

Abstract
Bacterial inclusion bodies are aggregations of mostly inactive and misfolded proteins. However, previously the in vivo self-assembly of green fluorescent protein (GFP) fusions into fluorescent particles which displayed specific binding sites suitable for applications in bioseparation and diagnostics was demonstrated. Here, the suitability of GFP particles for enzyme immobilization was assessed. The enzymes tested were a thermostable α-amylase from Bacillus licheniformis, N-acetyl-d-neuraminic acid aldolase (NanA) from Escherichia coli, and organophosphohydrolase (OpdA) from Agrobacterium radiobacter. Respective GFP particles were isolated and could be stably maintained outside the cell. These enzyme-bearing GFP particles exhibited considerable stability across a range of temperature, pH, and storage conditions and could be recycled. The α-amylase-bearing particles retained activity after treatments at 4 to 85°C and at pHs 4 to 10, were stable for 3 months at 4°C, and could be recycled up to three times. OpdA-bearing particles retained degradation activity after treatments at 4 to 45°C and at pHs 5 to 10 and were able to be recycled up to four times. In contrast, the performance of NanA-bearing particles rapidly declined (>50% loss) after each recycling step and 3 months storage at 4°C. However, they were still able to convert N-acetylmannosamine and pyruvate to N-acetylneuraminic acid after treatment at 4 to 85°C and at pHs 4 to 11. Fluorescent GFP fusion particles represent a novel method for the immobilization and display of enzymes. Potential applications include diagnostic assays, biomass conversion, pharmaceutical production, and bioremediation.
INTRODUCTION
A wide range of industries currently use enzymes in their processes, including food production, detergent manufacture, agriculture, chemical and pharmaceutical manufacture, textiles, leather, paper, diagnostics, medicine, and bioremediation (1, 2). Although the demand for enzymes is increasing, the equipment and reagents required for their isolation and purification are expensive. To counter this, various immobilization methods have been developed that enhance the utility of enzymes in a wider range of environments, including nonaqueous media, as well as improve enzyme performance via improved stability and reuse (3–5). Examples of enzymes immobilized to particulate support materials include cross-linked enzyme aggregates, cross-linked enzyme crystals, and Spherezymes (6–8). However, of particular interest are the methods that employ recombinant DNA technology to allow the production, purification, and immobilization of the enzyme(s) of interest in a single step. Examples include enzyme-bearing nanotubes, fibrils, and polyhydroxyalkanoate (PHA) granules (9–13).
In this study, the design and recombinant production of green fluorescent protein (GFP) particles were investigated in view of their potential to display active enzymes. Previously, GFP particles were engineered to display functional binding proteins including maltose binding protein (MalE), the IgG binding domain of protein A (ZZ), and the IgG binding domain of protein G (GB1) (14). These functional GFP particles exhibited binding activity and in many cases outperformed commercially available bead-based bioseparation resins. The formation of GFP particles is triggered by a short N-terminal extension of GFP (AVTS, LAVG, or FHKP) and the presence of a large protein linker (C-terminal GFP extension) as the central domain of the fusion protein while a desirable protein function is fused to the C terminus of the overall fusion protein (14). This modular arrangement enables the customization of GFP particles that exhibit different properties via the C terminus of the respective fusion protein. The ability to immobilize active enzyme on GFP particles would present an alternative enzyme immobilization method that is potentially more efficient, cost-effective, and easier to use than current methods.
To demonstrate the ability of GFP particles to display active enzymes exhibiting different quaternary structures, a monomeric thermostable α-amylase (BLA) variant (Termamyl) from Bacillus licheniformis (EC 3.2.1.1), a homotetrameric N-acetyl-d-neuraminic acid aldolase (NanA) from Escherichia coli (EC 4.1.3.3), and a dimeric organophosphohydrolase (OpdA) from Agrobacterium radiobacter were fused to the C termini of engineered GFP fusion proteins, respectively. Respective fusion proteins self-assembled to fluorescent GFP particles inside Escherichia coli BL21(DE3) cells. These particles were isolated and assessed for activity, stability, and reusability.
MATERIALS AND METHODS
Strains, plasmids, and primers.
The bacterial strains, plasmids, and primers used in this study are listed in Table S1 in the supplemental material.
Cultivation conditions.
E. coli XL1-Blue was grown in Luria-Bertani medium at 37°C and 200 rpm for 16 h. E. coli BL21(DE3) was grown at 25°C and 200 rpm for 48 h supplemented with 1% (wt/vol) glucose. Ampicillin (75 μg/ml) and chloramphenicol (50 μg/ml) were added as required.
Plasmid construction.
General cloning procedures and DNA isolation were performed as described elsewhere (15). Primers were purchased from Integrated DNA Technologies (Coraville, IA) while Taq and Platinum Pfx polymerases were purchased from Invitrogen (Carlsbad, CA). The nanA gene was synthesized by Genscript Corporation (Piscataway, NJ) and inserted in pUC57 vectors with flanking restriction endonuclease sites for subsequent cloning.
The construction of the plasmids pET14b-ext(AVTS)gfp-phaC(C319A)-linker-nanA (pET14b-GiCLN) and pET14b-ext(AVTS)gfp-nanA-linker-nanA (pET14b-GNLN) was based upon the plasmids pET14b-ext(AVTS)gfp-phaC(C319A)-linker-ZZ (pET14b-GiCLZ) and pET14b-ext(AVTS)gfp-nanA-linker-ZZ (pET14b-GNLZ) as described previously (14). It must be noted that the abbreviation ext(AVTS)gfp is used to describe the N-terminal extension of the enhanced gfp gene using bases encoding the amino acids alanine, valine, threonine, and serine. The plasmids were hydrolyzed with XhoI and BamHI restriction enzymes, cleaving the zz region from the vector. The resulting backbones were independently ligated to the purified nanA fragment creating the two new plasmids. The protein linker sequence is VLAVIDKRGGGGG (14, 16).
The construction of pET14b-ext(AVTS)gfp-phaC(C319A)-linker-opdA (pET14b-GiCLO) (where GiCLO indicates GiCL fused with OpdA) was similarly based upon pET14b-GiCLZ. The opdA gene was hydrolyzed using XhoI and BamHI restriction enzymes from the plasmid pET14b-phaC-linker-opdA (10), purified, and ligated into the hydrolyzed pET14b-ext(AVTS)gfp-phaC(C319A)-linker-backbone. Bla(−ss) (where −ss indicates the absence of the signal sequence) was obtained from the plasmid pET14b-bla(−ss)-phaC (13) using PCR to amplify the fragment of interest with the addition of XhoI and BamHI restriction sites. It was then purified and ligated into the hydrolyzed pET14b-ext(AVTS)gfp-phaC(C319A)-linker-backbone to yield the plasmid pET14b-ext(AVTS)gfp-phaC(C319A)-linker-bla(−ss) (pET14b-GiCLB) (where GiCLB indicates GiCL fused to BLA).
Protein particle isolation.
Expression of GFP fusion proteins was induced in E. coli BL21(DE3) with the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.3 to 0.4. After a 48-h cultivation at 25°C, cell pellets (6,000 × g at 4°C for 20 min) were washed with 50 mM potassium phosphate buffer (pH 7.5) and mechanically disrupted at 20,000 lb/in2 using a Z Plus 1.1-kW Benchtop Cell Disrupter (Constant Cell Disruption Systems, United Kingdom). GFP particles in the lysate were recovered by centrifugation (8,000 × g at 4°C for 20 min). Final separation of the particles from bacterial lysate was performed using ultracentrifugation (150,000 × g at 10°C for 2 h) on a WX Ultra 80 ultracentrifuge (Thermofisher Scientific, USA) and employing a gradient with layers of 49.75% and 99.5% glycerol. Recovered particles were washed with 50 mM potassium phosphate buffer (pH 7.5) to remove glycerol and stored at 4°C in aliquots supplemented with 20% ethanol until required.
Fluorescence microscopy.
Images of recombinant E. coli BL21(DE3) cells containing GFP particles and of GFP particles postextraction were obtained using an Olympus BX51 fluorescent light microscope (Olympus Optical Co., Ltd., Japan). Images were captured under ×100 and ×1,000 magnifications using a U-MWIBA2 filter (blue excitation, 460 nm to 490 nm; green emission, 510 nm to 550 nm), an Optronics camera (Optronics, USA), and MagnaFire, version 2.1C, application software (Optronics, USA).
Protein analysis.
GFP particles and particle-associated proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described elsewhere (17). Bis-Tris polyacrylamide gels were used which had acrylamide concentrations of 8% (resolving gel) and 4% (stacking gel). Gels were stained with 0.1% (wt/vol) Coomassie blue for at least 20 min and destained overnight. Protein concentration was determined by densitometry after gel staining comparing the GFP fusion protein to known quantities of bovine serum albumin (BSA). Protein bands of interest were excised and subjected to tryptic digest and mass spectrometry.
Neu5Ac production.
For the synthesis of N-acetylneuraminic acid (Neu5Ac), 1 M sodium pyruvate and 200 mM N-acetylmannosamine (ManNAc) (≥98% purity) were reacted with particle slurries containing 25 μg or 50 μg of fusion protein in 50 mM potassium phosphate buffer (pH 7.5) at 50°C for up to 72 h. The total reaction volume was 0.5 ml. Following incubation, the samples were centrifuged (16,000 × g for 10 min), and the supernatants were removed. Neu5Ac, ManNAc, and pyruvate were quantified by high-performance liquid chromatography (HPLC).
HPLC for Neu5Ac.
Neu5Ac quantification was performed using high-performance liquid chromatography (HPLC) on a Dionex Ultimate 3000 System (Dionex Softron, Germany) using a Phenomenex Rezex RHM-Monosaccharide H+ column (300 by 7.80 mm). The mobile phase was 6 mM H2SO4 (0.5 ml/min). Column temperature was maintained at 60°C, the sample injection volume was 25 μl, and the UV detector was set at 205 nm. The software used was Chromeleon Chromatography Management system, version 6.80 (Dionex, USA).
α-Amylase activity assay.
The α-amylase activity was assessed using as the substrate 1% (wt/vol) starch dissolved in 20 mM Na-phosphate buffer containing 6.7 mM NaCl (pH 6.9) at 25°C for up to 72 h. The total reaction volume was 1 ml. The assay measures the release of maltose and was conducted as previously described (13) utilizing particle slurries containing 50 μg of fusion protein.
OpdA activity.
Degradation of 200 μM methyl parathion by OpdA was assessed in 50 mM HEPES buffer (pH 8.0) containing 20% (vol/vol) methanol at 25°C as previously described (10). Reactions were conducted for up to 72 h, and the total volume in each reaction mixture was 1 ml. Particle slurries containing 50 μg of fusion protein were used. The extinction coefficient of paranitrophenol was taken as 19,454 M−1 cm−1 (18). It must be noted that there were difficulties experienced in the dissolution of methyl parathion in the reaction buffer. Although a concentration of 200 μM was the aim for every assay, the actual concentration of the chemical fluctuated below this threshold due to various amounts of the chemical dissolving in the buffer each time the substrate was prepared. This makes it impossible to compare activity data between different assays as different batches of methyl parathion substrate were used; however, comparison of samples within each assay is still possible.
Temperature stability.
To assess the temperature stability of enzyme-bearing PHA beads/GFP particles, samples were preincubated at 4°C and at 15 to 95°C (at 10°C intervals) for 10 min in a Digital Dry Bath (Labnet International, Inc., USA). Enzyme activity was then assessed as described above by monitoring substrate conversion as a function of time.
pH stability.
To assess the stability of enzyme-bearing PHA beads/GFP particles at different pH values, samples were preincubated for 10 min at room temperature (RT) in the following 20 mM solutions/buffers: potassium chloride (pH 2.00), sodium citrate (pH 3.00), sodium acetate (pHs 4.00, 5.00, and 6.00), tris-HCl (pHs 7.00 and 8.00), sodium tetraborate (pHs 9.00 and 10.00), and disodium hydrogen orthophosphate (pHs 11.00 and 12.00). After pH treatment, the samples were centrifuged at 16,000 × g for 10 min and suspended in the respective reaction buffer. Enzyme activity was assessed as described above by monitoring substrate conversion as a function of time.
Long-term storage.
To determine the effect of storage conditions on GFP particles, aliquots of each type of enzyme displaying GFP particles were stored at room temperature, at 4°C, and at −80°C. Samples stored at room temperature and at 4°C were suspended in 20% ethanol in 50 mM potassium phosphate buffer, while samples stored at −80°C were suspended in 25% glycerol in 50 mM potassium phosphate buffer. BLA- and NanA-containing samples were stored for up to 3 months, while OpdA-containing samples were stored for up to 4.5 months. At the end of the storage period, the samples were assayed as described above by monitoring substrate conversion as a function of time.
Reusability of enzyme-bearing GFP particles.
To assess the reusability of GFP particles displaying enzyme, samples were prepared and assayed as described above for the relevant enzyme. At the end of the incubation period, samples were centrifuged at 16,000 × g for 5 to 10 min in a Heraeus Pico 17 centrifuge, and the supernatant was removed and analyzed. The GFP particle pellets were then resuspended in fresh substrate solution as described above and reincubated. This cycle was repeated four times.
RESULTS
Engineering a GFP-NanA fusion which self-assembles inside E. coli.
N-terminally extended GFP, which was previously shown to self-assemble (14), was used as a scaffold for the immobilization of various industrially relevant enzymes. For extended GFP to self-assemble and display a foreign protein, a large C-terminal extension was required which could be further C-terminally extended by a protein of interest (14). Here, various enzymes representing different quaternary structures (monomer, dimer, and tetramer) were targeted as proteins of interest (see Fig. 4). Both PhaC and NanA were used as C-terminal extensions of GFP in addition to the target enzyme NanA, a 33-kDa tetrameric aldolase from E. coli.
FIG 4.

Schematic diagrams of the hybrid genes used in this study. Green fluorescent protein (GFP) was N-terminally extended by (M)AVTS and fused to a large linker, either inactive PhaC or NanA, which was also fused to genes encoding functions of interest. The hybrid genes were then expressed in E. coli. The resulting fluorescent protein particles were able to display active enzyme as demonstrated by N-acetyl neuraminic acid aldolase (NanA), organophosphohydrolase (OpdA), and thermostable α-amylase (BLA). L, linker.
The fusion proteins GiCLZ, GNLZ, GiCLN, and GNLN (where G is GFP, iC is an inactive site-specific mutant of PhaC, N is NanA, L is linker, and Z is the ZZ IgG binding domain) all mediated the formation of fluorescent GFP particles, as shown by fluorescence microscopy (Fig. 1), at a yield of approximately 11 to 23% of cellular wet weight (CWW) as calculated by mass. Fluorescence microscopy showed that GiCLZ, GNLZ, and GiCLN particles were similar in terms of their irregular shapes and size distribution, while GNLN particles appeared smaller and had a tendency to aggregate postextraction (data not shown). All particle-producing E. coli strains accumulated 2 to 4 particles per cell. The particles were isolated and examined by SDS-PAGE. Theoretical molecular masses of GiCLZ, GNLZ, GiCLN, and GNLN were 111, 79, 127, and 95 kDa, respectively, and they appeared as predominant proteins with the corresponding apparent molecular masses (Fig. 2). The identity of each fusion protein was confirmed by tryptic peptide fingerprinting using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (see Table S2 in the supplemental material).
FIG 1.

Fluorescence microscopy analysis of E. coli BL21(DE3) cells containing GFP particles. Images were obtained at ×1,000 magnification using a U-MWIBA2 Blue excitation filter cube fitted to an Olympus BX51 fluorescent light microscope (Olympus Optical Co., Japan), an Optronics camera (Optronics, USA), and MagnaFire, version 2.1C, application software (Optronics, USA). Panels are as follows: A, GiCLZ; B, GNLZ; C, GiCLN; D, GNLN; E, GiCLB; F, GiCLO. G, GFP; iC, inactive PhaC; L, linker; N, NanA; B, BLA; O, OpdA; Z, ZZ.
FIG 2.
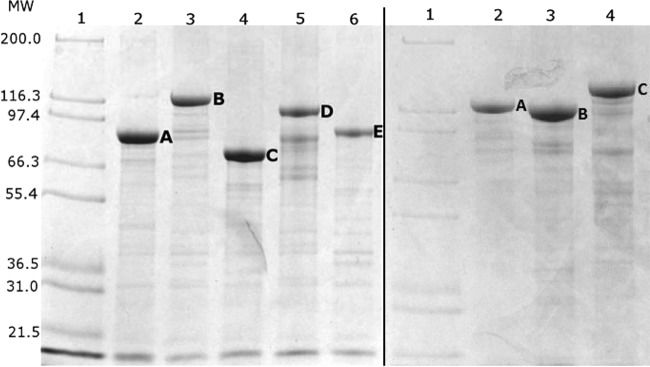
Protein profiles of isolated GFP particles as shown by SDS-PAGE analysis. (Left) Lane 1, Mark 12 Unstained Standard Protein Ladder (Invitrogen, USA); lane 2, GNLN (A); lane 3, GiCLN (B); lane 4, GNLZ (C); lane 5, GiCLZ (D); lane 6, NanA-PhaC (E). (Right) Lane 1, Mark 12 Unstained Standard Protein Ladder (Invitrogen, USA); lane 2, GiCLO (A); lane 3, GiCLN (B); lane 4, GiCLB (C) The letters A, B, C, D, and E correspond to the fusion proteins of interest overproduced in their respective lanes. G, GFP; iC, inactive PhaC; L, linker; N, NanA; Z, ZZ; B, BLA; O, OpdA. MW, molecular weights in thousands.
Assessment of NanA activity when immobilized to self-assembling GFP particles.
NanA-displaying GFP particles were assessed with respect to NanA activity using 16-h and 48-h reaction times for the synthesis of Neu5Ac from ManNAc and pyruvate (Fig. 3). The amount of each particle sample used was standardized at 50 μg of fusion protein as calculated by densitometry. GiCLZ (negative control) produced no detectable Neu5Ac after 16 h, while after 48 h a conversion of 1.7% was observed. This was confirmed as background noise conversion by analyzing synthesis reactions in the absence of any GFP particles (data not shown). Using NanA as the central domain (GNLZ) resulted in particles that produced a 22% conversion yield of Neu5Ac after 48 h. Placing NanA at the C terminus of the GFP fusion particle (GiCLN) mediated a conversion of 41% after 16 h, with an increase to 75% at 48 h. Particles with NanA as both the central and C-terminal domains (GNLN) had only 3% Neu5Ac conversion after 48 h. For comparative purposes and as a positive control, PHA beads displaying NanA were included (11). This system produced a 90% conversion yield in the first 16 h, confirming previously published results (11). The above finding indicated that the GFP fusion protein scaffold should be based on the GiCLN fusion protein design, i.e., that NanA should be replaced by other enzymes of interest to test the versatility of this enzyme immobilization concept.
FIG 3.
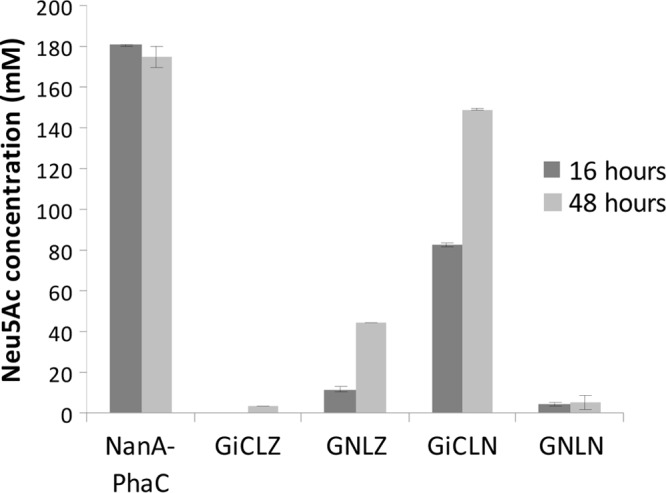
Neu5Ac synthesis catalyzed by particles displaying NanA. Enzyme-bearing particles containing 50 μg of NanA fusion protein were reacted with 0.2 M N-acetyl-d-mannosamine and 1 M sodium pyruvate at 50°C. Samples were taken at 16 h and 48 h. Production of Neu5Ac was measured using HPLC and detection at 205 nm. Error bars are ±1 standard deviation (n = 3). G, GFP; iC, inactive PhaC; L, linker; N, NanA; Z, ZZ.
Bioengineering of GFP particles with various immobilized enzymes.
In total three enzymes were chosen to assess the versatility of the GFP particle display system: (i) tetrameric NanA, an aldolase used in synthesis of Neu5Ac; (ii) BLA, a thermostable variant of the α-amylase from Bacillus licheniformis, a monomeric 55-kDa protein; and (iii) OpdA, a dimeric enzyme from Agrobacterium radiobacter with potential application in the bioremediation of organophosphate pesticides. Hybrid genes were constructed encoding GFP fusion proteins containing the respective enzyme of interest at the C terminus of the fusion protein ext(AVTS)GFP-PhaC(C319A) (GiCL) (Fig. 4). Both fusion proteins, GiCLB and GiCLO, mediated self-assembly of fluorescent GFP particles in vivo as visualized by fluorescence microscopy (Fig. 1). The particles shared similar irregular shapes and size distributions in comparison to GiCLZ, GNLZ, and GiCLN particles, and the E. coli strains also accumulated 2 to 4 particles per cell. Theoretical molecular masses of GiCLB and GiCLO were 150 and 131 kDa, respectively, and corresponding dominant protein bands with the respective apparent molecular masses were demonstrated by SDS-PAGE (Fig. 2). The identity of each fusion protein was confirmed by tryptic peptide fingerprinting using MALDI-TOF mass spectrometry (see Table S2 in the supplemental material).
Comparative analysis between the GFP particle method and the PHA bead display of the same amount of fusion protein revealed in all cases that the initial substrate turnover rate was less for the GFP particles than for the respective PHA beads (Fig. 5). Based on the initial rates of these reactions (Fig. 5) and the mass of the fusion protein as calculated by densitometry, the NanA-containing GFP particle had a specific activity of 76 mU/mg of NanA fusion protein. GFP-OpdA particles had 2.1 U/mg of OpdA fusion protein, and GFP-BLA had 210 mU/mg of BLA fusion protein specific activity (in all cases, 1 U is defined as 1 μmol of product per min). Accordingly, the NanA particle showed after 72 h only 34% conversion compared to 82% conversion of the PHA beads when 25 μg of fusion protein was used (Fig. 5A). The BLA-displaying PHA beads reached complete substrate turnover in 4 h, whereas GiCLB particles reached the endpoint at 48 h (Fig. 5B). PhaC-OpdA beads catalyzed complete substrate turnover in 2 h while GiCLO beads required 24 h for completion of the reaction (Fig. 5C).
FIG 5.

Reaction time courses for all enzymes immobilized to GFP particles compared to the same enzyme immobilized to PHA granules. Each reaction proceeded for 72 h with samples taken at 1 h, 2 h, 3 h, 4 h, 6 h, 12 h, 24 h, 48 h, and 72 h. (A) Twenty-five micrograms of NanA-PhaC or GiCLN fusion protein reacted with 0.2 M N-acetyl-d-mannosamine and 1 M sodium pyruvate at 50°C. The production of N-acetyl neuraminic acid was quantified by HPLC. (B) Fifty micrograms of Bla(−ss)-PhaC, GiCLZ, or GiCLB fusion protein reacted with 1% starch solution at 25°C. The release of maltose was quantified by the α-amylase colorimetric assay. (C) Fifty micrograms of PhaC-OpdA, GiCLZ, or GiCLO fusion protein reacted with 200 μM methyl parathion at 25°C. The release of paranitrophenol was quantified by spectroscopy. Error bars are ±1 standard deviation (n = 3). G, GFP; iC, inactive PhaC; L, linker; N, NanA; B, BLA; O, OpdA; Z, ZZ.
Reusability of GFP particles.
The ability of the enzyme-displaying GFP particles to be reused was examined over four reaction cycles (Fig. 6). The yield of Neu5Ac for the GiCLN particles declined rapidly by ∼50% each cycle (Fig. 6A). The duration of each cycle was 48 h. After four cycles the final production of Neu5Ac was only 3 mM compared with 32 mM in the initial cycle. The GiCLB particles maintained their activity for the first three cycles while the fourth cycle showed only 8% of the initial conversion yield (Fig. 6B). The GiCLO construct delivered consistently high performance across all four cycles, maintaining 92% of its initial-cycle performance in the fourth reaction (Fig. 6C). The duration of each cycle for GiCLB and GiCLO reactions was 12 h. The reusability of GiCLO particles was extended to seven reaction cycles, under the same conditions. The construct delivered high performance across the first four cycles, as seen previously, and retained 81% of the initial-cycle performance in the seventh reaction (data not shown).
FIG 6.

Reusability of the immobilized enzyme GFP particles over four cycles. (A) Twenty-five micrograms of GiCLN fusion protein reacted with 0.2 M N-acetyl-d-mannosamine and 1 M sodium pyruvate at 50°C for 48 h per cycle. The production of N-acetyl neuraminic acid was quantified by HPLC. (B) Fifty micrograms of GiCLB fusion protein reacted with 1% starch solution at 25°C for 12 h per cycle. The release of maltose was quantified by the α-amylase colorimetric assay. (C) Fifty micrograms of GiCLO fusion protein reacted with 200 μM methyl parathion at 25°C for 12 h per cycle. The release of paranitrophenol was quantified by spectroscopy. Error bars are ±1 standard deviation (n = 3). G, GFP; iC, inactive PhaC; L, linker; N, NanA; B, BLA; O, OpdA.
Effect of preincubation temperature on GFP particle enzyme performance.
After exposure to a range of temperatures, the different enzyme-displaying particles were assessed for their activity levels (Fig. 7). Both immobilization systems performed similarly with NanA, showing consistent performance across all preincubation temperatures up to 95°C. At this temperature the activity of both materials declined, with 44% of initial product formation in the case of GFP particles and 37% of maximum production in the case of PHA granules (Fig. 7A). BLA immobilized to PHA granules performed consistently throughout the preincubation temperature range. In contrast, the performance of GFP-immobilized BLA declined slowly to 73% of its maximum at a preincubation temperature of 95°C (Fig. 7B). Both immobilization materials also performed similarly for OpdA. Degradation efficiency began to decline after treatment at 65°C, and by 95°C both systems were able to degrade only 5% of the initially degraded methyl parathion (Fig. 7C). However, in the case of OpdA, the GFP particles showed more activity loss at temperatures between 65 and 95°C than the PHA granules. GFP particles showed structural integrity and fluorescence up to a temperature of 75°C, as assessed by fluorescence microscopy (see Fig. S1 in the supplemental material).
FIG 7.

Temperature stability of enzymes immobilized to GFP particles. The particles were preincubated at 4°C, 15°C, 25°C, 35°C, 45°C, 55°C, 65°C, 75°C, 85°C, and 95°C for 10 min and then subjected to their respective reactions. Activity is relative to the PhaC bead at 25°C. (A) Twenty-five micrograms of NanA-PhaC or GiCLN fusion protein reacted with 0.2 M N-acetyl-d-mannosamine and 1 M sodium pyruvate at 50°C. The production of N-acetyl neuraminic acid was quantified by HPLC. (B) Fifty micrograms of Bla(−ss)-PhaC or GiCLB fusion protein reacted with 1% starch solution at 25°C. The release of maltose was quantified by the α-amylase colorimetric assay. (C) Fifty micrograms of PhaC-OpdA or GiCLO fusion protein reacted with 200 μM methyl parathion at 25°C. The release of paranitrophenol was quantified by spectroscopy. Error bars are ±1 standard deviation (n = 3). G, GFP; iC, inactive PhaC; L, linker; N, NanA; B, BLA; O, OpdA.
Effect of preincubation pH on GFP particle enzyme performance.
Enzyme-displaying particles were also exposed to a range of pH conditions and then assessed for activity (Fig. 8). NanA was functional on both GFP particles and PHA granules after treatment at pHs of 4 to 11 but declined to minimal levels at the extremes of pH (Fig. 8A). BLA was similar, with a slight decline at pH 11 when immobilized to GFP particles (Fig. 8B). OpdA showed a decline in activity around pH 4; however, PHA beads showed no decline in activity at pHs of 11 to 12, whereas GFP particles did (Fig. 8C). The GFP particles began to aggregate at the extremes of pH, possibly accounting for their loss of enzyme function (see Fig. S2 in the supplemental material).
FIG 8.

The pH stability of enzymes immobilized to GFP particles. The particles were preincubated at the indicated pH (2 to 12) for 10 min and then subjected to their respective reactions. Activity is relative to the PhaC bead at pH 7.0. (A) Twenty-five micrograms of NanA-PhaC or GiCLN fusion protein reacted with 0.2 M N-acetyl-d-mannosamine and 1 M sodium pyruvate at 50°C. The production of N-acetyl neuraminic acid was quantified by HPLC. (B) Fifty micrograms of Bla(−ss)-PhaC or GiCLB fusion protein reacted with 1% starch solution at 25°C. The release of maltose was quantified by the α-amylase colorimetric assay. (C) Fifty micrograms of PhaC-OpdA or GiCLO fusion protein reacted with 200 μM methyl parathion at 25°C. The release of paranitrophenol was quantified by spectroscopy. Error bars are ±1 standard deviation (n = 3). G, GFP; iC, inactive PhaC; L, linker; N, NanA; B, BLA; O, OpdA.
Long-term storage of enzyme-immobilized GFP particles.
After 97 days of storage at RT, both NanA-PhaC and GiCLN showed little remaining NanA activity. The amount of Neu5Ac produced by GiCLN particles after storage at RT was undetectable compared with 23 and 24 mM Neu5Ac produced after storage at 4°C and −80°C, respectively. Long-term storage at these lower temperatures resulted in an approximately 50% decline compared to the initial production level of 50.6 mM Neu5Ac. The storage temperature had little impact on BLA particles, with slight declines seen in both the RT and −80°C GiCLB samples to 92% and 94% of initial activity, respectively, while the 4°C samples showed a 4% increase. Although comparisons to initial activity could not be made for the OpdA particles (see Materials and Methods), the temperature of storage and suspension buffer used had little effect on OpdA activity after 132 days of storage, with the −80°C samples exhibiting slightly higher activity (see Table S3 in the supplemental material). PhaC-OpdA and GiCLO particles were also stored for 30 days at 37°C, 4°C, and −80°C. Both particles exhibited consistent activity after storage at these temperatures (see Table S3).
DISCUSSION
Previous research has shown that engineered GFP fusion proteins can self-assemble into binding domain-displaying fluorescent GFP particles inside recombinant E. coli (14). In this study, GFP fusion proteins were investigated in view of their potential to serve as scaffolds for covalent enzyme immobilization. Enzymes exhibiting various quaternary structures were translationally fused to the C termini of engineered GFP fusion proteins, and the ability of the GFP fusion proteins to self-assemble into fluorescent GFP particles was assessed. Following the successful isolation of GFP particles, their activity, stability, and reusability were examined.
Initially, three different types of NanA GFP fusion proteins were assessed. Within these fusion proteins, NanA was situated as the central domain (GNLZ), as the C terminus (GiCLN), or as a central domain/C terminus double fusion (GNLN). All types of fusion proteins mediated the formation of fluorescent GFP particles (Fig. 1) which could be stably maintained outside the cell. Furthermore, the NanA GFP particles also exhibited NanA activity (Fig. 3). A small amount of background activity of up to 1.7% Neu5Ac production was observed both for the negative control (GiCLZ) and for a reaction mixture containing no GFP particles (data not shown). The formation of fluorescent particles and display of NanA activity are interesting due to the strict conformational requirements for GFP fluorescence and also the complexity involved in the folding and assembly of a tetrameric enzyme, NanA, into an active conformation (19, 20). Only two to three particles were found in each cell (Fig. 1), resembling other types of inclusion bodies (21).
GiCLN fusion protein-based GFP particles showed the highest activity as assessed by Neu5Ac product formation from ManNAc and pyruvate (Fig. 3). This difference in NanA activities of the various fusion proteins could be due to differences in the exposure and orientation of the enzyme at the particle surface, i.e., the accessibility of the enzyme active sites (20, 22) to the substrate, as well as the ability of the enzyme to correctly fold and oligomerize at each position in the multidomain fusion protein. Interestingly, NanA in the central domain of the fusion protein was still active, suggesting flexibility of this region to enable protein folding and to form tetrameric structures in the particle itself. GNLZ particles have already been shown to exhibit IgG binding activity via the ZZ domain (14). That it also showed NanA activity demonstrated the possibility of incorporating multiple protein functionalities into a single GFP particle suitable for diagnostic applications where targeting or binding specificity and enzyme function in combination with a fluorescent label are beneficial.
Other enzymes, OpdA and BLA, were chosen to demonstrate the versatility of GFP particles in displaying enzymes that catalyze different types of reactions while exhibiting different quaternary structures. As the NanA C-terminal fusion led to the best activity, the same arrangement was used for OpdA and BLA (Fig. 4). Both enzymes were fused to GiCL (GiCLO and GiCLB, respectively) and mediated the formation of fluorescent protein particles inside recombinant E. coli (Fig. 1). Additionally, GiCLO and GiCLB particles could be isolated and stably maintained outside the cell, as well as show the activity of their constituent enzymes (Fig. 5).
A feature shared by all enzyme-bearing GFP particles used in this study is their weak activity in comparison to that of PHA bead controls when activity is standardized by the amount of fusion protein (Fig. 5). These differences in activity could be attributable to differences in the way in which PHA beads and GFP particles form and the resultant display of enzyme, i.e., to the access of the active sites to the substrate. Any protein that is fused to PhaC is also displayed on the surface of a hydrophobic polyester bead (23, 24). In contrast, the GFP particles consisted of self-assembled GFP fusion protein. Therefore, enzymes might be buried inside the GFP particle with limited access to substrate.
The enzyme-bearing GFP particles were found to be reusable and stable when exposed to a range of temperatures (4 to 95°C) and pH values (2 to 12) (Fig. 6 to 8). GiCLB and GiCLN particles were able to maintain reasonably constant levels of activity after treatment at the temperatures assessed and were resistant to low- and high-pH treatment. In contrast, GiCLO particles were the most sensitive to heat treatment but showed resistance to extremes of pH. This is consistent with previous research on the soluble form of these enzymes (10, 25–30). Native soluble BLA has been shown to be stable up to 75°C and between pH 6.0 and 8.5 (29). The BLA variant used in this study has been modified to increase thermostability by approximately 13°C (26). Soluble NanA is stable up to 60°C, increasing to 75°C in the presence of pyruvate, and between pH 6.0 and 9.0 (30). Soluble OpdA has been shown to have an apparent melting temperature of 62°C; however, activity rapidly decreased after 55°C (10). No structural or stability data are currently available for OpdA from A. radiobacter; however, other similar phosphotriesterases have been shown to be stable up to 50°C and between pH 6.0 and 10.0 (31, 32). The isoelectric points of BLA and NanA are 6.9 and 4.5, respectively (27, 29), while the isoelectric point of a well-characterized homologous phosphotriesterase from Brevundimonas diminuta is 8.3 (33). Therefore, OpdA is proposed to be more stable at alkaline pH. In comparison to the studies on soluble enzyme, immobilization to GFP particles resulted in increased thermostability and stability throughout a broader range of pH values. The consistency of activity of each GFP particle varied after consecutive cycles of use, with GiCLN particles showing the least consistency while GiCLO particles showed the most. This could be due to the higher reaction temperature and reaction time used for the GiCLN particle assay (50°C and 48 h in comparison to 25°C and 12 h) and an inherent ability of OpdA to reset to an active conformation.
All enzyme-bearing GFP particles retained fluorescence up to 75°C (see Fig. S1 in the supplemental material), which correlated with the thermostability of GFP (34, 35). Furthermore, individual fluorescent particles can be observed between pH 5.0 and 12.0 (see Fig. S2), which correlates with the fluorescence observed for wild-type (wt) GFP and enhanced GFP (EGFP) (36, 37). Large fluorescent aggregates can be observed at 85°C and the extremes of pH; however, the GFP molecules are presumably shielded from denaturation within the aggregates and have been shown to regain fluorescence after denaturation (37). Furthermore, the aggregation behavior of the GFP particles at extreme pH might explain changes in enzyme activity. At pH values where aggregation occurred, low levels of enzyme activity were observed. However, at pH values where single GFP particles could be observed, high levels of enzyme activity were detected. GiCLO particles displayed higher activity at pH 11 and 12 than GiCLB and GiCLN particles, which corresponded with the presence of individual fluorescent particles as observed by fluorescence microscopy (see Fig. S2 in the supplemental material). These data suggested that the C-terminal fusion partner, the enzyme, impacted on pH stability with respect to GFP particle aggregation.
PHA beads and GFP particles displaying the relevant enzymes were assessed for storage stability at various temperatures and in 50 mM potassium phosphate buffer (pH 7.5) containing either 20% ethanol or 25% glycerol (see Table S3 in the supplemental material). The GiCLB particles were stable at all tested temperatures for at least 2 months without significant decline in activity. All GiCLN samples were stored for 3 months; however, while particles stored at 4°C and −80°C retained about 50% of initial activity, particles stored at RT lost their activity. All GiCLO samples, whether stored for 30 days or 4.5 months, exhibited similar levels of activity.
Fluorescence microscopy analysis of GFP particles obtained after long-term storage showed that the enzyme-bearing GFP particles retained their fluorescence, suggesting that GFP remained intact (data not shown). These results indicated that variation in storage stability is due to the stability of the respective enzymes under the various conditions. The use of 20% (vol/vol) ethanol in 50 mM potassium phosphate buffer or 25% (vol/vol) glycerol in 50 mM potassium phosphate buffer appeared to have no significant effects as samples stored at 4°C and −80°C exhibited similar activity for all types of GFP particles tested (see Table S3 in the supplemental material).
In this study, a one-step process of immobilization of enzymes translationally fused to self-assembling engineered GFP fusion proteins was investigated. Recombinant E. coli overproduced active enzyme-bearing GFP particles. This self-assembly-based enzyme immobilization approach proved to be versatile as different enzyme classes exhibiting various quaternary structures (monomer, dimer, and tetramer) were successfully implemented. Enzyme-bearing GFP particles were found to be stable and reusable, suggesting their suitability for applications in food processing and/or biotechnological industries.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the New Zealand Foundation for Research Science and Technology and the Institute of Fundamental Sciences at Massey University. D.O.H. received a Ph.D. scholarship from Massey University. The purchase of the Phenomenex RHM HPLC columns was funded by New Zealand Pharmaceuticals, Ltd. (Palmerston North, New Zealand).
Footnotes
Published ahead of print 7 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00323-14.
REFERENCES
- 1.Sanchez S, Demain AL. 2011. Enzymes and bioconversions of industrial, pharmaceutical, and biotechnological significance. Org. Process Res. Dev. 15:224–230. 10.1021/op100302x [DOI] [Google Scholar]
- 2.Scott C, Pandey G, Hartley CJ, Jackson CJ, Cheesman MJ, Taylor MC, Pandey R, Khurana JL, Teese M, Coppin CW, Weir KM, Jain RK, Lal R, Russell RJ, Oakeshott JG. 2008. The enzymatic basis for pesticide bioremediation. Indian J. Microbiol. 48:65–79. 10.1007/s12088-008-0007-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady D, Jordaan J. 2009. Advances in enzyme immobilisation. Biotechnol. Lett. 31:1639–1650. 10.1007/s10529-009-0076-4 [DOI] [PubMed] [Google Scholar]
- 4.Sheldon RA. 2007. Enzyme immobilization: the quest for optimum performance. Adv. Synth. Catal. 349:1289–1307. 10.1002/adsc.200700082 [DOI] [Google Scholar]
- 5.Hanefeld U, Gardossi L, Magner E. 2009. Understanding enzyme immobilisation. Chem. Soc. Rev. 38:453–468. 10.1039/b711564b [DOI] [PubMed] [Google Scholar]
- 6.Brady D, Jordaan J, Simpson C, Chetty A, Arumugam C, Moolman FS. 2008. Spherezymes: a novel structured self-immobilisation enzyme technology. BMC Biotechnol. 8:8. 10.1186/1472-6750-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheldon RA, Schoevaart R, Van Langen LM. 2005. Cross-linked enzyme aggregates (CLEAs): a novel and versatile method for enzyme immobilization (a review). Biocatal. Biotransformation 23:141–147. 10.1080/10242420500183378 [DOI] [Google Scholar]
- 8.St. Clair NL, Navia MA. 1992. Cross-linked enzyme crystals as robust biocatalysts. J. Am. Chem. Soc. 114:7314–7316. 10.1021/ja00044a064 [DOI] [Google Scholar]
- 9.Guglielmi F, Monti DM, Arciello A, Torrassa S, Cozzolino F, Pucci P, Relini A, Piccoli R. 2009. Enzymatically active fibrils generated by the self-assembly of the ApoA-I fibrillogenic domain functionalized with a catalytic moiety. Biomaterials 30:829–835. 10.1016/j.biomaterials.2008.10.036 [DOI] [PubMed] [Google Scholar]
- 10.Blatchford PA, Scott C, French N, Rehm BH. 2012. Immobilization of organophosphohydrolase OpdA from Agrobacterium radiobacter by overproduction at the surface of polyester inclusions inside engineered Escherichia coli. Biotechnol. Bioeng. 109:1101–1108. 10.1002/bit.24402 [DOI] [PubMed] [Google Scholar]
- 11.Hooks DO, Blatchford PA, Rehm BHA. 2013. Bioengineering of bacterial polymer inclusions catalyzing the synthesis of N-acetyl neuraminic acid. Appl. Environ. Microbiol. 79:3116–3121. 10.1128/AEM.03947-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters V, Rehm BH. 2006. In vivo enzyme immobilization by use of engineered polyhydroxyalkanoate synthase. Appl. Environ. Microbiol. 72:1777–1783. 10.1128/AEM.72.3.1777-1783.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasiah IA, Rehm BH. 2009. One-step production of immobilized alpha-amylase in recombinant Escherichia coli. Appl. Environ. Microbiol. 75:2012–2016. 10.1128/AEM.02782-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jahns AC, Maspolim Y, Chen S, Guthrie JM, Blackwell LF, Rehm BHA. 2013. In vivo self-assembly of fluorescent protein microparticles displaying specific binding domains. Bioconjug. Chem. 24:1314–1323. 10.1021/bc300551j [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 16.Jahns AC, Rehm BH. 2009. Tolerance of the Ralstonia eutropha class I polyhydroxyalkanoate synthase for translational fusions to its C terminus reveals a new mode of functional display. Appl. Environ. Microbiol. 75:5461–5466. 10.1128/AEM.01072-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 18.Chow J, Kovacic F, Antonia YD, Krauss U, Fersini F, Schmeisser C, Lauinger B, Bongen P, Pietruszka J, Schmidt M, Menyes I, Bornscheuer UT, Eckstein M, Thum O, Liese A, Mueller-Dieckmann J, Jaeger K-E, Streit WR. 2012. The metagenome-derived enzymes LipS and LipT increase the diversity of known lipases. PLoS One 7:e47665. 10.1371/journal.pone.0047665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmer M. 2002. Green fluorescent protein (GFP)—applications, structure, and related photophysical behaviour. Chem. Rev. 102:759–781. 10.1021/cr010142r [DOI] [PubMed] [Google Scholar]
- 20.Izard T, Lawrence MC, Malby RL, Lilley GG, Colman PM. 1994. The three-dimensional structure of N-acetylneuraminate lyase from Escherichia coli. Structure 2:361–369. 10.1016/S0969-2126(00)00038-1 [DOI] [PubMed] [Google Scholar]
- 21.Peternel S, Komel R. 2011. Active protein aggregates produced in Escherichia coli. Int. J. Mol. Sci. 12:8275–8287. 10.3390/ijms12118275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence MC, Barbosa JA, Smith BJ, Hall NE, Pilling PA, Ooi HC, Marcuccio SM. 1997. Structure and mechanism of a sub-family of enzymes related to-acteylneuraminate lyase. J. Mol. Biol. 266:381–399. 10.1006/jmbi.1996.0769 [DOI] [PubMed] [Google Scholar]
- 23.Grage K, Jahns AC, Parlane N, Palanisamy R, Rasiah IA, Atwood JA, Rehm BHA. 2009. Bacterial polyhydroxyalkanoate granules: biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules 10:660–669. 10.1021/bm801394s [DOI] [PubMed] [Google Scholar]
- 24.Peters V, Rehm BH. 2005. In vivo monitoring of PHA granule formation using GFP-labeled PHA synthases. FEMS Microbiol. Lett. 248:93–100. 10.1016/j.femsle.2005.05.027 [DOI] [PubMed] [Google Scholar]
- 25.Vihinen M, Mantsala P. 1989. Microbial amylolytic enzymes. Crit. Rev. Biochem. Mol. Biol. 24:329–418. 10.3109/10409238909082556 [DOI] [PubMed] [Google Scholar]
- 26.Machius M, Declerck N, Huber R, Wiegand G. 2003. Kinetic stabilization of Bacillus licheniformis alpha-amylase through introduction of hydrophobic residues at the surface. J. Biol. Chem. 278:11546–11553. 10.1074/jbc.M212618200 [DOI] [PubMed] [Google Scholar]
- 27.Aisaka K, Igarashi A, Yamaguchi K, Uwajima T. 1991. Purification, crystallisation and characterisation of N-acetylneuraminate lyase from Escherichia coli. Biochem. J. 276:541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu X, Gao C, Zhang X, Che B, Ma C, Qiu J, Tao F, Xu P. 2011. Production of N-acetyl-d-neuraminic acid by use of an efficient spore surface display system. Appl. Environ. Microbiol. 77:3197–3201. 10.1128/AEM.00151-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanova VN, Dobreva EP, Emanuilova EI. 1993. Purification and characterization of a thermostable alpha-amylase from Bacillus licheniformis. J. Biotechnol. 28:277–289. 10.1016/0168-1656(93)90176-N [DOI] [Google Scholar]
- 30.Uchida Y, Tsukada Y, Sugimori T. 1984. Purification and properties of N-acetylneuraminate lyase from Escherichia coli. J. Biochem. 96:507–522 [DOI] [PubMed] [Google Scholar]
- 31.Ekkhunnatham A, Jongsareejit B, Yamkunthong W, Wichitwechkarn J. 2012. Purification and characterization of methyl parathion hydrolase from Burkholderia cepacia capable of degrading organophosphate insecticides. World J. Microbiol. Biotechnol. 28:1739–1746. 10.1007/s11274-011-0985-y [DOI] [PubMed] [Google Scholar]
- 32.Rowland SS, Speedie MK, Pogell BM. 1991. Purification and characterization of a secreted recombinant phosphotriesterase (parathion hydrolase) from Streptomyces lividans. Appl. Environ. Microbiol. 57:440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dumas DP, Caldwell SR, Wild JR, Raushel FM. 1989. Purification and properties of the phosphotriesterase from Pseudomonas diminuta. J. Biol. Chem. 264:19659–19665 [PubMed] [Google Scholar]
- 34.Ward WW. 1981. Properties of the coelenterate green-fluorescent protein, p 235–242 In DeLuca MA, McElroy WD. (ed), Bioluminescence and chemiluminescence. Academic Press, New York, NY [Google Scholar]
- 35.Ward WW, Prentice HJ, Roth AF, Cody CW, Reeves SC. 1982. Spectral perturbations of the Aequorea green-fluorescent protein. Photochem. Photobiol. 35:803–808. 10.1111/j.1751-1097.1982.tb02651.x [DOI] [Google Scholar]
- 36.Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. 1997. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 73:2782–2790. 10.1016/S0006-3495(97)78307-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bokman SH, Ward WW. 1981. Renaturation of Aequorea green fluorescent protein. Biochem. Biophys. Res. Comm. 101:1372–1380. 10.1016/0006-291X(81)91599-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.