chem. 280:3426–3432. 10.1074/jbc.M411210200 [DOI] [PubMed] [Google Scholar]
- 39.Greene NP, Porcelli I, Buchanan G, Hicks MG, Schermann SM, Palmer T, Berks BC. 2007. Cysteine scanning mutagenesis and disulfide mapping studies of the TatA component of the bacterial twin arginine translocase. J. Biol. Chem. 282:23937–23945. 10.1074/jbc.M702972200 [DOI] [PubMed] [Google Scholar]
- 40.Yen MR, Tseng YH, Nguyen EH, Wu LF, Saier MH., Jr 2002. Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Arch. Microbiol. 177:441–450. 10.1007/s00203-002-0408-4 [DOI] [PubMed] [Google Scholar]
- 41.Jongbloed JD, van der Ploeg R, van Dijl JM. 2006. Bifunctional TatA subunits in minimal Tat protein translocases. Trends Microbiol. 14:2–4. 10.1016/j.tim.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 42.Jongbloed JD, Grieger U, Antelmann H, Hecker M, Nijland R, Bron S, van Dijl JM. 2004. Two minimal Tat translocases in Bacillus. Mol. Microbiol. 54:1319–1325. 10.1111/j.1365-2958.2004.04341.x [DOI] [PubMed] [Google Scholar]
- 43.Jongbloed JD, Martin U, Antelmann H, Hecker M, Tjalsma H, Venema G, Bron S, van Dijl JM, Muller J. 2000. TatC is a specificity determinant for protein secretion via the twin-arginine translocation pathway. J. Biol. Chem. 275:41350–41357. 10.1074/jbc.M004887200 [DOI] [PubMed] [Google Scholar]
- 44.Tjalsma H, Bolhuis A, Jongbloed JD, Bron S, van Dijl JM. 2000. Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 64:515–547. 10.1128/MMBR.64.3.515-547.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monteferrante CG, Baglieri J, Robinson C, van Dijl JM. 2012. TatAc, the third TatA subunit of Bacillus subtilis, can form active twin-arginine translocases with the TatCd and TatCy subunits. Appl. Environ. Microbiol. 78:4999–5001. 10.1128/AEM.01108-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dilks K, Rose RW, Hartmann E, Pohlschroder M. 2003. Prokaryotic utilization of the twin-arginine translocation pathway: a genomic survey. J. Bacteriol. 185:1478–1483. 10.1128/JB.185.4.1478-1483.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goosens VJ, Otto A, Glasner C, Monteferrante CC, van der Ploeg R, Hecker M, Becher D, van Dijl JM. 2013. Novel twin-arginine translocation pathway-dependent phenotypes of Bacillus subtilis unveiled by quantitative proteomics. J. Proteome Res. 12:796–807. 10.1021/pr300866f [DOI] [PubMed] [Google Scholar]
- 48.Pop O, Martin U, Abel C, Muller JP. 2002. The twin-arginine signal peptide of PhoD and the TatAd/Cd proteins of Bacillus subtilis form an autonomous Tat translocation system. J. Biol. Chem. 277:3268–3273. 10.1074/jbc.M110829200 [DOI] [PubMed] [Google Scholar]
- 49.Thomas JD, Daniel RA, Errington J, Robinson C. 2001. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol. Microbiol. 39:47–53. 10.1046/j.1365-2958.2001.02253.x [DOI] [PubMed] [Google Scholar]
- 50.Albiniak AM, Matos CF, Branston SD, Freedman RB, Keshavarz-Moore E, Robinson C. 2013. High-level secretion of a recombinant protein to the culture medium with a Bacillus subtilis twin-arginine translocation system in Escherichia coli. FEBS J. 280:3810–3821. 10.1111/febs.12376 [DOI] [PubMed] [Google Scholar]
- 51.van der Ploeg R, Monteferrante CG, Piersma S, Barnett JP, Kouwen TR, Robinson C, van Dijl JM. 2012. High-salinity growth conditions promote Tat-independent secretion of Tat substrates in Bacillus subtilis. Appl. Environ. Microbiol. 78:7733–7744. 10.1128/AEM.02093-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerlach R, Pop O, Muller JP. 2004. Tat dependent export of E. coli phytase AppA by using the PhoD-specific transport system of Bacillus subtilis. J. Basic Microbiol. 44:351–359. 10.1002/jobm.200410423 [DOI] [PubMed] [Google Scholar]
- 53.Yasbin RE, Young FE. 1974. Transduction in Bacillus subtilis by bacteriophage SPP1. J. Virol. 14:1343–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konkol MA, Blair KM, Kearns DB. 2013. Plasmid-encoded ComI inhibits competence in the ancestral 3610 strain of Bacillus subtilis. J. Bacteriol. 195:4085–4093. 10.1128/JB.00696-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patrick JE, Kearns DB. 2008. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol. 70:1166–1179. 10.1111/j.1365-2958.2008.06469.x [DOI] [PubMed] [Google Scholar]
- 56.Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336. 10.1016/0378-1119(95)00652-4 [DOI] [PubMed] [Google Scholar]
- 57.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6:343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 58.Kearns DB, Losick R. 2005. Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev. 19:3083–3094. 10.1101/gad.1373905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hulett FM, Bookstein C, Jensen K. 1990. Evidence for two structural genes for alkaline phosphatase in Bacillus subtilis. J. Bacteriol. 172:735–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeigler DR, Pragai Z, Rodriguez S, Chevreux B, Muffler A, Albert T, Bai R, Wyss M, Perkins JB. 2008. The origins of 168, W23, and other Bacillus subtilis legacy strains. J. Bacteriol. 190:6983–6995. 10.1128/JB.00722-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu XC, Nathoo S, Pang AS, Carne T, Wong SL. 1990. Cloning, genetic organization, and characterization of a structural gene encoding bacillopeptidase F from Bacillus subtilis. J. Biol. Chem. 265:6845–6850 [PubMed] [Google Scholar]
- 62.Sloma A, Rufo GA, Jr, Theriault KA, Dwyer M, Wilson SW, Pero J. 1991. Cloning and characterization of the gene for an additional extracellular serine protease of Bacillus subtilis. J. Bacteriol. 173:6889–6895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang MY, Ferrari E, Henner DJ. 1984. Cloning of the neutral protease gene of Bacillus subtilis and the use of the cloned gene to create an in vitro-derived deletion mutation. J. Bacteriol. 160:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stahl ML, Ferrari E. 1984. Replacement of the Bacillus subtilis subtilisin structural gene with an in vitro-derived deletion mutation. J. Bacteriol. 158:411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong SL, Price CW, Goldfarb DS, Doi RH. 1984. The subtilisin E gene of Bacillus subtilis is transcribed from a sigma 37 promoter in vivo. Proc. Natl. Acad. Sci. U. S. A. 81:1184–1188. 10.1073/pnas.81.4.1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruckner R, Shoseyov O, Doi RH. 1990. Multiple active forms of a novel serine protease from Bacillus subtilis. Mol. Gen. Genet. 221:486–490 [DOI] [PubMed] [Google Scholar]
- 67.Sloma A, Ally A, Ally D, Pero J. 1988. Gene encoding a minor extracellular protease in Bacillus subtilis. J. Bacteriol. 170:5557–5563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Margot P, Karamata D. 1996. The wprA gene of Bacillus subtilis 168, expressed during exponential growth, encodes a cell-wall-associated protease. Microbiology 142(Pt 12):3437–3444. 10.1099/13500872-142-12-3437 [DOI] [PubMed] [Google Scholar]
- 69.Rufo GA, Jr, Sullivan BJ, Sloma A, Pero J. 1990. Isolation and characterization of a novel extracellular metalloprotease from Bacillus subtilis. J. Bacteriol. 172:1019–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sloma A, Rudolph CF, Rufo GA, Jr, Sullivan BJ, Theriault KA, Ally D, Pero J. 1990. Gene encoding a novel extracellular metalloprotease in Bacillus subtilis. J. Bacteriol. 172:1024–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Strauch EM, Georgiou G. 2007. Escherichia coli tatC mutations that suppress defective twin-arginine transporter signal peptides. J. Mol. Biol. 374:283–291. 10.1016/j.jmb.2007.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caspers M, Brockmeier U, Degering C, Eggert T, Freudl R. 2010. Improvement of Sec-dependent secretion of a heterologous model protein in Bacillus subtilis by saturation mutagenesis of the N-domain of the AmyE signal peptide. Appl. Microbiol. Biotechnol. 86:1877–1885. 10.1007/s00253-009-2405-x [DOI] [PubMed] [Google Scholar]
- 73.Hinsley AP, Stanley NR, Palmer T, Berks BC. 2001. A naturally occurring bacterial Tat signal peptide lacking one of the “invariant” arginine residues of the consensus targeting motif. FEBS Lett. 497:45–49. 10.1016/S0014-5793(01)02428-0 [DOI] [PubMed] [Google Scholar]
- 74.DeLisa MP, Samuelson P, Palmer T, Georgiou G. 2002. Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J. Biol. Chem. 277:29825–29831. 10.1074/jbc.M201956200 [DOI] [PubMed] [Google Scholar]
- 75.Harwood CR. 1992. Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol. 10:247–256. 10.1016/0167-7799(92)90233-L [DOI] [PubMed] [Google Scholar]
- 76.Schallmey M, Singh A, Ward OP. 2004. Developments in the use of Bacillus species for industrial production. Can. J. Microbiol. 50:1–17. 10.1139/w03-076 [DOI] [PubMed] [Google Scholar]
- 77.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. 2007. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 104:1506–1509. 10.1073/pnas.0610503104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. 2011. Tracing the domestication of a biofilm-forming bacterium. J. Bacteriol. 193:2027–2034. 10.1128/JB.01542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stanley NR, Lazazzera BA. 2005. Defining the genetic differences between wild and domestic strains of Bacillus subtilis that affect poly-γ-dl-glutamic acid production and biofilm formation. Mol. Microbiol. 57:1143–1158. 10.1111/j.1365-2958.2005.04746.x [DOI] [PubMed] [Google Scholar]
- 80.Kearns DB, Chu F, Rudner R, Losick R. 2004. Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol. Microbiol. 52:357–369. 10.1111/j.1365-2958.2004.03996.x [DOI] [PubMed] [Google Scholar]
- 81.Parashar V, Konkol MA, Kearns DB, Neiditch MB. 2013. A plasmid-encoded phosphatase regulates Bacillus subtilis biofilm architecture, sporulation, and genetic competence. J. Bacteriol. 195:2437–2448. 10.1128/JB.02030-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kobayashi K. 2007. Gradual activation of the response regulator DegU controls serial expression of genes for flagellum formation and biofilm formation in Bacillus subtilis. Mol. Microbiol. 66:395–409. 10.1111/j.1365-2958.2007.05923.x [DOI] [PubMed] [Google Scholar]
- 83.Yang M, Ferrari E, Chen E, Henner DJ. 1986. Identification of the pleiotropic sacQ gene of Bacillus subtilis. J. Bacteriol. 166:113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.