

ABSTRACT
Dengue viruses (DENV) are endemic pathogens of tropical and subtropical regions that cause significant morbidity and mortality worldwide. To date, no vaccines or antiviral therapeutics have been approved for combating DENV-associated disease. In this paper, we describe a class of tricyclic small-molecule compounds—dihydrodibenzothiepines (DHBTs), identified through high-throughput screening—with potent inhibitory activity against DENV serotype 2. SKI-417616, a highly active representative of this class, displayed activity against all four serotypes of DENV, as well as against a related flavivirus, West Nile virus (WNV), and an alphavirus, Sindbis virus (SINV). This compound was characterized to determine its mechanism of antiviral activity. Investigation of the stage of the viral life cycle affected revealed that an early event in the life cycle is inhibited. Due to the structural similarity of the DHBTs to known antagonists of the dopamine and serotonin receptors, we explored the roles of two of these receptors, serotonin receptor 2A (5HTR2A) and the D4 dopamine receptor (DRD4), in DENV infection. Antagonism of DRD4 and subsequent downstream phosphorylation of epidermal growth factor receptor (EGFR)-related kinase (ERK) were found to impact DENV infection negatively, and blockade of signaling through this network was confirmed as the mechanism of anti-DENV activity for this class of compounds.
IMPORTANCE The dengue viruses are mosquito-borne, reemerging human pathogens that are the etiological agents of a spectrum of febrile diseases. Currently, there are no approved therapeutic treatments for dengue-associated disease, nor is there a vaccine. This study identifies a small molecule, SKI-417616, with potent anti-dengue virus activity. Further analysis revealed that SKI-417616 acts through antagonism of the host cell dopamine D4 receptor and subsequent repression of the ERK phosphorylation pathway. These results suggest that SKI-417616, or other compounds targeting the same cellular pathways, may have therapeutic potential for the treatment of dengue virus infections.
INTRODUCTION
The dengue viruses (DENV) are mosquito-borne viruses of the family Flaviviridae that comprise four antigenically distinct serotypes (DENV1 to -4). DENV is an emerging pathogen that is endemic in tropical and subtropical regions. Estimates of the annual number of DENV infections range from 50 million to more than 230 million, resulting in approximately 500,000 to 2 million cases of dengue hemorrhagic fever (DHF) per year (1–3; http://www.cdc.gov/dengue/). Increased frequency of travel, environmental changes, and expansion of human populations into regions where the primary DENV vector, Aedes aegypti, is prevalent have contributed to the emerging nature of this pathogen (4). Primary infection by a single serotype can result in a range of disease severities, which can include asymptomatic infection; dengue fever, a flu-like illness characterized by prolonged fever and severe joint pain; and severe dengue, or DHF/dengue shock syndrome (DSS), which often presents with hemorrhagic symptoms and thrombocytopenia and can be fatal. It is generally believed that while one is protected from secondary infection with a strain of the same serotype, secondary infection with a heterologous strain predisposes one to the more severe forms of dengue disease (5). Currently, no approved vaccine is available, and treatment for DENV infection consists primarily of supportive therapy.
Transmission of DENV to a human host is initiated through the bite of an infected mosquito. Uptake by skin-resident Langerhans cells promotes transport to draining lymph nodes, where the virus then infects dendritic cells, monocytes, and macrophages, allowing amplification of the virus and dissemination throughout the body via the lymphatic and circulatory systems (6). In situ hybridization and immunocytochemistry analyses of samples from naturally infected humans have demonstrated that infection occurs in a range of organs in vivo, including the liver, spleen, and kidneys (7). Although robust replication is believed to occur primarily in cells of myeloid origin, the characteristic hemorrhagic symptoms of severe dengue infection suggest a role for endothelial cells in contributing to pathogenesis (5, 6).
Completion of the DENV life cycle is heavily dependent on host cell factors. The identities of initial attachment and entry receptors for DENV remain unclear, although the heparan sulfates and DC-SIGN (dendritic-cell-specific intercellular adhesion molecule 3-grabbing nonintegrin) have been demonstrated to contribute to the internalization of DENV particles into human cell types (8, 9). Receptor-mediated endocytosis proceeds through clathrin-coated vesicles, followed by low-pH-induced fusion of the viral particle with the late-endosomal membrane, resulting in the release of the capsid-bound positive-sense RNA genome (10, 11). Initial translation from the incoming genome occurs to produce viral factors required for subsequent replication. Genome amplification occurs in membranous pockets formed within the endoplasmic reticulum (ER) membrane, and translation of the viral polypeptide and subsequent cleavage to the 10 individual viral proteins occur in tight association with the ER membrane as well (12). Following the assembly of the viral nucleocapsid, particles bud through the ER and are transported through the secretory pathway before release at the cell surface. Throughout these processes, multiple classes of cellular factors play critical roles in both supporting and restricting the completion of the viral life cycle, including, but not limited to, endocytic proteins, cellular proteases, the ubiquitin proteasome system, factors of the autophagosome, and ER/Golgi factors regulating the secretory pathway (reviewed in reference 13). Although the development of antivirals that directly target viral proteins, such as proteases or polymerases, has proved to be successful for the treatment of infections with other viruses, the strategy of examining host cell factors as targets for limiting flavivirus infection has recently gained traction. Targeting host proteins, in contrast to relying on direct binding to viral factors, can help avoid some of the pitfalls commonly encountered during antiviral development, such as the promotion of viral escape mutants (reviewed in reference 14). Furthermore, compounds that affect host proteins required for viral replication may include previously characterized drugs with defined side effects, in vivo toxicity, and pharmacology, providing the potential for rapid development of antiviral applications.
In this study, we report on the discovery of a group of small-molecule compounds sharing a common dihydrodibenzothiepine (DHBT) scaffold with potent anti-DENV2 activity. This antiviral activity was found to extend to all representative strains of the four DENV serotypes, as well as to West Nile virus (WNV) and Sindbis virus (SINV), but to have no effect against two DNA viruses, herpes simplex virus 1 (HSV-1) and vaccinia virus (VACV). We demonstrate that the antiviral effects of a highly active representative compound, 7-fluoro-11-(4-methylpiperazin-1-yl)-10,11-dihydrodibenzo[b,f]thiepin-2-ol (referred to here as SKI-417616), take place early during the viral life cycle, at the step of virus internalization. Based on the structural similarity of the DHBTs to known inhibitors of serotonin and dopamine receptors, we investigate the roles of these receptors in the action of DHBTs against DENV infection. Here we demonstrate that signaling through dopamine receptor 4 (DRD4), and subsequent activation of epidermal growth factor receptor (EGFR)-responsive kinase (ERK), is the mechanism of the observed antiviral activity of DHBTs.
MATERIALS AND METHODS
Cell culture and reagents.
HEK293 and HeLa cells were grown in modified Eagle medium (MEM; Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin G sodium, 100 μg/ml streptomycin sulfate (Invitrogen), and 1× nonessential amino acids (Gibco). THP1 cells were grown in RPMI medium (Gibco) supplemented with 10% FBS (HyClone), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin G sodium, 100 μg/ml streptomycin sulfate (Invitrogen), and 1× nonessential amino acids (Gibco). Differentiation of THP1 cells was carried out by treatment with 50 ng/ml phorbol myristate acetate (PMA; Sigma), followed by infection at 24 h posttreatment.
Virus strains.
DENV1 (strain TH-Sman), DENV2 (New Guinea C [NGC]), DENV3 (H87), DENV4 (H241), and Sindbis virus (strain AR339) were obtained from the ATCC. West Nile virus (strain 385-99) has been described previously (15). DENV and WNV strains were passaged twice on C6/36 cells and were purified by centrifugation as described previously (16). Virus titers were determined by a focus-forming assay as described previously (17). Herpes simplex virus 1 (strain F1) was a kind gift from A. Hill (Oregon Health and Sciences University). Vaccinia virus (strain Western Reserve) was a kind gift from M. Slifka (Oregon Health and Sciences University).
Luciferase-based secondary screening.
The DENV2 (strain 16681)-based luciferase reporter virus (DENV2-Luc) was a kind gift from A. Gamarnik (18). HEK293 cells were seeded in 96-well plates and were infected at 24 h postseeding with 10 μl per plate (∼0.1 μl/well) of DENV2-Luc plus 312.5 nM to 20 μM compound in triplicate. At 72 h postinfection (p.i.), luciferase activity was measured by using the Renilla-Glo luciferase assay system (Promega) according to the manufacturer's instructions. Values were normalized to those for dimethyl sulfoxide (DMSO)-treated controls and were fit to a sigmoidal dose-response curve, and 50% and 80% inhibitory concentrations (IC50 and IC80, respectively) were calculated using GraphPad Prism software.
Toxicity assays.
HEK293 cells were incubated in cell media containing increasing concentrations of compounds in constant 1% DMSO (vol/vol). After 48 h, CellTiter-Glo reagent (Promega) was added and luminescence measured according to the manufacturer's instructions. Fifty percent cytotoxic concentrations (CC50) were calculated using GraphPad Prism software.
Fold reduction.
HEK293 cells were infected with DENV2 (NGC) at a multiplicity of infection (MOI) of 0.1 focus-forming unit (FFU)/cell in MEM–2% FBS plus 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate (PSG) (Invitrogen) in the presence of 10 μM compound or DMSO. At 72 h postinfection, supernatants were collected, and the number of focus forming units was determined as described previously (17). Fold reduction (log10) was calculated as log10(FFU/ml in the presence of DMSO) − log10(FFU/ml in the presence of the compound).
Multistep growth curves.
Low-multiplicity infections were performed in triplicate in HEK293 cells. The virus was diluted in 2% FBS–MEM plus PSG at an MOI of 0.1 PFU/cell in the presence of DMSO or SKI-417616 (1 µM or 10 µM) and was incubated with rocking in a low volume for 1 h at 37°C. Unattached virus was removed, and cells were refed with normal growth medium plus DMSO or the compound. At the times postinfection indicated on Fig. 1D and E and 2A through E, supernatants were collected, and the virus was quantitated by focus-forming assays.
FIG 1.

DHBTs are potent inhibitors of DENV2 infection. (A) Structure of shared DHBT scaffold of DENV2 inhibitors. R groups R1 and R2 may be located anywhere on their respective rings. (B) Structure of SKI-417616. (C) Dose-response curve for SKI-417616 and DENV-Luc in HEK293 cells. (D) HEK293 cells were infected with DENV2 at an MOI of 0.1 FFU/cell in the presence of 1 μM or 10 μM SKI-417616 or DMSO (control). Supernatants were collected at the indicated times p.i., and titers were determined on Vero cells. Significant (P, <0.05 by a two-tailed t test) inhibition by 1 μM and 10 μM SKI-417616 (compared to virus levels with the DMSO control) was detected at days 2 and 3. (E) Differentiated THP1 cells were infected with DENV2 at an MOI of 10 FFU/cell in the presence of 10 μM SKI-417616. Supernatants were collected at the indicated times p.i., and titers were determined on Vero cells. *, P < 0.05; **, P < 0.01.
FIG 2.

SKI-417616 inhibits multiple positive-sense RNA viruses. HEK293 cells were infected at an MOI of 0.1 FFU/cell in the presence of 1 μM or 10 μM SKI-417616 or DMSO. Supernatants were collected at the indicated times p.i., and infectious virus was measured by a focus-forming assay (A to D) or a plaque-forming assay (E to G) on Vero cells. (A) DENV1; (B) DENV3; (C) DENV4; (D) WNV; (E) SINV; (F) VACV; (G) HSV-1.
Time-of-addition assay.
HEK293 cells were infected with DENV2 at an MOI of 3 FFU/cell in a low volume with rocking. At the times postinfection indicated on Fig. 1 and 2, the medium was removed and was replaced with 5 μM SKI-417616 or DMSO diluted in medium. Supernatants were collected at 48 h postinfection, and the virus was quantitated by a focus-forming assay.
Infectious-center assay.
HEK293 cells were prechilled to 4°C for 10 min. The cells were then fed with a medium containing 4 μg/ml carrageenan, 20 μM chlorpromazine, or 10 μM SKI-417616, followed by infection with DENV2 (NGC) at 20 FFU/cell. The cells were incubated at 4°C for 1 h, followed by incubation at 37°C for 1 h. The culture medium was removed, and unattached/uninternalized virus was then inactivated by incubation with citrate acid buffer (40 mM Na citrate, 135 mM NaCl, 10 mM KCl [pH 3.2]) for 1 min, followed by 2 washes with phosphate-buffered saline (PBS). The cells were treated with 0.05% trypsin-EDTA for 3 min and were then counted, and serial dilutions ranging from 100 to 10,000 cells were added to a monolayer of Vero cells. Twenty-four hours later, the cultures were overlaid with a medium containing 0.5% carboxymethyl cellulose. At 72 h postplating, the cells were fixed and were stained for foci as described above, and infectious centers were quantified.
Transferrin microscopy.
HEK293 cells seeded onto glass coverslips were pretreated for 1 h at 37°C with either DMSO, 20 μM chlorpromazine, or 10 μM SKI-417616 and were then incubated with Alexa Fluor 594-conjugated transferrin (T13343; Molecular Probes) in the presence of the drug for 1 h at 37°C. The cells were fixed in 4% paraformaldehyde, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were obtained on a Leica SP5 acousto-optical beam splitter (AOBS) confocal microscope.
RNA replication assay.
HEK293 cells were infected with DENV2 at an MOI of 3 FFU/cell, and 5 μM SKI-417616 or DMSO (control) was added. At 24 h postinfection, total RNA was isolated and was analyzed by reverse transcription-quantitative PCR (RT-qPCR) using a universal primer/probe set designed to amplify regions of the viral genome within the 3′ untranslated region (3′ UTR) (19) or the envelope protein (E) coding sequence (20).
Luciferase replicon.
BHK-21 cells stably expressing a DENV2 luciferase replicon (a kind gift from M. Diamond, Washington University [21]) were treated with increasing concentrations of SKI-417616 (612.5 nM to 10 μM) or ribavirin (3.75 μM to 60 μM) in triplicate. At 48 h posttreatment, cells were lysed and were analyzed for luciferase activity by using the Luciferase Assay System (Promega) according to the manufacturer's instructions.
siRNA transfections.
HeLa cells were transfected with 50 nM small interfering RNA (siRNA) (s4290 for DRD4; s7041 for 5HTR2A; Applied Biosystems) by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. At 48 h posttransfection, the cells were infected with DENV2 at an MOI of 5 FFU/cell. The cells were fixed at 72 h postinfection and were analyzed by immunofluorescence for envelope protein expression.
ERK phosphorylation.
HEK293 cells were serum starved for 24 h before treatment with 5 μM dopamine plus DMSO or 5 μM dopamine plus 5 μM SKI-417616 diluted in serum-free MEM. Total protein was isolated at 10 min posttreatment and was subjected to SDS-PAGE and Western blot analysis with an anti-phosphorylated ERK (anti-phospho-ERK) antibody (4370S; Cell Signaling Technologies) and an anti-ERK antibody (sc-93; Santa Cruz Biotechnology).
Cyclic AMP (cAMP) signaling.
HEK293 cells were serum starved for 24 h before treatment with 5 μM dopamine plus DMSO or 5 μM dopamine plus 5 μM SKI-417616 diluted in serum-free MEM. Total RNA was isolated at 2 h posttreatment. RT-qPCR using the TaqMan One-Step RT-PCR kit (catalog no. 4313803; Life Technologies) was performed according to the manufacturer's instructions to quantitate c-Fos (Life Technologies assay identification [ID] Hs04194186_s1) and β-actin (Hs99999903_m1). Normalization was carried out, and the fold change was calculated using the comparative threshold cycle (ΔΔCT) method.
RESULTS
Identification of DHBTs as potent anti-DENV compounds.
Previous screening efforts through a collaboration with the HTS Core Facility at the Memorial Sloan-Kettering Cancer Center, New York, NY, developed a robust high-content assay for screening small-molecule libraries (obtained from CRL, BioFocus, and SPECS) to identify inhibitors of DENV2 infection (17). During this study and subsequent studies, several compounds with a common scaffold, DHBT (Fig. 1A), were identified as potent inhibitors of DENV2 infection. DHBTs with DENV2-inhibitory activity were further characterized using a luciferase reporter virus (18) (Fig. 1C) and were assessed for cytotoxicity using a CellTiter-Glo assay and for activity against wild-type DENV2 replication by measurement of released infectious particles (see Table S1 in the supplemental material). In order to determine if antiviral activity is due to the activation of canonical cellular antiviral pathways, all compounds were also assayed for their abilities to activate NF-κB or JAK/STAT signaling by using HEK293 cells expressing luciferase reporter constructs under the control of an NF-κB or interferon-stimulated response element (ISRE)-responsive promoter. None of the compounds tested displayed significant activation of either of these pathways (data not shown). 7-Fluoro-11-(4-methylpiperazin-1-yl)-10,11-dihydrodibenzo[b,f]thiepin-2-ol, referred to here as SKI-417616 (Fig. 1B), displayed the strongest inhibition of viral progeny production combined with low toxicity (IC50, 1.2 μM; CC50, 43.2 μM) and thus was used as an active representative for compounds of this class in further studies. Multistep growth curves in both HEK293 cells (Fig. 1D) and differentiated monocytic THP1 cells (Fig. 1E) demonstrated that SKI-417616 displays anti-DENV2 activity in a range of cell types.
DHBTs inhibit infection by flaviviruses and alphaviruses.
Given the potent inhibitory activities of SKI-417616 and similar compounds against DENV2, we sought to determine the effect of this compound against other DENV types. Multistep growth curves were performed in the presence of 1 μM and 10 μM SKI-417616 or DMSO (control). Inhibition of the replication of representative strains of DENV1 (Fig. 2A), DENV3 (Fig. 2B), and DENV4 (Fig. 2C) by SKI-417616 was observed. Furthermore, SKI-417616 displayed inhibitory activity against a related flavivirus, West Nile virus (WNV) (Fig. 2D). To determine whether the antiviral activity of SKI-417616 is restricted to flaviviruses or extends to other types of viruses, we examined the effects of this compound on the replication of Sindbis virus (SINV). SINV is a member of the genus Alphavirus. Like the flaviviruses, alphaviruses are enveloped and possess a positive-sense RNA genome. Similar to the effects observed against flaviviruses, SINV infection was dramatically reduced by treatment with SKI-417616 (Fig. 2E). Finally, the effects of SKI-417616 on infection by two enveloped viruses with DNA genomes, vaccinia virus (VACV) (Fig. 2F) and herpes simplex virus 1 (HSV-1) (Fig. 2G), were examined. In neither case did SKI-417616 display significant inhibition of virus growth.
We also attempted to select for DENV2 mutants resistant to the effects of SKI-417616 via multiple passages in the presence of the drug. Notably, we were unable to elicit such mutants through 4 passages in the presence of 5 μM drug (data not shown). This inability to elicit resistance suggests the targeting of a host cell factor by SKI-417616, rather than direct interaction with a viral protein, as its mechanism of inhibition.
DHBTs inhibit DENV infection at an early stage in the life cycle.
To define the effect of SKI-417616 on viral entry, a time-of-addition experiment was performed. DENV2 was incubated with cells in the presence of SKI-417616 or DMSO (control). The cells were then washed at a low pH to remove/inactivate all uninternalized virus and were refed with a normal growth medium. At the times postinfection indicated in Fig. 3A, supernatants were removed and were replaced with a medium containing SKI-417616 or DMSO. The addition of SKI-417616 as early as 2 h p.i. resulted in decreased inhibitory activity compared with that when SKI-417616 was added concurrently with infection, while addition at 6 h p.i. displayed negligible effects on infection (Fig. 3A), suggesting that SKI-417616 acts at an early stage of the virus replication cycle.
FIG 3.

DHBTs inhibit DENV2 infection at an early stage in the life cycle. (A) HEK293 cells were incubated with DENV2 at an MOI of 3 FFU/cell for 1 h. Uninternalized virus was removed by an acid wash, and cells were refed with a normal growth medium. At the indicated times postattachment, the medium was removed and replaced with a medium containing DMSO or 5 μM SKI-417616. Supernatants were collected at 48 h p.i., and titers were determined on Vero cells. (B) HEK293 cells were infected with DENV2 at an MOI of 3 FFU/cell in the presence of DMSO or 5 μM SKI-417616. Total RNA was collected at 24 h postinfection, and genome equivalents were detected by RT-qPCR using primer and probe sets specific for the 3′ UTR (dark shaded bars) or E (light shaded bars) region of the viral genome. (C) Cells were treated with carrageenan (4 μg/ml), chlorpromazine (20 μM), or SKI-417616 (10 μM) and were then infected for 1 h with DENV2 (MOI, 20 FFU/cell). External virus was removed and infectious centers quantified as described in Materials and Methods. ***, P < 0.001. (D) Cells were treated with 20 μM chlorpromazine, 10 μM SKI-417616, or DMSO (control), followed by incubation with fluorescently labeled transferrin. After 1 h, cells were fixed, and transferrin internalization was analyzed by confocal microscopy. (E) BHK21-DVrep cells were treated with 2-fold-increasing concentrations of ribavirin (3.75 μM to 60 μM) or SKI-417616 (612.5 nM to 10 μM). Cells were lysed at 48 h posttreatment, and firefly luciferase activity was measured by luminescence. Values were normalized to those for DMSO-treated controls.
In order to examine the replication of viral RNA in the presence of SKI-417616, HEK293 cells were infected with DENV2 in the presence of SKI-417616, and total RNA was isolated from infected cells at 24 h p.i. This time point and MOI were used to ensure the measurement of a single round of infection and genome replication. Viral genomes were detected using qRT-PCR with primer/probe sets that that amplify a region within the DENV 3′ UTR or envelope protein (E) coding sequence. A 2-log reduction in viral genome production was detected in SKI-417616-treated cells with either probe set (Fig. 3B).
Additionally, we tested the ability of SKI-417616 to inhibit the generation of infectious centers. Cells cultured in the presence of the drugs indicated in Fig. 3C were infected at 4°C for 1 h to allow virus attachment and were then shifted to 37°C for 1 h to allow internalization. Virus remaining outside the cells was inactivated by a low-pH wash, followed by trypsin treatment to obtain a single-cell suspension. Infectious centers were quantified by plating defined numbers of cells onto uninfected Vero cell monolayers and immunostaining the resultant foci. As shown, both carrageenan, an inhibitor of viral attachment and internalization, and chlorpromazine, an inhibitor of clathrin-dependent endocytosis, significantly inhibited infectious-center formation (Fig. 3C). This effect was also observed in cells treated with SKI-417616. Taken together, the results in Fig. 3 are consistent with SKI-417616 inhibiting an early stage of the viral life cycle, which may include attachment, internalization, translation of the genomic RNA, or establishment of the RNA replication complex.
Certain cationic amphiphilic drugs, such as chlorpromazine, have been shown to inhibit viral entry through interference with the formation of clathrin-coated pits and subsequent clathrin-mediated endocytosis (22). We examined the effect of SKI-417616 on this process by monitoring its effect on the internalization of the transferrin receptor. As shown in Fig. 3D, fluorescently labeled transferrin was efficiently internalized when added to control cells, and internalization was blocked in the presence of 20 μM chlorpromazine, so that transferrin remained largely associated with the peripheries of the cells. In contrast, cells treated with SKI-417616 continued to internalize transferrin comparably to control cells. These results demonstrate that SKI-417616 does not act as a general inhibitor of endocytosis.
We next sought to separate the effects of SKI-417616 on viral RNA replication from its effect on entry. A cell line containing a constitutively replicating DENV2 subgenomic replicon bearing a Renilla luciferase reporter was treated with increasing concentrations of the compound (612.5 nM to 10 μM). The cells were also treated with ribavirin, a known inhibitor of DENV replication (23), as a positive control. As expected, dose-dependent inhibition of luciferase activity was observed following treatment with ribavirin. Interestingly, no effect on luciferase expression was observed in SKI-417616-treated cells (Fig. 3E), even at concentrations previously demonstrated to reduce viral infection dramatically. This result, together with the results shown in Fig. 3A and B, demonstrate that SKI-417616 does not inhibit DENV2 translation or RNA replication but rather acts to inhibit events prior to these steps.
DHBTs antagonize dopamine receptor D4 to inhibit DENV infection.
Small-molecule compounds with scaffolds and structures similar to those of DHBTs have been researched extensively for their effects on activation and signaling through serotonin and dopamine receptors (24–28). To investigate the possibility that SKI-417616 is acting through antagonism of serotonin or dopamine receptors, we first consulted a microarray data set to determine the identities of any serotonin or dopamine receptors expressed in HEK293 cells. Significant expression of mRNAs encoding only two receptors was found: serotonin receptor 2A (5HTR2A) and the D4 dopamine receptor (DRD4) (data not shown). Therefore, two approaches were used to determine whether antagonism of either of these receptors had inhibitory effects on DENV2 infection. First, selective antagonists of each receptor that are structurally unrelated to DHBTs were analyzed for their effects on DENV2 infection. Treatment with L741,742 dihydrochloride, a potent and selective antagonist of DRD4 (29), dramatically reduced the production of DENV2 infectious progeny. In contrast, antagonism of 5HTR2A with 4-(4-fluorobenzoyl)-1-(4-phenylbutyl)piperidine (4F-4PP) oxalate (30) had no effect on DENV2 infection (Fig. 4A). Further, the addition of exogenous dopamine rescued viral replication in cultures treated with L741,742 or SKI-417616. Depletion of each receptor using siRNA knockdown (Fig. 4B) was used to further investigate the requirement for each receptor during DENV2 infection. In agreement with the biochemical antagonist studies, only siRNA knockdown of DRD4 inhibited DENV infection, while siRNA to 5HTR2A had no effect (Fig. 4C and D). Collectively, these results demonstrate an important role for DRD4 in DENV infection.
FIG 4.

Inhibition of DRD4 activity inhibits DENV2 infection. (A) HEK293 cells were infected with DENV2 at an MOI of 0.1 FFU/cell in the presence of either DMSO, 10 μM 4F-4PP oxalate (a 5HTR2A antagonist), L741,742 dihydrochloride (a DRD4 antagonist), or SKI-417616. Additionally, samples were treated with DMSO or 10 μM dopamine, as indicated. Supernatants were collected at 48 h p.i., and titers were determined on Vero cells. ***, P < 0.001; **, P < 0.01. (B) HeLa cells were transfected with siRNAs targeting 5HTR2A or DRD4. At 48 h posttransfection, total RNA was isolated and was analyzed by RT-qPCR for 5HTR2A (left) or DRD4 (right) expression; results were normalized to β-actin levels. (C) siRNA-transfected HeLa cells were infected with DENV2 at an MOI of 5 FFU/cell and were fixed at 72 h p.i. Envelope protein was detected by immunofluorescence. (D) Integrated density was quantitated for three independent experiments using ImageJ software.
DHBTs antagonize signaling through DRD4 to affect viral entry.
The identity of the flavivirus entry receptor(s) still remains unclear, and given the wide range of cell types, both mammalian and invertebrate, into which flaviviruses enter and in which they replicate, it is likely that multiple attachment and entry factors are utilized. However, based on the relatively specific expression profile and activity of dopamine receptors (31), we hypothesized that DRD4 does not represent an entry receptor but more likely affects a signaling event important for viral internalization. Several signaling events downstream of DRD4 activation have been described, including phosphorylation and subsequent activation of ERK, as well as repression of cAMP signaling (reviewed in reference 31). To determine whether DHBT treatment affects either of these signaling events, HEK293 cells were treated with dopamine to activate DRD4 in the presence of SKI-417616 or DMSO (control). Phosphorylation of ERK was measured by Western blot analysis, and both SKI-417616 and L741,742 were observed to inhibit dopamine-induced activation of ERK efficiently (Fig. 5A). In contrast, neither SKI-417616 nor L741,742 had any effect on dopamine-dependent repression of expression from a cAMP-responsive gene, c-fos (Fig. 5B). Because SKI-417616 blocks the phosphorylation of ERK, we hypothesized that this activity may contribute to the inhibition of DENV replication.
FIG 5.
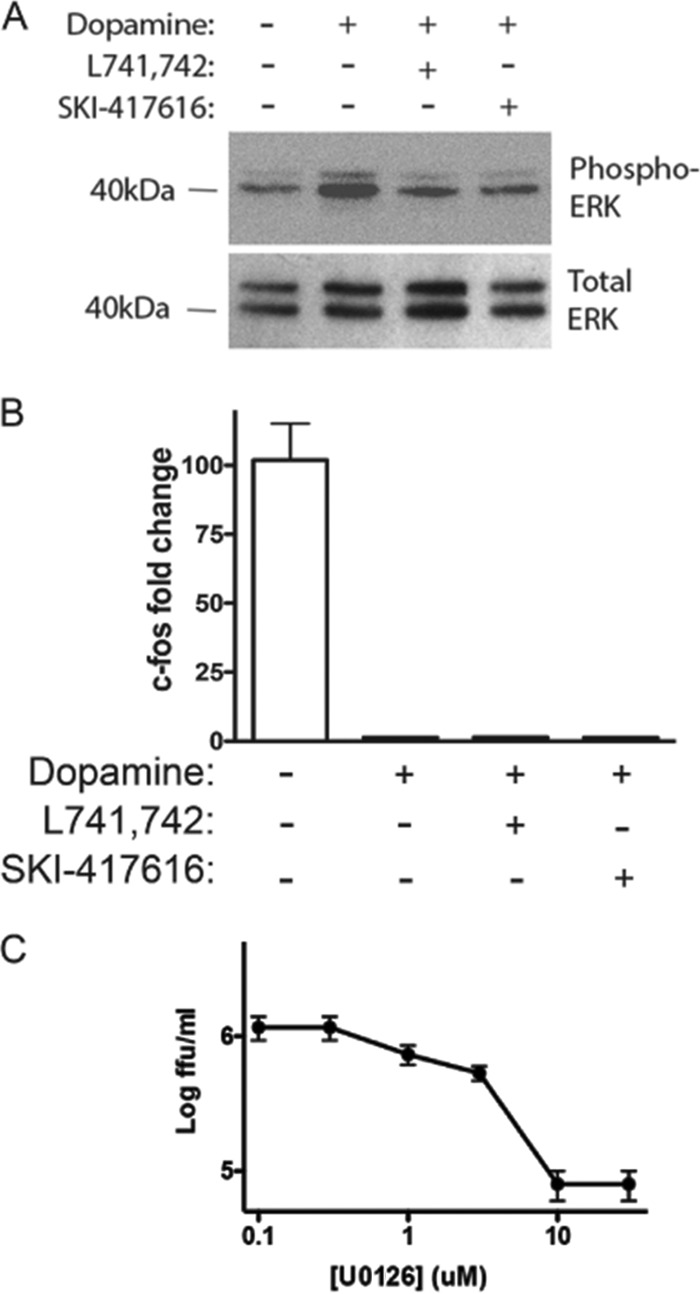
DHBTs inhibit DENV2 infection through antagonism of ERK signaling via DRD4. (A and B) Serum-starved HEK293 cells were treated with the indicated compound at 5 μM. Total protein (A) or total RNA (B) was isolated at 10 min (A) or 2 h (B) posttreatment. (A) SDS-PAGE and Western blot analysis were performed to detect phospho-ERK (top) and total ERK (bottom). (B) c-fos and β-actin levels were determined by RT-qPCR. The fold change in the c-fos level was calculated by ΔΔCT analysis. (C) HEK293 cells were infected with DENV2 at an MOI of 0.1 FFU/cell with increasing concentrations of U0126 (1 to 10 μM). Supernatants were collected at 48 h p.i., and titers were determined on Vero cells.
To further determine whether ERK signaling downstream of DRD4 activation affects DENV infection, direct antagonism of ERK signaling was examined using the well-characterized pharmacological inhibitor of ERK phosphorylation U0126. Inhibition of ERK phosphorylation resulted in dose-dependent inhibition of DENV2 (Fig. 5C), suggesting that DHBTs affect DENV replication through antagonism of DRD4 signaling and subsequent inhibition of downstream ERK signaling.
DISCUSSION
Dengue viruses are important pathogens; approximately 2.5 billion people live in countries where DENV is endemic and are at risk of being infected (3). Alarmingly, both the range and the severity of dengue-related disease appear to be expanding (4). Currently, no effective vaccines or antivirals are in use, making the development of DENV control strategies a critical worldwide health priority. We have previously reported the development of a high-content immunofluorescent assay to monitor small-molecule libraries for compounds with anti-DENV activity (17), and in this report, we expand this research to describe a group of related compounds with a common scaffold, DHBT, that exhibit potent antiviral properties. Characterization of a representative compound from this group, SKI-417616, demonstrated inhibition of infection not only with DENV2 but also with other serotypes of DENV, as well as another flavivirus, WNV, and the alphavirus SINV. Interestingly, no inhibitory effects against HSV-1 or VACV were observed. This relative specificity suggests restricted targeting, making the DHBTs attractive compounds for antiviral development.
Similarity in structure to well-characterized tricyclic antipsychotic compounds, which exert their effects through antagonism of serotonin and dopamine receptors, led us to investigate the involvement of these receptors in the inhibitory activity of SKI-417616. siRNA-mediated knockdown of dopamine receptor D4, but not of the 5HTR2A serotonin receptor, resulted in reduced DENV replication. Furthermore, addition of exogenous dopamine was able to rescue DENV replication in the presence of SKI-417616. Notably, SKI-417616 inhibits the phosphorylation of ERK in response to dopamine but does not affect the repression of cAMP signaling, a second arm of DRD4 downstream signaling. The expression and activity of dopamine receptors are typically considered to be specific to neurons; however, several recent reports have demonstrated expression of dopamine receptors on rodent and human macrophages (32, 33), a primary target cell of DENV infection. Although the exact mechanism has not yet been elucidated, dopamine receptors expressed on macrophages appear to play an important role in regulating cytokine production and modulating immune function (32). Whether this function of macrophage-expressed dopamine receptors is related to the effect of DRD4 inhibition by DHBTs on DENV infection is unclear and warrants further investigation. Nevertheless, our findings indicate a requirement for ERK phosphorylation for the completion of early events in the DENV life cycle. ERK signaling has been demonstrated to occur early during flavivirus infection (34–37), and this signaling pathway has been implicated in the life cycles of a range of DNA and RNA viruses (reviewed in references 38 and 39). The role of the ERK cascade during viral infection appears to differ depending on the virus studied and includes effects on viral ribonucleoprotein trafficking (influenza viruses), viral protein synthesis and the production of infectious progeny (coxsackievirus B3), the production of cytokines, and pathogenesis. The downstream consequences of this signaling event during DENV infection are the subject of current investigation. Furthermore, because inhibition of ERK signaling alone does not recapitulate the full inhibitory effect of SKI-417616, we are currently investigating other signaling events that are antagonized by treatment with the DHBTs and contribute to their inhibition of DENV2 infection. Agonists and antagonists of dopamine receptors have multiple clinical applications (40, 41). Antagonists (largely to the D2 dopamine receptors) have been used to treat schizophrenia, bipolar disorder, and depression, while antagonists of DRD4 are promising candidates for the treatment of drug addiction. Use in the clinic suggests the intriguing possibility of repurposing such compounds for use as anti-DENV therapeutics, provided their pharmacological properties allow in vivo distribution to areas of DENV replication at sufficiently high concentrations to be effective.
The role of host signaling pathways in supporting viral internalization and replication has been demonstrated extensively. The most commonly described role of host signaling networks in virus internalization involves reorganization of the actin cytoskeleton. These rearrangements, which occur downstream of both phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) activation, can support virus internalization through several mechanisms, including breakdown of the cortical actin skeleton to allow the intracellular passage of endocytic vesicles (42–46) and promotion of the transport of extracellular virions to areas of high endocytic activity (47, 48). Signaling can contribute to the formation of clathrin-coated vesicles under extracellular bound virions (49, 50), as well as to the transport of virions to lipid rafts, cholesterol-rich regions of the plasma membrane high in signaling molecules, where caveola-mediated internalization is initiated (44, 51, 52). Endocytosis itself, whether through clathrin-coated vesicles or via caveolae, often requires signaling through protein kinase C or tyrosine kinases (44, 53). Additionally, the entry of a range of viruses through macropinocytosis, which is highly dependent on signaling as well, has been demonstrated recently (54–58). Once the virus is internalized, signaling events can also contribute to directing the trafficking of virions to appropriate intracellular locales to support the progression of the viral life cycle (reviewed in reference 59). Thus, cellular signaling during viral entry represents a critical host process supporting virus infection and an attractive target for the development of therapeutic interventions.
In summary, we have identified a class of compounds with potent inhibitory activity against a range of flaviviruses. Further investigation of these compounds using structure-activity relationship analysis and exploration of their activity in vivo represent a promising strategy for the development of antivirals against the important human pathogen DENV.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIAID Pacific Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (grant U54 AI 081680). J.L.S. and M.A.F. were supported, respectively, by OHSU training grants T32 AI07472 and T32 A1074494. The confocal microscopy studies described here were carried out in the ONPRC Imaging and Microscopy Core and were supported by grant S10RR024585 from the National Center for Research Resources.
The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Published ahead of print 5 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00365-14.
REFERENCES
- 1.Gubler DJ. 2012. The economic burden of dengue. Am. J. Trop. Med. Hyg. 86:743–744. 10.4269/ajtmh.2012.12-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martínez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. 2010. Dengue: a continuing global threat. Nat. Rev. Microbiol. 8:S7–S16. 10.1038/nrmicro2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Special Programme for Research & Training in Tropical Diseases (TDR). 2007. Report of the Scientific Working Group Meeting on Dengue, Geneva, 1–5 October 2006. World Health Organization, Geneva, Switzerland: http://www.who.int/tdr/publications/documents/swg_dengue_2.pdf?ua=1 [Google Scholar]
- 4.Kyle JL, Harris E. 2008. Global spread and persistence of dengue. Annu. Rev. Microbiol. 62:71–92. 10.1146/annurev.micro.62.081307.163005 [DOI] [PubMed] [Google Scholar]
- 5.Pang T, Cardosa MJ, Guzman MG. 2007. Of cascades and perfect storms: the immunopathogenesis of dengue haemorrhagic fever-dengue shock syndrome (DHF/DSS). Immunol. Cell Biol. 85:43–45. 10.1038/sj.icb.7100008 [DOI] [PubMed] [Google Scholar]
- 6.Gubler D, Kuno G, Markoff L. 2007. Flaviviruses, p 1153–1253 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, ]?> 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 7.Jessie K, Fong MY, Devi S, Lam SK, Wong KT. 2004. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 189:1411–1418. 10.1086/383043 [DOI] [PubMed] [Google Scholar]
- 8.Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Virelizier JL, Arenzana-Seisdedos F, Despres P. 2003. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 4:723–728. 10.1038/sj.embor.embor866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, Eller MA, Pattanapanyasat K, Sarasombath S, Birx DL, Steinman RM, Schlesinger S, Marovich MA. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197:823–829. 10.1084/jem.20021840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acosta EG, Castilla V, Damonte EB. 2008. Functional entry of dengue virus into Aedes albopictus mosquito cells is dependent on clathrin-mediated endocytosis. J. Gen. Virol. 89:474–484. 10.1099/vir.0.83357-0 [DOI] [PubMed] [Google Scholar]
- 11.van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. 2008. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 4:e1000244. 10.1371/journal.ppat.1000244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. 10.1016/j.chom.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischl W, Bartenschlager R. 2011. Exploitation of cellular pathways by dengue virus. Curr. Opin. Microbiol. 14:470–475. 10.1016/j.mib.2011.07.012 [DOI] [PubMed] [Google Scholar]
- 14.Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res. 85:450–462. 10.1016/j.antiviral.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 15.Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB. 2001. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg. Infect. Dis. 7:714–721. 10.3201/eid0704.017420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medigeshi GR, Hirsch AJ, Brien JD, Uhrlaub JL, Mason PW, Wiley C, Nikolich-Zugich J, Nelson JA. 2009. West Nile virus capsid degradation of claudin proteins disrupts epithelial barrier function. J. Virol. 83:6125–6134. 10.1128/JVI.02617-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shum D, Smith JL, Hirsch AJ, Bhinder B, Radu C, Stein DA, Nelson JA, Früh K, Djaballah H. 2010. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev. Technol. 8:553–570. 10.1089/adt.2010.0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mondotte JA, Lozach PY, Amara A, Gamarnik AV. 2007. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J. Virol. 81:7136–7148. 10.1128/JVI.00116-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurukumar KR, Priyadarshini D, Patil JA, Bhagat A, Singh A, Shah PS, Cecilia D. 2009. Development of real time PCR for detection and quantitation of dengue viruses. Virol. J. 6:10. 10.1186/1743-422X-6-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito M, Takasaki T, Yamada K-I, Nerome R, Tajima S, Kurane I. 2004. Development and evaluation of fluorogenic TaqMan reverse transcriptase PCR assays for detection of dengue virus types 1 to 4. J. Clin. Microbiol. 42:5935–5937. 10.1128/JCM.42.12.5935-5937.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitby K, Pierson TC, Geiss B, Lane K, Engle M, Zhou Y, Doms RW, Diamond MS. 2005. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 79:8698–8706. 10.1128/JVI.79.14.8698-8706.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang LH, Rothberg KG, Anderson RG. 1993. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 123:1107–1117. 10.1083/jcb.123.5.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koff WC, Elm JL, Jr, Halstead SB. 1982. Antiviral effects of ribavirin and 6-mercapto-9-tetrahydro-2-furylpurine against dengue viruses in vitro. Antiviral Res. 2:69–79. 10.1016/0166-3542(82)90027-4 [DOI] [PubMed] [Google Scholar]
- 24.Campiani G, Nacci V, Bechelli S, Ciani SM, Garofalo A, Fiorini I, Wikstrom H, de Boer P, Liao Y, Tepper PG, Cagnotto A, Mennini T. 1998. New antipsychotic agents with serotonin and dopamine antagonist properties based on a pyrrolo[2,1-b][1,3]benzothiazepine structure. J. Med. Chem. 41:3763–3772. 10.1021/jm9706832 [DOI] [PubMed] [Google Scholar]
- 25.Niemegeers C, Awouters F, Heylen S, Gelders Y. 1991. 5-HT receptor antagonists in schizophrenia: preclinical and clinical considerations, p 535–537 In Racagni G, Brunello N, Fukuda T. (ed), Biological psychiatry, vol 1 Elsevier Science Publishers, Amsterdam, Netherlands [Google Scholar]
- 26.Liljefors T, Bogeso KP. 1988. Conformational analysis and structural comparisons of (1R,3S)-(+)- and (1S,3R)-(−)-tefludazine, (S)-(+)- and (R)-(−)-octoclothepin, and (+)-dexclamol in relation to dopamine receptor antagonism and amine-uptake inhibition. J. Med. Chem. 31:306–312. 10.1021/jm00397a006 [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson LO, Middlemiss DN. 1992. Metitepine distinguishes two receptors mediating inhibition of [3H]-5-hydroxytryptamine release in guinea pig hippocampus. Naunyn Schmiedebergs Arch. Pharmacol. 345:696–699. 10.1007/BF00164585 [DOI] [PubMed] [Google Scholar]
- 28.Monachon MA, Burkard WP, Jalfre M, Haefely W. 1972. Blockade of central 5-hydroxytryptamine receptors by methiothepin. Naunyn Schmiedebergs Arch. Pharmacol. 274:192–197. 10.1007/BF00501854 [DOI] [PubMed] [Google Scholar]
- 29.Rowley M, Broughton HB, Collins I, Baker R, Emms F, Marwood R, Patel S, Ragan CI, Freedman SB, Leeson PD. 1996. 5-(4-Chlorophenyl)-4-methyl-3-(1-(2-phenylethyl)piperidin-4-yl)isoxazole: a potent, selective antagonist at human cloned dopamine D4 receptors. J. Med. Chem. 39:1943–1945. 10.1021/jm960072u [DOI] [PubMed] [Google Scholar]
- 30.Herndon JL, Ismaiel A, Ingher SP, Teitler M, Glennon RA. 1992. Ketanserin analogues: structure-affinity relationships for 5-HT2 and 5-HT1C serotonin receptor binding. J. Med. Chem. 35:4903–4910. 10.1021/jm00104a017 [DOI] [PubMed] [Google Scholar]
- 31.Huff RM. 1996. Signal transduction pathways modulated by the D2 subfamily of dopamine receptors. Cell. Signal. 8:453–459. 10.1016/S0898-6568(96)00074-5 [DOI] [PubMed] [Google Scholar]
- 32.Gaskill PJ, Carvallo L, Eugenin EA, Berman JW. 2012. Characterization and function of the human macrophage dopaminergic system: implications for CNS disease and drug abuse. J. Neuroinflammation 9:203. 10.1186/1742-2094-9-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Färber K, Pannasch U, Kettenmann H. 2005. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol. Cell. Neurosci. 29:128–138. 10.1016/j.mcn.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 34.Menzel N, Fischl W, Hueging K, Bankwitz D, Frentzen A, Haid S, Gentzsch J, Kaderali L, Bartenschlager R, Pietschmann T. 2012. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog. 8:e1002829. 10.1371/journal.ppat.1002829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scherbik SV, Brinton MA. 2010. Virus-induced Ca2+ influx extends survival of West Nile virus-infected cells. J. Virol. 84:8721–8731. 10.1128/JVI.00144-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katsarou K, Lavdas AA, Tsitoura P, Serti E, Markoulatos P, Mavromara P, Georgopoulou U. 2010. Endocytosis of hepatitis C virus non-enveloped capsid-like particles induces MAPK-ERK1/2 signaling events. Cell. Mol. Life Sci. 67:2491–2506. 10.1007/s00018-010-0351-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shyu HW, Lin YY, Chen LL, Wang YF, Yeh TM, Su SJ, Cheng WC, Chen CY, Lin KH, Chou MC. 2010. The dengue virus envelope protein induced PAI-1 gene expression via MEK/ERK pathways. Thromb. Haemost. 104:1219–1227. 10.1160/TH10-05-0302 [DOI] [PubMed] [Google Scholar]
- 38.Greber UF. 2002. Signalling in viral entry. Cell. Mol. Life Sci. 59:608–626. 10.1007/s00018-002-8453-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pleschka S. 2008. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 389:1273–1282. 10.1515/BC.2008.145 [DOI] [PubMed] [Google Scholar]
- 40.Beaulieu JM, Gainetdinov RR. 2011. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63:182–217. 10.1124/pr.110.002642 [DOI] [PubMed] [Google Scholar]
- 41.Di Ciano P, Grandy DK, Le Foll B. 2014. Dopamine D4 receptors in psychostimulant addiction. Adv. Pharmacol. 69:301–321. 10.1016/B978-0-12-420118-7.00008-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 22:4346–4355. 10.1093/emboj/cdg439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breau WC, Atwood WJ, Norkin LC. 1992. Class I major histocompatibility proteins are an essential component of the simian virus 40 receptor. J. Virol. 66:2037–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelkmans L, Puntener D, Helenius A. 2002. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296:535–539. 10.1126/science.1069784 [DOI] [PubMed] [Google Scholar]
- 45.Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. 2003. Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 163:283–293. 10.1083/jcb.200301084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollard TD, Borisy GG. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112:453–465. 10.1016/S0092-8674(03)00120-X [DOI] [PubMed] [Google Scholar]
- 47.Smith JL, Lidke DS, Ozbun MA. 2008. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Virology 381:16–21. 10.1016/j.virol.2008.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 170:317–325. 10.1083/jcb.200503059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rust MJ, Lakadamyali M, Zhang F, Zhuang X. 2004. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 11:567–573. 10.1038/nsmb769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591–605. 10.1016/j.cell.2004.08.017 [DOI] [PubMed] [Google Scholar]
- 51.Pietiäinen V, Marjomaki V, Upla P, Pelkmans L, Helenius A, Hyypia T. 2004. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol. Biol. Cell 15:4911–4925. 10.1091/mbc.E04-01-0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PW, Liao CL, Chang W. 2012. Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 86:6677–6687. 10.1128/JVI.06860-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Upla P, Marjomaki V, Kankaanpaa P, Ivaska J, Hyypia T, Van Der Goot FG, Heino J. 2004. Clustering induces a lateral redistribution of α2β1 integrin from membrane rafts to caveolae and subsequent protein kinase C-dependent internalization. Mol. Biol. Cell 15:625–636. 10.1091/mbc.E03-08-0588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakraborty S, Veettil MV, Bottero V, Chandran B. 2012. Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. U. S. A. 109:E1163–E1172. 10.1073/pnas.1119592109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M. 2002. Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J. Virol. 76:6689–6700. 10.1128/JVI.76.13.6689-6700.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meier O, Boucke K, Hammer SV, Keller S, Stidwill RP, Hemmi S, Greber UF. 2002. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J. Cell Biol. 158:1119–1131. 10.1083/jcb.200112067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sánchez EG, Quintas A, Perez-Nunez D, Nogal M, Barroso S, Carrascosa AL, Revilla Y. 2012. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 8:e1002754. 10.1371/journal.ppat.1002754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 6:e1001110. 10.1371/journal.ppat.1001110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pálfy M, Remenyi A, Korcsmaros T. 2012. Endosomal crosstalk: meeting points for signaling pathways. Trends Cell Biol. 22:447–456. 10.1016/j.tcb.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.