

Abstract
Saliva from arthropod vectors facilitates blood feeding by altering host inflammation. Whether arthropod saliva counters inflammasome signaling, a protein scaffold that regulates the activity of caspase-1 and cleavage of interleukin-1β (IL-1β) and IL-18 into mature molecules, remains elusive. In this study, we provide evidence that a tick salivary protein, sialostatin L2, inhibits inflammasome formation during pathogen infection. We show that sialostatin L2 targets caspase-1 activity during host stimulation with the rickettsial agent Anaplasma phagocytophilum. A. phagocytophilum causes macrophage activation and hemophagocytic syndrome features. The effect of sialostatin L2 in macrophages was not due to direct caspase-1 enzymatic inhibition, and it did not rely on nuclear factor κB or cathepsin L signaling. Reactive oxygen species from NADPH oxidase and the Loop2 domain of sialostatin L2 were important for the regulatory process. Altogether, our data expand the knowledge of immunoregulatory pathways of tick salivary proteins and unveil an important finding in inflammasome biology.
INTRODUCTION
The inflammasome is a key element in many inflammatory processes (1–3). The canonical paradigm in inflammasome activation establishes that Nod-like receptors (NLRs) or absent in melanoma 2 (AIM2) recruits the adaptor molecule apoptosis-associated speck-like protein (ASC) to activate the enzyme caspase-1 (1–3). Caspase-1 then cleaves inactive cytokine precursors, such as pro-interleukin-1β (pro-IL-1β) and pro-IL-18, leading to the release of mature IL-1β and IL-18 forms. Four classical inflammasomes have been described. NLRP3 is activated by a wide range of stimuli with diverse physicochemical structures (4). The NLRC4 inflammasome is activated mainly in response to cytosolic flagellin or bacterial type III and IV secretion systems of Gram-negative bacteria (5). The AIM2 inflammasome directly binds viral and bacterial cytosolic DNAs, whereas the NLRP1 inflammasome confers susceptibility to the Bacillus anthracis lethal toxin (1–3).
Because the inflammasome plays a critical role in host defense, it is not surprising that pathogens have evolved strategies to disrupt this molecular scaffold. Poxvirus members encode a protein that interacts with the adaptor molecule ASC of the inflammasome (6). Influenza virus uses nonstructural protein 1 (NS1) for inflammasome evasion (7), whereas bacterial effector molecules bind caspase-1 to arrest inflammasome assembly (1–3). Previously, we demonstrated that caspase-1-deficient mice were more susceptible than wild-type animals to Anaplasma phagocytophilum infection (8). A. phagocytophilum causes human granulocytic anaplasmosis and colonizes neutrophils during infection (9–12). However, macrophages are responsible for disease pathology. Clinical and histopathological features in patients suggest classical macrophage activation (13), and animal models show increased macrophage infiltration and hemophagocytosis during infection (10, 14, 15).
Arthropod vector saliva counteracts host-derived inflammation by impairing the complement system (16), the function of macrophages and dendritic cells, and T cell biology (17–19). How disease vectors inhibit inflammasome signaling during pathogen infection remains poorly understood (20). Because we uncovered that Ixodes scapularis saliva inhibits cytokine secretion during stimulation of NLRs in macrophages (21), we hypothesized that tick saliva manipulates inflammation through caspase-1 activity during A. phagocytophilum infection. Here we show that the tick salivary protein sialostatin L2 inhibits inflammasome signaling during A. phagocytophilum infection. These findings shed some light onto the fundamental basis of pathogen-vector-host interactions.
MATERIALS AND METHODS
Ethics statements.
All animal breeding and experiments were performed in strict compliance with guidelines set forth by the National Institutes of Health (Office of Laboratory Animal Welfare [OLAW] assurance number A3439-01). All animal and biosafety procedures were approved by the Institutional Animal Care and Use (IACUC numbers A-20110030BE, 0413017, 1104066, and 03759) and Biological Use Authorization (BUA numbers 20120020, 00002247, 130047, 20120020, and 130047) Committees at the University of California, Riverside, the University of Maryland, Baltimore, the University of Iowa, and Harvard Medical School. C57BL/6 (database number 000664) and nox2−/− (database number 002365) mice were purchased from Jackson Laboratories. cathepsin l−/− mice were previously described (22–24). Mice were at least 6 to 10 weeks of age and were gender matched throughout our analysis.
Chemical reagents.
Lipopolysaccharide (LPS) was obtained from Invivogen. Nigericin, diphenyleneiodonium chloride (DPI), and uric acid were purchased from Sigma-Aldrich. The proteasomal inhibitor MG-132 and a cathepsin L inhibitor were purchased from Millipore. Brefeldin A was purchased from Epicentre Biotechnologies.
A. phagocytophilum infection and gentamicin protection assays.
A. phagocytophilum strain HZ was grown in HL-60 cells (ATCC CCL-240). Cells were maintained in Iscove's modified Dulbecco's medium (IMDM) with l-glutamine, HEPES (Thermo Scientific), and 20% heat-inactivated fetal bovine serum (FBS) in 5% CO2 and humidified air at 37°C (25). A. phagocytophilum organisms multiply as microcolonies (morulae) inside cells. Therefore, it is impractical to accurately count individual organisms (26). The number of host-free A. phagocytophilum organisms was estimated as previously described (26, 27). We used the following formula: number of A. phagocytophilum organisms = total number of infected cells × average number of morulae in an infected cell (typically 5) × average number of A. phagocytophilum organisms in a morula (typically 19) × percentage of A. phagocytophilum recovered as host cell free (typically 50% as determined by using metabolically [35S]methionine-labeled A. phagocytophilum) (28). Cells were infected with A. phagocytophilum strain HZ at the indicated times and multiplicities of infection (MOIs). Cells were treated with gentamicin (50 μg/ml) for 90 min before being harvested from the culture plate and then washed with phosphate-buffered saline (PBS) by centrifugation at 200 × g for 10 min at room temperature.
C57BL/6 mice were infected by intraperitoneal injection with the A. phagocytophilum wild-type HZ strain (1 × 107 bacteria) in the presence or absence of sialostatin L2 (6.6 mg/kg of body weight). To quantify A. phagocytophilum loads in the peripheral blood, DNA from anticoagulated peripheral blood was extracted with a DNeasy Tissue kit (Qiagen) according to the manufacturer's recommendations. Quantitative reverse transcription-PCR (RT-PCR) was performed using iQ SYBR green Supermix and a Bio-Rad iQ5 optical system. Primer sequences for A. phagocytophilum were as follows: 16S-F, 5′-CAGCCACACTGGAACTGAGA-3′; and 16S-R, 5′-CCCTAAGGCCTTCCTCACTC-3′. Gene expression was normalized to β-actin by using the primers β-actin F (5′-CGCATCCTCTTCCTCCCT-3′) and β-actin R (5′-TGGAATCCTGTGGCATCC-3′) and the absolute quantification method (29). Total RNA was isolated by using TRIzol reagent (Invitrogen) and a PureLink RNA minikit (Life Technology). cDNA was produced by using a Verso cDNA synthesis kit (Thermo Scientific).
Cell culture generation and stimulation.
Bone marrow-derived macrophages (BMDMs) were generated as previously described, with minor modifications (30). Briefly, femurs and tibias were removed from mice and kept in PBS plus 1% penicillin-streptomycin-amphotericin (PSA) (Thermo Scientific). Marrow was flushed from the bone by using cold Dulbecco's modified Eagle medium (DMEM) (Invitrogen) and a 25-gauge needle. BMDMs were grown on petri dishes containing differentiation medium (DMEM supplemented with 10% FBS [Invitrogen], 30% L929 cell conditioning medium, and 1% PSA). BMDMs were grown in a humidified incubator at 37°C and 5% CO2 for 6 days prior to stimulation. On the 3rd day, differentiation medium was added to each dish. BMDMs were detached and plated on 24-well culture plates at 1 × 106 cells per well or on 96-well plates at 2 × 105 cells per well, unless otherwise stated, in DMEM plus 10% FBS. Chemicals, sialostatin L2, or polypeptides were added at the indicated concentrations and prior to A. phagocytophilum stimulation. Heat-killed pathogens were obtained by boiling A. phagocytophilum for 15 min. BMDMs were also stimulated with nigericin, LPS, uric acid, Pseudomonas aeruginosa PAK, and Francisella tularensis LVS. F. tularensis LVS was obtained from the ATCC (ATCC 29684) and grown on Difco cysteine heart agar supplemented with 9% sheep red blood cells for 48 h at 37°C (31). P. aeruginosa PAK was cultured in Luria-Bertani (LB) broth (32).
Sialostatin L2 and peptide preparation.
PyMOL (33) was used to generate a structural representation of sialostatin L2 (PDB entry 3LH3). Sialostatin L2 was produced as previously described (34–36). Briefly, sialostatin L2 cDNA was PCR amplified and subcloned into the pET17b bacterial expression vector. The expression vector was placed into Escherichia coli strain BL21(DE3)pLysS for expression. Cultures were grown and induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG). Inclusion bodies were dissolved in 6 M guanidine hydrochloride, 20 mm Tris-HCl, pH 8.0, and reduced with 10 mM dithiothreitol (DTT). Sialostatin L2 was refolded in a large volume of 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, and stirred overnight at 4°C. The refolded protein was concentrated with a tangential-flow filtration device and purified by gel filtration chromatography on Sephacryl S-100, followed by anion-exchange chromatography on Q-Sepharose. Dialysis was then performed against 20 mM Tris-HCl, pH 7.4, 150 mm NaCl. LPS contamination was removed by using the detergent-based method from Arvys Proteins. Endotoxin presence was estimated by using a sensitive fluorescence-based endotoxin assay from Lonza Biologics.
Three sialostatin L2 peptides were synthesized based on the resolved structure of sialostatin L2 (34). The amino acid sequences for the three synthesized peptides were as follows: (i) N terminus, ELALRGGYRERSNQDDPEY; (ii) Loop1, SAQQPGKTHFDTVVEVLKVETQTVAGT; and (iii) Loop2, TCELTSTYNKDTCQANANAAQRTCTTVIYRNLQGEKSISSFECAA. The sialostatin L2 protein sequences LELAHYATSTW and NYRLTLKVAES were not included in the synthesized peptides because, based on the resolved structure of sialostatin L2, these amino acids were not accessible to molecular interactions with potential effector molecules. Peptide synthesis and folding were performed by an external contractor (SynBioSci Corporation). All three peptides were verified as not containing any endotoxin contamination by using the Lonza Limulus amebocyte lysate QCL-1000 chromogenic assay (Lonza Group).
Enzymatic assays.
Enzymatic assays were previously described (37, 38). Briefly, recombinant sialostatin L2 and peptides were preincubated with cathepsin L (EMD Millipore) for 10 min before the addition of the corresponding substrates. Substrates were used at 0.25 mM throughout the studies. N-Carbobenzyloxy-Arg-Arg-7-amino-4-methylcoumarin (EMD Millipore) was used for cathepsin L. The composition of the assay buffer was 100 mM sodium acetate (NaAc), pH 5.5, 100 mM NaCl, 1 mM EDTA, 1 mg/ml cysteine, and 0.005% Triton X-100.
The observed substrate hydrolysis rate in the absence of the tested polypeptides was considered 100% and then compared with the remaining enzymatic activity in the presence of the different polypeptides tested. Enzymatic activity was reflected by the hydrolysis rate of the fluorescent substrate used in each assay; the substrate hydrolysis rate was estimated from the slope of the linear fit curve obtained by plotting the increase of the detected relative fluorescence units (RFU) as a function of time (relative fluorescence unit increase per second; for the linear fit to be acceptable, the r2 value must have been >0.95). The mean substrate hydrolysis rate for three experiments and the standard error of the mean (SEM) were calculated. The linear fit of the fluorescence increase as a function of time was verified with Magellan data analysis software (Tecan). The substrate hydrolysis rate was monitored in an Infinite M200 96-well-plate fluorescence reader (Tecan) using 365-nm excitation and 450-nm emission wavelengths. Assays were performed at 30°C in triplicate.
Confocal microscopy.
Cells were stimulated with A. phagocytophilum strains. After two washes with PBS, cells were stained with 1 μg/ml cholera toxin B (Molecular Probes) at 4°C for 10 min and then fixed with methanol. Cells were blocked with 5% bovine serum albumin (BSA)-PBS and stained for 60 min with A. phagocytophilum antibody (raised in rabbits at Yale University) (1:100) at room temperature. Cells were then washed with PBS and stained with fluorescence-conjugated secondary antibodies for 30 min at room temperature. Cells were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI). Confocal microscopy was done by using a Leica SP2 microscope.
Immunoblotting.
Cell lysates were extracted using RIPA lysis buffer (Boston Bioproducts) with Complete Mini protease inhibitor cocktail and PhosSTOP (Roche Applied Science). Protein concentration was determined via the Bradford protein assay method, using protein assay dye reagent concentrate and an iMark reader (Bio-Rad). An SDS-polyacrylamide gel was made and run at 200 V for 1 h. Protein transfer was done under wet conditions with polyvinylidene difluoride (PVDF) membranes for 60 min at 100 V. Membranes were blocked in 5% nonfat dry milk (LabScientific, Inc.). Western blot antibodies for β-actin (Neomarker-Thermo Scientific) (1:500 to 1:1,000), IκBα (Cell Signaling) (1:1,000), IL-1β (R&D Systems) (1:1000), caspase-1 (Millipore) (1:1,000), caspase-1 (Santa Cruz) (1:100 to 1:1,000), and IL-18 (MBL) (0.5 to 4 μg/ml), as well as anti-goat–horseradish peroxidase (HRP) (Santa Cruz) (1:7,500 to 1:10,000), anti-rabbit–HRP (Santa Cruz) (1:7,500 to 1:10,000), and anti-mouse–HRP (Santa Cruz) (1:7,500 to 1:10,000), were used. In some experiments, supernatants were concentrated with centrifugal filter units (3,000-molecular-size cutoff; Amicon), and caspase-1 immunoblots were performed. An enhanced chemiluminescence (ECL) Western blotting substrate and Super Signal West Pico chemiluminescence substrate were used (Pierce Thermo Scientific). Densitometry was performed by using ImageQuant TL software (GE Healthcare Life Sciences).
ROS and FLICA.
We detected reactive oxygen species (ROS) by using the fluorescence probe 2′,7′-dichlorofluorescin diacetate (H2DCFDA) (Invitrogen), as described previously (25). Fluorescence in 96-well plates was recorded over time by use of a Spectra MAX Gemini EM microplate reader (Molecular Devices) using a fluorescein isothiocyanate (FITC) filter (excitation, 485 nm; and emission, 538 nm). A green fluorescent labeled inhibitor of caspase-1 (FLICA) assay kit (Immunochemistry) was used to detect active caspase-1 in macrophages. Measuring was done with excitation at 490 nm and emission at 520 nm.
Enzyme-linked immunosorbent assay (ELISA).
Mouse tumor necrosis factor alpha (TNF-α), IL-1β, and IL-6 were measured with a BD OptEIA set from BD Biosciences. Mouse IL-12p40 was measured with capture and detection antibodies from eBiosciences. IL-18 capture and detection antibodies were purchased from MBL. Supernatants were collected, and absorbance was measured using a Bio-Rad iMark instrument at 450 nm, with a 595-nm correction wavelength.
Binding assays.
Wild-type C57BL/6 and cathepsin l−/− BMDMs were lysed as described above. Cell lysates (0.5 μg) were resuspended in 0.5 M carbonate-bicarbonate (pH 9.5) and used to coat a 96-well standard microtiter plate (Corning) at 4°C overnight. After washing, wells were blocked with assay diluent (1× PBS, 10% FBS) followed by binding of either BSA or sialostatin L2 at the indicated concentrations. In-house sialostatin L2 polyclonal antibodies derived from rabbits (1:200) and goat anti-rabbit IgG–HRP (1:10,000) (Abcam) were used. Detection was obtained by using a 1:1 mixture of 3,3′,5,5′-tetramethylbenzidine (TMB) with HRP (BD Biosciences). A Bio-Rad iMark instrument was used to measure the absorbance at 450 nm, with a 595-nm correction wavelength.
Histopathology.
To study the effects of sialostatin L2 on local inflammation, back-shaved C57BL/6 mice received intradermal injections with the following, on separate sites of dorsal skin: (i) 20 μl of PBS, (ii) sialostatin L2 (20 μg), (iii) A. phagocytophilum strain Hz (1 × 104 cells), (iv) sialostatin L2 plus A. phagocytophilum strain Hz, and (v) LPS (40 μg). Sialostatin L2 was injected 5 h prior to, at the start of, and 5 h after bacterial infection. The skin surrounding injected sites was excised from each mouse 24 h after infection. The skin was fixed with 10% neutral buffered formalin for histopathologic study. To study the effects of sialostatin L2 on A. phagocytophilum-induced splenomegaly, C57BL/6 mice were infected by intraperitoneal injection with A. phagocytophilum strain Hz (1 × 107 cells) in the presence or absence of sialostatin L2 (6.6 mg/kg). The spleens were removed at day 14, normalized to the body weight, and compared to those of noninfected mice (PBS). Measurement of A. phagocytophilum loads was done by using quantitative RT-PCR as described above.
Formalin-fixed skin was sectioned longitudinally and stained with hematoxylin and eosin. A pathologist blinded to the groups scored sections for inflammation and determined the degree of inflammation. Four skin sections from each animal per treatment were evaluated and scored for dermal and subcutaneous adipose/skeletal muscle inflammation. Subcutaneous adipose/skeletal muscle inflammation was graded on a scale of 0 to 3 based on granulocyte number, as follows: 0, <5 granulocytes; 1, 5 to 10 granulocytes; 2, 10 to 50 granulocytes; and 3, >50 granulocytes. The scores from three 400× fields were averaged to determine the subcutaneous adipose/skeletal muscle score per tissue section. Dermal inflammation was scored as either absent (0) or present (1). The scores were added to determine the skin inflammation score.
Statistical analysis.
Data were expressed as means ± SEM. Gaussian distribution was determined by the D'Agostino and Pearson normality test. For data points that followed a Gaussian distribution, the following parametric analyses were used: unpaired Student's t test (two-group comparisons) and one-way analysis of variance (ANOVA) (comparisons of three or more groups). Kruskal-Wallis ANOVA was used for data points that did not follow a Gaussian distribution. Bonferroni or Tukey (parametric) and Dunn's (nonparametric) post hoc multiple-comparison tests were used following ANOVA. All statistical calculations and graphs were made by using GraphPad Prism, version 5.04. P values of <0.05 were considered statistically significant.
RESULTS
A. phagocytophilum survives transiently inside macrophages.
Because macrophages are important for immunopathology during A. phagocytophilum infection (10, 13–15), we tested whether this infectious agent would survive inside these cells. A. phagocytophilum infection was monitored under gentamicin protection assay conditions, as previously described (25, 39). By using confocal microscopy, we verified that A. phagocytophilum formed small microcolonies (morulae) inside cells (Fig. 1A). We confirmed pathogen infection by performing RT-PCR, using primers specific for the A. phagocytophilum 16S rRNA gene. While we did not observe the presence of A. phagocytophilum in noninfected cells, macrophages infected with A. phagocytophilum were positive for the 16S rRNA gene (Fig. 1B). Indeed, quantitative RT-PCR provided convincing evidence that A. phagocytophilum survives transiently inside macrophages. Using a time point series, we detected small numbers of bacteria at 2 and 6 h postinfection, followed by an increased load at 18 h (Fig. 1C). Bacterial infection was reduced at 48 h, which led to complete elimination of A. phagocytophilum in macrophages after 72 h of infection. These findings show that macrophages temporarily sustain A. phagocytophilum infection.
FIG 1.

A. phagocytophilum survives transiently inside macrophages. (A) BMDMs (1 × 106) from C57BL/6 mice were stimulated for 24 h with the wild-type A. phagocytophilum strain HZ (MOI = 50) and stained with cholera toxin subunit B (CTB; green), DAPI (blue), and an antibody against A. phagocytophilum (red; white arrows). (B) BMDMs (1 × 106) from C57BL/6 mice were stimulated for 24 h with the wild-type A. phagocytophilum strain HZ (MOI = 30). Ninety minutes after infection, gentamicin (50 μg/ml) was added to the medium to eliminate extracellular bacteria. Cells were washed extensively with PBS, and the A. phagocytophilum load was measured by PCR, as judged by the 16S rRNA gene. Three representative samples of infected (+) and noninfected (−) cells are shown. (C) Time course series showing A. phagocytophilum loads in macrophages (MOI = 30) after gentamicin protection assays, as judged by the 16S rRNA/mouse actin ratio from quantitative RT-PCR. One-way ANOVA with the Tukey test was used to compare noninfected and infected cells. *, P < 0.05; NS, not significant; (−), nonstimulated cells.
Sialostatin L2 inhibits caspase-1 maturation and diminishes IL-1β and IL-18 secretion by macrophages during A. phagocytophilum stimulation.
Because A. phagocytophilum survival inside macrophages suggested that these cells could mount an immune response against this pathogen, we ascertained whether macrophages responded to infection. Pro-IL-1β (p31) is induced by stimulation of pattern recognition receptors via NF-κB signaling. This signal is referred to as priming and is commonly initiated in vitro by stimulating macrophages with LPS. A second signal is mediated by NLR-dependent caspase-1 activation, which cleaves pro-IL-1β into its mature p17 form (1–3). A. phagocytophilum induced the secretion of IL-1β by macrophages when two MOIs were used: 10 and 50 (Fig. 2A). Moreover, a dose-dependent inhibition of IL-1β secretion was observed in the presence of the I. scapularis tick protein sialostatin L2.
FIG 2.

Sialostatin L2 inhibits A. phagocytophilum-induced caspase-1-mediated cytokine secretion. (A) BMDMs (5 × 105) were stimulated with A. phagocytophilum (MOIs of 10 and 50) in the presence or absence of sialostatin L2 (SL2) for 18 h at the indicated concentrations. IL-1β was measured by ELISA. One-way ANOVA with the Tukey test was used to compare nontreated and SL2-treated cells. *, P < 0.05; NS, not significant. (B) IL-18 (p18) and mature caspase-1 (p20) in supernatants were measured by Western blotting (IB). β-Actin and pro-caspase-1 were used as loading controls. (C) BMDMs (2 × 105) were stimulated with A. phagocytophilum (MOI = 10) for 18 h in the presence or absence of SL2 (3 μM). The fluorescence inhibitor probe FAM-YVAD-FMK was used to label active caspase-1 in macrophages. The signal is shown as a function of relative fluorescence units (RFU). (D and E) BMDMs (5 × 105) were stimulated with LPS (100 ng/ml) plus nigericin (10 μM) in the presence or absence of SL2 (5 μM). (D) IL-1β was measured by ELISA. Unpaired Student's t test was used to compare nontreated and SL2-treated cells. (E) IL-18 (p18) and mature caspase-1 (p20) in supernatants were measured by Western blotting (IB). β-Actin and pro-caspase-1 were used as loading controls. (−), nonstimulated cells. Numbers below the Western blot panels show quantitation by densitometry and indicate fold differences compared to non-SL2-treated samples.
Importantly, sialostatin L2 affected neither NF-κB signaling nor the secretion of NF-κB-derived cytokines IL-6 and IL-12p40 by macrophages after A. phagocytophilum stimulation (see Fig. S1 in the supplemental material), which suggested that sialostatin L2 only targeted inflammasome signaling. Indeed, we observed that sialostatin L2 inhibited caspase-1 activation (p20) and IL-18 (p18) maturation during pathogen stimulation of macrophages (Fig. 2B). We obtained similar results when caspase-1 activation was measured by using a fluorescence assay (Fig. 2C) (40). A. phagocytophilum stimulation of macrophages did not require LPS priming for IL-1β secretion. These results corroborated the notions that A. phagocytophilum lacks genes for LPS synthesis in the genome (41) and that molecules other than LPS prime macrophages for production of pro-IL-1β during infection (21). The effect of sialostatin L2 on inflammasome signaling also appeared to be specific for A. phagocytophilum, because caspase-1 activation and IL-1β and IL-18 secretion were not inhibited when macrophages were stimulated by known NLRP3 (e.g., nigericin), NLRC4 (e.g., P. aeruginosa), and AIM2 (e.g., F. tularensis LVS) agonists (Fig. 2D and E; see Fig. S2).
Sialostatin L2 inhibits caspase-1 maturation and mitigates IL-1β and IL-18 secretion by macrophages via NADPH ROS during A. phagocytophilum stimulation.
We did not obtain any evidence that sialostatin L2 could bind A. phagocytophilum or caspase-1 directly (data not shown). These results suggested that sialostatin L2 inhibition of caspase-1 activation was indirect. Therefore, we investigated the mechanisms by which A. phagocytophilum triggers caspase-1 activation in macrophages. We first observed that A. phagocytophilum induction of IL-1β secretion in macrophages required proteasomal degradation and the transport of proteins from the endoplasmic reticulum to the Golgi apparatus. Brefeldin A and MG-132 pretreatment of macrophages completely abrogated caspase-1 activation and IL-1β and IL-18 but not IL-6 secretion during A. phagocytophilum stimulation (Fig. 3A to C).
FIG 3.

Caspase-1-mediated cytokine secretion during A. phagocytophilum stimulation of macrophages is dependent on protein transport from the endoplasmic reticulum to the Golgi apparatus, the proteasome, and ROS generation. (A and B) BMDMs (1 × 106) were pretreated for 1 h with brefeldin A (20 μg/ml) or MG-132 (1 μM) prior to A. phagocytophilum stimulation (MOI = 50) for 18 h. IL-1β (A) and IL-6 (B) were measured by ELISA. (C) IL-18 (p18) and mature caspase-1 (p20) in supernatants were measured by Western blotting (IB). β-Actin and pro-caspase-1 were used as loading controls. (D and E) BMDMs (1 × 106) were stimulated with heat-killed (HK) or live A. phagocytophilum (MOI = 50) in the presence or absence of DPI (10 μM) for 18 h. BMDMs were pretreated with DPI for 1 h prior to stimulation. IL-1β (D) and IL-6 (E) were measured by ELISA. (F) IL-18 (p18) and mature caspase-1 (p20) in supernatants were measured by Western blotting (IB). β-Actin and pro-caspase-1 were used as loading controls. Cytokine measurements were taken in triplicate and presented as means and SEM. For panels A and B, one-way ANOVA with the Bonferroni test was used to compare nontreated and chemically treated cells. For panels D and E, Student's t test was used to compare nontreated and DPI-treated cells. (−), nonstimulated cells; *, P < 0.05; NS, not significant. Numbers below the Western blot panels show quantitation by densitometry and indicate fold differences compared to non-chemically treated samples.
Previously, we showed that A. phagocytophilum did not affect mitochondrial ROS in macrophages (25). Thus, we investigated whether NADPH ROS were involved in caspase-1 function in the A. phagocytophilum model. DPI is a competitive inhibitor of flavin-containing cofactors and is an important inhibitor of NADPH-dependent ROS (42). We stimulated macrophages with heat-killed and live A. phagocytophilum in the absence or presence of DPI. Heat-killed A. phagocytophilum induced lower levels of IL-1β secretion (Fig. 3D), while live A. phagocytophilum induced more IL-1β secretion by macrophages. Moreover, the presence of DPI significantly reduced IL-1β secretion during A. phagocytophilum stimulation. This effect seemed specific for IL-1β, because secretion of IL-6, a cytokine that is not directly regulated by caspase-1, was not affected (Fig. 3E). The effect of DPI was also noticeable for caspase-1 and IL-18 maturation (Fig. 3F).
DPI may produce off-target effects at high concentrations (42). Therefore, we used nox2−/− mice, which do not produce ROS from NADPH oxidase (25). We measured A. phagocytophilum-induced ROS production in macrophages (25). Although it was not completely abrogated, we observed that nox2−/− macrophages stimulated with A. phagocytophilum produced fewer ROS than those in wild-type cells (Fig. 4A). Sialostatin L2 completely inhibited ROS production by wild-type macrophages during A. phagocytophilum stimulation. These results implicated the regulation of ROS by sialostatin L2 via NADPH-dependent and, possibly, -independent pathways. These findings were surprising because, in neutrophils, A. phagocytophilum actively suppresses NADPH oxidase assembly and ROS production (9, 11).
FIG 4.

Sialostatin L2 inhibits IL-1β and IL-18 secretion via NADPH ROS during A. phagocytophilum stimulation of macrophages. (A) BMDMs (2 × 105) from WT and nox2−/− mice were stimulated with A. phagocytophilum (MOI = 10) in the presence or absence of sialostatin L2 (SL2; 3 μM). ROS production was monitored using the fluorescent probe H2DCFDA at the indicated time points. (B) BMDMs (1 × 106) from WT and nox2−/− mice were stimulated with A. phagocytophilum (MOI = 50) in the presence or absence of SL2 (3 μM) for 18 h. Pro-IL-1β (p31) was measured by Western blotting (IB). β-Actin was used as a loading control. (C and E) BMDMs (1 × 106) from WT and nox2−/− mice were stimulated with A. phagocytophilum (MOIs of 10 and 20) in the presence or absence of SL2 (3 μM) for 18 h. IL-1β (C) and IL-6 (E) were measured by ELISA. (D and F) BMDMs (2 × 105) from WT and nox2−/− mice were stimulated with A. phagocytophilum (MOI = 50) for 18 h in the presence or absence of SL2 (5 μM). (D) IL-18 secretion was measured by ELISA. (F) The fluorescent inhibitor probe FAM-YVAD-FMK was used to label active caspase-1 in macrophages. The signal was corrected for the background, and the percentage of caspase-1 activation in macrophages is shown as a function of relative fluorescence units. Cytokine measurements were taken in triplicate and are presented as means and SEM. Experiments for panels A, B, D, and E were repeated twice, whereas those for panels C and F were repeated five times. *, P < 0.05. For panel A, ANOVA (with post hoc Bonferroni test) was performed. For panels C to F, Student's t test was performed. (−), nonstimulated cells; NS, not significant. Numbers below the Western blot panel show quantitation by densitometry and indicate fold differences compared to WT, non-SL2-treated macrophages.
The effect of NADPH-dependent ROS or sialostatin L2 on IL-1β secretion was not due to differential IL-1β translation (p31), because nox2−/− macrophages produced similar amounts of pro-IL-1β in the presence and absence of sialostatin L2 compared to wild-type macrophages (Fig. 4B). Conversely, we noticed a dose-dependent effect for sialostatin L2 and NADPH-mediated ROS production on IL-1β and IL-18 secretion during A. phagocytophilum stimulation (Fig. 4C and D). IL-6 secretion by macrophages was not affected in these assays (Fig. 4E). Supporting our findings, caspase-1 activation was inhibited by sialostatin L2 (Fig. 4F).
To confirm the phenotype of the nox2−/− mice, we stimulated primed wild-type and nox2−/− macrophages with uric acid. Uric acid crystals are a known inflammasome agonist that leads to IL-1β secretion by macrophages, independent of NADPH-mediated ROS production (43, 44). As expected, uric acid induced the secretion of IL-1β in wild-type macrophages (Fig. 5A). Importantly, lack of NADPH oxidase activity affected neither IL-1β, IL-18, and IL-6 secretion nor caspase-1 activation by nox2−/− macrophages after uric acid stimulation (Fig. 5B and C). Moreover, sialostatin L2 did not affect inflammasome signaling in macrophages after crystal stimulation (Fig. 5). It has been suggested that changes in the redox microenvironment modulate the activation potential of the inflammasome (45). To exclude any extraneous oxidative artifacts during experimentation, we stimulated wild-type and nox2−/− macrophages with a low A. phagocytophilum MOI. Similar to our findings observed at a higher A. phagocytophilum MOI (Fig. 4), ROS from NADPH oxidase and the tick salivary protein sialostatin L2 reduced IL-1β secretion by macrophages during microbial stimulation (Fig. 5D).
FIG 5.

Sialostatin L2 does not inhibit caspase-1-mediated cytokine secretion during uric acid stimulation of macrophages. (A to C) BMDMs (1 × 106) from WT and nox2−/− mice were primed with LPS (100 ng/ml) overnight and stimulated with uric acid (50 μg/ml) for 4 h in the presence or absence of SL2 (5 μM). SL2 was added to the cell culture 30 min before uric acid stimulation. IL-1β (A) and IL-6 (C) were measured by ELISA. (B) IL-18 (p18) and mature caspase-1 (p20) in the supernatants of LPS-primed cells were measured by Western blotting (IB). β-Actin, pro-IL-1β, and caspase-1 were detected in the cell lysate. (D) BMDMs (1 × 106) from wild-type (WT) and nox2−/− mice were stimulated with A. phagocytophilum (MOI = 5) for 18 h in the presence or absence of sialostatin L2 (SL2; 5 μM). IL-1β secretion was measured by ELISA. Cytokine measurements were taken in triplicate and presented as means ± SEM. *, P < 0.05 by Student's t test comparing nontreated and SL2-treated cells. (−), nonstimulated cells; NS, not significant. Experiments were repeated twice. Numbers below the Western blot panels show quantitation by densitometry and indicate fold differences compared to WT, non-SL2-treated samples.
The Loop2 domain of sialostatin L2 contributes to inhibition of caspase-1 activity and mitigation of IL-1β and IL-18 secretion by macrophages during A. phagocytophilum stimulation.
The crystal structure of sialostatin L2 was recently published (34). The interaction surface is formed by an N-terminal peptide extending to a glycine residue, which acts as a hinge. The second structural component consists of the loop between strands 2 and 3 of the β-sheet (Loop1), and the third region is the loop between β-strands 4 and 5 (Loop2) (Fig. 6A) (34). Based on the resolved structure of sialostatin L2, we synthesized peptides for the N-terminal, Loop1, and Loop2 domains, aiming to determine the domain that contributes to caspase-1 inhibition during A. phagocytophilum stimulation of macrophages. In contrast to sialostatin L2, which was previously shown to inhibit cathepsin L function (34, 36), none of the three peptides showed an effect on cathepsin L activity (Fig. 6B). Additionally, we did not observe any difference in sialostatin L2 binding when cathepsin l−/− and wild-type lysates were compared (Fig. 6C). Finally, the lack of cathepsin L activity did not affect IL-1β secretion during A. phagocytophilum stimulation of macrophages (see Fig. S3 in the supplemental material). Next, we stimulated macrophages with A. phagocytophilum in the absence or presence of these polypeptides. We noticed that the Loop2 domain of sialostatin 2 partially inhibited caspase-1 activation and secretion of IL-1β and IL-18 (Fig. 6D and E). IL-6, a cytokine that is not directly regulated by caspase-1, did not show any inhibitory effect by the Loop2 peptide during A. phagocytophilum stimulation of macrophages (Fig. 6F).
FIG 6.

Loop2 of sialostatin L2 inhibits caspase-1-induced cytokine secretion during A. phagocytophilum stimulation of macrophages. (A) Sialostatin L2 (SL2) structure (PDB entry 3LH4) depicting the N terminus, Loop1, and Loop2. The synthesized Loop2 peptide is highlighted in green, with the yellow ball-and-stick format representing conserved disulfide bonds. (Inset) Loop2 residues RNLQGE are depicted in gray, with neighboring polar contacts shown as black dashed lines. (B) Inhibition assay showing the percentage of remaining cathepsin L activity in the presence or absence of SL2 or the N-terminal, Loop1, or Loop2 peptide (6 μM). Cathepsin L was used at 100 pM. Comparisons were done with the enzymatic activity in the absence of polypeptides, which was considered 100%. (C) BMDMs (5 × 106) from wild-type (WT) and cathepsin l−/− mice were obtained, and 0.5 μg of cell lysate was used for SL2 binding at the indicated concentrations (0.4 to 3.2 μg). Polyclonal SL2 antibody (1:200 dilution) was used. (D and F) BMDMs (1 × 106) were stimulated with A. phagocytophilum (MOI = 5) for 18 h in the presence or absence of the peptides representing the N terminus, Loop1, or Loop2. Peptides (5 μM) were added 30 min prior to A. phagocytophilum stimulation. IL-1β (D) and IL-6 (F) were measured by ELISA. (E) IL-18 (p18) and mature caspase-1 (p20) in supernatants were measured by Western blotting (IB). β-Actin, pro-IL-1β, and pro-caspase-1 were detected in the cell lysate at an MOI of 50. Experiments for panels B and C were repeated twice, while those for panels D to F were repeated seven times. Data are shown in triplicate and presented as means ± SEM. *, P < 0.05. For panels B, D, and F, one-way ANOVA with the Bonferroni test was used to compare nontreated and peptide-treated cells. For panel C, Student's t test was used to compare WT and cathepsin l−/− cells. (−), nonstimulated cells; NS, not significant. Numbers below the Western blot panels show quantitation by densitometry and indicate fold differences compared to non-peptide-treated samples.
Sialostatin L2 inhibits IL-1β-induced inflammation by A. phagocytophilum in vivo.
To determine whether sialostatin L2 could inhibit caspase-1 in vivo, we performed intradermal injections of sialostatin L2 in the presence of A. phagocytophilum. Similar to the case for the positive control, LPS, intradermal injection of A. phagocytophilum into mice led to caspase-1 activation (p20) and IL-1β maturation (Fig. 7A). Consistent with our results observed for macrophages, sialostatin L2 impaired caspase-1 activation and IL-1β maturation triggered by A. phagocytophilum at the skin site (Fig. 7A). Furthermore, IL-1β translation was not affected by sialostatin L2 in vivo, suggesting that NF-κB signaling was not affected. Interestingly, sialostatin L2 did not inhibit the constitutive expression of IL-18 during pathogen injection (Fig. 7A). We also did not observe any effect of A. phagocytophilum injection on IL-18 maturation at the skin site (data not shown).
FIG 7.

Sialostatin L2 inhibits A. phagocytophilum-induced inflammation at the skin site. Intradermal injection of C57BL/6 mice was performed with PBS (−), sialostatin L2 (20 μg), A. phagocytophilum (1 × 104 cells), sialostatin L2 (20 μg) plus A. phagocytophilum (1 × 104 cells), and LPS (40 μg). (A) Proteins from the injection sites were extracted from skin homogenates, and Western blotting (IB) was performed to detect caspase-1 (p45 and p20), IL-1β (p31 and p17), and pro-IL-18 (p24). β-Actin was used as a loading control. (B) Skin inflammation was characterized by infiltration of neutrophils, eosinophils, and a few macrophages in the dermis, subcutaneous adipose tissue, and underlying skeletal muscle of mice (arrows). Hematoxylin and eosin staining was performed. Magnification, ×200. Bars = 100 μm. (C) Inflammation score, determined as described in Materials and Methods. (D) Spleen weights for C57BL/6 mice infected with A. phagocytophilum (n = 7) or treated with sialostatin L2 (20 μg) plus A. phagocytophilum (n = 7), normalized to the animal's body weight and contrasted to those for noninfected mice (n = 7) (PBS) at day 14 post-intraperitoneal infection. *, P < 0.05. For panel C, the Kruskal-Wallis test (with post hoc Dunn's test) was performed. For panel D, ANOVA (with post hoc Bonferroni test) was performed. NS, not significant.
Next, we determined whether sialostatin L2 could affect inflammation (as judged by neutrophil and macrophage infiltration). Our results demonstrate that similar to LPS, A. phagocytophilum induced neutrophil infiltration within the dermis, subcutaneous adipose tissue, and skeletal muscle of host skin (Fig. 7B and C). Importantly, sialostatin L2 alone stimulated minimal cell infiltration at the skin site. Moreover, sialostatin L2 inhibited inflammation at the skin during A. phagocytophilum infection. Injection of sialostatin L2 together with A. phagocytophilum reduced inflammation to background levels (Fig. 7B and C). Splenomegaly is an overt clinical symptom of aberrant inflammation in both mice and humans during A. phagocytophilum infection (11). To test whether reduced inflammation due to sialostatin L2 would affect enlargement of the spleen during pathogen infection, we infected mice by intraperitoneal injection. The spleen size of A. phagocytophilum-infected mice was increased compared to that of naive mice (Fig. 7D). Conversely, mice injected with A. phagocytophilum in the presence of sialostatin L2 did not show any spleen enlargement upon infection. Spleen sizes were comparable to those of noninfected mice.
Although host inflammation is mediated by innate immunity during A. phagocytophilum infection, pathogen eradication is obtained by adaptive immunity (11, 46). To determine whether sialostatin L2 also favored pathogen infection, we tested the effect of sialostatin L2 on microbial colonization. After 24 h, bacterial numbers in the skin did not differ significantly after intradermal injection of either the vehicle alone or the vehicle containing sialostatin L2 (Fig. 8A). Similar results were obtained for intraperitoneal injection of A. phagocytophilum (Fig. 8B).
FIG 8.
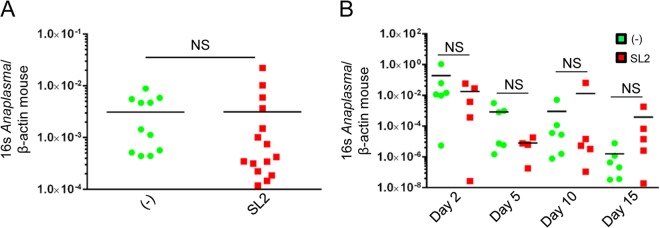
Sialostatin L2 does not affect A. phagocytophilum load during infection. (A) Bacterial loads following intradermal infection of A. phagocytophilum in the presence of sialostatin L2 (SL2) (10 μg/injection). Sialostatin L2 was injected 5 h prior to, during, and 5 h after pathogen inoculation. Bacterial loads were measured at 24 h postinjection by quantitative RT-PCR. (B) Bacterial loads in the peripheral blood of mice following intraperitoneal infection of A. phagocytophilum in the presence of SL2 (10 μg/injection). *, P < 0.05 (Student's t test); NS, not significant.
DISCUSSION
In this study, we demonstrated that the tick salivary protein sialostatin L2 inhibited inflammasome-mediated inflammation during stimulation with the rickettsial pathogen A. phagocytophilum. More specifically, sialostatin L2 mitigated caspase-1 activation and subsequent IL-1β and IL-18 release by inhibiting ROS production from NADPH oxidase in macrophages. A. phagocytophilum did not induce IL-18 secretion at the skin site. This is an interesting result, because for certain stimuli, IL-18 regulation may be decoupled from IL-1β secretion. For instance, in syk−/− macrophages, IL-18 secretion and pro-IL-18 levels were elevated after LPS priming followed by silica stimulation, whereas the opposite behavior was observed for IL-1β secretion (47). Leptin also increased IL-18 secretion by human monocytes, while the same effect was not observed for IL-1β secretion (48). Whether these in vivo findings are mechanistically analogous to A. phagocytophilum infection remains unclear.
We observed that the Loop2 domain of sialostatin L2 partially contributes to the modulation of caspase-1 activity and IL-1β and IL-18 secretion during A. phagocytophilum stimulation of macrophages. Based on the structure of sialostatin L2 (34), we speculate that this could be due to the equal numbers of positive and negative amino acids, making Loop2 of sialostatin L2 less charged in the physiological setting. The greater isoelectric point of Loop2 favors protein-protein interactions in the cell membrane. Moreover, Loop2 of sialostatin L2 has a larger percentage of amino acids that can serve as donors for hydrogen bonding (28.9%) than the Loop1 (22.2%) and N-terminal (15.8%) domains. Alternatively, because the Loop2 domain of sialostatin L2 does not contain the conserved type 2 cystatin residues PW observed in other cystatins (34, 49), we posit that these structural differences may permit a more favorable biophysical interaction with an as yet unidentified molecule in macrophages.
We reason that sialostatin L2 may not be the only vector salivary protein that inhibits inflammasome activation and regulates inflammation during pathogen infection. Biologically active proteins in tick saliva are commonly used as a strategy for immune evasion during feeding, and it is estimated that hematophagy has evolved independently in more than 14,000 arthropod species (50, 51). Ticks also have large genomes and carry many gene paralogs (52). These gene paralogs may act redundantly to provide inhibition of protein scaffolds, such as the inflammasome, in the mammalian host. For instance, Ramachandra and Wikel showed that salivary gland extracts from the tick Dermacentor andersoni reduced IL-1 levels during the early phases of tick feeding (53), while another group determined that human IL-1β secretion was mitigated when cells were treated with LPS and salivary gland extracts from partially fed adult female Rhipicephalus appendiculatus ticks (54).
We did not observe any A. phagocytophilum load differences in mouse skin when sialostatin L2 was coinjected with A. phagocytophilum and compared to the control treatment. These findings were in contrast to a study showing that sialostatin L2 facilitates the growth of the Lyme disease agent, Borrelia burgdorferi (34). It is difficult to apply an overly simplified model for A. phagocytophilum, as this rickettsial agent infects myeloid and nonmyeloid cells, and macrophages are important for disease pathology (10, 11). Thus, we speculate that the redundancy of the immune system during pathogen infection, the unique manner by which A. phagocytophilum colonizes the mammalian host, and the mode of action of sialostatin L2 may explain these seemingly contrasting findings. It is also possible that the mechanism of A. phagocytophilum immunopathology is distinct from bacterial colonization. This dichotomy is supported if one considers uncoupling the regulation of IL-1β and IL-18 secretion during pathogen infection in vivo.
Testing the effect of sialostatin L2 during A. phagocytophilum colonization by I. scapularis ticks in vivo is technically unfeasible. First, RNA interference (RNAi) silencing and vaccination against sialostatin L2 impair the feeding ability of I. scapularis (34, 35). Thus, a reliable comparison of pathogen colonization in control and RNAi-silenced ticks or immunized and control groups is not possible. Second, mice are the natural hosts of ticks. Therefore, they do not typically develop immunity against salivary proteins (50, 55). Third, many sialostatin L2 paralogues are present in the I. scapularis genome. Hence, these molecules may cross-react with antibodies, calling into question the validity of any assay that measures sialostatin L2 concentrations in tick saliva.
In conclusion, our studies show that the I. scapularis tick salivary protein sialostatin L2 reduces inflammasome activation and dampens inflammation in the mouse skin during A. phagocytophilum colonization. Further understanding of how sialostatin L2 inhibits caspase-1 assembly during A. phagocytophilum infection may have direct implications for uncovering basic aspects of inflammasome and vector biology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anthony Choy, Linda McCloud, and Rosanne Hearn for excellent technical assistance; invaluable colleagues for intellectual input and manuscript comments; and the Center for Disease Vector Research and the Institute for Integrative Genome Biology Core Facilities at the University of California-Riverside.
This work was supported by the Centers for Disease Control and Prevention (grant K01 CK000101), the National Institutes of Health (grant R01 AI093653), an initial complement provided by the University of California, start-up funds provided by the University of Maryland, Baltimore School of Medicine, and an international fellowship from the American Association of University Women.
Footnotes
Published ahead of print 31 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01679-14.
REFERENCES
- 1.Strowig T, Henao-Mejia J, Elinav E, Flavell R. 2012. Inflammasomes in health and disease. Nature 481:278–286. 10.1038/nature10759 [DOI] [PubMed] [Google Scholar]
- 2.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. 2013. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 31:73–106. 10.1146/annurev-immunol-032712-095944 [DOI] [PubMed] [Google Scholar]
- 3.Rathinam VA, Vanaja SK, Fitzgerald KA. 2012. Regulation of inflammasome signaling. Nat. Immunol. 13:333–342. 10.1038/ni.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamkanfi M. 2011. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 11:213–220. 10.1038/nri2936 [DOI] [PubMed] [Google Scholar]
- 5.Miao EA, Rajan JV. 2011. Salmonella and caspase-1: a complex interplay of detection and evasion. Front. Microbiol. 2:85. 10.3389/fmicb.2011.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnston JB, Barrett JW, Nazarian SH, Goodwin M, Ricciuto D, Wang G, McFadden G. 2005. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 23:587–598. 10.1016/j.immuni.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 7.Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM. 2013. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 9:e1003256. 10.1371/journal.ppat.1003256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pedra JH, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, Flavell RA, Fikrig E. 2007. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-γ axis during Anaplasma phagocytophilum infection. J. Immunol. 179:4783–4791 [DOI] [PubMed] [Google Scholar]
- 9.Rikihisa Y. 2010. Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nat. Rev. Microbiol. 8:328–339. 10.1038/nrmicro2318 [DOI] [PubMed] [Google Scholar]
- 10.Dumler JS. 2012. The biological basis of severe outcomes in Anaplasma phagocytophilum infection. FEMS Immunol. Med. Microbiol. 64:13–20. 10.1111/j.1574-695X.2011.00909.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Severo MS, Stephens KD, Kotsyfakis M, Pedra JH. 2012. Anaplasma phagocytophilum: deceptively simple or simply deceptive? Future Microbiol. 7:719–731. 10.2217/fmb.12.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Truchan HK, Seidman D, Carlyon JA. 2013. Breaking in and grabbing a meal: Anaplasma phagocytophilum cellular invasion, nutrient acquisition, and promising tools for their study. Microbes Infect. 15:1017–1025. 10.1016/j.micinf.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumler JS, Barat NC, Barat CE, Bakken JS. 2007. Human granulocytic anaplasmosis and macrophage activation. Clin. Infect. Dis. 45:199–204. 10.1086/518834 [DOI] [PubMed] [Google Scholar]
- 14.Lepidi H, Bunnell JE, Martin ME, Madigan JE, Stuen S, Dumler JS. 2000. Comparative pathology, and immunohistology associated with clinical illness after Ehrlichia phagocytophila-group infections. Am. J. Trop. Med. Hyg. 62:29–37 [DOI] [PubMed] [Google Scholar]
- 15.Choi KS, Scorpio DG, Dumler JS. 2004. Anaplasma phagocytophilum ligation to Toll-like receptor (TLR) 2, but not to TLR4, activates macrophages for nuclear factor-κB nuclear translocation. J. Infect. Dis. 189:1921–1925. 10.1086/386284 [DOI] [PubMed] [Google Scholar]
- 16.Hovius JW, Levi M, Fikrig E. 2008. Salivating for knowledge: potential pharmacological agents in tick saliva. PLoS Med. 5:e43. 10.1371/journal.pmed.0050043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Silva AM, Tyson KR, Pal U. 2009. Molecular characterization of the tick-Borrelia interface. Front. Biosci. 14:3051–3063. 10.2741/3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu XY, Bonnet SI. 2014. Hard tick factors implicated in pathogen transmission. PLoS Neglect. Trop. Dis. 8:e2566. 10.1371/journal.pntd.0002566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kazimirova M, Stibraniova I. 2013. Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front. Cell Infect. Microbiol. 3:43. 10.3389/fcimb.2013.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakhon OS, Severo MS, Kotsyfakis M, Pedra JH. 2013. A Nod to disease vectors: mitigation of pathogen sensing by arthropod saliva. Front. Microbiol. 4:308. 10.3389/fmicb.2013.00308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen G, Severo MS, Sohail M, Sakhon OS, Wikel SK, Kotsyfakis M, Pedra JH. 2012. Ixodes scapularis saliva mitigates inflammatory cytokine secretion during Anaplasma phagocytophilum stimulation of immune cells. Parasit. Vectors 5:229. 10.1186/1756-3305-5-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang M, Zhang Y, Pan J, Sun J, Liu J, Libby P, Sukhova GK, Doria A, Katunuma N, Peroni OD, Guerre-Millo M, Kahn BB, Clement K, Shi GP. 2007. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat. Cell Biol. 9:970–977. 10.1038/ncb1623 [DOI] [PubMed] [Google Scholar]
- 23.Kitamoto S, Sukhova GK, Sun J, Yang M, Libby P, Love V, Duramad P, Sun C, Zhang Y, Yang X, Peters C, Shi GP. 2007. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation 115:2065–2075. 10.1161/CIRCULATIONAHA.107.688523 [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Sukhova GK, Zhang J, Chen H, Sjoberg S, Libby P, Xiang M, Wang J, Peters C, Reinheckel T, Shi GP. 2011. Cathepsin L activity is essential to elastase perfusion-induced abdominal aortic aneurysms in mice. Arterioscler. Thromb. Vasc. Biol. 31:2500–2508. 10.1161/ATVBAHA.111.230201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen G, Severo MS, Sakhon OS, Choy A, Herron MJ, Felsheim RF, Wiryawan H, Liao J, Johns JL, Munderloh UG, Sutterwala FS, Kotsyfakis M, Pedra JH. 2012. Anaplasma phagocytophilum dihydrolipoamide dehydrogenase 1 affects host-derived immunopathology during microbial colonization. Infect. Immun. 80:3194–3205. 10.1128/IAI.00532-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HY, Rikihisa Y. 2002. Roles of p38 mitogen-activated protein kinase, NF-κB, and protein kinase C in proinflammatory cytokine mRNA expression by human peripheral blood leukocytes, monocytes, and neutrophils in response to Anaplasma phagocytophila. Infect. Immun. 70:4132–4141. 10.1128/IAI.70.8.4132-4141.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HY, Rikihisa Y. 2000. Expression of interleukin-1β, tumor necrosis factor α, and interleukin-6 in human peripheral blood leukocytes exposed to human granulocytic ehrlichiosis agent or recombinant major surface protein P44. Infect. Immun. 68:3394–3402. 10.1128/IAI.68.6.3394-3402.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rikihisa Y, Zhang Y, Park J. 1994. Inhibition of infection of macrophages with Ehrlichia risticii by cytochalasins, monodansylcadaverine, and taxol. Infect. Immun. 62:5126–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 30.Johnson CR, Kitz D, Little JR. 1983. A method for the derivation and continuous propagation of cloned murine bone marrow macrophages. J. Immunol. Methods 65:319–332. 10.1016/0022-1759(83)90127-8 [DOI] [PubMed] [Google Scholar]
- 31.Ulland TK, Janowski AM, Buchan BW, Faron M, Cassel SL, Jones BD, Sutterwala FS. 2013. Francisella tularensis live vaccine strain folate metabolism and pseudouridine synthase gene mutants modulate macrophage caspase-1 activation. Infect. Immun. 81:201–208. 10.1128/IAI.00991-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204:3235–3245. 10.1084/jem.20071239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schrodinger LLC. 2010. The PyMOL molecular graphics system, version 1.3r1. Schrodinger LLC, San Diego, CA [Google Scholar]
- 34.Kotsyfakis M, Horka H, Salat J, Andersen JF. 2010. The crystal structures of two salivary cystatins from the tick Ixodes scapularis and the effect of these inhibitors on the establishment of Borrelia burgdorferi infection in a murine model. Mol. Microbiol. 77:456–470. 10.1111/j.1365-2958.2010.07220.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotsyfakis M, Anderson JM, Andersen JF, Calvo E, Francischetti IM, Mather TN, Valenzuela JG, Ribeiro JM. 2008. Immunity against a “silent” salivary antigen of the Lyme vector Ixodes scapularis impairs its ability to feed. J. Immunol. 181:5209–5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotsyfakis M, Karim S, Andersen JF, Mather TN, Ribeiro JM. 2007. Selective cysteine protease inhibition contributes to blood-feeding success of the tick Ixodes scapularis. J. Biol. Chem. 282:29256–29263. 10.1074/jbc.M703143200 [DOI] [PubMed] [Google Scholar]
- 37.Chmelar J, Oliveira CJ, Rezacova P, Francischetti IM, Kovarova Z, Pejler G, Kopacek P, Ribeiro JM, Mares M, Kopecky J, Kotsyfakis M. 2011. A tick salivary protein targets cathepsin G and chymase and inhibits host inflammation and platelet aggregation. Blood 117:736–744. 10.1182/blood-2010-06-293241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotsyfakis M, Sa-Nunes A, Francischetti IM, Mather TN, Andersen JF, Ribeiro JM. 2006. Anti-inflammatory and immunosuppressive activity of sialostatin L, a salivary cystatin from the tick Ixodes scapularis. J. Biol. Chem. 281:26298–26307. 10.1074/jbc.M513010200 [DOI] [PubMed] [Google Scholar]
- 39.Goodman JL, Nelson CM, Klein MB, Hayes SF, Weston BW. 1999. Leukocyte infection by the granulocytic ehrlichiosis agent is linked to expression of a selectin ligand. J. Clin. Invest. 103:407–412. 10.1172/JCI4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaRock CN, Cookson BT. 2012. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12:799–805. 10.1016/j.chom.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunning Hotopp JC, Lin M, Madupu R, Crabtree J, Angiuoli SV, Eisen JA, Seshadri R, Ren Q, Wu M, Utterback TR, Smith S, Lewis M, Khouri H, Zhang C, Niu H, Lin Q, Ohashi N, Zhi N, Nelson W, Brinkac LM, Dodson RJ, Rosovitz MJ, Sundaram J, Daugherty SC, Davidsen T, Durkin AS, Gwinn M, Haft DH, Selengut JD, Sullivan SA, Zafar N, Zhou L, Benahmed F, Forberger H, Halpin R, Mulligan S, Robinson J, White O, Rikihisa Y, Tettelin H. 2006. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2:e21. 10.1371/journal.pgen.0020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. 2011. Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187:613–617. 10.4049/jimmunol.1100613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–241. 10.1038/nature04516 [DOI] [PubMed] [Google Scholar]
- 44.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9:847–856. 10.1038/ni.1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubartelli A. 2012. Redox control of NLRP3 inflammasome activation in health and disease. J. Leukoc. Biol. 92:951–958. 10.1189/jlb.0512265 [DOI] [PubMed] [Google Scholar]
- 46.von Loewenich FD, Scorpio DG, Reischl U, Dumler JS, Bogdan C. 2004. Control of Anaplasma phagocytophilum, an obligate intracellular pathogen, in the absence of inducible nitric oxide synthase, phagocyte NADPH oxidase, tumor necrosis factor, Toll-like receptor (TLR)2 and TLR4, or the TLR adaptor molecule MyD88. Eur. J. Immunol. 34:1789–1797. 10.1002/eji.200425029 [DOI] [PubMed] [Google Scholar]
- 47.He Y, Varadarajan S, Munoz-Planillo R, Burberry A, Nakamura Y, Nunez G. 2014. 3,4-Methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 289:1142–1150. 10.1074/jbc.M113.515080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jitprasertwong P, Jaedicke KM, Nile CJ, Preshaw PM, Taylor JJ. 2014. Leptin enhances the secretion of interleukin (IL)-18, but not IL-1β, from human monocytes via activation of caspase-1. Cytokine 65:222–230. 10.1016/j.cyto.2013.10.008 [DOI] [PubMed] [Google Scholar]
- 49.Schwarz A, Valdes JJ, Kotsyfakis M. 2012. The role of cystatins in tick physiology and blood feeding. Ticks Tick Borne Dis. 3:117–127. 10.1016/j.ttbdis.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Francischetti IM, Sa-Nunes A, Mans BJ, Santos IM, Ribeiro JM. 2009. The role of saliva in tick feeding. Front. Biosci. 14:2051–2088. 10.2741/3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fontaine A, Diouf I, Bakkali N, Misse D, Pages F, Fusai T, Rogier C, Almeras L. 2011. Implication of haematophagous arthropod salivary proteins in host-vector interactions. Parasites Vectors 4:187. 10.1186/1756-3305-4-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pagel Van Zee J, Geraci NS, Guerrero FD, Wikel SK, Stuart JJ, Nene VM, Hill CA. 2007. Tick genomics: the Ixodes genome project and beyond. Int. J. Parasitol. 37:1297–1305. 10.1016/j.ijpara.2007.05.011 [DOI] [PubMed] [Google Scholar]
- 53.Ramachandra RN, Wikel SK. 1992. Modulation of host-immune responses by ticks (Acari: Ixodidae): effect of salivary gland extracts on host macrophages and lymphocyte cytokine production. J. Med. Entomol. 29:818–826 [DOI] [PubMed] [Google Scholar]
- 54.Fuchsberger N, Kita M, Hajnicka V, Imanishi J, Labuda M, Nuttall PA. 1995. Ixodid tick salivary gland extracts inhibit production of lipopolysaccharide-induced mRNA of several different human cytokines. Exp. Appl. Acarol. 19:671–676. 10.1007/BF00145255 [DOI] [PubMed] [Google Scholar]
- 55.Chmelar J, Calvo E, Pedra JH, Francischetti IM, Kotsyfakis M. 2012. Tick salivary secretion as a source of antihemostatics. J. Proteomics 75:3842–3854. 10.1016/j.jprot.2012.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.