

Abstract
Previous in vitro work demonstrated that Edwardsiella ictaluri produces an acid-activated urease that can modulate environmental pH through the production of ammonia from urea. Additional work revealed that expression of the E. ictaluri type III secretion system (T3SS) is upregulated by acidic pH. Both the urease and the T3SS were previously shown to be essential to intracellular replication. In this work, fluorescence microscopy with LysoTracker Red DND-99 (LTR) indicated that E. ictaluri-containing vacuoles (ECV) became acidified following ingestion by head kidney-derived macrophages (HKDM). In vivo ratiometric imaging demonstrated a lowered ECV pH, which fell to as low as pH 4 but subsequently increased to pH 6 or greater. Inhibition of vacuolar H+-ATPases by use of the specific inhibitor bafilomycin A1 abrogated both ECV acidification and intracellular replication in HKDM. Failure of an E. ictaluri urease knockout mutant to increase the ECV pH in the in vivo ratiometric assay suggests that ammonia produced by the urease reaction mediates the pH increase. Additionally, when the specific arginase inhibitor l-norvaline was used to treat E. ictaluri-infected HKDM, the ECV failed to neutralize and E. ictaluri was unable to replicate. This indicates that the HKDM-encoded arginase enzyme produces the urea used by the E. ictaluri urease enzyme. Failure of the ECV to acidify would prevent both upregulation of the T3SS and activation of the urease enzyme, either of which would prevent E. ictaluri from replicating in HKDM. Failure of the ECV to neutralize would result in a vacuolar pH too low to support E. ictaluri replication.
INTRODUCTION
Edwardsiella ictaluri, a Gram-negative rod-shaped bacterium, is the etiological agent of enteric septicemia of catfish (ESC) (1). This disease is a leading cause of mortality in channel catfish (Ictalurus punctatus) production facilities, with a substantial negative economic impact throughout the industry (2). Enteric septicemia of catfish presents most commonly as an acute septicemia with a propensity for infection of the head kidney, but also the liver, spleen, trunk kidney, and intestine (3). Infection of the head kidney is of particular relevance because E. ictaluri can enter, survive, and replicate in primary cell cultures of head kidney-derived macrophages (HKDM) (4), making it a facultative pathogen of professional phagocytes.
Using signature-tagged mutagenesis (STM), Thune et al. (5) identified several E. ictaluri virulence-related genes in an immersion challenge model. It was interesting that a urease operon was discovered, even though E. ictaluri is urease negative in standard biochemical tests. Subsequent characterization of the E. ictaluri urease showed that invasion and uptake of wild-type (WT) and urease mutant strains of E. ictaluri were not significantly different following immersion challenge of catfish. After initial invasion, however, the mutant was unable to increase in numbers (6). A gentamicin survival assay with HKDM also indicated that initial uptake was not significantly different but that the urease mutant was unable to replicate within HKDM. Further in vitro studies showed acid activation of the E. ictaluri urease, with activity increasing as the pH declined. Experiments in minimal media indicated that E. ictaluri survives very well at low pH but replicates poorly at pHs below 6. Booth et al. (6) also showed that the WT strain can increase the environmental pH from as low as 2.5 to over 6.0, and from pH 5 to over 7, by converting urea to ammonia in minimal media containing urea. Additional studies, however, demonstrated that urease activity is not required for acid resistance. Taken together, these data suggest that the urease pathogenicity island is involved in pH modulation of the E. ictaluri-containing vacuole (ECV) and that the ECV pH is either maintained near neutral or increased to a level that is permissive for replication following initial acidification by H+-ATPases in the vacuolar membrane. Although a source of urea for this activity is not readily apparent, the identification of arginine decarboxylase (adiA) as a potential virulence factor in the STM study led to speculation that AdiA could be involved in de novo synthesis of urea, which could be metabolized to ammonia by the urease enzyme (5). This was subsequently disproved using gene-specific mutants (unpublished results from our lab). Another possible source of urea is the macrophage-encoded arginase enzyme, a manganese metalloenzyme that hydrolyzes arginine to ornithine and urea in the host cell cytoplasm, regulating inducible nitric oxide synthase (iNOS) production of nitric oxide (NO) through modulation of the arginine supply (7, 8). Consequently, the primary objective of this study was to evaluate changes in the pH of the ECV during E. ictaluri infection of HKDM, using a urease knockout mutant; l-norvaline, a specific inhibitor of arginase; and bafilomycin A1, a specific inhibitor of vacuolar H+-ATPases.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Wild-type E. ictaluri 93-146 was isolated from moribund channel catfish in a natural outbreak of ESC on a commercial farm. The urease mutant strain (ΔureG::Km) carries a mini-Tn5Km insertion in the ureG gene of E. ictaluri 93-146, abrogating urease activity (6). Strains were grown in porcine brain heart infusion broth at 28°C. A defined minimal medium for E. ictaluri was also used (9), with minor modifications as indicated below. Antibiotics were used at the following concentrations, as needed: kanamycin (Km), 50 μg/ml; colistin (Col), 10 μg/ml; and ampicillin (Amp), 200 μg/ml. When necessary, numbers of E. ictaluri CFU were determined by making triplicate 10-fold serial dilutions in sterile phosphate-buffered saline and drop plating 20-μl aliquots on Trypticase soy agar plates supplemented with 5% defibrinated sheep blood (BA) for colony counting.
SPF channel catfish.
Channel catfish egg masses were obtained from a commercial producer with no history of ESC outbreaks. The eggs were disinfected with 100 ppm free iodine and hatched in closed recirculating systems in the specific-pathogen-free (SPF) laboratory at the Louisiana State University School of Veterinary Medicine. All experimental fish were maintained continuously in the SPF laboratory, fed commercial catfish feed, and reared to a weight of 1 to 1.5 kg. Holding systems consisted of four 350-liter round fiberglass tanks connected to a 45-liter biological bead filter (Aquaculture Systems Technologies, New Orleans, LA). Water temperature was maintained at 23 ± 2°C. Water quality parameters, including total ammonia nitrogen, total nitrate, pH, hardness, and alkalinity, were determined 3 times per week using a Hach water quality kit (Hach Company, Loveland, CO). Water quality was adjusted to maintain optimal conditions. All animal experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee and the Inter-Institutional Biological and Recombinant DNA Safety Committee of Louisiana State University.
Gentamicin survival assay.
A standard gentamicin survival assay using HKDM (4) was used to evaluate the ability of WT and mutant E. ictaluri to enter, survive, and replicate in HKDM under various conditions. Briefly, 1 to 1.5 kg channel catfish were euthanized with 200 mg/liter tricaine methanesulfonate and bled via phlebotomy to exsanguination. The head kidney, which is equivalent to mammalian bone marrow, was removed using sterile technique and macerated in a cell dissociation sieve (stainless steel mesh; 280 and 140 μm) (Sigma-Aldrich, St. Louis, MO) to collect a single-cell suspension. Cell viability was determined by trypan blue exclusion. Dissociated cells were suspended to a final concentration of 1 × 107 live cells/ml in channel catfish macrophage medium (CCMM), consisting of RPMI 1640 medium without phenol red or l-glutamine (Lonza, Walkersville, MD) but containing 1× glutamine substitute (GlutaMAX-I CTS; Gibco, Invitrogen Corporation, Carlsbad, CA), 15 mM HEPES buffer, 0.18% sodium bicarbonate (Gibco), 0.05 mM 2-mercaptoethanol (Sigma Chemical Co., St. Louis, MO), and 5% pooled channel catfish serum that was heat inactivated at 56°C. All media were diluted to the catfish tonicity of 240 mosmol/kg H2O by adding sterile deionized-distilled water at a 1:9 ratio. One milliliter of the cell suspension was added to each well of a 24-well Biocoat poly-d-lysine tissue culture plate (Becton, Dickinson Labware, Bedford, MA), allowed to adhere for 16 h at 28°C with 5% CO2, and returned to fresh CCMM for infection after three washes with catfish RPMI. Attached cells were infected at a multiplicity of infection (MOI) of 1 bacterium to 10 HKDM, using bacteria previously opsonized for 30 min in autologous serum. To synchronize infection, plates were centrifuged at 400 × g for 5 min and then incubated for 30 min, followed by the addition of 100 μg/ml gentamicin for 1 h to kill extracellular bacteria. Cells were washed once in catfish RPMI and placed in CCMM with a static dose of 0.5 μg/ml gentamicin. After 0, 4, 8, or 10 h of incubation at 28°C with 5% CO2, HKDM were washed with catfish RPMI and lysed by adding 100 μl of 1% Triton X-100 (Fisher Scientific, Fair Lawn, NJ). The number of CFU/ml was determined by dilution and drop plating, and the fold increase was calculated by dividing the number of bacteria present at 4, 8, or 10 h by the number present at 0 h.
Acidification of the ECV as evaluated using LTR.
Prior to infection, bacteria were stained with Oregon Green 514 carboxylic acid succinimidyl ester (OG) (Invitrogen, Carlsbad, CA). Infected HKDM were stained with 30 nM LysoTracker Red DND-99 (LTR) (Invitrogen). Cells were imaged by fluorescence microscopy at 510 and 577 nm for OG and LTR (Invitrogen), respectively, and by differential interference contrast (DIC) microscopy. Three groups of 100 bacteria each were counted from each treatment, and the average percentage of those that colocalized with the acidified LTR-stained vacuoles was determined for each period. Prior to the experiment, the gentamicin survival assay was used to demonstrate that OG staining of the bacteria did not affect intracellular replication of E. ictaluri in HKDM. For fluorescence microscopy, HKDM were prepared as described above and seeded on BioCoat poly-d-lysine-coated glass coverslips (Becton Dickinson) in 24-well tissue culture plates. Cells were incubated overnight at 28°C with 5% CO2, washed 3 times with RPMI medium, and used in the gentamicin exclusion assay as described previously, except that the MOI was 1 bacterium to 1 HKDM. At 30, 90, and 150 min postinfection (mpi), 30 nM LTR was added, and counts of acidified and neutral vacuoles were made 60 min later, at 90, 150, and 210 mpi. Additional experiments evaluated ECV acidification when LTR was maintained on the cells for the full 210 min of the assay.
In vivo ratiometric determination of ECV pH.
In order to further validate the LTR results, live-cell ratiometric imaging was used to directly measure intravacuolar pH. The differential pH sensitivity of OG fluorescence in the acidic range allows ratiometric estimation of the pH in cellular vacuoles. Staining of bacteria by OG was performed according to a previously described protocol (10). Dead wild-type bacteria (WTD) were produced by incubating stained bacteria at 70°C for 30 min, and killing was confirmed by plating on blood agar. When excited at 510 nm, OG fluorescence emission at 535 nm decreases as the pH decreases, while excitation at 450 nm does not vary as the pH changes. Excitation of OG-stained bacteria at both 510 nm and 450 nm in rapid succession allows calculation of the ratio of the measured fluorescence at 510 nm to that at 450 nm. The ratios calculated at different pH levels can then be used for comparative purposes to estimate the pH of infected vacuoles.
To conduct the ratiometric experiments, HKDM were harvested, cultured, and infected as described above for the gentamicin assay, except that 1 × 107 cells were plated onto 35-mm poly-d-lysine-coated no. 1.5 borosilicate German glass-bottom dishes (MatTek, Ashland, MA) and the cells were infected at an MOI of 10 bacteria to 1 HKDM with either live or killed OG-stained WT bacteria. For calibration, infected HKDM were incubated in ionophore calibration solutions containing 140 mM KCl, 1 mM MgCl2, 0.2 mM EGTA, and 20 mM morpholineethanesulfonic acid (MES), adjusted to pH 4, 5, and 6 with NaOH, and also containing 5 μg/ml nigericin and 5 μM monensin to collapse pH gradients across membranes (10). For each experiment, an in situ calibration curve was made, and each experiment was repeated three times on different days with different fish and bacterial cultures. Using the same dish for all pH calibrations, HKDM were sequentially bathed for 8 min in each solution and were visualized with a Zeiss Observer Z1 microscope with CO2 module S and TempModule S (Carl Zeiss MicroImaging GmbH, Jena, Germany), using a 63× oil objective and a stage insert (heating insert P; PeCon GmbH, Erbach, Germany), supplied with humidified CO2 at 5%, and maintained at a temperature of 28°C. Illumination was provided by a Lambda DG-4 175-W xenon arc lamp (Sutter Instrument Co., Novato, CA). Filter sets used for OG included a Chroma 71001 dual exciter (green; 440/20 and 495/10) with a single 535/25 emitter (Chroma Technology Corporation, Bellows Falls, VT). Digital images were captured using Zeiss AxioVision software, version 4.8.1, and the ratiometric data were analyzed with the AxioVision physiology acquisition module (Carl Zeiss, Germany). Edwardsiella ictaluri-infected HKDM used to measure experimental data were exposed to a 0.5-μg/ml static dose of gentamicin after the killing dose of gentamicin.
Effect of bafilomycin A1 on ECV acidification and E. ictaluri replication within HKDM.
Bafilomycin A1 is a known inhibitor of vacuolar H+-ATPases, but to confirm its activity in channel catfish HKDM, heat-killed E. ictaluri were used in a phagocytosis assay in 24-well plates. The HKDM were pretreated with 100 nM bafilomycin A1 for 15 min, after which killed bacteria were added at an MOI of 1:1. Following 90 min, bafilomycin A1 was removed from half of the wells. One hour later, LTR was added to all wells and incubated for another 60 min. Cells in all wells were fixed in 4% paraformaldehyde for 30 min and imaged with DIC and fluorescence microscopy at 577 nm.
To determine the potential effect of bafilomycin A1 on the intracellular replication of E. ictaluri, gentamicin exclusion assays were conducted as described previously, after first determining that bafilomycin A1 had no effect on the growth of E. ictaluri in CCMM without HKDM. Treatments included exposure to 100 nM bafilomycin A1 from 15 min prior to infection until the gentamicin killing dose was removed and exposure to 100 nM bafilomycin A1 from 15 min prior to infection until the macrophages were lysed and plated. Infected HKDM that were not treated with bafilomycin A1 served as controls. To determine if the effect of bafilomycin A1 actually occurred later in the infection, assays were also conducted where bafilomycin A1 was added at 110 and 210 min postinfection and remained throughout the assay.
Neutralization of the ECV.
To establish the role of the E. ictaluri acid-activated urease in the neutralization of the ECV, the urease knockout strain was compared to the WT in the live-cell ratiometric assay described above. To establish HKDM arginase activity as the source of urea for the urease enzyme, the ratiometric imaging assay was repeated using HKDM incubated overnight in 10 mM l-norvaline (Sigma, St. Louis, MO), a specific arginase inhibitor. Cells were processed as described above, except that 10 mM l-norvaline was maintained during both the pH calibration and experimental measurements. Finally, to evaluate E. ictaluri replication in HKDM when the ECV does not neutralize, gentamicin survival assays were conducted in the presence of 10 mM l-norvaline throughout the assay, until cells were lysed for bacterial enumeration at 0, 4, and 10 h postinfection (hpi). Infected HKDM without l-norvaline served as controls.
Arginase activity.
To evaluate the effect of infection of HKDM with both the WT and urease mutant strains on arginase activity, nitric oxide and urea concentrations were measured using a nitric oxide assay kit (Abcam) and a urea assay kit (Abcam) according to the manufacturer's instructions.
Statistical analysis.
Data for gentamicin survival assays in HKDM were determined by analysis of variance (ANOVA) using the general linear methods procedure (Proc GLM) (SAS 9.3; SAS Institute Inc.). The fold increase was determined by dividing the CFU/well from time points past time zero by the mean CFU/well at time zero for each strain. Mean fold replication was then calculated, along with the standard error of the mean. When the model indicated significance (P ≤ 0.05), a least-square mean procedure was used for pairwise comparison of interaction effects. For the ECV acidification assays using LTR, mean percent acidification was analyzed by ANOVA following an arcsine transformation of the percentage data.
RESULTS
Replication of OG-stained E. ictaluri in channel catfish macrophages.
Survival and replication of the OG-stained WT compared to the unstained WT, as determined by the gentamicin exclusion assay in channel catfish HKDM, are presented in Fig. 1. There was no significant difference in fold increase between the WT strain and the OG-stained WT at 4, 8, or 10 hpi.
FIG 1.

Intracellular survival and replication of wild-type E. ictaluri (WT) and the Oregon Green 514-stained WT (OG-WT) in channel catfish HKDM. Both WT and OG-WT levels increased approximately 20-fold by 10 hpi. Results for the WT and OG-WT were not significantly different from one another at any time point (P ≤ 0.05), indicating that OG staining of E. ictaluri does not affect replication in HKDM. Results are presented as means and standard errors of the means for three wells per treatment and are representative of three independent experiments.
Acidification of the ECV as evaluated using LTR.
LTR studies demonstrated colocalization of E. ictaluri within red-stained acidified vacuoles (Fig. 2), although not all E. ictaluri organisms were in acidified vacuoles at any given time (Fig. 3). When cells were exposed to LTR from 30 to 90 mpi, 20.8% of the bacteria were in acidified vacuoles. At 90 to 150 mpi, the number increased to 52.5%, and then declined to 13.0% when bacteria were exposed from 150 to 210 mpi, suggesting that a total of 86.3% of the vacuoles were acidified. This was confirmed by exposing the cells to LTR for a full 210 min, after which 86.0% of the vacuoles were acidified, indicating that acidification occurs primarily from 30 to 210 mpi. Because the green-to-red shift of LTR is not reversible if the pH neutralizes, LTR was added at three different times to demonstrate the reduction in acidified vacuoles over time. When the urease mutant was used, all of the vacuoles were acidified, even when LTR was added at 150 to 210 min postinfection.
FIG 2.

Fluorescence microscopy with HKDM stained with LysoTracker Red (LTR) and E. ictaluri stained with Oregon Green 514 (OG). (A) DIC image with a box visualizing the area depicted in frames B, C, and D. (B) E. ictaluri stained with OG. (C) E. ictaluri vacuoles stained red with LTR, indicating acidification. (D) Colocalization of OG-stained E. ictaluri and acidified vacuoles. Panel A also shows the location of E. ictaluri within the HKDM.
FIG 3.

Percentages of bacteria contained within acidified vacuoles in HKDM stained with LTR from 30 to 90, 90 to 150, and 150 to 210 mpi. Bars represent mean numbers of E. ictaluri bacteria colocalized in acidic vacuoles for three independent experiments plus standard errors. Asterisks indicate significant differences (P ≤ 0.05).
In vivo ratiometric determination of the ECV pH.
At 150 mpi, the majority of ECV containing the WT had a pH greater than pH 6 (Fig. 4). The relatively wide spread of data in the experimental group, in most cases well above pH 6, but in some cases as low as pH 4, is consistent with the wide time spread seen for initial acidification to occur in the LTR studies. At the same time point, the majority of WTD were digested, and the majority of vacuoles that contained remnants of WTD had a pH of about 4, which is in agreement with other studies concerning typical phagosome acidity (11).
FIG 4.
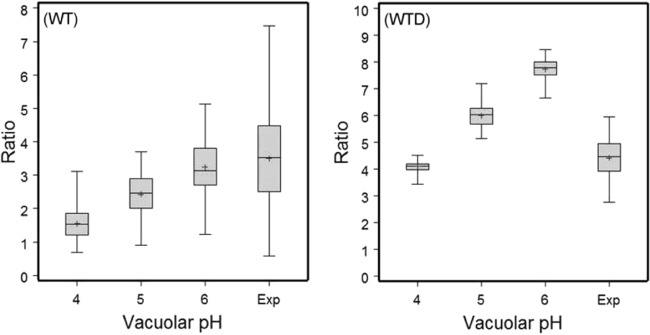
In vivo determination of the pH of the ECV in E. ictaluri-infected channel catfish HKDM at 150 mpi, using WT and heat-killed WT (WTD) bacteria. Plots show data ranges, interquartile ranges (middle 50% of data points; gray boxes), and means (+). Vacuolar pHs 4, 5, and 6 represent the vacuolar pHs established using ionophore calibration solutions to generate fluorescence ratio values. Exp, measurement of fluorescence ratios of the ECV when macrophages were bathed in neutral saline. Ratios represent the relative fluorescence of OG at 510 and 450 nm. Results from three independent experiments were combined.
Effect of bafilomycin A1 on ECV acidification and E. ictaluri replication within HKDM.
Using WTD in the LTR assay indicated that bafilomycin is an inhibitor of vacuolar ATPases in channel catfish, effectively preventing acidification of phagocytic vacuoles if maintained throughout the experiment (Fig. 5). Removal of bafilomycin A1 60 min prior to LTR exposure, however, resulted in acidification of more than 70% of the vacuoles, indicating that the effect of bafilomycin A1 is reversible if the drug is removed. When infected HKDM were exposed to bafilomycin A1 at 110 or 210 mpi for the remainder of the experiment, there was no significant effect on replication (Fig. 6), further suggesting that acidification is an early event in pathogenesis, not an effect of long-term exposure to bafilomycin A1 on the vacuolar H+-ATPases or some other physiological process later in the infection.
FIG 5.

Acidification of HKDM vacuoles containing heat-killed E. ictaluri and exposed to LTR from 150 to 210 min postinfection. The vacuolar H+-ATPase inhibitor bafilomycin A1 was either removed at 90 min postinfection or maintained throughout the 210 min of the experiment. The results show that bafilomycin A1 prevents vacuolar acidification and that the effect is reversible if bafilomycin is removed. Asterisks indicate a significant difference from cells in which the LTR was removed after 90 min (P ≤ 0.01).
FIG 6.

Fold increase of E. ictaluri in HKDM treated or not treated with the vacuolar H+-ATPase inhibitor bafilomycin A1. Treated HKDM were exposed to 100 nM bafilomycin A1 for the full duration of infection (Continual), or bafilomycin A1 was added at 110 mpi or 210 mpi for the remainder of the assay. Data represent mean fold increases at 10 h postinfection for 8 replicate wells plus standard errors. Asterisks indicate a significant difference from untreated cells (P < 0.01).
Short-term treatment of HKDM with 100 nM bafilomycin A1 did not reduce intracellular replication of E. ictaluri, but treatment of HKDM with bafilomycin A1 throughout the experiment significantly reduced intracellular replication (Fig. 7). Replication of E. ictaluri in the short-term-treated cells after removal of bafilomycin A1 demonstrates that bafilomycin A1 does not affect bacterial uptake or survival in HKDM.
FIG 7.

Replication of E. ictaluri in HKDM treated with the vacuolar H+-ATPase inhibitor bafilomycin A1. Head kidney-derived macrophages were exposed to 100 nM bafilomycin A1 until the gentamicin killing dose was removed or were exposed for the full duration of infection. The results indicate that acidification is required for intracellular replication of E. ictaluri. Bars represent mean fold increases at 10 h postinfection for three independent experiments plus standard errors. Asterisks indicate a significant difference from untreated cells (P ≤ 0.01).
Neutralization of the ECV.
WT E. ictaluri is able to neutralize the ECV following acidification, but the ECV of the urease knockout mutant remained acidified (Fig. 8), demonstrating that neutralization of the WT ECV is mediated by the bacterial urease. When HKDM arginase activity was specifically inhibited using l-norvaline, the ECV also remained acidified, indicating that arginase activity is the source of the urea for the urease reaction (Fig. 9). As predicted by the failure of the WT to neutralize the ECV when arginase activity was inhibited by l-norvaline, the WT was unable to replicate in norvaline-treated HKDM (Fig. 10), further confirming that ECV neutralization is required for replication.
FIG 8.

In vivo determination of vacuolar pH, comparing a urease mutant (ΔureG) to wild-type E. ictaluri at 150 to 180 mpi. Plots show data ranges, interquartile ranges (middle 50% of data points; gray boxes), and means (+). Vacuolar pHs 4, 5, and 6 represent vacuolar pHs obtained using ionophore calibration solutions to generate fluorescence ratio values. Exp, measurement of fluorescence ratios of the ECV when macrophages were bathed in neutral saline. Ratios represent the relative fluorescence of OG at 510 and 450 nm. Results were combined from two independent experiments and indicate that the E. ictaluri urease enzyme is required for ECV neutralization.
FIG 9.

In vivo determination of the pH of the ECV in channel catfish HKDM with and without 10 mM of the specific arginase inhibitor l-norvaline for 60 mpi. Plots showing data range (whiskers), interquartile range (the mid-50% of data points; box), and the mean (designated +). Vacuolar pH at 4, 5, and 6 represents vacuolar pH using ionophore calibration solutions to generate fluorescent ratio values. “Exp” represents the measurement of fluorescent ratios of the ECV when macrophages were bathed in neutral saline. “Ratio” represents the relative fluorescence of OG at 510 and 450 nm. Results are combined from 2 independent experiments and indicate that the HKDM arginase enzyme is required for ECV neutralization.
FIG 10.

Intracellular survival and replication of E. ictaluri in HKDM and l-norvaline-treated HKDM. The results show that the WT increased over 15-fold after 10 h, while norvaline treatment to block arginase activity and prevent ECV neutralization also prevented WT replication. l-Norvaline treatment did not affect WT survival. Results are presented as means plus standard errors for three wells per treatment per time point and are representative of three independent experiments. Asterisks indicate a significant difference from untreated cells (P ≤ 0.001).
Arginase activity.
Nitric oxide was not detected following infection with either the WT or the urease mutant. Urea concentrations, however, increased significantly for both strains after 30 mpi, declining to noninfected cell levels at 210 mpi for the urease mutant but remaining significantly elevated even at 210 mpi for the WT (Fig. 11).
FIG 11.

Urea concentrations in HKDM at 30 mpi and 3 h 30 mpi in uninfected HKDM and HKDM infected with the WT or urease knockout strain of E. ictaluri. Asterisks indicate a significant difference from uninfected cells: **, P ≤ 0.01; and ***, P ≤ 0.001.
DISCUSSION
When a bacterium enters a host, it is usually engulfed by phagocytic cells, including macrophages and dendritic cells, which internalize it into a vacuole known as a phagosome. Newly formed phagosomes are immature organelles that are unable to kill and degrade microorganisms. In macrophages and dendritic cells, phagosomes mature over time by fusing with endosomes and lysosomes, which deliver various hydrolases and proteases to the lumen of the phagosome (12, 13). As a central event in phagosome maturation and hydrolase/protease activation, the phagosomal lumen is acidified to pH 4 to 5 by membrane-embedded ATP complexes, the vacuolar H+-ATPases. The result is a harsh, lytic environment that kills and degrades most microbes.
Successful intracellular bacterial pathogens have evolved strategies to combat phagosomal maturation, including avoidance, prevention, and resistance. Listeria monocytogenes, Shigella flexneri, Francisella tularensis, and Rickettsia spp. disrupt the phagosomal membrane and escape to the cytoplasm (14–17). Brucella abortus and Legionella pneumophila evade macrophage killing by escaping from the endocytic pathway and developing a replicative vacuole derived from the endoplasmic reticulum (18, 19). Chlamydia spp. bypass the endocytic pathway and enter novel sphingomyelin-containing vacuoles derived from the Golgi apparatus (20, 21). Coxiella burnetii resists the vacuolar environment and is able to survive and replicate within a large, acidified phagolysosome-like vacuole (22, 23). Salmonella-containing vacuoles (SCV) acidify soon after formation and then progress through a modified maturation process that promotes Salmonella survival and replication in the acidified vacuole, primarily through the action of the Salmonella pathogenicity island 2 (SPI2)-encoded type III secretion system (T3SS), which is essential for virulence and intracellular replication (24, 25).
Like the SCV, the ECV initially acidifies, in a process required for intracellular replication. For Salmonella grown in bone marrow-derived macrophages (BMDM), which are homologous to HKDM, acidification of vacuoles to pH 4 occurs within 75 to 80 mpi (24), while ECV acidification is not as synchronous and occurs from 30 to 210 mpi. As in the development of the SCV, failure of E. ictaluri to replicate in the presence of bafilomycin A1 indicates that acidification is dependent on the action of the vacuolar H+-ATPases in the ECV membrane. Unlike the SCV, which remains acidified, the ECV subsequently requires neutralization for intracellular replication. In contrast, artificial neutralization of the SCV at 2 hpi results in a loss of viability (24).
Although structurally similar to the acid-activated ureases produced by Yersinia enterocolitica, Morganella morganii, and Klebsiella pneumoniae, which mediate acid tolerance, Booth et al. (6) demonstrated that the E. ictaluri-encoded urease is acid activated but is not required for acid tolerance. Failure of the ECV to neutralize when a urease knockout mutant is used indicates that the ammonia produced by the urease reaction acts as a proton sink for ECV neutralization. Treatment of E. ictaluri-infected HKDM with l-norvaline prevented neutralization of the ECV in the ratiometric, live-cell imaging assay (Fig. 9) and also prevented intracellular replication (Fig. 10). These results indicate that suppression of HKDM arginase activity by l-norvaline and the subsequent loss of urea production resulted in limited production of ammonia by the E. ictaluri urease enzyme and in failure of the ECV to neutralize. Based on previous in vitro data showing that E. ictaluri cannot replicate at pHs lower than pH 6 (6), replication was presumably limited by the low-pH environment of the ECV. Interestingly, the urea concentration remained elevated at 210 mpi, after neutralization of the ECV, suggesting that the vacuolar H+-ATPases remain active and that continued ammonia production by the urease enzyme is required. This further suggests that once activated, the E. ictaluri urease remains active, even if the environment is neutralized. In addition, the increase in urea concentration in HKDM infected with the urease mutant suggests that upregulation is not a function of urease activation.
Edwardsiella ictaluri is the only other known bacterial pathogen to encode a SPI2-like T3SS that is essential for virulence and intracellular replication, presumably by the action of effectors translocated through the ECV membrane into the host cell cytoplasm. Rogge and Thune (26) identified low pH and phosphate limitation as conditions conducive to expression of the E. ictaluri T3SS. Inhibition of the vacuolar H+-ATPases by bafilomycin A1 resulted in failure of the ECV to acidify, which would prevent activation of the urease enzyme and expression of the T3SS, either of which would preclude replication. Additionally, treatment with norvaline to prevent neutralization of the ECV precludes E. ictaluri replication due to the lack of urea production by the HKDM arginase. Failure of E. ictaluri to replicate if the ECV does not neutralize could also be an effect on translocation of the T3SS effectors. Assembly of the Salmonella SPI2 T3SS requires vacuole acidification, but translocation requires recognition of the neutral pH of the host cytosol by a complex of SPI2-encoded proteins: SsaM, SpiC, and SsaL (27). The E. ictaluri SPI2-like T3SS does not encode this complex, so recognition of the cytosolic pH probably does not trigger translocation. It is possible, however, that recognition of the neutralized environment of the ECV does.
In summary, E. ictaluri has evolved a unique approach for survival and replication in HKDM by modulating the pH environment of the ECV. Acidification occurs as normal, which is required for expression of the T3SS, but is not conducive to E. ictaluri replication. Concomitant activation of the acid-activated urease enzyme, using urea produced by the HKDM arginase enzyme, results in ammonia production and an increase in the pH of the ECV to a level permissive for E. ictaluri replication. The specific role of the T3SS in promoting further development of the ECV and E. ictaluri replication remains to be determined.
Footnotes
Published ahead of print 24 March 2014
REFERENCES
- 1. Hawke JP. 1979. A bacterium associated with disease of pond cultured catfish, Ictalurus punctatus. J. Fish. Res. Board Can. 36:1508–1512. 10.1139/f79-219 [DOI] [Google Scholar]
- 2.USDA. 2010. Catfish 2010 part I: reference of catfish health and production practices in the United States, 2009. USDA-APHIS-VS, CEAH, Ft. Collins, CO [Google Scholar]
- 3. Thune RL, Stanley LA, Cooper RK. 1993. Bacterial diseases of catfishes, p 511–520 In Stoskopf MK. (ed), Fish medicine. W B Saunders Company, Philadelphia, PA [Google Scholar]
- 4. Booth NJ, El Kamel A, Thune RL. 2006. Intracellular replication of Edwardsiella ictaluri in channel catfish macrophages. J. Aquat. Anim. Health 18:101–108. 10.1577/H05-025.1 [DOI] [Google Scholar]
- 5. Thune RL, Fernandez DH, Benoit JL, Kelly-Smith M, Rogge ML, Booth NJ, Landry CA, Bologna RA. 2007. Signature-tagged mutagenesis of Edwardsiella ictaluri identifies virulence-related genes, including a salmonella pathogenicity island 2 class of type III secretion systems. Appl. Environ. Microbiol. 73:7934–7946. 10.1128/AEM.01115-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Booth NJ, Beekman JB, Thune RL. 2009. Edwardsiella ictaluri encodes an acid-activated urease that is required for intracellular replication in channel catfish (Ictalurus punctatus) macrophages. Appl. Environ. Microbiol. 75:6712–6720. 10.1128/AEM.01670-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang CI, Liao JC, Kuo L. 1998. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. 274:H342–H348 [DOI] [PubMed] [Google Scholar]
- 8. Mori M. 2007. Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J. Nutr. 137:1616S–1620S [DOI] [PubMed] [Google Scholar]
- 9. Collins LA, Thune RL. 1996. Development of a defined minimal media for the growth of Edwardsiella ictaluri. Appl. Environ. Microbiol. 62:848–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Demaurex N, Grinstein S. 2006. Measurements of endosomal pH in live cells by dual-excitation fluorescence imaging, p 163–169 In Celis JE. (ed), Cell biology: a laboratory handbook, 3rd ed. Elsevier, Inc, San Diego, CA [Google Scholar]
- 11. Mukherjee EE, Maxfield FR. 2009. Acidification of endosomes and phagosomes, p 225–233 In Russell DG, Gordon S. (ed), Phagocyte-pathogen interaction. ASM Press, Washington, DC [Google Scholar]
- 12. Claus V, Jahraus A, Tjelle T, Berg T, Kirschke H, Faulstich H, Griffiths G. 1998. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J. Biol. Chem. 273:9842–9851 [DOI] [PubMed] [Google Scholar]
- 13. Yates RM, Hermetter A, Russell DG. 2005. The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic 6:413–420. 10.1111/j.1600-0854.2005.00284.x [DOI] [PubMed] [Google Scholar]
- 14. Chico-Calero I, Suarez M, Gonzalez-Zorn B, Scortti M, Slaghuis J, Goebel W, Vazquez-Boland JA. 2002. Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. U. S. A. 99:431–436. 10.1073/pnas.012363899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goebel W, Kuhn M. 2000. Bacterial replication in the host cell cytosol. Curr. Opin. Microbiol. 3:49–53. 10.1016/S1369-5274(99)00050-8 [DOI] [PubMed] [Google Scholar]
- 16. Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W. 2001. Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc. Natl. Acad. Sci. U. S. A. 98:12221–12226. 10.1073/pnas.211106398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti PJ, Cossart P. 1999. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J. Cell Sci. 112:1697–1708 [DOI] [PubMed] [Google Scholar]
- 18. Celli J, Gorvel JP. 2004. Organelle robbery: Brucella interactions with the endoplasmic reticulum. Curr. Opin. Microbiol. 7:93–97. 10.1016/j.mib.2003.11.001 [DOI] [PubMed] [Google Scholar]
- 19. Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR. 2001. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114:4637–4650 [DOI] [PubMed] [Google Scholar]
- 20. Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. 1996. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 15:964–977 [PMC free article] [PubMed] [Google Scholar]
- 21. Hackstadt T, Fischer ER, Scidmore MA, Rockey DD, Heinzen RA. 1997. Origins and functions of the chlamydial inclusion. Trends Microbiol. 5:288–293. 10.1016/S0966-842X(97)01061-5 [DOI] [PubMed] [Google Scholar]
- 22. Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. 2002. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 70:5816–5821. 10.1128/IAI.70.10.5816-5821.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 9:891–909. 10.1111/j.1462-5822.2006.00838.x [DOI] [PubMed] [Google Scholar]
- 24. Rathman M, Sjaastad MD, Falkow S. 1996. Acidification of phagosomes containing Salmonella typhimurium in murine macrophages. Infect. Immun. 64:2765–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Steele-Mortimer O. 2008. The Salmonella-containing vacuole: moving with the times. Curr. Opin. Microbiol. 11:38–45. 10.1016/j.mib.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rogge M, Thune R. 2011. The Edwardsiella ictaluri type III secretion system is regulated by pH and phosphate concentration through EsrA, EsrB, and EsrC. Appl. Environ. Microbiol. 77:4293–4302. 10.1128/AEM.00195-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu XJ, McGourty K, Liu M, Unsworth KE, Holden DW. 2010. pH sensing by intracellular Salmonella induces effector translocation. Science 328:1040–1043. 10.1126/science.1189000 [DOI] [PMC free article] [PubMed] [Google Scholar]